
Abstract
Bacillus species as fungal antagonistic agents have been widely used in the agriculture and considered as safe products for the management of plant pathogens. In this study, we reported the whole genome sequence of strain LJBV19 isolated from grapevine rhizosphere soil. Strain LJBV19 was identified as Bacillus velezensis through morphological, physicochemical, molecular analysis and genome comparison. Bacillus velezensis LJBV19 had a significant inhibitory effect on the growth of Magnaporthe oryzae with an inhibition ratio up to 75.55% and showed broad spectrum of activity against fungal phytopathogens. The 3,973,013-bp circular chromosome with an average GC content of 46.5% consisted of 3993 open reading frames (ORFs), and 3308 ORFs were classified into 19 cluster of orthologous groups of proteins (COG) categories. Genes related to cell wall degrading enzymes were predicted by Carbohydrate-Active enZYmes (CAZy) database and validated at the metabolic level, producing 0.53 ± 0.00 U/mL cellulose, 0.14 ± 0.01 U/mL chitinase, and 0.11 ± 0.01 U/mL chitosanase. Genome comparison confirmed the taxonomic position of LJBV19, conserved genomic structure, and genetic homogeneity. Moreover, 13 gene clusters for biosynthesis of secondary metabolites in LJBV19 genome were identified and two unique clusters (clusters 2 and 12) shown to direct an unknown compound were only present in strain LJBV19. In general, our results will provide insights into the antifungal mechanisms of Bacillus velezensis LJBV19 and further application of the strain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plant diseases are a major and long-term threat to crop yield and quality worldwide (Compant et al. 2005). Fungicides have been major method to control pathogen over the past decades. Continuous use of fungicides leads to fungicide resistance, causes environmental pollution, and brings risks to food safety and human health (Shafi et al. 2017). The use of natural antagonistic microorganisms as microbial inoculants is an ideal alternative or a supplemental way to control pathogens. Bacillus species are effective against a broad range of pathogenic microorganisms (Berg 2009). Bacillus velezensis with characteristics of fast growth and stability is widely distributed in nature and plays an increasingly important role in the fields of agriculture (Ye et al. 2018). B. velezensis as heterotypic synonyms of B. amyloliquefaciens subsp. plantarum is distinguished from B. amyloliquefaciens by secondary metabolite production, comparative genomics, and DNA-DNA relatedness calculations, and these two species are separated from B. subtilis (Dunlap et al. 2016; Fan et al. 2017; Andrić et al. 2020; Rabbee et al. 2019). More and more studies have investigated the potential of B. velezensis for curing or preventing plant diseases. For example, B. velezensis strain ZSY-1, isolated from Chinese catalpa, inhibited the growth of Alternaria solani and Botrytis cinerea by volatile organic compounds (Gao et al. 2017). B. velezensis strains 5YN8 and DSN012 controlled pepper gray mold disease by suppressing mycelium growth and spore formation (Jiang et al. 2018). B. velezensis C2, isolated from the crown tissue of tomato, exhibited significant antifungal activity against Verticillium dahliae through secondary metabolites and lytic enzymes (Dhouib et al. 2019).
With the increasing number of Bacillus species isolated, antimicrobial substances and fungal antagonistic mechanisms have been gradually explored (Lopes et al. 2018). Bacillus spp. are able to control plant diseases through diverse mechanisms, including producing antimicrobial compounds, competition with pathogens for space and nutrients, stimulation the induced systemic resistance (ISR) of plant, and promotion of plant growth (Fan et al. 2018; Shafi et al. 2017). B. velezensis harbors a high genetic capacity for synthesizing secondary metabolites, playing important roles in pathogen suppression. For example, gene clusters encoding surfactin (srf), fengycin (fen), macrolactin (pks2), bacillaene (bae), difficidin (dfn), bacilysin (bac), and bacillibactin (dhb) were present in model fungal antagonistic bacterium B. velezensis FZB42 (Fan et al. 2018). Various metabolic substances exert fungal antagonistic effects through different mechanisms. For instance, B. velezensis RC 218 showed antagonist activity against Fusarium graminearum due to direct antagonism by secondary metabolites (Palazzini et al. 2016). Lipopeptides surfactin and fengycin can act as elicitors of induced systemic resistance in plants (Chen et al. 2020). Surfactin is also essential for root colonization and influenced the ecological fitness (Ongena and Jacques 2008). Siderophore bacillibactin is involved in regulation of ferric ion (Khan et al. 2018). Nevertheless, the fungal antagonistic mechanisms still need to be further elucidated. Genome sequencing, genome annotation, and comparative genome analysis are important approaches to provide insight into the biology of fungal antagonistic strains.
In this study, Bacillus strain LJBV19 was isolated from rhizosphere soil of grapevine and evaluated against 12 phytopathogens such as Magnaporthe oryzae, Colletotrichum gloeosporioides, and Fusarium solani. In view of morphological, physicochemical, molecular analysis and genome comparison, strain LJBV19 belonged to B. velezensis. The whole genome of LJBV19 was sequenced and annotated to explore its fungal antagonistic mechanisms. LJBV19 genome was compared with three close strains B. velezensis FZB42, B. amyloliquefaciens DSM7T, and B. subtilis 168T. Genome comparison showed common and unique gene clusters related to the biosynthesis of secondary metabolites in LJBV19 genome. Overall, our data indicated that LJBV19 had the potential for protecting plant health against a broad range of pathogens.
Materials and methods
Isolation of bacteria
In October 2019, rhizosphere soil sample was collected from vineyard in Wujing town, Minhang district, Shanghai, China. Sample was dissolved in NaCl solution (0.85% w/v) and vibrated violently for 2 min (Santana et al. 2008). One milliliter of soil solution was incubated at 80 °C for 30 min, and then was diluted tenfold, 100-fold, and 1000-fold. After the solution cooled on ice, 0.1 mL liquid from each dilution was uniformly coated on Luria–Bertani (LB) agar medium (5.0 g/L yeast extract, 10.0 g/L peptone, 10.0 g/L NaCl, and 15.0 g/L agar, pH neutral), and was incubated at 37 °C for 12 h. Single clones with biofilm were randomly selected for streaking purification according to Sari et al. (2019). One clone, named as LJBV19, which obviously inhibited the hyphae growth of C. gloeosporioides, Coniothyrium diplodiella, and B. cinereal, was maintained on LB slants at 4 °C and stored with glycerol at −20 °C for further study.
Morphological, physiological, and biochemical analysis
After incubating on LB agar plate at 37 °C for 24 h, the colony characters of LJBV19 were recorded. Gram staining and spore staining were performed using Gram Stain Kit (Beijing Solarbio Science & Technology Co., Ltd) and Spore Stain Kit (Solarbio), respectively. The biochemical characteristics were identified using traditional approaches according to the Bergey’s Manual of Systematic Bacteriology (Anonymous 2001). LB liquid medium fermentation broth was centrifuged after incubated at 37 °C for 48 h, and the supernatant was used for determining defense-related enzyme activities. Cellulase, chitinase, and chitosanase activities were measured using 3,5-dinitro salicylic acid (DNS) method (Zhu et al. 2007). One unit (U) of chitinase, chitosanase, and cellulase resulted in 1 μmol of D-glucose, N-acetyl-D-glucosamine, and D-glucosamine per min, respectively.
Antifungal spectrum analysis
The antifungal spectrum of LJBV19 against plant pathogens was performed through plate assays on potato dextrose agar (PDA) according to the previous method (Wang et al. 2021). Briefly, LJBV19 was streaked horizontally in the center of the 9-cm-diameter PDA agar (boil chopped potatoes at 200 g/L for 30 min then filter with gauze and discard the residue, add 20 g/L glucose, and 20 g/L agar, pH neutral) plate. Mycelial plugs from the margin of the pathogen colony were placed on the left and right sides 2.2 cm from the center of plate. One mycelial plug was placed on each side of the center of plate, and the two plugs were removed from the same pathogen. LB medium was used as the control. The plates were incubated in the dark for 7 days for fungal growth. The inhibition activity was defined as the percentage of mycelial growth inhibition and calculated using the following formula: inhibition (%) = ((R1 − R2)/R1) × 100% (Zhang et al. 2016). R1 and R2 were the radius of the mycelium in the control and treatment, respectively.
Genome sequencing and assembly
The genome of LJBV19 was sequenced by Personalbio Technology Co., Ltd., Shanghai, China. TIANamp Bacteria DNA Kit provided by Tiangen Biotech (Beijing) Co., Ltd. was used to extract the genomic DNA of LJBV19. The qualified genomic DNA of LJBV19 was fragmented with G-tubes for Oxford Nanopore Technologies (ONT) Library. Then, DNA fragmentations were treated for damage repair and end repair, adaptor ligation, and size selection with a BluePippin system to prepare ONT libraries. ONT Library quality was detected by Qubit and sequencing was performed by ONT platform according to standard protocols. The reads of the ONT were assembled de novo using Hierarchical Genome Assembly Process (HGAP) (Chin et al. 2016). Libraries for Illumina PCR-free paired-end genome sequencing were constructed according to Illumina TruSeq DNA Sample Preparation Guide. The genomic DNA was fragmented using Covaris. DNA fragments were treated for double-end repair and sequencing adapter ligation. After quality control, the PCR-free libraries were sequenced using paired-end sequencing by Illumina NovaSeq platform. Utilizing Illumina short reads, software Pilon (Walker et al. 2014) was used to correcting the errors in ONT long-read assembly and improve the accuracy of the sequence.
Genomic feature prediction and annotation
The ORF of LJBV19 genome was predicted using GeneMarkS (Besemer et al. 2001). tRNA genes were predicted by tRNAscan-SE (Lowe and Eddy 1997), and rRNA genes were carried out by RNAmmer (Lagesen et al. 2007). Other non-coding RNA, such as small nuclear RNAs (snRNAs), was predicted by BLAST searching against the Rfam database (Kalvari et al. 2018). The functions of genes were predicted through comparisons against diverse protein databases, including Gene Ontology (GO) (Ashburner et al. 2000), Swiss-Prot (Boeckmann et al. 2003), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2016), the enhanced Cluster of Orthologous Groups of proteins (eggNOG) (Jensen et al. 2008), and Non-Redundant Protein Database (NR) (Li et al. 2002). CGView was used to generate the graphical view of LJBV19 genome (Chin et al. 2016). SignalP (Bendtsen et al. 2004) was used to annotate signal peptides, and TMHMM (Chen et al. 2003) was used to annotate proteins with transmembrane structure. Proteins containing the signal peptide structure without transmembrane structure were secreted proteins. The CAZy database was used to further analyze carbohydrate active enzymes (CAZymes) (Lombard et al. 2014). Additional annotations were performed by the following software: IslandViewer (Bertelli et al. 2017), hmmscan (Choo et al. 2004), and CRISPR finder (Grissa et al. 2008).
Phylogenetic analysis and comparative genomic analysis
27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-ACGGCTACCTTGTTACGACTT-3′) primers were used to amplify 16S rRNA. The PCR product was sequenced by Shanghai Sunny Biotechnology Co., Ltd. The 16S rRNA sequence of LJBV19 was deposited in GenBank (Accession No. MZ157279) and compared in the EZBiocloud (https://www.ezbiocloud.net/) database. MEGA 7.0 software was used for phylogenetic analysis using neighbor joining method. The average nucleotide identity (ANI) and digital DNA: DNA hybridization (dDDH) were analyzed by Jspecies (http://jspecies.ribohost.com/jspeciesws/) and Genome-to-Genome Distance Calculator (GGDC) (https://ggdc.dsmz.de/ggdc.php) (Richter and Rossello-Mora 2009), respectively. Four closely related Bacillus species with released complete genomes, including B. velezensis FZB42 (GenBank: NC_009725.2) (Chen et al. 2007), B. amyloliquefaciens DSM7T (GenBank: NC_014551.1) (Borriss et al. 2011), and B. subtilis 168T (GenBank: NC_000964.3) (Borriss et al. 2018), were selected for genome comparation by Mauve using the progressive alignment, and the LJBV19 genome served as the reference genome (Darling et al. 2010). R package was used to generate Venn diagram (Richter and Rossello-Mora 2009). Furthermore, the putative secondary metabolite clusters were identified using the antiSMASH v6.0 (https://antismash.secondarymetabolites.org) program with default parameters (Blin et al. 2021). Comparative analyses of gene clusters identified in B. velezensis LJBV19, B. velezensis FZB42, B. amyloliquefaciens DSM7T, and B. subtilis 168T were performed by KEGG (Kanehisa et al. 2016) and the GenBank database.
Nucleotide sequence accession numbers
The complete genome sequence of B. velezensis LJBV19 strain was deposited in the GenBank database under the accession number CP072563. This strain is available with the “China General Microbiological Culture Collection Center” (CGMCC), Beijing, China, under the accession number CGMCC No. 21804.
Results and discussion
Organism information
LJBV19 was a gram-positive, endospore-forming, rod-shaped, and aerobic bacterium. This strain grew on LB agar at 37 °C for 24 h, producing nearly round and creamy white colonies with irregular margins and dry wrinkles on the surface (Fig. S1). LJBV19 could grow in 8.5% (w/v) NaCl and over a wide pH range (4.5–10.0). LJBV19 was positive for catalase, Voges-Proskauer, nitrate reduction, gelatin liquefaction, and hydrogen sulfate test (Table S1). Besides, strain LJBV19 could utilize diverse carbon sources, including starch, citrate, xylose, sucrose, and multiple monosaccharides (Table S1). All tests indicated that the morphology, physiological, and biochemical characteristics of LJBV19 were similar to Bacillus species. Minimum information about the genome sequence (MIGS) of LJBV19 was listed in Table S2.
Taxonomic position of LJBV19
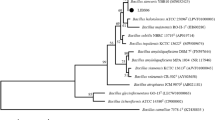
Compared to sequences of the type strains of B. velezensis, B. subtilis, and B. amyloliquefaciens, the 16S rRNA identity of LJBV19 were 100, 99.70, and 99.70%, respectively. To understand the systematic classification of LJBV19, a phylogenetic tree was constructed based on16S rRNA gene (Fig. S2). However, it was difficult to differentiate B. velezensis, B. amyloliquefaciens, and B. subtilis according to traditional phenotypic and similarity analysis of 16S rRNA (Rooney et al. 2009). With the development of sequencing technology and bioinformatics, many approaches such as ANI and silico DDH analyses have been used to differentiate and re-categorized species in Bacillus taxa (Cai et al. 2017). For example, B. velezensis FZB42 was previously grouped as B. amyloliquefaciens (Adeniji et al. 2019).
To further clarify the taxonomic position of LJBV19, ANI and DDH analyses were performed (Table S3). ANI and dDDH values between strains LJBV19 and B. velezensis NRRL B-41580 were 98.89 and 92.1%, respectively. Similarly, ANI and dDDH value were 98.17 and 85.5% compared to B. velezensis FZB42, respectively. The ANI and dDDH values between LJBV19 and B. amyloliquefaciens DSM7T were 93.39 and 56.0%, respectively. In addition, there were lower ANI and dDDH values when compared with B. subtilis 168T. According to ≥ 95% similarity in ANI and ≥ 70% homology in dDDH belonging to the same species (Chun et al. 2018), LJBV19 was affiliated with B. velezensis. Overall, results of ANI and DDH consistent with the result in the 16S rRNA tree showed that LJBV19 should be classified as B. velezensis.
Data information about the public accessibility of all material
Bacillus sp. LJBV19 genome assembly ASM1779784v1, submitted by Shanghai Jiao Tong University. April, 2021. RefSeq: GCF_017797845.1, GenBank: GCA_017797845.1.
Fungal antagonistic effect of LJBV19
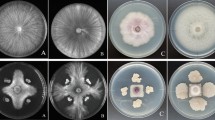
There were diverse genera of microorganisms in rhizosphere, which had important effects on plant growth, pathogen defense, and resistance (Sasse et al. 2018). In the study, we have screened LJBV19 with broad spectrum antagonistic activities from rhizosphere soil to enhance plant disease resistance. Plate co-culture assay showed that LJBV19 could inhibit the mycelia growth of diverse pathogens (Fig. 1). The growth of M. oryzae was significantly suppressed with an inhibition ratio up to 75.55%. Strain LJBV19 showed good inhibition (> 30%) towards C. gloeosporioides, F. solani, Verticiltium dahlia, Exserohilum rostratum, Phytophthora capsica, and F. graminearum (Table S4). In addition, the mycelial growth of B. cinerea, Fusarium equiseti, C. diplodiella, Fusarium oxysporum, and Rhizoctonia solani were influenced by LJBV19 in varying degrees (Table S4). Importantly, the mycelia on the antagonistic plate, such as B. cinerea, were significantly enlarged, twisted, and broken under microscopic observation (Fig. S3B and C). The mycelia in the control grew normally (Fig. S3A). These results indicated that strain LJBV19 had broad spectrum antimicrobial activity to fungal phytopathogens. Meanwhile, most researches supported the fungal antagonistic potential of B. velezensis, such as B. velezensis CC09 inhibiting wheat powdery mildew, B. velezensis BAC03 as an effective antagonist of Streptomyces scabies, and B. velezensis BS87 and RK1 as bioprotection agents of strawberries against F. oxysporum (Adeniji et al. 2019).

Effect of B. velezensis LJBV19 on growth of 12 plant pathogens. A and a Magnaporthe oryzae. B and b Colletotrichum gloeosporioides. C and c Fusarium solani. D and d Verticiltium dahlia. E and e Exserohilum rostratum. F and f Phytophthora capsica. G and g Fusarium graminearum. H and h Botrytis cinerea. I and i Fusarium equiseti. J and j Coniothyrium diplodiella. K and k Fusarium oxysporum. L and l Rhizoctonia solani. Uppercase letter indicated the treatment and lowercase letter indicated the control
Genome features of LJBV19
The complete genome of LJBV19 contained a circular 3,973,013 bp chromosome with 43.96% GC, which were within the genome size range of 3.81–4.24 Mbp and 45.9–46.8% GC content reported for this species (Mullins et al. 2020). A graphical circular map of the genome showing the genome structure and functions was presented in Fig. 2. There were 3993 open reading frames (ORFs) predicted by GeneMarkS in the genome of LJBV19. In addition, 27 rRNA genes, 86 tRNA genes, and 50 pseudogenes were contained in LJBV19 genome. Using the SignalP, and TMHMM databases, 210 (5.26%) and 1,012 (25.34%) of ORFs were divided into encoding signal peptides and transmembrane helices, respectively. One hundred and five (2.63%) proteins contained the structure of signal peptides without transmembrane helices, secreted proteins, were predicted. Besides, 248 genomics islands (GI), 13 virulence factors of pathogenic bacteria (VFDB), and 8 prophage regions were present in the genome of LJBV19. The functions of genes that were predicted using various databases showed that 3836 (96.07%), 3308 (82.84%), 2181 (54.62%), 3493 (87.48%), and 2728 (68.32%) ORFs matched in the NR, eggNOG, KEGG, SwissProt, and GO databases, respectively. In the eggNOG database, 3308 ORFs were classified into 19 COG categories, including 2.08% related to secondary metabolites biosynthesis, transport, and catabolism (Q); 5.84% to carbohydrate transport and metabolism (G); 6.69% to amino acid transport and metabolism (E); and 6.71% to transcription (K) (Table 1).

The graphical circular genomic map of LJBV19 using the CGview server. Circles represented, from inner to outer: scale marks; GC skew (green, positive skew; purple, negative skew); GC content; reverse COG annotated coding sequences; protein-coding genes on reverse strand; protein-coding genes on forward strand; forward COG annotated coding sequences
CAZymes analysis
There were 138 putative CAZymes-coding genes in the LJBV19 genome, including 48 glycoside hydrolases (GHs), 40 glycosyl transferases (GTs), 3 polysaccharide lyases (PLs), 26 carbohydrate esterases (CEs), 7 auxiliary activities (AAs), and 14 carbohydrate-binding modules (CBMs) (Fig. 3A). Moreover, 13 (9.42%) CAZymes with amino-terminal signal peptides for guiding through cytoplasmic membrane as secreted enzymes were crucial for LJBV19 biological activity (Chen et al. 2021). The genome of LJBV19 had 5, 4, 3, and 1 secreted CAZymes in the GHs, CEs, CBMs, and PL families, respectively (Fig. 3A). CAZymes degrade plant polysaccharides by enzymatic reaction (Chen et al. 2021). There were genes encoding for possible antifungal CAZymes, including 6-phospho-β-galactosidase (GH1), 6-phospho-glucosidase (GH4), endo-1,4-β-glucanase (GH5), β-glucanase (GH16), and endoglucanase (GH51) for cellulose degradation, chitinase (GH18), and chitosanase (GH46) (Fig. 3B). The functions of annotated genes involved in hydrolases were validated at the metabolic level, showing that LJBV19 could produce cellulose (0.53 ± 0.00 U/mL), chitinase (0.14 ± 0.01 U/mL), and chitosanase (0.11 ± 0.01 U/mL). These CAZymes in the genome LJBV19 can degrade the cell wall components of pathogens, which played an important role in fungal antagonism (Shafi et al. 2017). For example, β-chitinase or glucanase had the ability to inhibit infection by B. cinerea and C. gloeosporioides (Hamaoka et al. 2021).

Distribution of carbohydrate active enzymes (CAZymes) families in the genome of B. velezensis LJBV19. A The classification of CAZymes in the LJBV19 genome. B Functional characterization of glycoside hydrolase family
Comparative genomics analysis
Comparative analysis among the genome sequences of three closely related strains B. velezensis FZB42, B. amyloliquefaciens DSM7T, and B. subtilis 168T with the LJBV19 were performed (Table 2). Genome features of the four strains were annotated based on NCBI to ensure the same annotation conditions. Comparative results revealed that the genome size of LJBV19 (3,973,013 bp) was similar to DSM7T (3,980,199 bp) and FZB42 (3,918,596 bp), but smaller than 168T (4,215,606 bp). The GC content of LJBV19 (46.50%) was the same as FZB42 (46.50%), was approximately equal to DSM7T (46.1%), and was higher than 168T (43.5%). In addition, there was no plasmid in the four Bacillus genomes.
To evaluate the evolutionary distance among the four strains, their whole genome sequences were compared by Mauve program with default parameters (Fig. 4A). The alignments revealed no significant insertion of large regions or large local collinear block (LCB) inversion between LJBV19 and FZB42. Compared to DSM7T and 168T, a number of gene insertions or deletions and LCB inversions were present in LJBV19. More LCB inversions were occurred when LJBV19 compared with 168T showing that the LJBV19 genome was more similar to DSM7T than to 168T. The synteny plot of the pairwise alignments from Mauve program was consistent with taxonomic position of LJBV19.

Comparison of B. velezensis LJBV19 genome sequences against B. velezensis FZB42, B. amyloliquefaciens DSM7T, and B. subtilis 168T. A Mauve progressive alignment of the LJBV19, FZB42, DSM7T, and 168T. LJBV19 genome was used as the reference. Boxes with the same color indicated syntenic regions, and colored lines connected homologous regions. Boxes above the center line were forward regions and below the center line were reverse regions. The scale was in nucleotides. B Venn diagram showing the numbers of shared and unique clusters of orthologous genes
LJBV19 genome sequences were compared with above three genome sequences in order to identify the specific genes of LJBV19 (Fig. 4B). There were 1199 conserved genes shared with LJBV19, FZB42, DSM7T, and 168T. Comparison of orthologous genes showed that there were 2956 genes in common with average 85.51% identity between LJBV19 and FZB42, 2709 genes in common with average 78.36% identity between LJBV19 and DSM7T, and 1527 genes in common with average 44.17% identity between LJBV19 and 168T. Moreover, a total of 179 unique genes were present in the genome of LJBV19, and the functions of most of these genes need further confirmation. The result showed that the four strains had a conserved genomic structure and genetic homogeneity with some inversion events during evolution.
Comparison of gene clusters related to secondary metabolites
Bacillus species can secrete secondary metabolites with broad biological activities, such as antimicrobial, antiviral, and nematocidal action, protecting the plant against pathogens (Keswani et al. 2020). There were 13 gene clusters involved in the synthesis of secondary metabolites in the LJBV19, covering 18.93% (752.05 kb) of its genome (Table 3; Fig. S4). These gene clusters were consisted of three non-ribosomal peptide synthetase (NRPS) clusters, three trans-acyl transferase polyketide synthetase (transAT-PKS) clusters, two terpene clusters, one other unspecified ribosomally synthesized and post-translationally modified peptide (RiPP-like) cluster, one type 3 polyketide synthetase (T3PKS) cluster, one lanthipeptide-class-ii cluster, and one “other” type of gene cluster. Eight clusters corresponding with the production of identified secondary metabolites, including surfactin, butirosin, macrolactin H, bacillaene, fengycin, difficidin, bacillibactin, and bacilysin, matched to 82, 7, 100, 100, 100, 100, 100, and 100% of the known gene clusters, respectively. Moreover, five gene clusters with a total length of 116.62 kb encoding potential novel secondary metabolite-related proteins without previously known description.
The locations and products of secondary metabolite gene clusters in LJBV19, FZB42, DSM7T, and 168T were compared (Fig. 5). Interestingly, the core biosynthetic genes in the four strains were similar and the products of core genes exhibited very high homologues at the amino acid level (Fig. 6). The results showed that eight (clusters 1, 4, 6, 7, 8, 9, 11, and 13) involved in the biosynthesis of secondary metabolites in LJBV19 also existed in FZB42, DSM7T, and 168T strains. Among the eight clusters, five gene clusters were identified and specifically involved in the synthesis of surfactin (srfAA, srfAB, srfAC), bacillaene (baeCDEGJLMNR), fengycin (fen, myc, and yng), bacillibactin (dhbF), and bacilysin (bacD). However, B. amyloliquefaciens DSM7T lacked the genes fenA, fenB, and fenC for fengycin biosynthesis, whereas B. subtilis 168T lacked fenF, mycA, mycB, and mycC compared to B. velezensis LJBV19, suggesting that LJBV19 with the ability of fengycin synthesis may have stronger antimicrobial activity than DSM7T and 168T. Surfactin, bacillaene, fengycin, bacillibactin, and bacilysin were also observed in fungal antagonistic Bacillus spp. and proved to be antimicrobial compounds (Ravi et al. 2021). Surfactin and fengycin as non-ribosomal synthesis of cyclic lipopeptides (cLPs) have been proved to enhance plant defense response to pathogens. For example, the supernatant with surfactin and fengycin produced by B. subtilis GLB191 had direct antifungal effect and induced plant defense response, protecting grape against Plasmopara viticola (Li et al. 2019). Surfactin and fengycin exhibited antimicrobial and antiviral activities by altering membrane integrity of pathogens (Chen et al. 2015; Olishevska et al. 2019). Moreover, surfactin was related to quorum-sensing, biofilm formation, and root colonization (Anckaert et al. 2021). Bacillaene, a linear molecule with two amide bonds, was synthesized by PKS and selectively inhibited protein biosynthesis of prokaryotes (Moldenhauer et al. 2010). Siderophore bacillibactin with higher affinity for ferric ion possessed antimicrobial properties through depriving essential iron to alter the fitness and aggressiveness of pathogens (Khan et al. 2018). For example, B. velezensis FZB42 with the ability of bacillibactin production inhibited the growth of phytopathogens (Rabbee et al. 2019). Bacilysin, a dipeptide influencing biosynthesis of microbial cell wall by inhibiting the glucosamine-6-phosphate synthase, showed a broad range of antagonistic activity against phytopathogens, such as F. oxysporum, Erwinia amylovora, and Microcystis aeruginosa (Rabbee et al. 2019; Nannan et al. 2021). The other three (clusters 4, 8, and 9) were involved in unknown secondary metabolite-related proteins encoding terpene, terpene, and T3PKS, respectively.

Comparation of the location and products of secondary metabolite gene clusters in B. velezensis LJBV19, B. velezensis FZB42, B. amyloliquefaciens DSM7T and B. subtilis 168T

Comparisons of secondary metabolite clusters in LJBV19, FZB42, DSM7T, and 168T. Red, pink, blue, green, and gray indicated core biosynthetic genes, additional biosynthetic genes, transport-related genes, regulatory genes, and other genes, respectively. The core biosynthetic genes (red) were marked
Cluster 3 present in three strains excluding 168T encoding PKS-like was involved in the synthesis of butirosin. Butirosin was as a 2-deoxystreptamine (DOS)-containing aminoglycoside antibiotic, and the key part of biosynthetic gene clusters (ydhFR and pksF) was identified in B. circulans SANK 72073 (Kudo et al. 2005). Importantly, cluster 3 had only 7% similarity with known gene cluster of butirosin indicating that B. velezensis LJBV19 may be able to produce a structurally novel antibiotic compound. Clusters 5 and 10 were shared between LJBV19 and FZB42 encoding transAT-PKS. Cluster 5 had the core genes pks2A, pks2B, pks2C, pks2D, pks2E, pks2F, and pks2G related to the biosynthesis of macrolactin H (Fig. 6). Macrolactin belonging to nonribosomal synthesis of polyketides exhibited antimicrobial activity against gram-negative (i.e., Escherichia coli) and gram-positive (i.e., Staphylococcus aureus) bacteria and fungi (i.e., C. gloeosporioides, B. cinerea, and R. solani) (Ton That Huu et al. 2021). The genes difAFJHIJKL involved in the synthesis of difficidin were found in cluster 10 (Fig. 6). Difficidin, an antibaterial polyketides synthesized by NRPS, showed a broad spectrum of activity against aerobic (i.e., Pseudomonas aeruginosa and Salmonella typhimurium) and anaerobic (i.e., Clostridium difficile) bacteria (Rabbee et al. 2019). Two clusters (clusters 2 and 12) were only present in strain LJBV19: clusters 2 and 12 were shown to direct unknown compounds, encoding RiPP-like and class II lantipeptide, respectively. RiPP with a wide variety of structural features had antifungal, antibacterial, and antiviral activities (Ortega and van der Donk 2016). For example, thioamitides belonging to RiPP caused mitochondrial dysfunction and triggered apoptosis by inhibiting mitochodrial ATP synthase of pathogenic microorganisms (Eyles et al. 2021). Lantipeptide as antibacterial peptides synthesized by the ribosome destroyed the cell walls or membranes of pathogens, resulting in the outflow of small molecules and the dissipation of membrane potential (Dufour et al. 2007). For example, Streptomyces griseus S4-7 producing a class II lantipeptide called grisin suppressed wilt of strawberry caused by F. oxysporum (Kim et al. 2019). These results revealed that the secondary metabolite clusters in the LJBV19 genome were highly similar to gene clusters in fungal antagonistic strain B. velezensis FZB42, which was one of most important commercially available agents, such as RhizoVital®, RhizoPlus®, and Taegro® (Rabbee et al. 2019). Moreover, two unique clusters in LJBV19 genome would encode potential novel metabolites with unknown description. All results indicated that LJBV19 was expected to become a natural antagonist of plant pathogens.
Conclusions
Bacillus velezensis LJBV19, isolated for grapevine rhizosphere soil, showed a broad-spectrum antimicrobial activity against 12 plant pathogens. Whole genome sequencing, annotation, and genomic analysis revealed the structure and function of LJBV19 genome. Among 3993 ORFs in LJBV19 genome, 3308 ORFs were classified into 19 COG categories, such as secondary metabolites biosynthesis, transport and catabolism (Q), and carbohydrate transport and metabolism (G). Hydrolases were predicted by CAZy database and validated at the metabolic level, including cellulose (0.53 ± 0.00 U/mL), chitinase (0.14 ± 0.01 U/mL), and chitosanase (0.11 ± 0.01 U/mL). There were 13 gene clusters related to the biosynthesis of secondary metabolites in LJBV19 genome. Comparative genomic analysis confirmed the taxonomic position of LJBV19 and two unique clusters (clusters 2 and 12) in strain LJBV19. Taken together, these findings showed that LJBV19 possessed the necessary genetic machinery as fungal antagonistic agent and promoted its application.
Availability of data and material
The GenBank accession number for the 16S rRNA gene of Bacillus velezensis LJBV19 is MZ157279. The complete sequence of the LJBV19 has been deposited in GenBank database under the accession number CP072563. The strain B. velezensis LJBV19 was publicly available from China General Microbiological Culture Collection Center (CGMCC) under the accession number CGMCC No. 21804. Morphological characteristics, phylogenetic tree, effect of LJBV19 on the mycelia of Botrytis cinerea, the location and products of secondary metabolite gene clusters, physiological and biochemical characteristics, minimum information about the genome sequence, genome-to-genome comparison (ANI and dDDH), and inhibition radius of B. velezensis LJBV19 against 12 pathogens are available as Supplementary Materials.
References
Adeniji AA, Loots DT, Babalola OO (2019) Bacillus velezensis: phylogeny, useful applications, and avenues for exploitation. Appl Microbiol Biotechnol 103(9):3669–3682. https://doi.org/10.1007/s00253-019-09710-5
Anckaert A, Arias AA, Hoff G, Calonne-Salmon M, Declerck S, Ongena M (2021) The use of Bacillus spp. as bacterial biocontrol agents to control plant diseases. Microbial bioprotectants for plant disease management. Burleigh Dodds Series in Agricultural Science. Burleigh Dodds Science Publishing Limited, pp 247–300. https://doi.org/10.19103/as.2021.0093.10
Andrić S, Meyer T, Ongena M (2020) Bacillus responses to plant-associated fungal and bacterial communities. Front Microbiol 11:1350. https://doi.org/10.3389/fmicb.2020.0135
Garrity G (2001) Bergey's manual of systematic bacteriology 38(4):443–491
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene Ontology C (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340(4):783–795. https://doi.org/10.1016/j.jmb.2004.05.028
Berg G (2009) Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Appl Microbiol Biotechnol 84(1):11–18. https://doi.org/10.1007/s00253-009-2092-7
Bertelli C, Laird MR, Williams KP, Lau BY, Hoad G, Winsor GL, Brinkman FSL, Simon Fraser Univ Res Comp G (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45(W1):W30–W35. https://doi.org/10.1093/nar/gkx343
Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29(12):2607–2618. https://doi.org/10.1093/nar/29.12.2607
Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, Weber T (2021) antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res 49(W1):W29–W35. https://doi.org/10.1093/nar/gkab335
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31(1):365–370. https://doi.org/10.1093/nar/gkg095
Borriss R, Chen X-H, Rueckert C, Blom J, Becker A, Baumgarth B, Fan B, Pukall R, Schumann P, Sproeer C, Junge H, Vater J, Puehler A, Klenk H-P (2011) Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7(T) and FZB42(T): a proposal for Bacillus amyloliquefaciens subsp amyloliquefaciens subsp nov and Bacillus amyloliquefaciens subsp plantarum subsp nov based on complete genome sequence comparisons. Int J Syst Evol Microbiol 61:1786–1801. https://doi.org/10.1099/ijs.0.023267-0
Borriss R, Danchin A, Harwood CR, Medigue C, Rocha EPC, Sekowska A, Vallenet D (2018) Bacillus subtilis, the model Gram-positive bacterium: 20 years of annotation refinement. Microb Biotechnol 11(1):3–17. https://doi.org/10.1111/1751-7915.13043
Cai XC, Liu CH, Wang BT, Xue YR (2017) Genomic and metabolic traits endow Bacillus velezensis CC09 with a potential biocontrol agent in control of wheat powdery mildew disease. Microbiol Res 196:89–94. https://doi.org/10.1016/j.micres.2016.12.007
Chen K, Tian Z, He H, Long C-A, Jiang F (2020) Bacillus species as potential biocontrol agents against citrus diseases. Biol Control. https://doi.org/10.1016/j.biocontrol.2020.104419
Chen W-C, Juang R-S, Wei Y-H (2015) Applications of a lipopeptide biosurfactant, surfactin, produced by microorganisms. Biochem Eng J 103:158–169. https://doi.org/10.1016/j.bej.2015.07.009
Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, Junge H, Voigt B, Jungblut PR, Vater J, Suessmuth R, Liesegang H, Strittmatter A, Gottschalk G, Borriss R (2007) Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol 25(9):1007–1014. https://doi.org/10.1038/nbt1325
Chen YJ, Yu P, Luo JC, Jiang Y (2003) Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome 14(12):859–865. https://doi.org/10.1007/s00335-003-2296-6
Chen ZY, Abuduaini X, Mamat N, Yang QL, Wu MJ, Lin XR, Wang R, Lin RR, Zeng WJ, Ning HC, Zhao HP, Li JY, Zhao HX (2021) Genome sequencing and functional annotation of Bacillus sp. strain BS-Z15 isolated from cotton rhizosphere soil having antagonistic activity against Verticillium dahliae. Arch Microbiol 203(4):1565–1575. https://doi.org/10.1007/s00203-020-02149-7
Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer GR, Delledonne M, Luo C, Ecker JR, Cantu D, Rank DR, Schatz MC (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13(12):1050–1054. https://doi.org/10.1038/nmeth.4035
Choo KH, Tong JC, Zhang L (2004) Recent applications of hidden Markov models in computational biology. Genomics Proteomics Bioinformatics 2(2):84–96
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, Rooney AP, Yi H, Xu XW, De Meyer S, Trujillo ME (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68(1):461–466. https://doi.org/10.1099/ijsem.0.002516
Compant S, Duffy B, Nowak J, Clement C, Barka EA (2005) Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl Environ Microbiol 71(9):4951–4959. https://doi.org/10.1128/aem.71.9.4951-4959.2005
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5(6):e11147. https://doi.org/10.1371/journal.pone.0011147
Dhouib H, Zouari I, Ben Abdallah D, Belbahri L, Taktak W, Triki MA, Tounsi S (2019) Potential of a novel endophytic Bacillus velezensis in tomato growth promotion and protection against Verticillium wilt disease. Biol Control 139:11. https://doi.org/10.1016/j.biocontrol.2019.104092
Dufour A, Hindre T, Haras D, Le Pennec JP (2007) The biology of lantibiotics from the lacticin 481 group is coming of age. FEMS Microbiol Rev 31(2):134–167. https://doi.org/10.1111/j.1574-6976.2006.00045.x
Dunlap CA, Kim SJ, Kwon SW, Rooney AP (2016) Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and ‘Bacillus oryzicola’ are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int J Syst Evol Microbiol 66(3):1212–1217. https://doi.org/10.1099/ijsem.0.000858
Eyles TH, Vior NM, Lacret R, Truman AW (2021) Understanding thioamitide biosynthesis using pathway engineering and untargeted metabolomics. Chem Sci 12(20):7138–7150. https://doi.org/10.1039/d0sc06835g
Fan B, Blom J, Klenk H-P, Borriss R (2017) Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. amyloliquefaciens” within the B-subtilis species complex. Front Microbiol 8:22. https://doi.org/10.3389/fmicb.2017.00022
Fan B, Wang C, Song X, Ding X, Wu L, Wu H, Gao X, Borriss R (2018) Bacillus velezensis FZB42 in 2018: The Gram-positive model strain for plant growth promotion and biocontrol. Front Microbiol 9:2491. https://doi.org/10.3389/fmicb.2018.02491
Gao Z, Zhang B, Liu H, Han J, Zhang Y (2017) Identification of endophytic Bacillus velezensis ZSY-1 strain and antifungal activity of its volatile compounds against Alternaria solani and Botrytis cinerea. Biol Control 105:27–39. https://doi.org/10.1016/j.biocontrol.2016.11.007
Grissa I, Vergnaud G, Pourcel C (2008) CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 36:W145–W148. https://doi.org/10.1093/nar/gkn228
Hamaoka K, Aoki Y, Suzuki S (2021) Isolation and characterization of endophyte Bacillus velezensis KOF112 from grapevine shoot xylem as biological control agent for fungal diseases. Plants 10(9):1815. https://doi.org/10.3390/plants10091815
Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, Bork P (2008) eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36:D250–D254. https://doi.org/10.1093/nar/gkm796
Jiang C-H, Liao M-J, Wang H-K, Zheng M-Z, Xu J-J, Guo J-H (2018) Bacillus velezensis, a potential and efficient biocontrol agent in control of pepper gray mold caused by Botrytis cinerea. Biol Control 126:147–157. https://doi.org/10.1016/j.biocontrol.2018.07.017
Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, Petrov AI (2018) Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics 62(1):e51–e51. https://doi.org/10.1002/cpbi.51
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44(D1):D457–D462. https://doi.org/10.1093/nar/gkv1070
Keswani C, Singh HB, Garcia-Estrada C, Caradus J, He Y-W, Mezaache-Aichour S, Glare TR, Borriss R, Sansinenea E (2020) Antimicrobial secondary metabolites from agriculturally important bacteria as next-generation pesticides. Appl Microbiol Biotechnol 104(3):1013–1034. https://doi.org/10.1007/s00253-019-10300-8
Khan A, Singh P, Srivastava A (2018) Synthesis, nature and utility of universal iron chelator - siderophore: a review. Microbiol Res 212–213:103–111. https://doi.org/10.1016/j.micres.2017.10.012
Kim DR, Jeon CW, Shin JH, Weller DM, Thomashow L, Kwak YS (2019) Function and distribution of a lantipeptide in strawberry Fusarium wilt disease-suppressive soils. Mol Plant Microbe Interact 32(3):306–312. https://doi.org/10.1094/mpmi-05-18-0129-r
Kudo F, Numakura M, Tamegai H, Yamamoto H, Eguchi T, Kakinuma K (2005) Extended sequence and functional analysis of the butirosin biosynthetic gene cluster in Bacillus circulans SANK 72073. J Antibiot 58(6):373–379. https://doi.org/10.1038/ja.2005.47
Lagesen K, Hallin P, Rodland EA, Staerfeldt H-H, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9):3100–3108. https://doi.org/10.1093/nar/gkm160
Li WZ, Jaroszewski L, Godzik A (2002) Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18(1):77–82. https://doi.org/10.1093/bioinformatics/18.1.77
Li Y, Heloir MC, Zhang X, Geissler M, Trouvelot S, Jacquens L, Henkel M, Su X, Fang XW, Wang Q, Adrian M (2019) Surfactin and fengycin contribute to the protection of a Bacillus subtilis strain against grape downy mildew by both direct effect and defence stimulation. Mol Plant Pathol 20(8):1037–1050. https://doi.org/10.1111/mpp.12809
Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42(D1):D490–D495. https://doi.org/10.1093/nar/gkt1178
Lopes R, Tsui S, Goncalves PJRO, de Queiroz MV (2018) A look into a multifunctional toolbox: endophytic Bacillus species provide broad and underexploited benefits for plants. World J Microbiol Biotechnol 34(7):94. https://doi.org/10.1007/s11274-018-2479-7
Lowe TM, Eddy SR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964. https://doi.org/10.1093/nar/25.5.955
Moldenhauer J, Goetz DCG, Albert CR, Bischof SK, Schneider K, Suessmuth RD, Engeser M, Gross H, Bringmann G, Piel J (2010) The final steps of Bacillaene biosynthesis in Bacillus amyloliquefaciens FZB42: direct evidence for beta, gamma dehydration by a trans-acyltransferase polyketide synthase. Angew Chem Int Ed 49(8):1465–1467. https://doi.org/10.1002/anie.200905468
Mullins AJ, Li Y, Qin L, Hu X, Xie L, Gu C, Mahenthiralingam E, Liao X, Webster G (2020) Reclassification of the biocontrol agents Bacillus subtilis BY-2 and Tu-100 as Bacillus velezensis and insights into the genomic and specialized metabolite diversity of the species. Microbiology 166(12):1121–1128. https://doi.org/10.1099/mic.0.000986
Nannan C, Vu HQ, Gillis A, Caulier S, Nguyen TTT, Mahillon J (2021) Bacilysin within the Bacillus subtilis group: gene prevalence versus antagonistic activity against Gram-negative foodborne pathogens. J Biotechnol 327:28–35. https://doi.org/10.1016/j.jbiotec.2020.12.017
Olishevska S, Nickzad A, Deziel E (2019) Bacillus and Paenibacillus secreted polyketides and peptides involved in controlling human and plant pathogens. Appl Microbiol Biotechnol 103(3):1189–1215. https://doi.org/10.1007/s00253-018-9541-0
Ongena M, Jacques P (2008) Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol 16(3):115–125. https://doi.org/10.1016/j.tim.2007.12.009
Ortega MA, van der Donk WA (2016) New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem Biol 23(1):31–44. https://doi.org/10.1016/j.chembiol.2015.11.012
Palazzini JM, Dunlap CA, Bowman MJ, Chulze SN (2016) Bacillus velezensis RC 218 as a biocontrol agent to reduce Fusarium head blight and deoxynivalenol accumulation: Genome sequencing and secondary metabolite cluster profiles. Microbiol Res 192:30–36. https://doi.org/10.1016/j.micres.2016.06.002
Ravi S, Nakkeeran S, Saranya N, Senthilraja C, Renukadevi P, Krishnamoorthy AS, El Enshasy HA, El-Adawi H, Malathi VG, Salmen SH, Ansari MJ, Khan N, Sayyed RZ (2021) Mining the genome of Bacillus velezensis VB7 (CP047587) for MAMP genes and non-ribosomal peptide synthetase gene clusters conferring antiviral and antifungal activity. Microorganisms 9(12):2511. https://doi.org/10.3390/microorganisms9122511
Rabbee MF, Ali MS, Choi J, Hwang BS, Jeong SC, Baek KH (2019) Bacillus velezensis: a valuable member of bioactive molecules within plant microbiomes. Molecules 24(6):1046. https://doi.org/10.3390/molecules24061046
Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106(45):19126–19131. https://doi.org/10.1073/pnas.0906412106
Rooney AP, Price NP, Ehrhardt C, Swezey JL, Bannan JD (2009) Phylogeny and molecular taxonomy of the Bacillus subtilis species complex and description of Bacillus subtilis subsp. inaquosorum subsp. nov. Int J Syst Evol Microbiol 59(Pt 10):2429–2436. https://doi.org/10.1099/ijs.0.009126-0
Santana MA, Moccia-V CC, Gillis AE (2008) Bacillus thuringiensis improved isolation methodology from soil samples. J Microbiol Methods 75(2):357–358. https://doi.org/10.1016/j.mimet.2008.06.008
Sari GL, Trihadiningrum Y, Ni’matuzahroh (2019) Isolation and identification of native bacteria from total petroleum hydrocarbon polluted soil in Wonocolo public oilfields, Indonesia. J Ecol Eng 20(8):60–64. https://doi.org/10.12911/22998993/110816
Sasse J, Martinoia E, Northen T (2018) Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci 23(1):25–41. https://doi.org/10.1016/j.tplants.2017.09.003
Shafi J, Tian H, Ji M (2017) Bacillus species as versatile weapons for plant pathogens: a review. Biotechnol Biotechnol Equip 31(3):446–459. https://doi.org/10.1080/13102818.2017.1286950
Ton That Huu D, Nguyen Thi Kim C, Pham Viet C, Smidt H, Sipkema D (2021) Diversity and antimicrobial activity of Vietnamese sponge-associated bacteria. Marine Drugs 19(7):353. https://doi.org/10.3390/md19070353
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9(11):e112963. https://doi.org/10.1371/journal.pone.0112963
Wang B, Peng H, Wu W, Yang B, Chen Y, Xu F, Peng Y, Qin Y, Lu J, Fu P (2021) Genomic insights into biocontrol potential of Bacillus stercoris LJBS06. 3 Biotech 11:458. https://doi.org/10.1007/s13205-021-03000-6
Ye M, Tang X, Yang R, Zhang H, Li F, Tao F, Li F, Wang Z (2018) Characteristics and application of a novel species of Bacillus: Bacillus velezensis. ACS Chem Biol 13(3):500–505. https://doi.org/10.1021/acschembio.7b00874
Zhang M, Li J, Shen A, Tan S, Yan Z, Yu Y, Xue Z, Tan T, Zeng L (2016) Isolation and Identification of Bacillus amyloliquefaciens IBFCBF-1 with potential for biological control of Phytophthora blight and growth promotion of pepper. J Phytopathol 164(11–12):1012–1021. https://doi.org/10.1111/jph.12522
Zhu XF, Zhou Y, Feng JL (2007) Analysis of both chitinase and chitosanase produced by Sphingomonas sp. CJ-5. J Zhejiang Univ Sci B 8(11):831–838. https://doi.org/10.1631/jzus.2007.B0831
Funding
This work was supported by Shanghai Municipal Agricultural Commission (grant no. 2021–02-08–00-12-F00751), National Key R & D Program of China (2019YFD1002501 and 2018YFD1000301), Shanghai Municipal Commission for Science and Technology (18391900400), Yunnan Province Science and Technology Department (202005AF150023 to J. Lu), and China Agriculture Research System (grant no. CARS-29-yc-2).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Bo Wang, Bohan Yang, Hang Peng, and Peining Fu. The first draft of the manuscript was written by Bo Wang and Bohan Yang; all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
All authors approved the manuscript.
Consent for publication
Written informed consent for publication was obtained from all participants.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Bo Wang and Bohan Yang contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, B., Yang, B., Peng, H. et al. Genome sequence and comparative analysis of fungal antagonistic strain Bacillus velezensis LJBV19. Folia Microbiol 68, 73–86 (2023). https://doi.org/10.1007/s12223-022-00996-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-022-00996-z