
Abstract
Information on the sex- and individual-specific space use by a species elucidates demography, resource selection and individual life history. However, traditional field surveys often lack information on sex and individual identity, thereby not maximizing the potential of the effort put in. Recent advances in genetic non-invasive sampling provide cost-effective approaches to determine identity and sex from faecal DNA with high accuracy, which are advantageous for tracking individuals compared to field observations. Therefore, we describe the first single multiplex-based sex and individual identification protocol using faecal samples of the wild Asian elephant (Elephas maximus) collected from the vicinity of Rajaji Tiger Reserve, Uttarakhand, India. We co-amplified fluorescence-labelled microsatellites (n = 5) and a Y chromosome-linked sex marker in four replicates from faecal DNA extracts (n = 149). The mean per genotype allelic drop-out rate was 0.11 ± 0.02, while the false allele rate was 0.05 ± 0.01. The mean null allele frequency across the markers was 0.15 ± 0.02. We obtained 74.1% consensus genotypes across microsatellites and dropped samples with more than one-locus missing genotype from further analyses. The remaining dataset comprised 105 samples, 30.5% of which were females. We identified 51 unique individuals (25 males and 26 females) with a maximum of one-locus mismatch. With low genotyping error rates and adequate misidentification probabilities (PID = 4.2 × 10−4; PIDSib = 3.0 × 10−2), the described panel provides a cost-effective method (US$ 18/sample) for molecular sexing and individual identification. Hence, the suggested multiplex panel would provide a thorough understanding of individual and sex-specific differences in habitat use across heterogeneous landscapes, facilitating effective conservation strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Developing an effective conservation strategy for long-ranging mammalian species, often traversing through a human-modified landscape, requires fine-scale information on resource requirements (Sanderson et al. 2002). Multiple studies in recent times have concluded the existence of individualistic as well as sex-linked variability in patterns of resource utilization across various taxa to account for differential physiological requirements at various life-history stages (Sprogis et al. 2018; Ofstad et al. 2019; Carrasco et al. 2020; Johnson and Derocher 2020; Rehnus and Bollmann 2020). Individual-level studies on animals with distinct morphological marks or tags can monitor their life history while also providing information such as abundance and survivorship at the population level, often requiring ample resources (Galvis et al. 2014; Sadhu et al. 2017). For species lacking such markings, invasive physical tags, e.g. coloured bands or collars, ear tags, transmitters and skin brands are used that may be lost over time and are cost- and time-intensive (Woods et al. 1999).
With the recent advances, permanent genetic tags such as multi-locus microsatellite genotypes and molecular sexing from non-invasive faecal samples of individuals overcome these limitations and hence, are useful for long-term monitoring across large landscapes and over the focal species’ lifespan. Molecular tracking and faecal DNA-based abundance estimation have been used as cost-effective techniques alternative to physical tagging or camera trapping with comparable accuracy and precision (Ernest et al. 2000; Janečka et al. 2011; Hedges et al. 2013; Caniglia et al. 2014; Gray et al. 2014).
However, some key parameters (e.g. age of the individual, the decay rate of the faeces deposited) for obtaining reliable demographic estimates from non-invasive sampling have been calibrated for only a limited number of species (Eggert et al. 2003; Flagstad et al. 2012; Hedges et al. 2013; Poutanen et al. 2019). Additionally, molecular tagging poses a substantial challenge in obtaining reliable genotypes from the inferior quality faecal DNA due to intrinsic errors, i.e. allelic drop-outs and false alleles (Fernando et al. 2003a; Scandura et al. 2006; Sethi et al. 2014). Too few microsatellites lacking optimal resolution produce ‘shadow’ genotypes caused by merging individual identities, thereby underestimating the actual count (Mills et al. 2000; Sethi et al. 2014). Conversely, ‘ghost’ individuals are produced if samples from the same individual generate non-identical genotypes due to the accumulation of genotyping errors (Creel et al. 2003; Lampa et al. 2013; Sethi et al. 2014). The proportion of ‘ghost’ individuals is positively correlated with the number of microsatellite loci used for individual identification, incurring up to five-fold positive bias in enumerating unique individuals (Creel et al. 2003). Multiple studies (Wang et al. 2012; Rothstein et al. 2016; Wang 2016) have developed algorithms incorporating genotyping error rates to minimise ‘ghost’ errors.
The Asian elephant (Elephas maximus) has been obliterated from 95% of the historical stronghold, whereas, in India, its geographic distribution has shrunk by 70% since the 1960s (Sukumar 2006). Additionally, the loss of a quarter of the elephant habitat in India within the last century (Padalia et al. 2019) underscores the importance of regular monitoring of elephant populations to understand habitat use, movement patterns, estimation of abundance and dynamics of human-elephant interactions. Monitoring elephants at the individual level based on variables such as body shape, ear, tail, and tusk require enormous efforts to maintain such a database over the species’ lifespan and across the vast landscape it inhabits (Morley and van Aarde 2007; de Silva et al. 2011; Goswami et al. 2019).
For Asian elephants, non-invasive genetic sampling (gNIS) has been employed to address a broad range of research topics, i.e. population monitoring (Vidya et al. 2007; Flagstad et al. 2012; Hedges et al. 2013; Chakraborty et al. 2014; Gray et al. 2014; Zhang et al. 2015), social organization (Vidya and Sukumar 2005a; Ahlering et al. 2011a), population and landscape genetics (Fernando et al. 2003b; Vidya et al. 2005; Goossens et al. 2016; De et al. 2021; Parida et al. 2022), demographic history (Sharma et al. 2018), and phylogeography (Vidya et al. 2009). However, the sets of markers used by different research groups are often non-overlapping, rendering the data difficult to compare, as is the case with microsatellite data of most other species (Garner et al. 2005; Li and Kimmel 2013). It is critical to generate harmonized, comparable data, especially for threatened taxa having a wide distribution range for conservation planning at the landscape level (de Groot et al. 2016).
Molecular sexing in elephants has been carried out using either of the following strategies: restriction fragment length polymorphism (RFLP) (Fernando and Melnick 2001; Vidya et al. 2003; Ahlering et al. 2011a; Hedges et al. 2013), polymerase chain reaction (PCR) followed by gel-based evaluation (Gupta et al. 2006; Vidya et al. 2007; Munshi-South et al. 2008; Ahlering et al. 2011b; Chakraborty et al. 2014), PCR and subsequent fragment analysis of fluorescent dye labelled product or quantitative PCR (qPCR) based assay (Aznar-Cormano et al. 2021).
A gNIS based method for cost-effective identification of unique individuals and their sex is crucial for augmenting information on elephant ecology, demography, and understanding the patterns of human-elephant interactions at the landscape scale (Chiyo et al. 2011; Chakraborty et al. 2014). Therefore, we aimed to propose a microsatellite panel with sufficient resolution while using a minimum number of microsatellites to avoid compounding genotyping errors that inflate unique individual counts (Creel et al. 2003). We prioritized co-amplification of the microsatellite markers along with a sexing marker in a single multiplex, thereby minimizing reagent and plasticware consumption and chances of human errors during handling. The specific objectives of this study using degraded elephant faecal DNA were: (i) to standardize a co-amplifiable multiplex panel consisting of a sex-linked marker and microsatellite markers that would provide low misidentification probability and genotyping error rates, and (ii) empirical validation of the proposed microsatellite panel using a larger set of published markers.
Materials and methods
Study area
The sampling for this study was conducted in the human-dominated landscape around Rajaji Tiger Reserve (RTR) in the Shivalik Elephant Reserve (SER), with the majority of the samples being from areas under Haridwar and Dehradun Forest Divisions (FD), Uttarakhand, north-west India.
Field sampling and DNA extraction
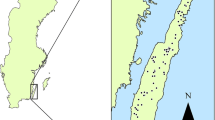
We opportunistically collected spatially segregated elephant faecal samples (n = 149, Fig. 1a) from the landscape between August 2014 and May 2018. We placed the samples in sterile containers (50 ml) over silica gel following Wasser et al. (1997). The samples were oven-dried at 56 °C placed ~ 15cm apart from one another (~ 10 samples per rack) < 30 days post-collection. We set the airflow velocity of the oven at a minimum to reduce the chance of cross-contamination. The dried samples were capped and stored at room temperature away from sunlight for up to 32 months until DNA extraction.

Locations of the a elephant faecal samples collected (n = 149) and b samples used in analyses (n = 105) along with their identified sex from the vicinity of the Rajaji Tiger Reserve, Uttarakhand, India
Faecal samples of multiple mammalian species were routinely dried together in the same oven, and we assessed cross-contamination possibilities based on > 1000 DNA sequences from samples processed using this protocol. Downstream Sanger sequencing of amplicons (~ 400 bp) using universal mammalian primers consistently yielded distinct sequences of the target species (Goyal et al. unpublished data) based on nucleotide BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Upon scrutiny of the raw chromatograms (Q > 20), none of the samples displayed any ambiguous peaks across the variable sites. Similar oven drying protocols have been used in non-invasive genotyping studies for sample desiccation before long-term storage (Murphy et al. 2000; Borthakur et al. 2011, 2013; De et al. 2021, 2022). Therefore, we believe that the chance of the results being affected by cross-contamination during oven drying of elephant faecal samples is negligible.
We scraped the top layer of the dung boluses with a sterile blade into 2 ml polypropylene centrifuge tubes. We used QIAamp DNA Stool Mini Kit (Qiagen GmbH, Hilden, Germany) to isolate DNA from the faecal scrapings using the manufacturer-specified stool DNA extraction protocol after overnight digestion in stool lysis buffer at 56 °C. We carried out all the DNA extractions in a separate low DNA isolation facility with negative controls to track contaminations.
PCR amplification
Selection of microsatellite markers
We initially selected a published multiplex microsatellite panel (EMU13, EMU17, LafMS02 and LafMS05; Nyakaana and Arctander 1998; Kongrit et al. 2008) exhibiting high amplification success with faecal DNA (De et al. 2021). We modified the combination to exclude a marker (EMU17) producing inconsistent ‘stutter’ bands with degraded samples during initial testing (unpublished data) and replaced with two more loci (EMU07 and EMU12; Kongrit et al. 2008) of higher success and low error rates (De et al. 2021). Finally, the selected panel consisted of five dinucleotide microsatellite loci, i.e. EMU07, EMU12, EMU13, LafMS05 and LafMS02. This combination of microsatellite markers could assign the highest number of individuals to their respective identities among several panels tested using a blind test approach (De et al. 2022).
Molecular sexing
For molecular identification of sex from faecal DNA, we selected an elephant-specific Y-chromosome linked Amelogenin marker (Ahlering et al. 2011b) for co-amplification with the microsatellite panel. The sex marker, AMELY2, produces a 121 bp amplicon in Asian elephant males and does not amplify in females (Ahlering et al. 2011b). The criteria for assigning a sample of female origin were non-amplification of the AMELY2 fragment and a minimum of four microsatellites co-amplifying. For males, co-amplification of AMELY2 and at least four microsatellites were required.
Reaction composition and conditions
The forward primers of the microsatellite panel (n = 5) and the sexing marker (n = 1) were labelled with fluorescent G5 dyes (Applied Biosystems, CA, USA). Each reaction consisted of 5 µl Qiagen Multiplex PCR Master-mix, 10µg bovine serum albumin (BSA), 2 µl DNA template and 1.0 µl labelled primer cocktail containing equal proportions of 10 µM microsatellite and sexing markers and nuclease-free water to make the reaction volume up to 10 µl. The thermal cycling profile (Veriti thermocycler, Applied Biosystems) consisted of initial denaturation at 95 °C for 15 min, 45 cycles of denaturation at 95 °C for 30 s, touchdown annealing at 62 to 52 °C for 1 min—a drop of 1 °C every two cycles up to 20th cycle and 52 °C for rest of the 25 cycles, extension at 72 °C for 40 s; final extension at 60 °C for 30 min and finally, hold at 4 °C. To screen for PCR performance, we visualized the products in 2% w/v agarose gel stained with ethidium bromide.
We followed a multi-tube approach (Taberlet et al. 1996) to restrict genotyping errors with minor modifications. Instead of a two-step protocol to amplify up to seven replicates (Taberlet et al. 1996), we replicated the multiplex reaction a total of four times (Ruiz-González et al. 2013; Bhatt et al. 2020; De et al. 2021) with each DNA isolate (n = 149).
All sets of PCRs included positive and negative controls to track PCR failure and reagent contamination. We dissolved 1 µl of each PCR product in 8.93 µl HiDi formamide and 0.07 µl Gene Scan 500 Liz size standard (Applied Biosystems) and denatured at 95 °C for 5 min before loading in an ABI 3500xl automated genetic analyser (Applied Biosystems) for fragment analysis.
Data analysis
The resulting electropherograms were visualized using GENEMAPPER 5.0 (Applied Biosystems). Automated allele scoring was performed subsequently while we manually verified each call. We only considered alleles that produced sharp, clear peaks without any ambiguity caused due to stuttering or + A peaks (Matsumoto et al. 2004). Additionally, we carefully reviewed any peak under a relative fluorescence unit (RFU) value of 1000. We removed data with RFU < 500 from all further analyses as the probability of allelic drop-out (ADO), inversely proportional to peak heights, has been demonstrated to be ≤ 5% at RFU ≥ 487 (Gill et al. 2015). We used a lower threshold peak height ratio (PHR; signal intensity ratio of the larger allele in terms of fragment length to the smaller allele) of 40% (Mäck et al. 2021) for heterozygous genotypes to minimize false alleles in the dataset. Considering samples with high RFU ensured a greater quality of data with unambiguous profiles, falling in the top two categories of ‘SEQ-score’ 1 and 2 as suggested in the literature (Scandura et al. 2006).
We binned the resulting raw fragment length data using AUTOBIN v0.9 Excel Macro (Salin 2010). The criteria for a consensus homozygous genotype was the amplification of the same allele in a minimum of three replicates, while a consensus heterozygous genotype was recorded in the case at least two replicates produced the exact same sets of alleles (Sawaya et al. 2011; Morin et al. 2016). Any replicate genotypes that did not pass the criteria for consensus were recorded as missing data. We considered a sample of male origin when the AMELY2 fragment co-amplified with at least four microsatellites, while for a female individual, non-amplification of AMELY2 and amplification of four microsatellites were required. We used GIMLET v1.3.3 (Valière 2002) to compute ADO and false allele (FA) rates per heterozygote and homozygote genotypes, respectively, based on the four sets of multi-tube repeat genotypes.
We proceeded for individual identification with all samples with a minimum of 4-loci consensus microsatellite data using the program CERVUS 3.0.7 (Kalinowski et al. 2007). A threshold of at least four-loci match (i.e., exact matches across five-loci and four-loci) along with the molecular sexing data was set as the criteria to assign unique genotypes. We used the R package ‘diveRsity’ (Keenan et al. 2013; R Core Team 2019) to compute the diversity statistics for the microsatellite markers from the identified individual data. We tested for linkage disequilibrium (LD) using GENEPOP 4.7 (Rousset 2008) with 1000 dememorization, 100 batches and 1000 iterations per batch. Null allele frequencies were calculated using the EM algorithm (Dempster et al. 1977) as implemented by the software FreeNA (Chapuis and Estoup 2007). We used GenAlEx 6.5.0.1 (Peakall and Smouse 2006, 2012) to calculate the probability of misidentifying two different random individuals as a single individual (PID) and two full siblings as an individual (PIDSib) for the chosen panel of markers.
Empirical validation of panel resolution
We amplified and scored additional microsatellite markers (n = 9; EMU03, EMU04, EMU09, EMU10, EMU11, EMU14, EMU15, EMU17 and LafMS03) (Nyakaana and Arctander 1998; Kongrit et al. 2008) with the samples meeting evaluation criteria using reaction conditions described in De et al. (2021). We used the five-loci and 14-loci (including the original five microsatellite loci) panels separately to identify first-order relatives (parent-offspring and full siblings) using the software COLONY v2.0.6.6 (Jones and Wang 2010) under a full-likelihood framework with medium run lengths and weak sib-ship prior for three independent runs. We screened for the potential ‘shadow’ error by comparing the incongruity between the two datasets. Thereafter, we manually quantified the sample pairs assigned as first-order relatives with the 14-loci panel but merged as same individuals using 5-loci genotypes.
Results and discussion
Marker amplification and genotyping errors
We recorded a 74.1% average success rate (calculated as the proportion of successful consensus genotypes out of the total attempted genotypes) across the five microsatellite loci, ranging between 59.7 and 83.2% successful amplification (Table 1). The PCRs yielded microsatellite amplicon sizes between 101 and 157 bp, while AMELY2 fragments were 121 bp (Fig. 2). All positive controls amplified successfully, along with accurate identification of sex. Neither of the negative controls showed any amplification. Most alleles (95.6%) produced a peak height of > 1000 RFU (Supplementary Fig. S1). Allele frequencies across the markers ranged from 0.01 to 0.62 (Supplementary Fig. S2). The mean ADO rate across loci was 0.11 ± 0.02, and the FA rate was 0.05 ± 0.01. Null allele frequencies varied between 0.12 and 0.22 with a mean of 0.15 ± 0.02. None of the pairs of the loci showed any evidence for linkage disequilibrium (LD) at a 95% level of significance. The locus LafMS02 produced allelic peaks followed by a ‘+A’ peak, one bp apart (Fig. 2). We retained the allelic peak and ignored the trailing +A despite having comparable peak height. Accurate allele scoring from faecal samples has been a significant limitation in getting reliable data from non-invasive samples. However, characterizing each observed allele across loci using stutter-to-peak and first-to-second allele ratios (Table 1), as suggested by Matsumoto et al. (2004), may aid accurate allele scoring by providing a quantitative criterion.

Electropherograms of the microsatellites and the Y-linked sex marker co-amplified from faecal DNA extracts of a male and b female elephant individuals
〹
Individual identification
We dropped samples with lower than four-loci microsatellite consensus genotypes (n = 44) from further analyses. There were 148 missing genotypes for the 44 samples discarded. The majority of the gaps (n = 127; 85.8%) within the dropped samples were caused by non-amplification, while consensus could not be reached for the rest 14.2% (Supplementary Fig. S3).
The remaining samples (n = 105) contained only 4.6% missing genotypes across the five-microsatellite panel. Complete five-locus data were available for 81 samples (77.1%), while the rest of the 24 samples (22.9%) had one missing genotype each. Based on the threshold set for microsatellite similarity, CERVUS algorithm identified 151 pairs of matching genotypes (putative recaptures) out of the 5460 pair-wise comparisons possible between the samples under analysis (n = 105). Out of these matching sample pairs (n = 151), 47% matched exactly across all five microsatellite loci, whereas 53% had genotypes that matched for four loci, with the remaining one locus data either missing or mismatching. We identified a minimum of 51 unique genotypes or individuals from the samples analyzed (n = 105). There were 39 individuals (76.5%) appearing in the dataset only once (single genetic captures), while 23.5% of individuals (n = 12) showed multiple captures each, thereby accounting for 62.9% of the total samples analyzed. We observed five individuals with two to three captures, one individual with four captures, four individuals with six to seven captures and one individual each with nine and fifteen captures, respectively.
Molecular sexing
Molecular sexing was successful for all 105 samples. We identified 73 samples of male and 32 samples of female origin (Fig. 1b). Out of 51 individuals identified using microsatellite data, 25 (49.1%) were identified as males using molecular sexing, while 26 (50.9%) were females.
Marker characteristics
The number of alleles per locus (Na) varied from four to seven (Mean Na = 4.80 ± 0.58) (Table 1). For the 51 individuals, estimates of expected heterozygosity (HE) across loci were in the range of 0.54 to 0.77 (Mean HE = 0.63 ± 0.04). The mean observed heterozygosity was HO = 0.40 ± 0.05 (0.22–0.56). PID and PIDSib varied between 0.09 and 0.27 and 0.38–0.55 across the loci (Table 1).
We designed this microsatellite panel to reliably identify Asian elephant individuals under the worst-case scenario of using degraded DNA for genotyping as we used dry-stored samples kept at room temperature up to ~ 3 years after field collection. We achieved a mean success rate (74.1%) and genotyping error rate (Table 1; allelic dropout: 0.11 ± 0.02, false alleles: 0.05 ± 0.01) comparable to other non-invasive studies on Asian elephants (Vidya and Sukumar 2005b; Flagstad et al. 2012; Hedges et al. 2013; Chakraborty et al. 2014; Gray et al. 2014; Goossens et al. 2016). Therefore, adopting a similar methodology of a single multiplex PCR with four replicates using elephant faecal DNA would be useful for a rapid sweeping survey to identify individuals and their sex under financial and logistic constraints. The efficacy of the prescribed panel with dried samples stored in room temperature indicates that post-facto analyses can also be taken up using this approach where faecal samples have already been collected for nutritional, parasitological or endocrinological studies.
The five-loci microsatellite panel suggested in this study has a probability of misidentification rate of 0.04% for random individuals (recommended range < 1%; Waits et al. 2001) and 3% between full siblings (Supplementary Fig. S4). It is possible to achieve even lower probabilities of ‘shadow’ genotypes (Mills et al. 2000) dataset by scrutiny of matching genotypes for differences in molecular sexing results, bolus morphometry, location and time of sampling.
Validation of the suggested panel
It has been established that ‘ghost’ individuals, directly proportional to the number of markers used for individual identification, may incur significant positive bias while estimating wildlife abundance (Creel et al. 2003; Lampa et al. 2013, 2015; Winiarski and McGarigal 2016; De et al. 2022). Due to the prevalence of genotyping errors in non-invasive microsatellite data, several studies suggest using the minimum number of markers to obtain sufficient resolution (i.e. reasonably low PID) for identifying unique individuals (Waits et al. 2001; Creel et al. 2003; Wang 2016). The typical social organization of elephants warrant validation of the suggested panel for its ability to distinguish between closely-related individuals, as the theoretical PIDsib obtained may be considered sub-optimal.
We recorded 86 dyads of first-order relatives using the 14-loci data set generated for the purpose of validation. In comparison, the suggested 5-loci panel could correctly differentiate 83 dyads (96.5%) as individuals. Discrepant merging of three dyads of first-order relatives while using the five-loci panel translated into a ‘shadow’ error of only 0.2% across 1275 possible dyads (51 individuals). Therefore, the empirical validation suggests that the multiplex five-loci panel, along with the sexing marker, is sufficient for identifying unique elephant individuals for monitoring purposes. Using the additional nine markers did not add any significant information towards individual identity. This is clearly indicated by the cumulative PID and PIDsib values plotted against the number of markers used for individual identification (Supplementary Fig. S4).
Cost-effectiveness of the described protocol
We propose collecting 10 to 15 g of faecal samples from the outer surface of an intact elephant dung bolus as a DNA source during routine field surveys or patrolling activities. Following the protocol outlined in this study, the cost of laboratory analyses to obtain sex and individual identity is US$ 18/sample, including reagents and laboratory staff (Supplementary Table S1). We excluded the cost of permanent laboratory equipment from the calculation. In comparison, Hedges et al. (2013) reported a cost of US$ 55/sample (reagents and human resources) for laboratory analyses to estimate demographic parameters of the Asian elephant in Laos, including microsatellite markers (n = 8), mtDNA sequence and RFLP-based molecular sexing data, including sequencing costs (US$ 14). While acknowledging intrinsic differences such as age of the samples being collected, impact of environmental conditions on DNA quality, skill of the human resources involved, we believe the protocol suggested in the current study provide a cheap alternative for faecal DNA based monitoring of the Asian elephant.
Conclusion
We suggest harmonized use of the multiplex panel described in this study for multi-locus genotyping during future status surveys undertaken for Asian elephants across its range. Identifying elephant individuals and their sex using the optimized single multiplex panel provides a high potential to reveal additional information on (i) sex-specific spatio-temporal distribution patterns, abundance, habitat use (Fig. 1b), (ii) population estimation from periodic surveys, and (iii) understanding human-elephant interaction including individual-based crop-raiding behaviour. Based on the requirement, additional markers (n = 9) can be supplemented as described in a previous pan-India study using non-invasive faecal sampling of the Asian elephant (De et al. 2021) for information such as population and landscape genetics and kinship patterns. Detailed information on the fine-scale spatio-temporal resource utilization patterns is lacking for most of the elephant populations in India (Vijayakrishnan et al. 2020). Hence, we believe that an effort to understand the demography and individual-level distribution and ranging patterns using the described protocol would provide additional insight, which is critical to formulating successful elephant conservation strategies.
Data availability
Raw genotypes will be made available upon reasonable request.
References
Ahlering MA, Hedges S, Johnson A, Tyson M, Schuttler SG, Eggert LS (2011a) Genetic diversity, social structure, and conservation value of the elephants of the Nakai Plateau, Lao PDR, based on non-invasive sampling. Conserv Genet 12:413–422. https://doi.org/10.1007/s10592-010-0148-y
Ahlering MA, Hailer F, Roberts MT, Foley C (2011b) A simple and accurate method to sex savannah, forest and Asian elephants using noninvasive sampling techniques. Mol Ecol Resour 11:831–834. https://doi.org/10.1111/j.1755-0998.2011.03030.x
Aznar-Cormano L, Bonnald J, Krief S, Guma N, Debruyne R (2021) Molecular sexing of degraded DNA from elephants and mammoths: a genotyping assay relevant both to conservation biology and to paleogenetics. Sci Rep 11:1–11. https://doi.org/10.1038/s41598-021-86010-x
Bhatt S, Biswas S, Karanth K, Pandav B, Mondol S (2020) Genetic analyses reveal population structure and recent decline in leopards (Panthera pardus fusca) across the indian subcontinent. PeerJ 8:e8482. https://doi.org/10.7717/peerj.8482
Borthakur U, Barman RD, Das C, Basumatary A, Talukdar A, Ahmed MF, Talukdar BK, Bharali R (2011) Noninvasive genetic monitoring of tiger (Panthera tigris tigris) population of Orang National Park in the Brahmaputra floodplain, Assam, India. Eur J Wildl Res 57:603–613. https://doi.org/10.1007/s10344-010-0471-0
Borthakur U, Saini RP, Gupta SS, Jakher R, Das C, Das AK, Sarma PK, Talukdar BK, Bharali R (2013) Noninvasive genetic assessment of population status of tigers (Panthera tigris tigris) in Buxa Tiger Reserve, West Bengal, India. Int J Biodivers Conserv 5:27–32. https://doi.org/10.5897/IJBC12.067
Caniglia R, Fabbri E, Galaverni M, Milanesi P, Randi E (2014) Noninvasive sampling and genetic variability, pack structure, and dynamics in an expanding wolf population. J Mammal 95:41–59. https://doi.org/10.1644/13-MAMM-A-039
Carrasco TS, de Lima RC, Botta S, Machado R, Simões-Lopes PC, Ott PH, Secchi ER (2020) Temporal and individual variation in the diet of the neotropical otter, Lontra longicaudis (Olfers, 1818) (Carnivora, Mustelidae), as revealed by stable isotope analysis of vibrissae. Mamm Biol 100:505–520. https://doi.org/10.1007/s42991-020-00060-8
Chakraborty S, Boominathan D, Desai AA, Vidya TNC (2014) Using genetic analysis to estimate population size, sex ratio, and social organization in an Asian elephant population in conflict with humans in Alur, southern India. Conserv Genet 15:897–907. https://doi.org/10.1007/s10592-014-0587-y
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chiyo PI, Moss CJ, Archie EA, Hollister-Smith JA, Alberts SC (2011) Using molecular and observational techniques to estimate the number and raiding patterns of crop-raiding elephants. J Appl Ecol 48:788–796. https://doi.org/10.1111/j.1365-2664.2011.01967.x
R Core Team (2019) R: a language and environment for statistical computing. Available at: http://www.R-project.org/. Accessed 29 Nov 2019
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009. https://doi.org/10.1046/j.1365-294X.2003.01868.x
De R, Sharma R, Davidar P et al (2021) Pan-India population genetics signifies the importance of habitat connectivity for wild Asian elephant conservation. Glob Ecol Conserv 32:e01888. https://doi.org/10.1016/j.gecco.2021.e01888
De R, Nigam P, Williams AC, Goyal SP (2022) Beyond consensus genotyping: a case study on the Asian elephant Elephas maximus. Conserv Genet Resour. https://doi.org/10.1007/s12686-022-01287-y
de Groot GA, Nowak C, Skrbinšek T et al (2016) Decades of population genetic research reveal the need for harmonization of molecular markers: the grey wolf Canis lupus as a case study. Mamm Rev 46:44–59. https://doi.org/10.1111/mam.12052
de Silva S, Ranjeewa ADG, Weerakoon D, de Silva S, Ranjeewa ADG, Weerakoon D (2011) Demography of Asian elephants (Elephas maximus) at Uda Walawe National Park, Sri Lanka based on identified individuals. Biol Conserv 144:1742–1752. https://doi.org/10.1016/j.biocon.2011.03.011
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc 39:1–38. https://doi.org/10.2307/2984875
Eggert LS, Eggert JA, Woodruff DS (2003) Estimating population sizes for elusive animals: the forest elephants of Kakum National Park, Ghana. Mol Ecol 12:1389–1402. https://doi.org/10.1046/j.1365-294X.2003.01822.x
Ernest HB, Penedo MCT, May BP, Boyce MSyvanen WM (2000) Molecular tracking of mountain lions in the Yosemite Valley region in California: genetic analysis using microsatellites and faecal DNA. Mol Ecol 9:433–441. https://doi.org/10.1046/j.1365-294X.2000.00890.x
Fernando P, Melnick DJ (2001) Molecular sexing eutherian mammals. Mol Ecol Notes 1:350–353. https://doi.org/10.1046/j.1471-8278
Fernando P, Vidya TNC, Rajapakse C, Dangolla A, Melnick DJ (2003a) Reliable noninvasive genotyping: fantasy or reality? J Hered 94:115–123. https://doi.org/10.1093/jhered/esg022
Fernando P, Vidya TNC, Payne J, Stuewe M, Davison G, Alfred RJ, Andau P, Bosi E, Kilbourn A, Melnick DJ (2003b) DNA analysis indicates that Asian elephants are native to Borneo and are therefore a high priority for conservation. PLoS Biol 1:110–115. https://doi.org/10.1371/journal.pbio.0000006
Flagstad Ø, Pradhan NMB, Kvernstuen LG, Wegge P (2012) Conserving small and fragmented populations of large mammals: non-invasive genetic sampling in an isolated population of Asian elephants in Nepal. J Nat Conserv 20:181–190. https://doi.org/10.1016/j.jnc.2012.01.006
Galvis N, Link A, Di Fiore A (2014) A novel use of camera traps to study demography and life history in wild animals: a case study of spider monkeys (Ateles belzebuth). Int J Primatol 35:908–918. https://doi.org/10.1007/s10764-014-9791-3
Garner A, Rachlow JL, Hicks JF (2005) Patterns of genetic diversity and its loss in mammalian populations. Conserv Biol 19:1215–1221. https://doi.org/10.1111/j.1523-1739.2005.00105.x
Gill P, Haned H, Bleka O, Hansson O, Dørum G, Egeland T (2015) Genotyping and interpretation of STR-DNA: Low-template, mixtures and database matches—twenty years of research and development. Forensic Sci Int Genet 18:100–117. https://doi.org/10.1016/j.fsigen.2015.03.014
Goossens B, Sharma R, Othman N, Kun-Rodrigues C, Sakong R, Ancrenaz M, Ambu LN, Jue NK, O’Neill RJ, Bruford MW, Chikhi L (2016) Habitat fragmentation and genetic diversity in natural populations of the Bornean elephant: implications for conservation. Biol Conserv 196:80–92. https://doi.org/10.1016/j.biocon.2016.02.008
Goswami VR, Yadava MK, Vasudev D, Prasad PK, Sharma P, Jathanna D (2019) Towards a reliable assessment of Asian elephant population parameters: the application of photographic spatial capture–recapture sampling in a priority floodplain ecosystem. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-019-44795-y
Gray TNE, Vidya TNC, Potdar S, Bharti DK, Sovanna P (2014) Population size estimation of an Asian elephant population in eastern Cambodia through non-invasive mark-recapture sampling. Conserv Genet 15:803–810. https://doi.org/10.1007/s10592-014-0579-y
Gupta SK, Thangaraj K, Singh L (2006) A simple and inexpensive molecular method for sexing and identification of the forensic samples of elephant origin. J Forensic Sci 51:805–807. https://doi.org/10.1111/j.1556-4029.2006.00154.x
Hedges S, Johnson A, Ahlering MA, Tyson M, Eggert LS (2013) Accuracy, precision, and cost-effectiveness of conventional dung density and fecal DNA based survey methods to estimate Asian elephant (Elephas maximus) population size and structure. Biol Conserv 159:101–108. https://doi.org/10.1016/j.biocon.2012.12.010
Janečka JE, Munkhtsog B, Jackson RM, Naranbaatar G, Mallon DP, Murphy WJ (2011) Comparison of noninvasive genetic and camera-trapping techniques for surveying snow leopards. J Mammal 92:771–783. https://doi.org/10.1644/10-mamm-a-036.1
Johnson AC, Derocher AE (2020) Variation in habitat use of Beaufort Sea polar bears. Polar Biol 43:1247–1260. https://doi.org/10.1007/s00300-020-02705-3
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodöhl PA (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kongrit C, Siripunkaw C, Brockelman WY, Akkarapatumwong V, Wright TF, Eggert LS (2008) Isolation and characterization of dinucleotide microsatellite loci in the Asian elephant (Elephas maximus). Mol Ecol Resour 8:175–177. https://doi.org/10.1111/j.1471-8286.2007.01916.x
Lampa S, Henle K, Klenke R, Hoehn M, Gruber B (2013) How to overcome genotyping errors in non-invasive genetic mark-recapture population size estimation—a review of available methods illustrated by a case study. J Wildl Manage 77:1490–1511. https://doi.org/10.1002/jwmg.604
Lampa S, Mihoub J-B, Gruber B, Klenke R, Henle K (2015) Non-invasive genetic mark-recapture as a means to study population sizes and marking behaviour of the elusive Eurasian otter (Lutra lutra). PLoS ONE 10:e0125684. https://doi.org/10.1371/journal.pone.0125684
Li B, Kimmel M (2013) Factors influencing ascertainment bias of microsatellite allele sizes: impact on estimates of mutation rates. Genetics 195:563–572. https://doi.org/10.1534/genetics.113.154161
Mäck K, Scharbert A, Schulz R, Sahm R (2021) A new approach combining forensic thresholds and a multiple-tubes approach to unravel false microsatellite profiles from cross-contaminated sample material. Conserv Genet Resour 13:89–95. https://doi.org/10.1007/s12686-020-01175-3
Matsumoto T, Yukawa W, Nozaki Y, Nakashige R, Shinya M, Makino S, Yagura M, Ikuta T, Imanishi T, Inoko H, Tamiya G, Gojobori T (2004) Novel algorithm for automated genotyping of microsatellites. Nucleic Acids Res 32:6069–6077. https://doi.org/10.1093/nar/gkh946
Mills LS, Citta JJ, Lair KP, Schwartz MK, Tallmon DA (2000) Estimating animal abundance using noninvasive DNA sampling: promise and pitfalls. Ecol Appl 10:283–294. https://doi.org/10.2307/2641002
Morin DJ, Kelly MJ, Waits LP (2016) Monitoring coyote population dynamics with fecal DNA and spatial capture–recapture. J Wildl Manage 80:824–836. https://doi.org/10.1002/jwmg.21080
Morley RC, van Aarde RJ (2007) Estimating abundance for a savanna elephant population using mark-resight methods: a case study for the Tembe Elephant Park, South Africa. J Zool 271:418–427. https://doi.org/10.1111/j.1469-7998.2006.00225.x
Munshi-South J, Tchignoumba L, Brown J, Abbondanza N, Maldonado JE, Henderson A, Alonso A (2008) Physiological indicators of stress in African forest elephants (Loxodonta africana cyclotis) in relation to petroleum operations in Gabon, Central Africa. Divers Distrib 14:995–1003. https://doi.org/10.1111/j.1472-4642.2008.00509.x
Murphy MA, Waits LP, Kendall KC (2000) Quantitative evaluation of fecal drying methods for brown bear DNA analysis. Wildl Soc Bull 28:951–957
Nyakaana S, Arctander P (1998) Isolation and characterization of microsatellite loci in the African elephant, Loxodonta africana. Mol Ecol 7:1436–1437
Ofstad EG, Herfindal I, Solberg EJ, Heim M, Rolandsen CM, Sæther BE (2019) Use, selection, and home range properties: complex patterns of individual habitat utilization. Ecosphere 10:e02695. https://doi.org/10.1002/ecs2.2695
Padalia H, Ghosh S, Reddy CS, Nandy S, Singh S, Kumar AS (2019) Assessment of historical forest cover loss and fragmentation in Asian elephant ranges in India. Environ Monit Assess 191:1–13. https://doi.org/10.1007/s10661-019-7696-5
Parida J, Sharma R, De R, Kalam T, Sedhupathy A, Digal DK, Reddy PA, Goyal SP, Puyravaud J-P, Davidar P (2022) Genetic characterisation of fragmented Asian elephant populations with one recent extinction in its eastern-central Indian range. Ecol Genet Genomics 24:100132. https://doi.org/10.1016/j.egg.2022.100132
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Poutanen J, Pusenius J, Wikström M, Brommer JE (2019) Estimating population density of the white-tailed deer in Finland using non-invasive genetic sampling and spatial capture–recapture. Ann Zool Fenn 56:1–16. https://doi.org/10.5735/086.056.0101
Rehnus M, Bollmann K (2020) Quantification of sex-related diet composition by free-ranging mountain hares (Lepus timidus). Hystrix 31:80–82. https://doi.org/10.4404/hystrix-00271-2019
Rothstein AP, McLaughlin R, Acevedo-Gutierrez A, Schwarz D, Acevedo-Gutiérrez A, Schwarz D (2016) Wisepair: a computer program for individual matching in genetic tracking studies. Mol Ecol Resour 17:267–277. https://doi.org/10.1111/1755-0998.12590
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Ruiz-González A, Madeira MJ, Randi E, Urra F, Gómez-Moliner BJ (2013) Non-invasive genetic sampling of sympatric marten species (Martes martes and Martes foina): assessing species and individual identification success rates on faecal DNA genotyping. Eur J Wildl Res 59:371–386. https://doi.org/10.1007/s10344-012-0683-6
Sadhu A, Jayam PPC, Qureshi Q, Shekhawat RS, Sharma S, Jhala YV (2017) Demography of a small, isolated tiger (Panthera tigris tigris) population in a semi-arid region of western India. BMC Zool 2:1–13. https://doi.org/10.1186/s40850-017-0025-y
Salin F (2010) Autobin v0.9. Available at: http://www.4.bordeaux-aquitaine.inra.fr/biogeco/Resources/Logiciels/Autobin. Accessed 26 April 2016
Sanderson EW, Redford KH, Vedder A, Coppolillo PB, Ward SE (2002) A conceptual model for conservation planning based on landscape species requirements. Landsc Urban Plan 58:41–56. https://doi.org/10.1016/S0169-2046(01)00231-6
Sawaya MA, Ruth TK, Creel S, Rotella JJ, Stetz JB, Quigley HB, Kalinowski ST (2011) Evaluation of noninvasive genetic sampling methods for cougars in Yellowstone National Park. J Wildl Manag 75:612–622. https://doi.org/10.1002/jwmg.92
Scandura M, Capitani C, Iacolina L, Marco A (2006) An empirical approach for reliable microsatellite genotyping of wolf DNA from multiple noninvasive sources. Conserv Genet 7:813–823. https://doi.org/10.1007/s10592-005-9106-5
Sethi SA, Cook GM, Lemons P, Wenburg J (2014) Guidelines for MSAT and SNP panels that lead to high-quality data for genetic mark-recapture studies. Can J Zool 92:515–526. https://doi.org/10.1139/cjz-2013-0302
Sharma R, Goossens B, Heller R, Rasteiro R, Othman N, Bruford MW, Chikhi L (2018) Genetic analyses favour an ancient and natural origin of elephants on Borneo. Sci Rep 8:1–11. https://doi.org/10.1038/s41598-017-17042-5
Sprogis KR, Christiansen F, Raudino HC, Kobryn HT, Wells RS, Bejder L (2018) Sex-specific differences in the seasonal habitat use of a coastal dolphin population. Biodivers Conserv 27:3637–3656. https://doi.org/10.1007/s10531-018-1618-7
Sukumar R (2006) A brief review of the status, distribution and biology of wild Asian elephants. Int Zoo Yearb 40:1–8. https://doi.org/10.1111/j.1748-1090.2006.00001.x
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194. https://doi.org/10.1093/nar/24.16.3189
Valière N (2002) GIMLET: a computer program for analysing genetic individual identification data. Mol Ecol Notes 2:377–379
Vidya TNC, Sukumar R (2005a) Social organization of the Asian elephant (Elephas maximus) in southern India inferred from microsatellite DNA. J Ethol 23:205–210. https://doi.org/10.1007/s10164-005-0144-8
Vidya TNC, Sukumar R (2005b) Amplification success and feasibility of using microsatellite loci amplified from dung to population genetic studies of the Asian elephant (Elephas maximus). Curr Sci 88:489–492
Vidya TNC, Kumar VR, Arivazhagan C, Sukumar R (2003) Application of molecular sexing to free-ranging Asian elephant (Elephas maximus) populations in southern India. Curr Sci 85:1074–1077
Vidya T, Fernando P, Melnick D, Sukumar R (2005) Population differentiation within and among Asian elephant (Elephas maximus) populations in southern India. Heredity 94:71–80. https://doi.org/10.1038/sj.hdy.6800568
Vidya T, Varma S, Dang NX, Van Thanh T, Sukumar R (2007) Minimum population size, genetic diversity, and social structure of the Asian elephant in Cat Tien National Park and its adjoining areas, Vietnam, based on molecular genetic analyses. Conserv Genet 8:1471–1478. https://doi.org/10.1007/s10592-007-9301-7
Vidya TNC, Sukumar R, Melnick DJ (2009) Range-wide mtDNA phylogeography yields insights into the origins of Asian elephants. Proc R Soc B Biol Sci 276:893–902. https://doi.org/10.1098/rspb.2008.1494
Vijayakrishnan S, Kumar MA, Sinha A (2020) The elephant in the room: methods, challenges and concerns in the monitoring of Asian elephant populations. Gajah 52:39–47
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256. https://doi.org/10.1046/j.1365-294X.2001.01185.x
Wang J (2016) Individual identification from genetic marker data: developments and accuracy comparisons of methods. Mol Ecol Resour 16:163–175. https://doi.org/10.1111/1755-0998.12452
Wang C, Schroeder KB, Rosenberg NA (2012) A maximum-likelihood method to correct for allelic dropout in microsatellite data with no replicate genotypes. Genetics 192:651–669. https://doi.org/10.1534/genetics.112.139519
Wasser SK, Houston CS, Koehler GM, Cadd GG, Fain SR (1997) Techniques for application of faecal DNA methods to field studies of ursids. Mol Ecol 6:1091–1097. https://doi.org/10.1046/j.1365-294X.1997.00281.x
Winiarski KJ, McGarigal K (2016) Effects of photo and genotype-based misidentification error on estimates of survival, detection and state transition using multistate survival models. PLoS ONE 11:e0145640. https://doi.org/10.1371/journal.pone.0145640
Woods JG, Paetkau D, Lewis D, McLellan BN, Proctor M, Strobeck C (1999) Genetic tagging of free-ranging black and brown bears. Wildl Soc Bull 27:616–627
Zhang L, Dong L, Lin L, Feng L, Yan F, Wang L, Guo X, Luo A (2015) Asian elephants in China: estimating population size and evaluating habitat suitability. PLoS ONE 10:e0124834. https://doi.org/10.1371/journal.pone.0124834
Acknowledgements
We sincerely thank the Director, the Dean, Faculty of Wildlife Sciences, the Research Coordinator, and the Nodal Officer, Wildlife Forensic and Conservation Genetics Cell, Wildlife Institute of India, Dehradun, for providing institutional support for the study. We appreciate Mr A Madhanraj, Technical Staff, Wildlife Institute of India, for his logistical support. The permission for the fieldwork was granted by the Uttarakhand Forest Department vide letter nos. 2477/5-6 dated 2nd May 2013 and 3143/5-6 dated 2nd May 2016. This study was a part of a research project funded by the Project Elephant Division, Ministry of Environment, Forests and Climate Change, New Delhi, India, Asian Rhino and Elephant Action Strategy, WWF—Nepal, Kathmandu, Nepal and Operation Eye of the Tiger, Dehradun, India. RD was supported by the University Grants Commission, New Delhi, India, during the tenure of his CSIR-UGC Junior Research Fellowship (National Eligibility Test).
Funding
Funding for this study was granted by the Project Elephant Division, Ministry of Environment, Forests and Climate Change, New Delhi, India, Asian Rhino and Elephant Action Strategy, WWF—Nepal, Kathmandu, Nepal, Operation Eye of the Tiger, Dehradun, India and University Grants Commission, New Delhi, India.
Author information
Authors and Affiliations
Contributions
Concept: RD, RS, SPG; Fieldwork: RD; Sample processing: RD; Experiments: RD; Data analysis: RD; Authored the draft manuscript: RD, RS; Reviews and comments on the draft manuscript: SPG, RS, PN, ACW, BH; Approved the final draft: RD, RS, PN, ACW, BH, SPG.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they do not have any competing interests.
Ethical approval
We obtained permission for the collection of non-invasive faecal samples from Uttarakhand Forest Department vide letter nos. 2477/5-6 dated 2nd May 2013 and 3143/5-6 dated 2nd May 2016.
Consent to participate
Not applicable.
Consent to publication
All authors approved and consented to the submission of the final draft manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
De, R., Sharma, R., Nigam, P. et al. Identifying sex and individual from faecal DNA of the Asian elephant using a single multiplex PCR for population monitoring. Conservation Genet Resour 15, 163–173 (2023). https://doi.org/10.1007/s12686-023-01316-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-023-01316-4