
Abstract
The Asian elephant is a flagship species for conservation in tropical Asia, but reliable population estimates are available only from a few populations. This is because the species can be elusive and occurs at low densities in dense habitat over a large part of its range. Phnom Prich Wildlife Sanctuary in the Eastern Plains, Cambodia, which is part of one of the largest protected area complexes in South-East Asia, is one such habitat that had not been systematically censused for elephants. We, therefore, used fecal-DNA based capture-mark-recapture sampling to estimate the population size for establishing a monitoring baseline. Five sampling sessions targeted all areas in and adjacent to Phnom Prich Wildlife Sanctuary believed to be used by elephants. Fresh dung was collected as the source of DNA and genotyping was carried out based on nine microsatellite loci. The 224 samples collected yielded 78 unique genotypes. Using model averaging of closed population capture-mark-recapture models, the elephant population in Phnom Prich Wildlife Sanctuary was estimated to number 136 ± 18 (SE) individuals. Our results suggest that eastern Cambodia supports a regionally important Asian elephant population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Asian elephants (Elephas maximus) are globally endangered and restricted to isolated populations across their ancestral range, where they are threatened by poaching, illegal capture for trade, habitat loss and fragmentation, and human-elephant conflict (Leimgruber et al. 2003; Sukumar 2003; Choudhury et al. 2008). Despite the species’ perceived status as a conservation flagship, there are few robust population estimates from the species’ range, hindering conservation efforts (Blake and Hedges 2004). This is especially true of South-East Asia, which is at the epicentre of the global conservation crisis. Here, biodiversity loss has been driven by the most rapid global rate of forest conversion and intense pressures on natural resources from high human population densities (see Sodhi et al. 2004; Nijman 2010). Despite chronic threats to the region’s biodiversity, few robust monitoring programmes exist within South-East Asia to assess the response of focal species to conservation interventions or management activities. Effective surveys of Asian elephant in South-East Asia based on direct sightings are hindered by the elusive nature of the species, its low density, and presence in dense, forest habitat that is difficult to survey.
Indirect methods of elephant population estimation may include the use of elephant signs such as dung (for example, Barnes 2002; Varma et al. 2008), acoustics (Payne et al. 2003), and genetic techniques (Eggert et al. 2003, 2008; Vidya et al. 2007). While the second requires specialized equipment to be deployed in the field, the first and third usually do not require this, and staff can be trained to carry out the fieldwork required. However, when elephant density is low, obtaining sufficient dung piles on transects to calculate densities can often be difficult. Genetic methods rely on fresh dung being collected but it might be possible to at least obtain a minimum population size with limited dung samples. When samples can be obtained repeatedly from the same area, capture-mark-recapture estimates of abundance, based on non-invasive sampling of genetic material, can be used for monitoring rare and elusive mammals (Kohn et al. 1999; Creel et al. 2003; Eggert et al. 2003; Bellemain et al. 2005; Prugh et al. 2005). There have been only a few studies using non-invasive sampling of dung, followed by genotyping, to obtain minimum population sizes of Asian elephant populations (Vidya et al. 2007; Ahlering et al. 2011; Chakraborty et al. unpublished data) or using capture-mark-recapture and non-invasive sampling to estimate Asian elephant population size (Pollard et al. 2008; Hedges et al. 2013).
The status of Asian elephants in Cambodia has been unclear, with estimates of 250–600 elephants (Sukumar 2003; Choudhury et al. 2008). While the Cardoman Mountains of southwestern Cambodia are believed to hold substantial elephant populations, the Eastern Plains landscape in eastern Cambodia is also thought to be significant for Asian elephant conservation (Pollard et al. 2008). Phnom Prich Wildlife Sanctuary (PPWS) forms part of the Eastern Plains landscape, one of the largest protected area complexes in South-East Asia, which includes Mondulkiri Protected Forest, Seima Protected Forest, Lumphat Wildlife Sanctuary, and Yok Don National Park (Vietnam). PPWS was recognized as a core area for elephants ranging in the landscape (Timmins and Ou 2001; Desai et al. 2002) and a minimum of 42 elephants was estimated from camera trap photographs (WWF, unpublished data). In the present study, we used capture-mark-recapture in concert with non-invasive sampling of dung to estimate the population size of elephants in PPWS.
Materials and methods
Study area
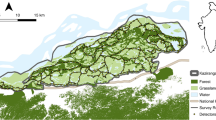
PPWS is located in the west of Mondulkiri Province, eastern Cambodia (centered on 12.8° N, 106.5° E), and covers 2,225 km2 (Fig. 1). It is part of a relatively undisturbed larger landscape as mentioned above. The climate is highly seasonal, with a cooler wet-season (July–November) and a hot dry-season, in which precipitation is extremely rare (December–June). PPWS is dominated by deciduous dipterocarp and mixed-deciduous forest, with smaller areas of semi-evergreen forest at higher elevations and along watercourses, and is home to many endangered species of mammals and birds.

Study area in eastern Cambodia indicating survey hot-spots and survey blocks used for dung sampling. Inset mainland South-East Asia with the location of the study area
Sampling design
Sampling design followed the principles of closed capture-mark-recapture studies with five sampling sessions, during which elephant fecal samples were collected. Sampling was conducted during the late dry-season, February–May, 2009. Each sampling session lasted 8–10 (mean 8.2) days, with 10–13 (mean 12) days between successive sampling sessions. Sampling was designed to ensure approximately equal survey coverage across all key dry-season elephant locations within the PPWS core area during each sampling session. To maximise the chances of demographic and geographic closure across the study period, the study was conducted during the dry-season when elephant movement was restricted due to limited water availability. The overall survey period (<90 days) was also kept to a minimum in order to satisfy requirements of demographic closure.
We obtained information about past elephant occurrence within PPWS from camera-trapping and enforcement ranger patrol data. Using this information, 11 ‘hot-spots’ of approximately 10–15 km2 each, where high concentrations of elephant signs had been recorded, and 3 ‘survey blocks’ of 30–60 km2 each, where elephants were known or believed to occur, were identified (Fig. 1). One of the three survey blocks was located largely within the boundaries of the adjacent Mondulkiri Protected Forest (Fig. 1); however this area surveyed is ecologically contiguous with PPWS and was surveyed by the same field teams as all other hot-spots and survey blocks. During each sampling session, each ‘hot-spot’ and ‘survey block’ was visited and searched for signs of elephant. Dung was collected by three teams comprising two Ministry of the Environment rangers, trained by the authors to collect samples, a local guide with knowledge of the protected area, and one of TNE Gray and P Sovanna in two of the teams. Each team was allocated between 3 and 4 ‘hot-spots’ and one ‘survey block’. Each ‘hot-spot’ and ‘survey block’ was accessed by motorbike or on foot and searched for 2 days (approx. 08h00–17h00) by one team.
Searches within ‘hot-spots’ and ‘survey blocks’ focussed around deep water pools in otherwise dry river channels, natural springs, salt-licks, animal tracks moving through the semi-evergreen forest, and the ecotone between semi-evergreen and deciduous dipterocarp forest. Upon encountering elephant signs, survey teams collected samples from all ‘fresh’ and ‘reasonably-fresh’ dung piles following the protocols of Hedges and Lawson (2006). When multiple dung piles were associated with a group of elephants, efforts were made to collect samples from all the different individuals present, based on bolus size and distribution of dung piles. Approximately 10 g of dung was placed in each case into sterile tubes filled with ~20 ml of Queen’s College Buffer (20 % DMSO, 100 mM Tris pH 7.5, 0.25 M EDTA, saturated with NaCl; Amos et al. 1992).
Genetic analysis
DNA was extracted from dung samples following Fernando et al. (2003). The purified DNA was eluted into 30–40 μl of elution buffer and stored at −20 °C. PCR amplification of nine microsatellite loci was carried out: EMU03, EMU04, EMU07, EMU10, EMU12, EMU14, EMU15, EMU17 (all from Kongrit et al. 2008), and FH94 (Comstock et al. 2000). PCRs were carried out in 12.5 μl volumes, using 0.25 μl of each 10 μM primer, 0.1 μl of 5 U/μl Taq DNA polymerase (Fermentas Life Sciences, Thermo Fisher Scientific Inc.), 2–2.9 μl DNA, and PCR mix with dNTPs, BSA, and buffer. The PCR consisted of an initial denaturation step at 95 °C for 3 min, followed by 40–45 cycles of denaturation at 93 °C for 1 min, annealing for 1 min (“touchdown PCRs” for some loci), and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 15 min. PCR products were electrophoresed, along with the internal size standard GS500LIZ, in an ABI PRISM 3730 DNA Analyzer (Applied Biosystems) in the JNCASR Sequencing Facility, and genotypes were scored using the GeneMapper software, version 4.0 (Applied Biosystems).
Every set of PCR reactions included a negative control, in which the DNA extract was replaced by water, and several sets of PCRs also included positive controls, which were DNA extracts from fresh dung samples (obtained upon observed defecation) from southern India, which we had previously amplified successfully. If two PCRs produced the same heterozygote with clear peaks in the electropherograms, no further PCRs were carried out for that sample at that locus. If both the PCRs produced the same homozygote but showed peaks of intensity <5,000 in the electropherograms (which was usually the case), a third repeat was run. If the first two PCRs did not work, or amplified with small peaks, or showed different results, they were re-run up to an additional six times. Care was taken to prevent contamination by using dedicated areas and instruments for pre- and post- amplification work and using aerosol-resistant barrier tips for pipettes. We used Pedant v 1.0 (Johnson and Haydon 2007) to calculate allelic dropout rates based on only the first two amplifications of a sample at a locus. Therefore, the subsequent amplifications would take care of most allelic dropout problems. We checked for the presence of null alleles using FreeNA (Chapuis and Estoup 2007).
We examined microsatellite genotypes using the Excel Microsatellite Toolkit (Park 2001) to check for duplications in this sample set, with the criterion that up to two alleles need not match, since it was possible to have allelic dropout. Pairs of samples that had two allele mismatches were then examined to determine whether the mismatches were of the heterozygote-homozygote type at two loci (for example, Sample A: 139/139, 217/219; Sample B: 139/141, 217/217) or heterozygote–heterozygote type, with no shared allele, at one locus (for example, Sample A: 217/219; Sample C: 215/221). Both the above combinations have two mismatches, but the probability of scoring heterozygotes as homozygotes at two loci because of allelic dropout is likely to be much higher than the probability of wrongly scoring one heterozygote as another with no alleles shared between them, repeatedly. Therefore, when we found two allele mismatches that corresponded to different heterozygotes (with no alleles shared), confirmed by repeated PCRs, the samples were treated as different individuals. In a previous study on African elephants using dung extracted DNA, differences in one or two loci were used as the cut-off to identify matching individuals (Eggert et al. 2008). Samples that differed at four or fewer alleles were re-examined, and those with up to two different alleles and the same sex were considered the same individual in an Asian elephant population (Ahlering et al. 2011). Our procedure was similar to those in the studies above, but included additional checks of heterozygote–homozygote versus heterozygote–heterozygote mismatches. The dataset of unique genotypes was then used to examine if the loci conformed to Hardy–Weinberg and linkage equilibria. These tests were done using Genepop v.3.1 (Raymond and Rousset 1995), with Markov chain parameters of 1,000 dememorization steps, 100 batches, and 1,000 iterations per batch. Type I errors were adjusted for by applying the Bonferroni test (Rice 1989). The expected Probability of Identity (PID) for each locus was calculated using Identity-4 (Wagner and Sefc 1999). The observed PID was calculated as the proportion of all possible pairs of identical genotypes based on an increasing number of loci. This was done for the locus with the highest heterozygosity first, and loci were added in decreasing order of heterozygosity and observed PIDs recalculated (see Waits et al. 2001).
Capture-mark-recapture analysis
Based on the observed genotypes, we developed capture histories in a standard ‘X-matrix’ format with rows representing the capture histories of each unique genotype (n = 78) and columns representing captures in each sampling session (n = 5). We formally tested for population closure during the sampling period using the Stanley and Burnham (1999) test in software CloseTest and open Pradel models in software MARK (White and Burnham 1999). For the Pradel models, we compared Akaike Information Criteria (AICc) scores, corrected for small sample size, between a model in which survival (θ) and recruitment (f) were constrained to 1 and 0, respectively (representing population closure), and an open model in which these parameters were estimated (Boulanger and McLellan 2001; Harihar et al. 2009).
The results of the closure tests suggested that abundance estimation within a closed capture-mark-recapture framework was reasonable (see Results). We, therefore, estimated the PPWS elephant population size using full closed captures with heterogeneity modeling, implementing Pledger mixture models with two mixtures of capture and recapture probabilities (White 2008). Models M-0, M-t, M-h and M-th (sensu White 2008) were run and model averaging (Burnham and Anderson 2002), based on AICc weights, used to estimate population size. These analyses were carried out using software MARK. Model M-b, the behaviour model in which capture and recapture probabilities differ, was not modeled as it was biologically unlikely given the non-invasive fecal sampling employed in the study.
Results
A total of 224 elephant fecal-DNA samples were collected, with 34–55 (mean 45) samples collected per sampling session (Table 1). The number of alleles per locus varied between 5 and 7, with observed heterozygosities not very different across loci, with the exception of EMU17, which had an unusually high heterozygosity of 0.929 (Table 2). Based on the nine loci used, the total exclusion probability was 0.995. The expected total PID across loci was 4.09 × 10−8 and the total PID(sib) was 1.47 × 10−3, which are sufficiently low to prevent different individuals from being wrongly identified as the same individual. The observed PID was much higher than the expected total PID as is usually the case (see Waits et al. 2001), but was sufficiently low to discriminate between individuals accurately (observed PID = 0.003 using six loci and 0.002 using eight loci) since the expected population size was not greater than a few hundred individuals at most (Supplementary Figure 1). Allelic dropout was found to be 13.2 % on average, based on only the first two amplifications of a sample. Three loci were not found to be in Hardy–Weinberg equilibrium after Bonferroni corrections: EMU10, FH94, and EMU14. EMU14 and FH94 did not show any departure from Hardy–Weinberg equilibrium in southern Indian elephant populations (Chakraborty et al. unpublished data; Nandini Shetty, Keerthipriya P, TNC Vidya, unpublished data) or in a population in Lao (Ahlering et al. 2011), but EMU10 showed departure from Hardy–Weinberg equilibrium in the Lao population (Ahlering et al. 2011). The same three loci were identified as having null alleles by FreeNA software (frequency of null alleles: EMU10: 0.11, FH94: 0.15, EMU14: 0.12). Null alleles are not expected to be a concern in elephants because mutation rates are very low, even in the relatively fast mutating mitochondrial DNA d-loop (see Vidya et al. 2009). Chapuis and Estoup (2007) showed that the prevalence of null alleles would be low for populations with Neμ <0.001 (Ne: effective population size, μ: mutation rate across the segment considered per generation) and that population processes would be more likely to result in signatures of null alleles. Based on mitochondrial DNA d-loop mutation rate in Asian elephants (Vidya et al. 2009), which would be orders of magnitude faster mutating than nuclear DNA sequence, μ = 4 × 10−6. Even if the census size (rather than Ne) in the larger landscape were considered (~300 elephants), Neμ ~0.001. If the true mutation rate and Ne were taken into account, Neμ would be two or three orders of magnitude smaller. Further, primers of microsatellites isolated from African elephants have been previously amplified successfully across different Asian elephant populations (Vidya et al. 2005, 2007; Chakraborty et al. unpublished data; Nandini Shetty, Keerthipriya P, TNC Vidya, unpublished data); hence it is unlikely that mutations in primer binding sites have occurred at three of the loci in only this Cambodian elephant population. It is much more plausible that historical population declines, fragmentation and the resulting non-random mating gave rise to the departure from Hardy–Weinberg equilibrium and signatures of null alleles observed. Since the removal of these highly polymorphic loci could have reduced discriminatory power and wrongly classified different individuals as the same individuals, these loci were initially included for assignment of identity. However, the analyses were also repeated after excluding the loci.
The 142 samples that could be genotyped successfully corresponded to 78 unique individuals (Table 1). Based on bolus size, 11 (14 %) of these were neonates/juveniles, 36 (46 %) were sub-adults, and 31 (40 %) were adults. Forty-seven of the unique genotypes were sampled on single occasions, 14 were sampled twice, six genotypes on three occasions, six genotypes on four occasions, two genotypes on six occasions and single individuals on five and eight occasions. Excluding within-session recaptures, a total of 55 individual elephants were caught during a single sampling session, 18 individuals during two sessions and 5 individuals during 3 sessions. CloseTest supported the assumption of population closure across the five sampling sessions (χ2 = 7.1; df = 6; P = 0.31). In MARK, the open Pradel model estimated survivorship (θ) at (mean ± 1.96 SE) 0.79 ± 0.196 and recruitment (f) at (mean ± 1.96 SE) 0.15 ± 0.059. However the constrained Pradel model, in which θ was set at 1.0 and f at 0.0 (the closed model), was better supported than the open model based on AICc scores (Δ AICc 0.84). Therefore the population size was estimated using full closed captures with heterogeneity models in program MARK. Model M-0 was the best supported model based on AICc scores, and this estimated the PPWS population at (mean ± 1.96 SE) 136 ± 35 individuals (Table 3). Additional models were ranked M-h > M-t > M-ht with mean population estimates of between 135 and 145 individuals (Table 3). The model averaged Asian elephant population estimate for PPWS was 136 ± 35 individuals. When the analyses were carried out excluding the loci EMU10, EMU14, and FH94, the model averaged population estimate was 103 ± 13 individuals.
Discussion
The most recent range-wide estimate, based on best guesses, of the Asian elephant population in South-East Asia suggested 250-600 individuals in Cambodia, 500–1,000 in Laos, 70–150 in Vietnam, and 2,500–3,200 in Thailand (Sukumar 2003). Our finding that PPWS harbours 136 ± 35 elephants and that the population has moderate-high heterozygosity at most of the loci examined indicates that PPWS is significant, nationally and at the landscape level, for Asian elephant conservation. PPWS forms part of the trans-boundary Eastern Plains landscape protected area complex, which also includes Mondulkiri and Seima Protected Forests, Lumphat Wildlife Sanctuary, and Yok Don National Park. Seima Protected Forest is estimated to have a population of 101–139 elephants based on a fecal DNA study (Pollard et al. 2008). We simultaneously recorded a minimum of 21 unique individuals in Mondulkiri Protected Forest based on genetic analysis (TNE Gray, TNC Vidya et al. unpublished data) and elephants are also known to occur in Yok Don National Park. Therefore, this landscape possibly harbours at least 300 elephants. Away from the Eastern Plains landscape, large Asian elephant populations in Cambodia are restricted to the Cardoman Mountains of south-western Cambodia, where the population has been estimated at a minimum of 175 individuals (M Maltby, Fauna and Flora International personal communication). In Laos, large elephant populations are restricted to the Nakai plateau, with approximately 140 individuals (Hedges et al. 2007, 2013), and Nam Pouy National Protected Area, estimated at <30 individuals (K Khounboline, WWF, personal communication). We, therefore, suggest that the Eastern Plains landscape Asian elephant meta-population is of high regional significance and that the landscape, which remains extensively forested and supports a mosaic of forest types, should be a priority for Asian elephant conservation in South-East Asia.
One individual elephant from PPWS, genotyped during this study, was subsequently sampled ~60-km to the north–east, in Mondulkiri Protected Forest during the period of our fifth sampling session. This demonstrates the large home-ranges of Asian elephant within the Eastern Plains Landscape and illustrates that elephant conservation requires collaboration between protected areas. We recommend radio or GPS telemetry studies to further understand Asian elephant movements across the landscape, including into Yok Don National Park, Vietnam, and thus facilitate effective landscape-wide Asian elephant conservation.
Although close test suggested population closure across the five sampling sessions, open Pradel models indicated some evidence of elephant movement in and out of the study area. This was confirmed with the re-capture of the individual mentioned above in Mondulkiri Protected Forest, outside our study area. Thus abundance estimates from closed population capture-mark-recapture models must be viewed with some caution and it is possible that the population estimate refers to the overall ‘super-population’ using the study area (sensu Boulanger and McLellan 2001) rather than an Asian elephant population restricted to the Phnom Prich Wildlife Sanctuary core, i.e. our study area. However when the assumption of geographic closure is violated, it is difficult to robustly estimate abundance, the key parameter of interest in conservation monitoring of rare and globally threatened species, using current capture-mark-recapture techniques. POPAN models (Schwarz and Arnason 1996) have been used for estimating population size in Asian elephant in India (Goswami et al. 2007) when populations are not closed. However this approach is constrained in that it only allows animals to enter or leave a population on one occasion (i.e. deaths/births and permanent emigration or immigration). Therefore, whilst a useful approach for estimating abundance in long-term studies where violations of the assumption of demographic closure are likely, POPAN models are likely to be less robust when populations are geographically open during the survey period.
Based on our results and those of Hedges et al. (2013), we recommend the use of non-invasive genetic sampling for monitoring Asian elephant populations in south-east Asia where elephant populations are relatively small and occur in low density in dense evergreen forest. The current baseline will be used for future monitoring of the population; we plan repeat surveys in the dry-season of 2013–14. Hedges et al. (2013) compared the costs and precision of population estimates based on fecal-DNA capture-mark-recapture with those obtained from dung density surveys. They concluded that DNA based methods provided more precise abundance estimates and were cheaper than dung density based estimates, provided that the cost of genetic analysis was less than the cost of dung decay rate monitoring. The cost of genetic analysis depends in part on the quality of samples collected, which determines the number of times amplifications have to be repeated.
The overall amplification success from our samples (63 %) was lower than that from previous work on Asian elephants, for example, 98 % amplification by Fernando et al. (2003) and Chakraborty et al. (unpublished data), and up to 97 % in Vidya et al. (2005), all of which used exactly the same laboratory protocols as in our study, and 81 % amplification from Pollard et al. (2008) and Hedges et al. (2007). The low amplification rates were a result of poor sample quality, as the electropherograms had lower intensities than those of positive controls, which were dung-extracted DNA samples collected within a few hours of observed defecation from elephants in southern India (Nandini Shetty, Keerthipriya P, TNC Vidya, unpublished data). Excluding loci that could possibly have null alleles led to similar population estimates as those including these loci, but the upper interval was higher when the loci were included. We obtained allelic dropout rates of about 13 %, which was much higher than that seen by Fernando et al. (2003), but similar to those seen in several other studies (see Broquet and Petit 2004). Since we usually carried out at least three PCRs per sample and genotyped nine loci, the allelic dropout rate per sample would be 0.021 on average (0.1323 × 9). We would, therefore, expect about five single locus errors in the 224 samples analysed. We, however, carried out multiple repeat amplifications for samples depending on the intensity of their amplification, which would have taken additional care of such error. We also allowed for mismatches between genotypes (as explained in the Methods) while obtaining unique individuals so that erroneous genotypes would not be recorded as new captures in the analysis, which would positively bias the population estimate (see Creel et al. 2003). Therefore, our approach is likely to have been conservative in estimating population size, given the allelic dropout rate. There was a large variation across sampling sessions (Table 1) and collection teams (97–50 %). Since low amplification rates, and hence low numbers of samples successfully genotyped, may lead to reduced precision in population estimates due to decreased individual capture and recapture probabilities (Gray and Prum 2011), future studies should aim to maximize successful genotyping rates through better field training for sample collection.
References
Ahlering MA, Hedges S, Johnson A, Tyson M, Schuttler SG et al (2011) Genetic diversity, social structure, and conservation value of the elephants of the Nakai Plateau, Lao PDR, based on non-invasive sampling. Conserv Gen 12:413–422
Amos W, Whitehead H, Ferrari MJ, Payne R, Gordon J (1992) Restrictable DNA from sloughed cetacean skin: its potential for use in population analyses. Mar Mamm Sci 8:275–283
Barnes RFW (2002) The problem of precision and trend detection posed by small elephant populations in West Africa. Afr J Ecol 40:179–185
Bellemain E, Swenson JE, Tallmon DA, Brunberg S, Taberlet P (2005) Estimating population size of elusive animals with DNA from hunter-collected feces: comparing four methods for brown bears. Conserv Biol 19:150–161
Blake S, Hedges S (2004) Sinking the flagship: the case of forest elephants in Asia and Africa. Conserv Biol 18:1191–1202
Boulanger J, McLellan B (2001) Closure violation in DNA-based mark–recapture estimation of grizzly bear populations. Can J Zool 79:642–651
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Burnham KP, Anderson DR (2002) Model selection and multi-model inference. Springer, New York
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Choudhury A, Choudhury LDK, Desai A, Duckworth JW, Easa PS et al (2008) Elephas maximus. In: IUCN 2011. IUCN red list of threatened species. Version 2011.1. www.iucnredlist.org
Comstock KE, Wasser SK, Ostrander EA (2000) Polymorphic microsatellite DNA loci identified in the African elephant (Loxodonta africana). Mol Ecol 9:1004–1006
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009
Desai AA, Chak S, Ou R, Lic V, Maxwell A (2002) Initial surveys to determine the distribution of wild Asian elephant populations in selected areas of Northeastern Cambodia, 2000–2001. WWF Cambodia, WWF Indochina Programme Office, Phnom Penh
Eggert LS, Eggert JA, Woodruff DS (2003) Estimating population sizes for elusive animals: the forest elephants of Kakum National Park, Ghana. Mol Ecol 12:1389–1402
Eggert LS, Patterson G, Maldonado JE (2008) The Knysna elephants: a population study conducted using faecal DNA. Afr J Ecol 46:19–23
Fernando P, Vidya TNC, Rajapakse C, Dangolla A, Melnick DJ (2003) Reliable non-invasive genotyping: fantasy or reality? J Hered 94:115–123
Goswami VR, Madhusudan MD, Karanth KU (2007) Application of photographic capture–recapture modelling to estimate demographic parameters for male Asian elephants. Anim Conserv 10:391–399
Gray TNE, Prum S (2011) Leopard density in a post-conflict landscape Cambodia: evidence from spatially explicit capture-mark-recapture. J Wildl Manag 76:163–169
Harihar A, Pandav B, Goyal SP (2009) Density of leopards (Panthera pardus) in the Chilla Range of Rajaji National Park, Uttarakhand, India. Mammalia 74:83–87
Hedges S, Lawson D (2006) Dung survey standards for the MIKE programme. CITES MIKE Programme, Central Coordinating Unit, Nairobi
Hedges S, Johnson A, Tyson M, Eggert L (2007) Determination of the size and distribution of populations of elephants, assessment of habitat and resource use, and assessment and management of human–elephant conflicts on the Nakai Plateau and surrounding areas. Wildlife Conservation Society Lao Program, Vientiane
Hedges S, Johnson A, Ahlering M, Tyson M, Eggert LS (2013) Accuracy, precision, and cost-effectiveness of conventional dung density and fecal DNA based survey methods to estimate Asian elephant (Elephas maximus) population size and structure. Biol Conserv 159:101–108
Johnson PCD, Haydon DT (2007) Maximum likelihood estimation of allelic dropout and false allele error rates from microsatellite genotypes in the absence of reference data. Genetics 175:827–842
Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM et al (1999) Estimating population size by genotyping faeces. Proc R Soc B 266:657–663
Kongrit C, Siripunkaw C, Brockelman WY, Akkarapatumwong V, Wright TF et al (2008) Isolation and characterization of dinucleotide microsatellite loci in the Asian elephant (Elephas maximus). Mol Ecol Res 8:175–177
Leimgruber P, Gagnon JB, Wemmer C, Kelly DS, Songer MA et al (2003) Fragmentation of Asia’s remaining wildlands: implications for Asian elephant conservation. Anim Conserv 4:347–359
Nijman V (2010) An overview of international wildlife trade from southeast Asia. Biodiv Conserv 19:1101–1114
Park SDE (2001) Trypanotolerance in West African cattle and the population genetic effects of selection. Ph.D. thesis, University of Dublin
Payne KB, Thompson M, Kramer L (2003) Elephant calling patterns as indicators of group size and composition: the basis for an acoustic monitoring system. Afr J Ecol 41:99–107
Pollard E, Eggert L, Cheur C, Hedges S (2008) The status and conservation of Asian elephant in the seima biodiversity conservation area. Wildlife Conservation Society Cambodia Program, Phnom Penh, Cambodia
Prugh LR, Ritland CE, Arthur SM, Krebs CJ (2005) Monitoring coyote population dynamics by genotyping faeces. Mol Ecol 14:1585–1596
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice W (1989) Analysing tables of statistical tests. Evolution 43:223–225
Schwarz CJ, Arnason AN (1996) A general methodology for the analysis of capture–recapture experiments in open populations. Biometrics 52:860–873
Sodhi NS, Koh LP, Brook BW, Ng PK (2004) Southeast Asian biodiversity: an impending disaster. Trends Ecol Evol 19:654–660
Stanley TR, Burnham KP (1999) A closure test for time-specific capture–recapture data. Environ Ecol Stat 6:197–220
Sukumar R (2003) The living elephants: evolutionary ecology, behaviour, and conservation. Oxford University Press, New York
Timmins RJ, Ou R (2001) The importance of Phnom Prich Wildlife Sanctuary and Adjacent areas for the conservation of tigers and other key species: a summary. Field survey report. WWF Cambodia Conservation Program, Phnom Penh
Varma S, Dang Nguyen Xuan, Van Thanh Tran, Sukumar R (2008) The elephants Elephas maximus of Cat Tien National Park, Vietnam: status and conservation of a vanishing population. Oryx 42:92–99
Vidya TNC, Fernando P, Melnick DJ, Sukumar R (2005) Population differentiation within and among Asian elephant (Elephas maximus) populations in southern India. Hered 94:71–80
Vidya TNC, Varma S, Dang NX, Van Thanh T, Sukumar R (2007) Minimum population size, genetic diversity, and social structure of the Asian elephant in Cat Tien National Park and its adjoining areas, Vietnam, based on molecular genetic analyses. Conserv Genet 8:1471–1478
Vidya TNC, Sukumar R, Melnick DJ (2009) Range-wide mtDNA phylogeography yields insights into the origins of Asian elephants. Proc R Soc B 276:893–902
Wagner HW, Sefc KM (1999) Identity 1.0. Centre for Applied Genetics, University of Agricultural Sciences, Vienna
Waits JL, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256
White GC (2008) Closed population estimation models and their extensions in program mark. Environ Ecol Stat 15:89–99
White GC, Burnham KP (1999) Program mark: survival estimation from populations of marked animals. Bird Study 46S:120–138
Acknowledgments
This study forms part of WWF Greater Mekong Cambodia Country Program’s Eastern Plains Landscape Project. Work in PPWS was carried out with permission of the Department for Wildlife and Nature Conservation of the Cambodian Ministry of the Environment, and support from His Excellency Chay Samith. Permits for exporting elephant fecal-DNA samples from Cambodia were obtained from the Ministry of Agriculture, Forestry and Fisheries of the Royal Cambodian Government. Permits for importing samples were obtained from the National Biodiversity Authority, Ministry of Environment and Forests, Government of India, and Additional Principal Chief Conservator of Forests (Wildlife), Karnataka State Forest Department. Sary Tre, Dong Kit, Sem Pros, Yet Sreng, Ngoeum Nareoun, Gkill, Kham Mephoeung, and Mel Trock assisted with data collection, whilst Christy Williams, Andrew Maxwell, Phan Channa, Craig Bruce, Barney Long, and Seng Teak provided support. Anitha CA, Nandini Shetty, and Kanika Mendiratta assisted with some laboratory analysis. We thank two anonymous reviewers for comments that helped improve the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Thomas N. E. Gray and T. N. C. Vidya have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gray, T.N.E., Vidya, T.N.C., Potdar, S. et al. Population size estimation of an Asian elephant population in eastern Cambodia through non-invasive mark-recapture sampling. Conserv Genet 15, 803–810 (2014). https://doi.org/10.1007/s10592-014-0579-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-014-0579-y