
Abstract
Alzheimer’s disease (AD) is an immutable neurodegenerative disease featured by the two hallmark brain pathologies that are the extracellular amyloid ß (Aß) and intraneuronal tau protein. People carrying the APOE4 allele are at high risk of AD concerning the ones carrying the ε3 allele, while the ε2 allele abates risk. ApoE isoforms exert a central role in controlling the transport of brain lipid, neuronal signaling, mitochondrial function, glucose metabolism, and neuroinflammation. Regardless of widespread indispensable studies, the appropriate function of APOE in AD etiology stays ambiguous. Existing proof recommends that the disparate outcomes of ApoE isoforms on Aβ accretion and clearance have a distinct function in AD pathogenesis. ApoE–lipoproteins combine diverse cell-surface receptors to transport lipids and moreover to lipophilic Aβ peptide, that is believed to begin deadly events that generate neurodegeneration in the AD. ApoE has great influence in tau pathogenesis, tau-mediated neurodegeneration, and neuroinflammation, as well as α-synucleinopathy, lipid metabolism, and synaptic plasticity despite the presence of Aβ pathology. ApoE4 shows the deleterious effect for AD while the lack of ApoE4 is defensive. Therapeutic strategies primarily depend on APOE suggest to lessen the noxious effects of ApoE4 and reestablish the protective aptitudes of ApoE. This appraisal represents the critical interactions of APOE and AD pathology, existing facts on ApoE levels in the central nervous system (CNS), and the credible active stratagems for AD therapy by aiming ApoE. This review also highlighted utmost ApoE targeting therapeutic tactics that are crucial for controlling Alzheimer’s pathogenesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the utmost common genre of dementia that often involves loss of memory and decline in other cognitive skills, which are required to maintain daily activities [1, 2]. Nearly 13% of people 65 or older and 45% of people older than 85 are projected to have Alzheimer’s that causes about 60–80% of dementia cases [1, 3]. Struggling with remembering recent events or simply short-term memory deficits is the most usual symptom of patients with AD. In later stages, symptoms including disorientation; language impairment; severe memory loss; mood and behavior changes; suspicions about family, friends; and difficulty speaking, swallowing, and walking are reported. As a patient’s condition deteriorates, they often isolate themselves from society and even from family. Progressively, when major physical functions are lost, a patient may even die. Though the promptness of progress of AD can differ, the ordinary life expectancy of the patients after diagnosis is usually 3 to 9 years [4].
Increasing evidence from various studies including pathological, genetic, and functional studies have demonstrated that accretion of Aβ can take place due to the disproportion, the production, and clearance of the Aβ peptides in the brain. Toxic Aβ aggregates can be found in the form of intraneuronal Aβ, Aβ oligomers (i.e., soluble), and amyloid plaques can eventually cause neurodegeneration and dementia by injuring the synapses [3, 5]. Conversely, the incidence of microtubule-linked protein tau can lead to Aβ toxicity [6], and neurofibrillary tangles (NFTs) are aggregates of hyperphosphorylated tau protein. Aβ is usually comprised of 40 to 42 amino acids and form via proteolytic degradation of the amyloid precursor protein (APP) [7].
The actual cause of AD is not well-known and around 70% of the risk is thought to be genetic [8]. The genetic heritability of AD is based on evaluations of twin, as well as family studies ranging from 49 to 79% [9]. About 0.1% of the cases are found in the familial sorts of autosomal-prevailing inheritance, which have an onset earlier of age 65, and this disease form is named as early-onset familial AD [10]. In most of the cases, the autosomal-prevailing AD can be ascribed due to the mutations in one of three genes including those encoding APP and presenilins (PSEN1 and PSEN2) [11]. Increased production of Aβ42, a small protein and main component of senile plaques, is seen with PSEN genes and most of the mutations in the APP [12]. Some of these mutations simply change the relation amid Aβ42 and Aβ40 without raising the levels of Aβ42 [13]. In most of the cases, AD does not show autosomal-prevailing inheritance and is characterized as the sporadic AD, wherein genetic and environmental differences may play roles as risk factors. The ε4 allele of the apolipoprotein E (APOE4) is considered as the most common inherited genetic risk aspect [14]. It has been found that 40 to 80% people with AD have at least one APOE4 [15], and it increases the risk of AD in heterozygotes and homozygotes by 3 and 15 times, respectively [16].
Although there is no remedy for AD, research is ongoing and symptomatic treatments are available [17]. While existing Alzheimer’s treatments cannot halt the progression, they can slow down the deterioration of the symptoms of dementia to some extent and improve AD patients’ quality of life. Currently, numerous efforts are ongoing to discover better treatment strategies based on ApoE4 to delay Alzheimer’s onset, treat the disease, and to stop its progression. Therefore, the objective of this appraisal is to explore the impact of APOE on AD pathology and promising ApoE4 target therapeutic strategy for abating the neurodegeneration for the management of Alzheimer’s pathogenesis.
APOE4 as a Strong Genetic Risk Factor for Alzheimer’s Pathogenesis
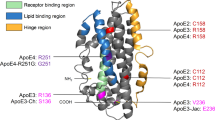
ApoE is a 299 amino acid protein and primary lipid transporter abundantly found in the brain. It is synthesized mainly by astrocytes within the blood-brain barrier (BBB) [18]. There are three major ApoE polymorphic alleles in humans, APOE2, APOE3, and APOE4. They encode three protein isoforms such as ApoE2, ApoE3, and ApoE4 that vary by merely two amino acids cysteine/arginine polymorphisms at positions 112 or 158 in the N-terminal domain. Furthermore, early-onset familial AD characteristically builds up before 65 years and is responsible for a small portion (< 1%) of AD cases. This form of AD is mainly generated by overproduction of Aβ on account of the mutations in either the APP gene or genes encoding presenilin 1 (PSEN1) or presenilin 2 (PSEN2). These genes are fundamental constituents of the γ-secretase complexes accountable for cleavage and release of Aβ [10, 19, 20]. On the other hand, late-onset AD (LOAD) is the most common form of AD that usually occurs at the later stage in life (> 65 years). APOE4 is the major risk factor for the pathogenesis of LOAD commonly present in 15% of people (Fig. 1) [21, 22].

The pathogenic effects of ApoE in Alzheimer’s disease. Proof recommends that ApoE increases the risk of Alzheimer’s by exerting its pathogenic effect on the production, aggregation, and clearance of Aβ that leads to Aβ deposition as well as other effects including tau hyperphosphorylation, α-synucleinopathy, neuroinflammation, lipid metabolism, and synaptic function also augment the disease propagation
In a study, Farrer et al. [23] based on the clinic- or autopsy-linked studies in Caucasian subjects reported that a copy or two copies of APOE4 was causative for increasing the risk of AD concerning people with the APOE3/APOE3 genotype. This study also stated a weak link amid APOE4 and AD for Hispanics, as well as African American people, but a stronger correlation was reported for Japanese. The researchers reported the proof of the APOE4 effect amid 40- to 90-year ages but abate afterward 70 years and that the risk of AD linked to a particular genotype differs concerning sex. Furthermore, Sepehrnia et al. [24] stated that Nigerian blacks have the highest frequency of APOE4 in world populations, but their adjusted mean cholesterol level is among the lowest reported in studies of cholesterol-APOE linkage. In another study, Hendrie et al. [25] reported AD is pretty rare amid Africans living in Africa than in African Americans. Mounting evidence from various studies suggested that this might be due to the low cholesterol levels present in these populations [26]. Conversely, Japanese and Caucasian people who carry APOE4 are found to have 10 to 30 times more risk of rising AD than people who are not any APOE4. Nonetheless, the precise mechanism of dramatic effects exerted by this allele is yet to be fully discovered; evidence from studies suggests the interaction with amyloid [27]. Though few patients with AD have minimum one copy of the ε4 allele, as this APOE4 is not the main contributing factors to the disease. Actually, APOE4 is absent for one third of Alzheimer’s patients. Interestingly, some APOE4 homozygotes even never develop AD. However, people with two copies of APOE4 have as high as 20 times the risk of rising AD [28]. In contrast, there is also evidence that supports the protective roles of APOE2 in patients with AD [29]. People with APOE2/APOE4, APOE3/APOE4, and APOE4/APOE4 are most likely to develop AD, but the odds ratios (ORs) were abated for people with genotypes APOE2/APOE2 and APOE2/APOE3 [23]. APOE4 has been found to significantly upsurge the odds that a person will develop AD. However, a study reported that in individuals several independent risk factors including high serum total cholesterol, any combination of APOE alleles, and high blood pressure in the midlife collectively could increase the risk by threefolds to develop AD at the later stage [26].
Effects of ApoE on Amyloid Pathways
ApoE and Aβ Production
Senile plaques composed primarily of the Aβ peptides are one of the neuropathological indicators of AD. Aβ is produced in an amyloidogenic path by the cleavage of β- and γ-secretases. Additionally, by influencing α-, β-, or γ-secretase activity, ApoE acts as a principal cholesterol carrier protein rises Aβ generation [30].
Previously, Ye et al. [31] inspected the effect of ApoE isoforms on APP processing and production of Aβ in rat neuroblastoma B103 cells firmly transfected with the human wild-sort APP695 (B103-APP). According to the statement of the researchers, ApoE4 seems to control the processing of APP and the generation of Aβ by using the pathway of low-density lipoprotein receptor-related protein (LRP) and domain interface. The outcomes give acumens into why ApoE4 is connected with high risk for AD and may signify a prospective therapeutic object for the drug development.
In another study, Hopkins et al. [32] reported that Aß generation rises on account of the induction of ApoE4 which could be interceded by a new ApoE-binding protein (TMCC2), recommended to expedite an interaction amid APP and the γ-secretase complex. In AD, it is clear that the interplay between TMCC2 and ApoE may also consequently make contributions to interrupt Aβ protein precursor metabolism and change Aβ genesis. Recently, using ES-cell-derived human neurons, Huang et al. [33] mentioned that ApoE isoforms (i.e., ApoE4 > ApoE3 > ApoE2) control APP transcription and Aβ production by triggering a non-canonical mitogen-activated protein kinase signaling pathway.
ApoE and Aβ Aggregation
ApoE exerts an imperative role in the Aβ levels, aggregation, and amyloid plaque loads. In the APP transgenic amyloid mice model, copious studies have demonstrated that ApoE is vital for Aβ deposition. When amyloid model PDAPP or Tg2576 mice have crossed with APOE knock-out (KO) mice, accumulation of Aβ in the form of amyloid plaques and the cerebral amyloid angiopathy (CAA) was drastically reduced [34, 35]. Occasionally increased accretion of Aβ in the form of diffused plaques and plaques of thioflavin-S-positive fibril were virtually lacking, especially, while there is quite substantial. Findings of these studies strongly suggest the important role of mouse ApoE in Aβ fibrillogenesis, fibrillar Aβ stabilization, and maturation of amyloid plaques [34, 35]. Nevertheless, the origin of APOE determines the effects of APOE on Aβ fibrillogenesis. In this regard, in PDAPP mice, reduced early Aβ deposition was observed by Holtzman et al. [36] with the expression of human APOE3 and APOE4 by astrocytes in the APOE-KO. Fryer et al. [37] reported that human APOE4 changes the ratio of Aβ40 and Aβ42, as well as stimulates the genesis of CAA in Tg2576 mice. Human APOE-targeted replacement (TR) mice had less accumulation of Aβ in Tg2576 mice concerning control mice expressing mouse APOE.
It has been found that when Aβ aggregates, soluble Aβ peptides can lead to change the conformation of these peptides into a β-sheet structure and can form nucleuses, which can also further speed up the process of fibrillogenesis to trigger the formation of insoluble fibrils with enriched β-sheet structures [38]. Several studies explored the roles of ApoE in Aβ aggregation. Nevertheless, the assumptions of the studies are debatable, where ApoE can either accelerate or inhibit Aβ aggregation. It has been found that high concentrations of ApoE can trigger the formation of enormous co-aggregates with Aβ [39], whereas ApoE4 is expected to stimulate Aβ aggregation greater than ApoE3 [40, 41]. Furthermore, it was also observed that ApoE upsurges the level of Aβ oligomers in an isoform-reliant way (i.e., ApoE4 > ApoE3 > ApoE2) [42]. Furthermore, ApoE4 steadies Aβ oligomers greater than ApoE3 [43]. These conclusions suggest that ApoE4 detrimentally triggers Aβ aggregation in AD (Fig. 2).

The impact of ApoE on Aβ aggregation in Alzheimer’s disease. Aβ monomers alter their conformation to generate oligomers and intermediate aggregates and then form large fibrils. The linkage of Aβ with ApoE generates aggregation of ApoE and Aβ as well as stimulates Aβ fibrillogenesis to form Aβ fibrils. The generated aggregates of ApoE and Aβ in conjunction with Aβ fibrils further accelerate to form larger co-aggregates. This co-aggregate deposited in the brain as amyloid plaques, Aβ fibrils also stimulate the formation of amyloid plaques [44]
In contrast, various studies have concluded that Aβ fibrillogenesis can be decreased by ApoE. Beginning of Aβ fibril formation can also be inhibited by ApoE, when investigated either with or without the addition of pre-formed Aβ aggregates as seeds [45, 46]. Since ApoE have preferences to interact with the β-sheet structure containing Aβ peptides [47], ApoE is also likely to capture Aβ nuclei and also prevents its scattering properties as well as trigger Aβ fibrillogenesis [46]. ApoE3 seems to interact with Aβ more than ApoE4 as mentioned; consequently, it is probable that Aβ fibril formation is less effectively inhibited by ApoE4. In this perspective, ApoE4 may not be that much effective to support the useful effects of ApoE to prevent Aβ fibrillation in AD. If the amount of the ApoE/Aβ complex increases as the only product of the reaction, they may form massive co-aggregates [46]. Hatters et al. [48] stated that ApoE could also aggregate with random protofilament-alike structure, where the aggregates form at extensively diverse rates which primarily depends on the isoform (i.e., ApoE4 > ApoE3 > ApoE2). Thus, through its self-aggregating tendency, ApoE4 may also be able to create more co-aggregates with Aβ. Furthermore, various experiments have also demonstrated that in the background of the amyloid model mice, APOE3-TR mice had less Aβ deposition than APOE4-TR mice [37, 49]. Instead, a more violent amyloid model mice known as 5xFAD [50], amyloid plaque deposition was far more in E4FAD mice, and E2/E3FAD mice have considerably higher diffuse plaques with E4FAD showing more dense plaques [51]. Altogether, these findings suggest that as compared to APOE2 or APOE3 and APOE4, it is either likely to stimulate Aβ fibrillogenesis or in case of prevention of Aβ aggregation it is less effective, or both. Moreover, the exact outcomes can be affected by copious factors including lipidation status, ApoE isoform, aggregation states, and the location and time of its existence during the process of disease. Furthermore, the investigation is also required to observe the effects of fibrillar plaques and diffuse on synaptic behaviors and functions in the presence of different ApoE isoforms [45]. In a current study, Hatami et al. [52] reported that most familial AD (FAD) mutations accelerated the rate of aggregation of Aβ. Moreover, declining of FAD mutations within the Aβ sequence is responsible for noticeable alterations in aggregation kinetics and finally impacts the capability of Aβ to generate immunologically and morphologically discrete amyloid assemblies.
ApoE and Aβ Clearance
The metabolism and transport of Aβ in the brain can be altered by ApoE. In cell culture systems, the roles of ApoE isoforms on Aβ production and processing of APP have been studied. Studies propose that lipid-free and lipid-poor ApoE4 increase LRP1- and ApoER2-reliant APP endocytosis by enhancing the production of Aβ [31, 53, 54]. Nonetheless, in other studies, no strong evidence was found to support isoform-specific effects on APP processing [55, 56]. Furthermore, there is no conclusive data to suggest that ApoE isoforms possess a different role in the Aβ production. Also, ApoE seems to exert an imperative role by numerous credible paths in the Aβ clearance [44] mentioned in Fig. 3. Sequestration of Aβ can take place via ApoE-comprising lipoprotein particles and modulation of the cellular uptake of an ApoE–Aβ complex can take place due to receptor-mediated endocytosis. Conversely, by transporting through the BBB, ApoE may control the elimination of Aβ from the brain cells to the systemic circulation. Through several sorts of neuronal cells, human ApoE helps the attachment as well as internalization of Aβ and this has been demonstrated in various studies [57,58,59]. No overall trend developed, although some of these studies observed ApoE isoform-reliant variances in the degree of the cellular uptake of Aβ. However, facilitation of cellular Aβ degradation was observed in few studies [60, 61], and further studies are required to establish whether ApoE helps in the uptake of Aβ into the several cell types present in the brain. Indeed, studies are also obligatory to establish and to clarify the mechanism whether this heightened uptake happens in an isoform-specific mode or not.

The impact of ApoE on Aβ clearance in Alzheimer’s disease. The Aβ is cleared from the brain by the proteolytic degradation, cellular clearance, and cerebrovascular system-linked clearance. ApoE likely facilitates Aβ clearance by triggering the aforementioned pathways. The Aβ clearance is also suppressed by ApoE through competing with either Aβ receptor or by blocking Aβ clearance [44]
It is still unclear that how exactly this denouement is related to the observed results, where deficiency of ApoE can lead to an intense decrease in a load of thioflavin-S-positive amyloid [34, 36, 62]. Fascinatingly, it has been found that in young PDAPP mice, before the onset of accretion of Aβ, soluble Aβ levels may be increased due to the lack of ApoE and this result is consistent with outcome obtained from cell culture data [63]. This finding was also confirmed in an experiment in which microdialysis was used to examine the Aβ level in interstitial fluid of the brain [64]. Some studies also support that cellular Aβ degradation and uptake are enhanced by ApoE, Indeed, it also needs to be considered that BBB can act as an effective pathway of clearance of Aβ in the brain and particularly by LRP1 [65].
The clearance of Aβ is yet to be extensively studied in the presence of human ApoE. Recent findings from a study have demonstrated that in the brain of the mouse, brain to blood clearance of lapidated ApoE4 is considerably lower than the ApoE3 and ApoE2 clearance [66]. However, this tendency is quite reversed for what is detected for the entire human ApoE levels in the brain especially when the APOE are expressed in knock-in (KI) mice. Thus, whether BBB shows any crucial part in case of controlling the brain’s ApoE levels yet needs to be established. Growing current findings strongly recommends that when a composite in between human ApoE and Aβ is formed, the brain to blood elimination of Aβ is essentially abated concerning free Aβ [66,67,68]. Moreover, Aβ composite to ApoE2 and ApoE3 is less more likely to be removed from the brain at a considerably quicker degree than Aβ composite to ApoE4 [66]. Ji et al. [69] observed at BBB transference of Aβ in transgenic mice for either human ApoE4 or ApoE3 did not exhibit any significant variances regarding Aβ removal from the brain. Lastly, in brain capillaries, an ApoE4–Aβ complex found in the periphery is far more sequestered as compared to the Aβ bound to ApoE2 or ApoE3. This denouement further suggests that ApoE4-facilitated blood to brain transportation of Aβ can play an important role in the accretion of amyloid in the brain [70]. In addition to these studies, further work is required to decipher the precise roles played by BBB in mediating clearance of Aβ, how exactly ApoE contributes in this process, and also whether isoform-explicit roles be existent or not. Presently, a study by Liu et al. [71] reported that astrocytic low-density lipoprotein receptor-related protein 1 (LRP1) exerts a pivotal role in Aβ metabolism. Moreover, it also accounts for restoring LRP1 expression and function in the brain could be a fruitful approach to expedite Aβ clearance and counter amyloid pathology in AD.
Effects of ApoE on Non-amyloid Pathways
ApoE and Tau
ApoE might also affect processes involved in neurodegeneration, in which, there is a link of tau pathology. Interestingly, overrepresentation of the APOE4 allele has been noticed in most of the clinical studies in case of both AD and frontotemporal dementia, whereas histopathologic examinations showed a substantial positive correlation between stage of neurofibrillary pathology and APOE genotype [72]. For instance, even following the correction of their levels of Aβ42, Alzheimer’s patients having ApoE4 allele normally contain more tau in the cerebrospinal fluid [73]. Agosta et al. [74] performed a study in 31 patients with behavioral and variant frontotemporal dementia and 51 patients with a probable AD, in comparison with 56 healthy controls, to explore the effect of ε4 allele carrier status on the disease severity pattern and atrophy of gray matter. It was found that the frequency of ε4 allele was notably higher in the patients with AD (P < 0.001) as compared to healthy controls, but not in the patients with variant frontotemporal dementia. No differences were noticed in terms of cognitive and demographic profiles between noncarriers and ε4 allele carriers within any of the diagnostic groups. Nonetheless, marked brain atrophy in disease-specific regions in comparison with noncarriers in both AD and variant frontotemporal dementia was linked with ε4 carrier status. AD ε4 carriers exhibited noticeable atrophy in the right hippocampus and bilateral parietal cortex. On the other hand, variant frontotemporal dementia ε4 carriers showed marked atrophy in the dorsolateral, bilateral medial, and frontal cortex, anterior insula and cingulate cortex with right predominance. This regional effect of ε4 is consistent with the hypothesis that in different neurodegenerative diseases ApoE may possibly alter the morphologic expression in a unique way. The patterns of atrophy in carriers of ε4 might specify that they are at significant danger for further clinical progression. However, Riemenschneider et al. [75] found that there is no noteworthy difference between the groups containing either ε2/ε4 allele frequency. Patients with the ε2/ε3 genotype (i.e., 61.3 years) showed the highest age at onset. In contrast, patients with the ε3/ε3 (i.e., 58.3 years) had the lower age at onset than the patients with the ε2/ε3 genotype and ε3/ε4 genotype (i.e., 56.4 years). Nonetheless, these differences had no statistical significance. Another study conducted by Srinivasan et al. [76] showed that frontotemporal dementia is not related to tau gene mutations. They also found that APOE4 allele possessions in men can approximately double the chances of developing the disease, while this type of possession has no impact upon disease risk in women.
Strittmatter et al. [77] stated the isoform-specific interactions of ApoE with microtubule-associated protein tau. They found in an in vitro binding assay that ApoE3 rather than ApoE4 mainly interacts with tau. Furthermore, isoform-specific interactions of ApoE with tau perhaps play a role in the regulation of intraneuronal tau metabolism in AD and also in the alteration of the rate of NFTs and paired helical filaments [77]. Phosphorylation of tau prevented the interaction between ApoE3 and tau, which suggests that ApoE3 binds preferably to non-phosphorylated tau. In another study, Chang et al., [78] mentioned that ApoE4 (1–272) was neurotoxic, however full-length ApoE4 (1–240) and ApoE4 (1–299) were not. These findings suggest that the lipid-binding region (i.e., amino acids 241–272) facilitates the neurotoxicity, besides that amino acids 273–299 are protective. The neurotoxicity of ApoE4 (1–272) was found to be abolished by a quadruple mutation in the lipid-binding regions mainly such as W264R, F257A, V269A, and I250A. In addition, neurotoxicity of full-length ApoE4 is associated with the single mutations in the amino acid regions 273–299 (i.e., Q284A, L279Q, or K282A). Study via immunofluorescence staining revealed that in some cells ApoE4 (1–272) made filamentous inclusions comprising phosphorylated tau and interacted with mitochondria in others, which can further lead to mitochondrial dysfunction as determined by flow cytometry and MitoTracker staining. Neurotoxicity or mitochondrial dysfunction was not caused by ApoE4 (241–272), which suggests that only the lipid-binding region is inadequate to cause neurotoxicity. Instead, neurotoxicity of ApoE4 (1–272) and the mitochondrial interaction were abolished upon truncation of N-terminal sequences (i.e., amino acids 1–170) having the receptor-binding region (i.e., amino acids 135–150) and triple mutations within that region (i.e., R142A, R147A, and K146A). Further studies revealed that the receptor-binding region is essential to escape from the secretory pathway and that the lipid-binding region facilitated mitochondrial interaction. Hence, the receptor- and lipid-binding regions in fragments of ApoE4 work together to facilitate neurotoxicity and dysfunction of mitochondria, which might be crucial in the pathogenesis of AD. A study conducted by Harris et al. [79] in ApoE4 transgenic mice showed that Erk activation was linked with the increased tau phosphorylation, and this might be modified by zinc, which suggests that zinc and ApoE4 work together to contribute to the AD pathogenesis. Hoe et al. [80] showed that a number of signaling cascades in neurons are affected by ApoE including increased level of disabled phosphorylation, ERK1/2 pathway activation pathway (i.e., reliant on calcium influx through the NMDA receptor) and the c-Jun N-terminal kinase 1/2 pathway inhibition (i.e., reliant on γ-secretase and G-proteins). Nevertheless, in another study, Huang et al. [81] stated that ApoE4 preferentially go through intracellular processing, generating a bioactive fragment that interacts with cytoskeletal components and induces NFT-like inclusions comprising phosphorylated neurofilaments and phosphorylated tau of high molecular weight in neurons.
Recently, Shi et al. [82] revealed a promising new role of APOE4 in the development of AD. A mouse model was designed by the researchers, in which the modified form of human tau was observed in the rodents, influencing them to form tangle. These researchers genetically modified the mice to contain human versions of the APOE genotypes (i.e., ε2, ε3, and ε4) instead of their mouse-specific APOE. They observed the mice for 9 months and in the meantime, the mice that had APOE2 showed the least neurodegeneration and the APOE4 showed the most neurodegeneration. The hippocampus and entorhinal cortex are the brain areas that play a pivotal role in memory were found to have atrophied in case of APOE variations containing mice. Furthermore, brain damage was also noticed in these mice with the significant number of dead brain cells. The study also showed that tau tangles were not that much harmful, particularly when APOE was absent. Conversely, no brain damage was observed in the mice that lacked APOE. The researchers also noticed a profound inflammatory response by observing the immune cells in the brains of mice with APOE4 were activated; in contrast, immune cell activation was not noticed in case of mice that lacked APOE4. To find out the functions of in human brains, these researchers also investigated autopsy samples, which were collected from the 79 dead people who primarily died due to tau pathologies and made a database of the ApoE variants that these dead people had. These studies also showed that people who had APOE4 experienced more severe damage concerning those without the variant. The denouements further recommend that APOE4 can interfere with neuroinflammation, tau pathogenesis as well as neurodegeneration mediated by tau which is independent of Aβ pathology [82].
ApoE and α-Synuclein
AD and synucleinopathies are found to share similar pathological mechanisms. ApoE4, which is known as the most common genetic risk factor for AD, also upsurges the risk for dementia in the context of pure synucleinopathies. However, the molecular mechanisms of the role of α-synuclein are yet to be fully revealed. On the other hand, pathologic effects of α-synuclein are generated by a gain-of-function toxic mechanism triggered by the accumulation of this molecule and this finding is found to have strong evidence. Nevertheless, it has also been recommended that loss of the normal α-synuclein physiological functions might play a crucial role [83]. Remarkably, up to 50% of patients with dementia and α-synucleinopathy also have Aβ plaques, whereas a smaller subset also has associated NFTs [84]. Gallardo et al. [85] using transgenic mice reported neurodegeneration induced by α-synuclein involving ubiquitin/proteasome system activation, the buildup of insoluble mouse Aβ, and an enormous upsurge in ApoE levels. ApoE was injurious and was not protective, since ApoE deletion caused the delay in neurodegeneration caused by α-synuclein and suppression of the Aβ accumulation. The results show a molecular link between central pathogenic mechanisms involved in AD and Parkinson’s disease. Furthermore, it also suggests that intracellular α-synuclein is pathogenic, at least partly due to the activation of ApoE involved extracellular signaling pathways.
In a recent study, Emamzadeh et al. [86] studied the effects of different isoforms of ApoE (i.e., ApoE2, ApoE3, ApoE4) on the α-synuclein aggregation. The results also showed that ApoE concentration influences α-synuclein aggregation. At low ApoE concentrations (< 15 nM), all of the isoforms were capable of increasing the α-synuclein aggregation (50 μM), among the isoforms ApoE4 showed the greatest stimulatory effect. On the contrary, a decrease in the α-synuclein aggregation was observed with the higher concentration (> 15 nM) of these isoforms. The denouements demonstrate that exceptionally low levels of ApoE may possibly seed α-synuclein aggregation, which could possibly lead to the pathogenesis of neurodegeneration induced by α-synuclein. Conversely, higher ApoE levels could possibly reduce the degree of aggregation of α-synuclein and confer protection. The differential effects observed with ApoE4 could clarify why ApoE4 results in onset for neurodegeneration in an earlier age [86].
Deficiency of α-synuclein can result in the shortfalls in the nigrostriatal system, impaired disrupted activity-dependent regulation of nondopaminergic and dopaminergic transmission [87], along with the synaptic proteins loss, for example synaptotagmin, in the course of aging [88]. In addition, impairment of the synaptic response to repetitive stimulation is initiated due to the deficiency of α-synuclein, which was linked with a noticeable decrease in the reserve pool of synaptic vesicles, especially reduced glutamate mobilization from the reserve pools [89]. Henceforth, these findings denote that α-synuclein binds precisely to different presynaptic proteins including Rab3 and vesicle-associated membrane protein 2 (VAMP2)/synaptobrevin, which suggests that an important role is played by α-synuclein in the function and maintenance of the release machinery and the nerve terminal [90]. Nerve terminals are predominantly found to be susceptible to ApoE4, which is revealed by the previous studies [91, 92].
Recently Bar et al. [93] stated that in TR mice, the pathologic effects of ApoE4 are heightened by the deficiency of α-synuclein and the ApoE4 effects are gene dose dependent and in the female the effects are more noticeable. In terms of accumulation of Aβ, it has been found that α-synuclein absence at old age can lead to the increased level of amyloid plaques, which suggests that α-synuclein possibly plays a role as a chaperone to help the cells to remove deposits of protein. This has been found to be consistent with the currently noticed Aβ42 accumulation in the ApoE4 mice lacking α-synuclein, which might be the synergistic outcome of two faulty mechanisms of clearance of Aβ. Ultimately, this may cause tau hyperphosphorylation, typically either by ApoE4-mediated mechanisms [81] or by the Aβ peptide [94].
Another probable mechanism is based on the interaction among α-synuclein and ApoE and their effects on microglial pro-inflammatory activation. In a study, Austin et al. [95] reported that, after stimulation, Scna−/− microglia secreted increased levels of interleukin 6 (IL-6), pro-inflammatory cytokines, and tumor necrosis factor alpha (TNFα), in comparison with wild-type. Nonetheless, Scna−/− cells exhibited impaired phagocytic ability, in spite of the reactive phenotype. These effects have been suggested to be facilitated by a subset of lipid-signaling-associated enzymes expression and through α-synuclein-mediated microglial secretory behavior regulation [96]. In addition, Li et al. [97] specified that the innate immune suppressor, triggering receptor expressed on myeloid cells 2, exhibited markedly reduced expression of microglia in ApoE4 cells in comparison with ApoE3. It was reported by Ouberai et al. [98] that the α-synuclein binding strength is associated with the specificity of the lipid environment such as the chemistry of lipid and steric properties inside a bilayer structure and to the capacity of the membranes to remodel and accommodate upon the interaction between lipid membranes and α-synuclein. In another study, Castagnet et al. [99] stated that disrupted uptake and trafficking of astrocyte fatty acid is related to the α-synuclein deficiency, with a significantly increased trafficking of fatty acid to triacylglycerols and cholesteryl esters and decreased phospholipids trafficking, as well as phosphatidylinositol. Nevertheless, ApoE is the foremost brain lipid transporter. Hu et al. [100] in a study described that ApoE4 can enhance accumulation of Aβ and decrease lipidation of ApoE, while ApoE2 has been found to have the opposite effects. These findings recommended that increasing ApoE2 in carriers of APOE4 could be a useful strategy in the treatment of AD, while increasing ApoE4 in carriers of APOE4 is likely to cause harm.
ApoE and Neuroinflammation
In the development and progression of AD, an inflammatory reaction in the brain due to glial activation plays a crucial role [101]. Increased ApoE has functional significance to limit the inflammatory response. Actually, in comparison with wild-type mice, glial cells cultured from ApoE KO mice show an increased production of several pro-inflammatory markers in response to treatment with Aβ and other activating stimuli [102]. Lynch et al. [103] stated in their study that ApoE can modulate the endogenous CNS inflammatory response and glial activation. Another study further recommended that animals who expressed the ε4 allele had considerably increased systemic and brain elevations of the pro-inflammatory cytokines IL-6 and TNFα in comparison with their ApoE3 counterparts, which suggest an isoform-specific effect of the immunomodulatory properties of ApoE [104]. Moreover, in mice, intravenous administration of a small ApoE mimetic peptide likewise suppressed both brain and systemic inflammatory responses following administration of lipopolysaccharide. Lowest levels of ApoE were observed with the APOE4 carriers. Ringman et al. [105] mentioned in their study that although young ε4 carriers possess increased inflammatory markers, that decrease with age. They confirmed altered inflammatory responses in ε4 carriers during young and middle adulthood, which may relate to Alzheimer’s risk later stages of life. Interestingly, Szekely et al. [106] stated that for nonsteroidal anti-inflammatory drugs (NSAIDs), the user has reduced the risk of AD, nonetheless this association was found only in the APOE4 allele and no specific advantage was found for Aβ (42)-lowering NSAIDs. APOE interactions particularly with molecules, which are significant for lipid endocytosis and lipid efflux, trigger effects of the APOE genotype on lipoprotein composition and neuroinflammation [72]. These effects suggest important targets for new therapies to reduce the risk of AD before the exhibition of any signs of pathogenesis.
ApoE and Lipid Metabolism
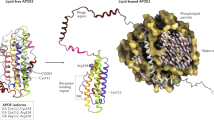
ApoE mediates neuronal delivery of cholesterol, as brain cholesterol levels are found to be considerably reduced in hippocampal and cortical areas in patients with Alzheimer’s in comparison with age-matched controls [107]. Riddell et al. [108] stated that astrocytes specifically damage ApoE4, which can lead to reduced secretion of ApoE4 and can ultimately reduce ApoE levels in the brain. Furthermore, the genotype-dependent decrease in ApoE levels in the CNS mirror the comparative risk in the development of AD and propose that low levels of total ApoE showed by APOE4 allele carriers might directly contribute to the progression of the disease, possibly by reducing the ApoE capacity to facilitate Aβ clearance and/or synaptic repair. A decreased ApoE4-bound cholesterol uptake was found by Rapp et al. [109] in hippocampal neurons. On the contrary, hippocampal astrocytes exhibit diminished internalization of ApoE2-bound cholesterol. Furthermore, lipidated ApoE4 is slightly related with neurites in hippocampal neurons in comparison with the other two isoforms. Hamanaka et al. [110] in their study stated altered lipid and cholesterol metabolism in human APOE4 KI mice. In a recent study, Moser and Pike [111] stated that obesity most likely accelerate AD-related pathology in APOE4 but not in the case of APOE3 mice.
ApoE and Synaptic Plasticity
Synaptic failure is considered as an early pathological feature of Alzheimer’s and variation in the regulation of synaptic plasticity is observed with ApoE isoforms [112]. Moreover, Buttini et al. [113] stated that old ApoE-deficient transgenic mice expressing in their brains human APP (hAPP)/ApoE3 and hAPP/ApoE4 had comparable cholinergic/synaptic deficits, and these deficits were observed not only in the hippocampus region but also in the neocortex, which in most mice did not contain any plaques. Another study conducted by Sen et al. [114] reported that ApoE3 but not ApoE4 provides protection against synaptic loss via greater expression of protein kinase Cε. Additionally, mean age-of-onset of dementia is significantly reduced by the ApoE4 isoform. Instead, Chen et al. [112] described that ApoE4 mediates reduction of synaptic plasticity and glutamate receptor function through specific impairment of ApoE receptor recycling. These observations implicate an isoform-specific role of ApoE in the intracellular trafficking and localization of glutamate receptors and lipoprotein, and thus suggest the existence of an alternative mechanism through which ApoE4 may accelerate the onset of neuronal degeneration and dementia by differential impairment of the maintenance of synaptic stability.
The effects of ApoE4 particularly on the long-term synaptic plasticity were examined by Qiao et al. [115]. The findings for the first time confirmed that ApoE4 could alter hippocampal late-phase long-term potentiation through the reduction of phosphorylated Ca2+/calmodulin-dependent protein kinase IIα (p-CaMKIIα) and phosphorylated cAMP response element-binding protein (p-CREB), which suggests that ApoE4 can induce the suppression of hippocampal long-term synaptic plasticity, and may possibly contribute to the cognitive impairments in genetic AD. Both of these CREB and CaMKIIα are vital intracellular targets of the neurotoxic ApoE4 [115]. In another study, Hwang et al. reported that acute treatment of PKR inhibitor can reinstate the shortfalls in long-term memory, synaptic plasticity, and long-term potentiation in case of both mouse models without disturbing the load of Aβ in the hippocampus [116].
ApoE Levels in the Central Nervous System of Alzheimer’s Disease
Studies examining the ApoE levels on AD suggest that APOE (Table 1) exerts a foremost role in developing AD. Moreover, the advancement of AD is also connected with the existence of APOE4 undoubtedly as a result of interactions with the Aß [123].
Copious analyses examined ApoE levels in the brain and cerebrospinal fluid (CSF) about the APOE genotypes has produced variable outcomes. Fukumoto et al. [124] stated that in APOE3/APOE4 people, the relation of ApoE4 to entire ApoE levels was 30 to 40% in plasma, signifying a reduced genesis or an augmented metabolism of ApoE4 concerning ApoE3. Astoundingly, the proportion in the CSF was opposing, with ApoE4 estimated for 60 to 70% of the entire ApoE. Nevertheless, a study by Bekris et al. [125] reported that it is esoteric to forecast the levels of CSF ApoE by APOE4. The APOE genotype, AD statement, gender, and race do not influence levels of CSF ApoE; however, normal CSF ApoE levels rise with age according to the study of Wahrle et al. [126]. A meta-analysis that comprised 1064 AD cases and 1338 non-demented control groups specified the perspective of ApoE levels of CSF as a hallmark of AD [127].
Controversial results were also reported for levels of ApoE isoforms in the parenchyma of the brain [128]. The contradictory findings from brain parenchyma researches might also stem from the comparatively minor number of sample size then heterogeneity in people, stages of the people as well as the period of the disease. Furthermore, the levels of ApoE may be affectedly changed by autopsy related delay [129]. Conversely, the amount of ApoE differs in diverse brain areas and numerous factors associated with the genesis and metabolism of ApoE may be fundamental in the pathogenesis of AD [130].
ApoE as a Therapeutic Target for Alzheimer’s Pathogenesis
ApoE is an important risk factor in AD pathogenesis; ApoE might suggest a smart target for AD therapy. In Table 2, ApoE-based therapeutic target in controlling AD is represented.
Cramer et al. [171] stated that the expression of ApoE is prompted transcriptionally by the act of the peroxisome proliferator-activated receptor gamma as well as LXRs that generate heterodimers in allocation to RXRs. In a mouse model of Alzheimer’s, bexarotene, an RXR agonist, caused a swift clearance of Aβ, especially soluble Aβ in ApoE-reliant mode within hours. In fact, more than 50% reduction of Aβ plaque was reported for 72 h [171]. Similarly, Riddell et al. [172] stated that the LXR agonist for example, TO901317, abates Aβ42 of hippocampal and expands memory in the Tg2576 mouse model of AD. Therefore, a surge in the levels of ApoE in the brain is probably advantageous in AD therapy. Nevertheless, huge caution is necessary for this concept since ApoE4 is also pathogenic. It also has been suggested that increasing ApoE lipidation perhaps be the key for ApoE-based therapy rather than focusing on increasing ApoE. The lipidation of ApoE is facilitated by ATP-binding cassette transporter A1, ABCA1 [132]. In case of PDAPP transgenic mouse model of AD, Wahrle et al. [173] stated that ABCA1 deletion could upsurge the deposition of Aβ peptide. The same researcher also reported that overexpression of ABCA1 suppresses Aβ deposition. These findings further advocate the deductions that increased ABCA1-mediated lipidation of ApoE in the CNS can reduce the load of amyloid. This enhancing the function of ABCA1 might have a beneficial effect on AD.
The dispute regarding the reduction in ApoE expression to treat AD is supported by the fact that Aβ deposition is primarily triggered by ApoE as mentioned above. On the other hand, Bien-Ly et al. [151] mentioned that abating human ApoE levels weaken the accumulation of age-dependent Aβ particularly in mutant human APP transgenic mice. Kim et al. [174] stated that in a mouse model of Aβ amyloidosis, haploinsufficiency of human APOE could reduce amyloid deposition. Further, immunotherapy for ApoE also reduces Aβ accumulation. In another study, Kim et al. [175] stated that anti-ApoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. These results suggest that decreasing ApoE levels has beneficial effects and that anti-ApoE immunization can be explored as a novel therapeutic tool, at least from the perspective of Aβ deposition. Furthermore, the interaction amid ApoE and Aβ can also be considered as therapy. ApoE can perform as a neurotic chaperone of Aβ, endorsing its morphological alteration from solvable Aβ into pathogenic aggregates. Sadowski et al. [176] reported that in the existence of ApoE, Aβ12-28P, a synthetic peptide, decreases the fibrillogenesis of Aβ and Aβ/ApoE toxicity in cell culture. This study avowed that 1-month treatment of transgenic mice by Aβ12-28P reduced 63.3 and 59.5% Aβ burden in the cortex and hippocampus, respectively, concerning control, transgenic mice. In another study, Shinohara et al. [177] found that a hydroxymethylglutaryl-CoA reductase inhibitor, for example, fluvastatin, abated the level of Aβ by an isoprenoid-reliant mode. Augmenting the roles of ApoE receptors can be an auspicious therapy for AD. In a current study, Luz et al. [178] examined the magnitude to which the roles of ApoE4 can be offset with an anti-ApoE4-specific monoclonal antibody (mAb), 9D11. The researcher reported that 9D11 stopped the ApoE4-focused accretion of Aβ in the hippocampus, caused the reverse of the cognitive deficiencies, and reversed the hyperphosphorylation of tau-mediated by ApoE4 as well as abated the expression of the ApoER2 receptor.
The tau protein mediates the transportation of nutrients and other necessary supplies to neurons in the brain of a healthy person. However, in case of a brain of an AD patient, this essential transport system does not function properly as tau forms tangles. Recently Shi et al. [82] reported that ApoE4 significantly worsens tau-facilitated neurodegeneration in a mouse model of tauopathy. The researchers avowed that the absence of ApoE is found to be protective, whereas ApoE4 shows noxious effects in AD. Therefore, targeting ApoE, particularly ApoE4, is an auspicious therapeutic tactic in controlling AD.
Conclusion
ApoE4 is the most powerful genetic risk factor for formation and propagation of the late-onset AD. Typically, ApoE4 enhances brain Aβ pathology concerning other ApoE isoforms. The existence of the APOE4 allele is related to more intense neurodegeneration in people with a sporadic primary tauopathy. Furthermore, in people with Aβ pathology with the symptomatic AD, as well as tau pathology ε4-carriers play a superior degree of disease progression. ApoE isoforms have diverse functions in regulating α-synuclein aggregation, neuroinflammation, lipid metabolism, and maintenance of synaptic plasticity. Exploring the exact impact of ApoE4 on AD pathogenesis is a great dispute, but ApoE4-targeted therapeutic strategies are an auspicious area of existing research in combating AD pathogenesis.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid β
- NFTs:
-
Neurofibrillary tangles
- ApoE:
-
Apolipoprotein E
References
Uddin MS, Mamun AA, Asaduzzaman M, Hosn F, Sufian MA, Takeda S et al (2018) Spectrum of disease and prescription pattern for outpatients with neurological disorders: an empirical pilot study in Bangladesh. Ann Neurosci 25(1):25–37.
Uddin M, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG et al (2018) Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci 10(4): 1–18.
Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9(2):106–118
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344
Uddin MS, Mamun AA, Kabir MT, Nasrullah M, Wahid F, Begum MM et al (2017) Neurochemistry of neurochemicals: messengers of brain functions. J Intell Dis-Diag Tre 5(4): 137–151.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T et al (2007) Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754.
Uddin MS, Mamun AA, Hossain MS, Akter F, Iqbal MA, Asaduzzaman M (2016) Exploring the effect of Phyllanthus emblica L. on cognitive performance, brain antioxidant markers and acetylcholinesterase activity in rats: promising natural gift for the mitigation of Alzheimer’s disease. Ann Neurosci 23(4):218–229
Burns A, Iliffe S (2009) Alzheimer’s disease. BMJ 338:b158
Waring SC, Rosenberg RN (2008) Genome-wide association studies in Alzheimer disease. Arch Neurol 65(3):329–334
Blennow KdeLeon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368(9533):387–403
Bekris LM, Yu CE, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23(4):213–227
Zhang YW, Thompson R, Zhang H, Xu H (2011) APP processing in Alzheimer’s disease. Mol Brain 4(1):3
Ryan NS, Rossor MN (2010) Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med 4(1):99–112
Uddin MS, Haque A, Mamun AA, Iqbal MA, Kabir MT, Rony RK et al (2016). Searching the linkage between high fat diet and Alzheimer’s disease: a debatable proof stand for ketogenic diet to alleviate symptoms of Alzheimer’s patient with APOE ε4 allele. J Neurol Neurophysiol 7(5):1–9.
Zhang L, Hong H (2015) Genomic discoveries and personalized medicine in neurological diseases. Pharmaceutics 7(4):542–553
Gureje O, Ogunniyi A, Baiyewu O, Price B, Unverzagt FW, Evans RM et al (2006) APOE ε4 is not associated with Alzheimer’s disease in elderly Nigerians. Ann Neurol 59(1):182–185.
Uddin MS, Asaduzzaman M, Mamun AA, Iqbal MA, Wahid F, Rony RK (2016) Neuroprotective activity of Asparagus racemosus Linn. against ethanol-induced cognitive impairment and oxidative stress in rats brain: auspicious for controlling the risk of Alzheimer’s disease. J Alzheimers Dis Parkinsonism 6(4):1–10
Mahley RW, Weisgraber KH, Huang Y (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 103(15):5644–5651
Hohman TJ, Dumitrescu L, Oksol A, Wagener M, Gifford KA, Jefferson AL et al (2017) APOE allele frequencies in suspected non-amyloid pathophysiology (SNAP) and the prodromal stages of Alzheimer’s disease. PLoS One 12(11):e0188501.
Xu X (2009) γ-Secretase catalyzes sequential cleavages of the AβPP transmembrane domain. J Alzheimers Dis 16(2):211–224
Myers RH, Schaefer EJ, Wilson PWF, d'Agostino R, Ordovas JM, Espino A et al (1996) Apolipoprotein E element 4 association with dementia in a population-based study: The Framingham Study. Neurology 46(3):673–677.
Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY et al (2017) Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun 8(1):1464.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 278(16):1349–1356.
Sepehrnia B, Kamboh MI, Adams-Campbell LL, Bunker CH, Nwankwo M, Majumder P. P et al (1989) Genetic studies of human apolipoproteins. X. The effect of the apolipoprotein E polymorphism on quantitative levels of lipoproteins in Nigerian blacks. Am J Hum Genet 45(4):586.
Hendrie HC, Ogunniyi A, Hall KS, Baiyewu O, Unverzagt FW, Gureje O et al (2001) Incidence of dementia and Alzheimer disease in 2 communities: Yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana. JAMA 285(6):739–747
Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K et al (2002) Apolipoprotein E ϵ4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 137(3):149–155
Wisniewski T, Frangione B (1992) Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 135(2):235–238
Hauser PS, Ryan RO (2013) Impact of apolipoprotein E on Alzheimer’s disease. Curr Alzheimer Res 10(8):809–817
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC et al (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7(2):180–184.
Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD et al (1998) Modulation of secreted β-amyloid precursor protein and amyloid β-peptide in brain by cholesterol. J Biol Chem 273(26):16576–16582.
Ye S, Huang Y, Müllendorff K, Dong L, Giedt G, Meng EC et al (2005) Apolipoprotein (apo) E4 enhances amyloid β peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc Natl Acad Sci U S A 102(51):18700–18705.
Hopkins PC, Sáinz-Fuertes R, Lovestone S (2011) The impact of a novel apolipoprotein E and amyloid-β protein precursor-interacting protein on the production of amyloid-β. J Alzheimers Dis 26(2):239–253
Huang Y-WA, Zhou B, Wernig M, Südhof TC (2017) ApoE2, ApoE3 and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168(3):427–441.e21
Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M et al (1997) Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet 17(3):263.
Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D et al (2000) Modulation of Aβ deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci 920(1):171–178.
Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM et al (1999) Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J Clin Invest 103(6):R15-R21.
Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM et al (2005) Human apolipoprotein E4 alters the amyloid-β 40: 42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25(11):2803–2810.
Harper JD, Lansbury PT Jr (1997) Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 66(1):385–407
Chan W, Fornwald J, Brawner M, Wetzel R (1996) Native complex formation between apolipoprotein E isoforms and the Alzheimer’s disease peptide Aβ. Biochemistry 35(22):7123–7130
Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C et al (1995) Fibrillogenesis in Alzheimer’s disease of amyloid β peptides and apolipoprotein E. Biochem J 306(2):599–604.
Ma J, Yee A, Brewer HB Jr, Das S, Potter H (1994) Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372(6501):92
Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO et al (2012) Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J Neurosci 32(43):15181–15192.
Cerf E, Gustot A, Goormaghtigh E, Ruysschaert JM, Raussens V (2011) High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J 25(5):1585–1595
Kanekiyo T, Xu H, Bu G (2014) ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81(4):740–754
Naiki H, Gejyo F, Nakakuki K (1997) Concentration-dependent inhibitory effects of apolipoprotein E on Alzheimer’s β-amyloid fibril formation in vitro. Biochemistry 36(20):6243–6250
Wood SJ, Chan W, Wetzel R (1996) An ApoE-Aβ inhibition complex in Aβ fibril extension. Chem Biol 3(11):949–956
Golabek AA, Soto C, Vogel T, Wisniewski T (1996) The interaction between apolipoprotein E and Alzheimers amyloid-peptide is dependent on-peptide conformation. J Biol Chem 271(18):10602–10606
Hatters DM, Zhong N, Rutenber E, Weisgraber KH (2006) Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol 361(5):932–944
Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C et al (2009) Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J Neurosci 29(21):6771–6779.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26(40):10129–10140.
Youmans KL. Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M et al (2012) APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem 287(50):41774–41786.
Hatami A, Monjazeb S, Milton S, Glabe CG (2017) Familial Alzheimer’s disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-β peptide. J Biol Chem 292(8):3172–3185
He X, Cooley K, Chung CH, Dashti N, Tang J (2007) Apolipoprotein receptor 2 and X11α/β mediate apolipoprotein E-induced endocytosis of amyloid-β precursor protein and β-secretase, leading to amyloid-β production. J Neurosci 27(15):4052–4060
Weisgraber KH, Pitas RF, Mahley RW (1994) Lipoproteins, neurobiology, and Alzheimer’s disease: structure and function of apolipoprotein E. Curr Opin Struct Biol 4(4):507–515
Biere AL, Ostaszewski B, Zhao H, Gillespie S, Younkin SG, Selkoe DJ (1995) Co-expression of β-amyloid precursor protein (βAPP) and apolipoprotein E in cell culture: analysis of βAPP processing. Neurobiol Dis 2(3):177–187
Irizarry MC, Deng A, Lleo A, Berezovska O, Von Arnim CA, Martin-Rehrmann M et al (2004) Apolipoprotein E modulates γ-secretase cleavage of the amyloid precursor protein. J Neurochem 90(5):1132–1143.
Beffert U, Aumont N, Dea D, Lussier-Cacan S, Davignon J, Poirier J (1998) β-amyloid peptides increase the binding and internalization of apolipoprotein E to hippocampal neurons. J Neurochem 70(4):1458–1466
Nielsen HM, Veerhuis R, Holmqvist BO, Janciauskiene S (2009) Binding and uptake of Aβ1-42 by primary human astrocytes in vitro. Glia 57(9):978–988
Yamauchi K, Tozuka M, Hidaka H, Nakabayashi T, Sugano M, Katsuyama T (2002) Isoform-specific effect of apolipoprotein E on endocytosis of fl-amyloid in cultures of neuroblastoma cells. Ann Clin Lab Sci 32(1):65–74
Jiang Q, Lee CD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N et al (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58(5):681–693.
Koistinaho M, Lin S, Wu XIN, Esterman M, Koger D, Hanson J et al (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med 10(7):719.
Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J et al (1999) Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 96(26):15233–15238.
Dodart JC, Bales KR, Johnstone EM, Little SP, Paul SM (2002) Apolipoprotein E alters the processing of the β-amyloid precursor protein in APPV717F transgenic mice. Brain Res 955(1–2):191–199
DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW et al (2004) ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron 41(2):193–202.
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57(2):178–201
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB et al (2008) apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118(12):4002–4013.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R et al (2007) Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27(5):909–918.
Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T (2007) Cerebral clearance of human amyloid-β peptide (1–40) across the blood–brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J Neurochem 103(6):2482–2490
Ji Y, Permanne B, Sigurdsson EM, Holtzman D, Wisniewski T (2001) Amyloid β40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimers Dis 3(1):23–30
Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W et al (1997) Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid β. J Neurochem 69(5):1995–2004.
Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR et al (2017) Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J Neurosci 37(15):4023–4031.
Rebeck GW (2017) The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res 58(8):1493–1499
Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K et al (2017) Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 133(5):839–856.
Agosta F, Vossel KA, Miller BL et al (2009) Apolipoprotein E ε4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci U S A 106(6):2018–2022.
Riemenschneider M, Diehl J, Muller U, Forstl H, Kurz A (2002) Apolipoprotein E polymorphism in German patients with frontotemporal degeneration. J Neurol Neurosurg Psychiatry 72(5):639–641
Srinivasan R, Davidson Y, Gibbons L et al (2006) The apolipoprotein E ε4 allele selectively increases the risk of frontotemporal lobar degeneration in males. J Neurol Neurosurg Psychiatry 77(2):154–158.
Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R et al (1994) Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 91(23):11183–11186.
Chang S, ran MT, Miranda RD, Balestra ME, Mahley RW, Huang Y (2005) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A 102(51):18694–18699
Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y (2004) Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J Biol Chem 279(43):44795–44801
Hoe HS, Harris DC, Rebeck GW (2005) Multiple pathways of apolipoprotein E signaling in primary neurons. J Neurochem 93(1):145–155
Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW (2001) Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98(15):8838–8843
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W et al (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549(7673):523.
Benskey MJ, Perez RG, Manfredsson FP (2016) The contribution of alpha synuclein to neuronal survival and function—implications for Parkinson’s disease. J Neurochem 137:331–359
Huynh TPV, Davis AA, Ulrich JD, Holtzman DM (2017) Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J Lipid Res 58(5):824–836
Gallardo G, Schlüter OM, Südhof TC (2008) A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat Neurosci 11(3):301
Emamzadeh FN, Aojula H, McHugh PC, Allsop D (2016) Effects of different isoforms of apoE on aggregation of the α-synuclein protein implicated in Parkinson’s disease. Neurosci Lett 618:146–151
Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25:239–252
Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL (2010) Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol Aging 31:796–804
Gureviciene I, Gurevicius K, Tanila H (2007) Role of alpha-synuclein in synaptic glutamate release. Neurobiol Dis 28:83–89
Chen RH, Wislet-Gendebien S, Samuel F, Visanji NP, Zhang G, Marsilio D (2013) Alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J Biol Chem 288:7438–7449
Teter B, Xu PT, Gilbert JR, Roses AD, Galasko D, Cole GM (2002) Defective neuronal sprouting by human apolipoprotein E4 is a gain-of-negative function. J Neurosci Res 68:331–336
Sen A, Alkon DL, Nelson TJ (2012) Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase C epsilon. J Biol Chem 287:15947–15958
Bar R, Boehm-Cagan A, Luz I, Kleper-Wall Y, Michaelson DM (2018) The effects of apolipoprotein E genotype, α-synuclein deficiency, and sex on brain synaptic and Alzheimer’s disease–related pathology. Alzheimers Dement (Amst) 10:1–11
Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S (2002) Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115:201–211
Austin SA, Floden AM, Murphy EJ, Combs CK (2006) Alpha-synuclein expression modulates microglial activation phenotype. J Neurosci 26(41):10558–10563
Austin SA, Rojanathammanee L, Golovko MY, Murphy EJ, Combs CK (2011) Lack of alpha-synuclein modulates microglial phenotype in vitro. Neurochem Res 36:994–1004
Li X, Montine KS, Keene CD, Montine TJ (2015) Different mechanisms of apolipoprotein E isoform-dependent modulation of prostaglandin E2 production and triggering receptor expressed on myeloid cells 2 (TREM2) expression after innate immune activation of microglia. FASEB J 29:1754–1762
Ouberai MM, Wang J, Swann MJ et al (2013) α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J Biol Chem 288(29):20883–20895.
Castagnet PI, Golovko MY, Barceló-Coblijn GC, Nussbaum RL, Murphy EJ (2005) Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J Neurochem 94(3):839–849
Hu J, Liu C-C, Chen X-F, Zhang Y, Xu H, Bu G (2015) Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol Neurodegener 10:6
Ardura-Fabregat A, Boddeke EWGM, Boza-Serrano A, Brioschi S, Castro-Gomez S, Ceyzériat K et al (2017) Targeting Neuroinflammation to treat Alzheimer’s disease. CNS Drugs, 31(12):1057–1082.
LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J et al (2001) Apolipoprotein E and apolipoprotein E receptors modulate Aβ-induced glial neuroinflammatory responses. Neurochem Int 39(5–6):427–434.
Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT (2001) Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol 114(1):107–113
Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM et al (2003) APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem 278(49):48529–48533.
Ringman JM, Elashoff D, Geschwind DH, Welsh BT, Gylys KH, Lee C et al (2012) Plasma signaling proteins in persons at genetic risk for Alzheimer disease: influence of APOE genotype. Arch Neurol 69(6):757–764.
Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH et al (2008) NSAID use and dementia risk in the cardiovascular health study role of APOE and NSAID type. Neurology 70(1):17–24.
Svennerholm L, Gottfries CG (1994) Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II). J Neurochem 62(3):1039–1047
Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J et al (2008) Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci 28(45):11445–11453.
Rapp A, Gmeiner B, Hüttinger M (2006) Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 88(5):473–483
Hamanaka H, Katoh-Fukui Y, Suzuki K, Kobayashi M, Suzuki R, Motegi Y et al (2000) Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet 9(3):353–361.
Moser VA, Pike CJ (2017) Obesity accelerates Alzheimer-related pathology in APOE4 but not APOE3 mice. eNeuro 4(3):ENEURO-0077
Chen Y, Durakoglugil MS, Xian X, Herz J (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107(26):12011–12016
Buttini M, Yu GQ, Shockley K, Huang Y, Jones B, Masliah E et al (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J Neurosci 22(24):10539–10548.
Sen A, Alkon DL, Nelson TJ (2012) Apolipoprotein E3 (apoE3) but not apoE4 protects against synaptic loss through increased expression of protein kinase Cϵ. J Biol Chem 287(19):15947–15958
Qiao F, Gao XP, Yuan L, Cai HY, Qi JS (2014) Apolipoprotein E4 impairs in vivo hippocampal long-term synaptic plasticity by reducing the phosphorylation of CaMKIIα and CREB. J Alzheimers Dis 41(4):1165–1176
Hwang KD, Bak MS, Kim SJ, Rhee S, Lee YS (2017) Restoring synaptic plasticity and memory in mouse models of Alzheimer’s disease by PKR inhibition. Mol Brain 10(1):57
Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I et al (2008) APOE ε4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from Central Norway. BMC Neurol 8(1):9.
Quiroga P, Calvo C, Albala C, Urquidi J, Santos J, Pérez H et al (1999) Apolipoprotein E polymorphism in elderly Chilean people with Alzheimer’s disease. Neuroepidemiology 18(1):48–52.
Kim KW, Jhoo JH, Lee KU, Lee DY, Lee JH, Youn JY et al (1999) Association between apolipoprotein E polymorphism and Alzheimer’s disease in Koreans. Neurosci Lett 277(3):145–148.
Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39(1):17
Raygani AV, Zahrai M, Raygani AV, Doosti M, Javadi E, Rezaei M et al (2005) Association between apolipoprotein E polymorphism and Alzheimer disease in Tehran, Iran. Neurosci Lett 375(1):1–6.
Rassas AA, Khiari HM, Fredj SH, Sahnoun S, Batti H, Zakraoui NO et al (2012) High APOE epsilon 4 allele frequencies associated with Alzheimer disease in a Tunisian population. Neurol Sci 33(1):33–37.
Kline A (2012) Apolipoprotein E, amyloid-ß clearance and therapeutic opportunities in Alzheimer’s disease. Alzheimers Res Ther 4(4):32
Fukumoto H, Ingelsson M, Gårevik N, Wahlund LO, Nukina N, Yaguchi Y et al (2003) APOE ε3/ε4 heterozygotes have an elevated proportion of apolipoprotein E4 in cerebrospinal fluid relative to plasma, independent of Alzheimer’s disease diagnosis. Exp Neurol 183(1):249–253.
Bekris LM, Millard SP, Galloway NM, Vuletic S, Albers JJ, Li G et al (2008) Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis 13(3):255–266.
Wahrle SE, Shah AR, Fagan AM, Smemo S, Kauwe JS, Grupe A et al (2007) Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener 2(1):7.
Talwar P, Sinha J, Grover S, Agarwal R, Kushwaha S, Srivastava MP et al (2016) Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer’s disease. J Neurol Sci 360:179–187.
Beffert U, Cohn JS, Petit-Turcotte C, Tremblay M, Aumont N, Ramassamy C et al (1999) Apolipoprotein E and β-amyloid levels in the hippocampus and frontal cortex of Alzheimer’s disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res 843(1–2):87–94.
Bray NJ, Jehu L, Moskvina V, Buxbaum JD, Dracheva S, Haroutunian V et al (2004) Allelic expression of APOE in human brain: effects of epsilon status and promoter haplotypes. Hum Mol Genet 13(22):2885–2892.
Pirttilä T, Soininen H, Heinonen O, Lehtimäki T, Bogdanovic N, Paljärvi L et al (1996) Apolipoprotein E (apoE) levels in brains from Alzheimer disease patients and controls. Brain Res 722(1–2):71–77.
Tachibana M, Shinohara M, Yamazaki Y, Liu CC, Rogers J, Bu G et al (2016) Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp Neurol 277:1–9.
Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S et al (2010) ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem 285(44):34144–34154.
Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC et al (2005) The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J Biol Chem 280(6):4079–4088.
Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK et al (2007) The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci 34(4):621–628.
Vanmierlo T, Rutten K, Dederen J, Bloks VW, van Vark-van der Zee LC, Kuipers F et al (2011) Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging 32(7):1262–1272.
Skerrett R, Pellegrino MP, Casali BT, Taraboanta L, Landreth GE (2015) Combined liver X receptor/peroxisome proliferator-activated receptor gamma agonist treatment reduces amyloid beta levels and improves behavior in amyloid precursor protein/Presenilin 1 mice. J Biol Chem 290(35):21591–21602
Hong C, Tontonoz P (2014) Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 13(6):433–444
Osei-Hwedieh DO, Amar M, Sviridov D, Remaley AT (2011) Apolipoprotein mimetic peptides: mechanisms of action as anti-atherogenic agents. Pharmacol Ther 130(1):83–91
Ghosal K, Stathopoulos A, Thomas D, Phenis D, Vitek MP, Pimplikar SW (2013) The apolipoprotein-E-mimetic COG112 protects amyloid precursor protein intracellular domain-overexpressing animals from Alzheimer’s disease-like pathological features. Neurodegener Dis 12(1):51–58
Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ et al (2012) APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis 10(1–4):122–126.
Minami SS, Cordova A, Cirrito JR, Tesoriero JA, Babus LW, Davis GC et al (2010) ApoE mimetic peptide decreases Abeta production in vitro and in vivo. Mol Neurodegener 5:16.
Handattu SP, Monroe CE, Nayyar G, Palgunachari MN, Kadish I, van Groen T et al (2013) In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J Alzheimers Dis 36(2):335–347.
Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE et al (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335(6075):1503–1506.
Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A et al (2016) Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther 8(1):4.
Boehm-Cagan A, Michaelson DM (2014) Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci 34(21):7293–7301
Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M et al (2013) Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am J Pathol 182(5):1750–1768.
Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D et al (2006) Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc Natl Acad Sci U S A 103(49):18787–18792.
Liu S, Breitbart A, Sun Y, Mehta PD, Boutajangout A, Scholtzova H et al (2014) Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J Neurochem 128(4):577–591.
Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM et al (2014) Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun 2:75.
Hao J, Zhang W, Zhang P, Liu R, Liu L, Lei G et al (2010) Abeta20-29 peptide blocking apoE/Abeta interaction reduces full-length Abeta42/40 fibril formation and cytotoxicity in vitro. Neuropeptides 44(4):305–313.
Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y (2012) Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci 32(14):4803–4811
Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H et al (2011) Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci 31(49):18007–18012.
Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE et al (2014) Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci ;34(21):7281–7292.
Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Stewart FR et al (2012) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med 209(12):2149–2156.
Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH (2008) Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement 4:179–192
Mahley RW, Huang Y (2012) Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem 55:8997–9008
Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG et al (2012) Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem 287:5253–5266.
Dow LE, Fisher J, O'Rourke KP, Muley A, Kastenhuber ER, Livshits G et al (2015) Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol 33:390–394.
Zhou W, Scott SA, Shelton SB, Crutcher KA (2006) Cathepsin D-mediated proteolysis of apolipoprotein E: possible role in Alzheimer’s disease. Neuroscience 143(3):689–701
Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T et al (2012) Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A 109:15502–15507.
Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM et al (2016) Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci Transl Med 8:332ra344.
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344
Kanekiyo T, Xu H, Bu G (2014) ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron 81(4):740–754
Getz GS, Reardon CA (2009) Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res 50(Suppl):S156–S161
Chan ES, Shetty MS, Sajikumar S, Chen C, Soong TW, Wong B-S (2016) ApoE4 expression accelerates hippocampus-dependent cognitive deficits by enhancing Aβ impairment of insulin signaling in an Alzheimer’s disease mouse model. Sci Rep 6:26119
Newport MT, VanItallie TB, Kashiwaya Y, King MT, Veech RL (2015) A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s. Alzheimers Dement 11(1):99–103
Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC, Clegg DJ (2012) Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol Aging 33(2):425.e19–425.e27
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485(7399):512–516.
Pankiewicz JE, Sadowski MJ (2017) APOE genotype and Alzheimer’s immunotherapy. Oncotarget 8(25):39941–39942
Pankiewicz JE, Baquero-Buitrago J, Sanchez S et al (2017) APOE genotype differentially modulates effects of anti-Aβ, passive immunization in APP transgenic mice. Mol Neurodegener 12:12.
Cramer PE, Cirrito JR, Wesson DW, Lee CD, Karlo JC, Zinn AE et al (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335(6075):1503–1506.
Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK et al (2007) The LXR agonist TO901317 selectively lowers hippocampal Aβ42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci 34(4):621–628.
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A et al (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 118(2):671–682.
Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H et al (2011) Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci 31(49):18007–18012.
Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J et al (2012) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med 209(12):2149–2156.
Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD et al (2004) A synthetic peptide blocking the apolipoprotein E/β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. Am J Pathol 165(3):937–948.
Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M et al (2010) Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Aβ clearance. J Biol Chem 285(29):22091–22102.
Luz I, Liraz O, Michaelson DM (2016) An anti-apoE4 specific monoclonal antibody counteracts the pathological effects of apoE4 in vivo. Curr Alzheimer Res 13(8):918–929
Acknowledgments
The authors are grateful to the Department of Pharmacy, Southeast University, Dhaka, Bangladesh. The authors wish to thank the anonymous reviewer(s)/editor(s) of this article for their constructive reviews.
Funding
The authors received no financial support for the research, authorship, and publication of this manuscript.
Author information
Authors and Affiliations
Contributions
This work was carried out in collaboration between all authors. MSU and GMA designed the study, wrote the protocol, and managed the analyses of the study. MSU, MTK, and AAM prepared the first draft of the manuscript. MMA-D, GEB, and GMA revised and improved the first draft. All authors read and approved the final submitted version of the manuscript.
Corresponding authors
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Uddin, M.S., Kabir, M.T., Al Mamun, A. et al. APOE and Alzheimer’s Disease: Evidence Mounts that Targeting APOE4 may Combat Alzheimer’s Pathogenesis. Mol Neurobiol 56, 2450–2465 (2019). https://doi.org/10.1007/s12035-018-1237-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1237-z