
Abstract
Purpose of Review
Chelation therapy is used for diseases causing an imbalance of iron levels (for example haemochromatosis and thalassaemia) or copper levels (for example Menkes’ and Wilson’s diseases). Currently, most pharmaceutical chelators are relatively simple but often have side effects. Some have been taken off the market.
This review attempts to find theory and knowledge required to design or find better chelators.
Recent Findings
Recent research attempting to understand the biological mechanisms of protection against iron and copper toxicity is reviewed. Understanding of molecular mechanisms behind normal iron/copper regulation may lead to the design of more sophisticated chelators. The theory of metal ion toxicity explains why some chelators, such as EDTA, which chelate metal ions in a way which exposes the ion to the surrounding environment are shown to be unsuitable except as a means of killing cancer cells.
The Lewis theory of acids and bases suggests which amino acids favour the attachment of the hard/intermediate ions Fe2+, Fe3+, Cu2+ and soft ion Cu+. Non-polar amino acids will chelate the ion in a position not in contact with the surrounding cellular environment. The conclusion is that only the soft ion binding cysteine and methionine appear as suitable chelators. Clearly, nature has developed proteins which are less restricted.
Recent research on naturally produced chelators such as siderophores and phytochemicals show some promise as pharmaceuticals.
Summary
Although an understanding of natural mechanisms of Fe/Cu regulation continues to increase, the pharmaceutical chelators for metal overload diseases remain simple non-protein molecules. Natural and synthetic alternatives have been studied but require further research before being accepted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metals are generally toxic to organisms. In higher organisms, these compounds interfere with functions of various organs, usually the central nervous system, haematopoietic system, liver and kidneys [1••]. However, many metals, including iron and copper in particular, are essential in trace amounts [2, 3]. It is well-known that transition metals have catalytic properties. Biological organisms combine one of two metal atoms to a protein to form important enzymes. Essential trace amounts of these metals are absorbed, usually in the diet, and are transported to the sites where the metal is added to the protein to make the functional enzyme. During transport, the metal is often tightly bound to chaperone proteins protecting the organism from the harmful effects of naked metal ions. Excess iron is safely stored as ferritin [4].
The Double Roles of Iron and Copper
Iron is necessary for the body for many iron-based enzymes such as cytochromes, catalase, hydrogenase, iron-responsive element-binding proteins (IRE-BPs), aconitase, as well as the essential oxygen storage molecule myoglobin and oxygen transporter haemoglobin [3]. Likewise, copper is an essential trace element for many biological functions such as removing superoxide radicals, formation of ATP, synthesis of important mediators and regulation of transcription [5–7]. It is the key component in many enzymes. Metalloproteins of copper include superoxide dismutases (SODs) such as human SOD1, human SOD3, plastocyanin, cytochrome c oxidase, laccase, nitrous-oxide-reductase, nitrite reductase and NADH dehydrogenase 2. Copper exists in lysyl oxidase (important in connective tissue). In particular, cytochrome oxidase is very important as an electron transport protein in respiration [8]. Copper is also important in mammalian gene expression, nerve myelation and endorphin action. Copper deficiency has been shown to impair immunity [2].
Unbound free iron and copper are toxic for reasons well known to researchers into the possible effects of ageing and free-radical damage during respiration. Hydrogen peroxide is produced both as a part of the respiration cycle and as a weapon to destroy pathogens by macrophages. Hydrogen peroxide is removed by catalase but not immediately. If free iron is present then hydrogen peroxide reacts with Fe(II) to produce hydroxyl radicals. This is known as the Fenton reaction. Equation (1) gives Fenton reaction for iron.
Hydroxyl radicals are very aggressive fast reacting species reacting with the nearest molecule which may be, in the worst cases, a protein, lipid or DNA molecule. Due to their highly reactive and transient nature, hydroxyl radicals cannot all be neutralised by antioxidants. Fe(II) is oxidised to Fe(III) which is insoluble. However, Fe(III) can be converted back to soluble Fe(II) and oxygen by reaction with the superoxide radical. This superoxide radical is also produced as an intermediate as part of the respiration cycle and is sufficiently long lived to be a problem. Thus, Fe(II) can potentially produce more hydroxyl radicals causing more damage to surrounding molecules [9]. This cycle is known as the Haber-Weiss reaction. This regeneration of Fe(II) is shown by Eq. (2).
Likewise, free copper is toxic as it can potentially take part in the Haber-Weiss reaction by moving between Cu(I) and Cu(II) generating hydroxyl radicals which can damage important surrounding biomolecules such as proteins, lipids, nucleic acids and interfering with iron-sulphur clusters. The corresponding equations for copper are (3) and (4).
Iron-sulphur clusters are essential for the activity of number of cellular enzymes with diverse essential functions [10, 11]. Copper can also displace zinc from cognate ligands in metalloproteins causing inappropriate protein structure/inhibition of enzymatic activity.
The unfortunate nature of iron, in relation to respiration intermediates, is superficially supported by the fact that during inflammation leads to a withdrawal of Fe from the body (hypoferric response) [12, 13]. While anaemia appears to be a natural response against iron toxicity, prolonged anaemia is harmful and a patient may become anaemic for other reasons.
Natural Regulation of Iron and Copper
The tight regulation of iron and copper levels and safe transport to sites of genesis of metalloproteins ensures that normally the toxic effects are minimised. Much progress in understanding iron and copper regulation has been made in recent years. A fuller account than given here can be found elsewhere [3, 14, 15].
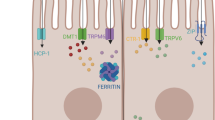
Briefly, iron and copper regulation involves sophisticated proteins such as ferritin, transferrin (Tf) for iron [16] and cytochrome c oxidase chaperone (COX17), copper chaperone for Zn, Cu superoxide dismutase (CCS) and antioxidant protein (ATOX) for copper [17]. In fact Rae et al. showed that the upper limit of any ‘free pools’ of intracellular copper was far less than one atom per cell in normal cases [18•]. A simplified model for the absorption of copper by intestinal enterocytes is shown in Fig. 1.

A simplified model for the absorption of copper by intestinal enterocytes. Ctr1 = copper transporter 1, DMT1 = divalent metal transporter 1, TGN = trans golgi network, MNK = Menkes’ protein (ATP7A), IREG1 = iron regulator transporter protein 1
The cellular import proteins for copper are copper transporter 1 (ctr1) and divalent metal transporter 1 (DMT1). The export proteins are ATPase copper transporting alpha protein (ATP7A) and ATPase copper transporting beta protein (ATP7B). Menkes’ disease is a result of a mutation of ATP7A resulting in a failure to mobilise dietary copper. Wilson’s disease is a result of a mutation of ATP7B and results in copper overload.
Copper also plays an important role in the mammalian immune system [2]. Macrophages, consequently, have to up-regulate copper in response to microbial pathogens when the body is infected.
The mechanism of iron homeostasis is particularly complex, compared to other metals, because iron cannot be simply eliminated as for other metals, at least in humans. Iron loss is through the shedding epithelial cells and menstruation. Consequently, iron must be absorbed from gut to keep balance.
The protein ferritin stores and transports iron. This remarkable protein can store up to approximately 4500 iron atoms. Transferrin (Tf) transports iron. Divalent transporter 1 (DMT1), as its name suggests, is a transporter of divalent metal ions and so transports Fe(II) as well as Cu(II).
In higher organisms, iron regulation is a systemic mechanism relying on a number of specialised cells with different functions. Iron uptake from the gut and release into the system is complex. Import proteins include DMT1 and heme carrier protein (HCP1). Export proteins are ferroportins (FPNs). Transferrin receptors (Tfrs) in association with ferroxidases as duodenal cytochrome B, ceruloplasmin (Cp) and heme carrier protein (HCP1) are involved in cellular membrane transportation of iron [14]. Iron is transported to the extracellular medium from certain cells by the protein ferroportin (FPN). This is important in the highly specialised cells such as neurons, erythrocytes, macrophages and enterocytes.
There are also iron regulatory proteins (IRPs), the most important being hepcidin (a hormone) which inhibits iron intake. Hepcidin levels are controlled by a gene regulation mechanism which is responsive to iron levels and demand. IRPs act on iron-responsive elements (IREs) finely tuning synthesis and suppression of the many proteins involved in iron regulation according to prevailing circumstances.
It is not fully understood in what form iron is present in the cytoplasm. It is believed that a small amount of iron forms a labile iron pool where Fe(II) is loosely bound to ligands of various kinds, as well being tightly and safely bound by chaperones. This labile iron pool is a very small fraction of the iron and must be tightly controlled to minimise participation in the redox reactions described earlier. However, the labile iron pool may be important as a ready available source of iron for incorporation into proteins. Mitochondria may obtain iron from this pool via the protein mitoferrin. There are several iron containing proteins made in the mitochondria as well as iron sulphur clusters.
Pharmaceutical chelators are a poor substitute to the natural mechanisms but necessary when the natural mechanisms are dysfunctional as in iron overload diseases (haemochromatosis, thalassaemia), anaemia (kidney malfunction, infection by parasitic worms, loss of blood), copper overload (Wilson’s disease) [19] and copper deficiency diseases (Menkes’ disease, occipital horn syndrome) [20]. Metal chelation is a simple way of shielding the metal ion to prevent biological damage mimicking the protein chaperones providing a therapy for metal poisoning. However, metal chelation may prevent the organism from obtaining the essential trace quantities of metals it needs. Many pharmaceutical chelators have side effects and some have been taken off the market.
General Factors to Consider in Drug Design of Chelators
Treatment of metal poisoning by a pharmaceutical involves an integrated drug design approach [1••]. The pharmaceutical ideally needs to remove the metal from its site of accumulation in the body. This means it has to reach the site, chelate the metal and be removed by the organism somehow. Both the pharmaceutical chelator and its resulting metal complex must not be toxic. There may be competition for the chelate by other metals. Competition may be because of a higher binding constant or a higher concentration. Predictability is limited [1••]. Sometimes a good candidate for a chelator in vitro turns out to be a bad choice in vivo. This may be because endogenous substances such as haemoglobin and cytochromes may compete with the chelator. The pharmaceutical has to have some resistance to biological transformation or breakdown.
In order to reach the site of the metal, the pharmaceutical may have to cross membrane barriers and so must be sufficiently lipophilic but not so much that it becomes stuck in the membrane. Likewise, the metal-pharmaceutical complex must be able to leave the site and be excreted by the body. If the complex is too bulky, its movement may be hindered. Most chelating agents are sensitive to pH, and this can have implications under pathological conditions like acidosis or alkalosis [1••]. If the chelating agent only gets into extracellular fluid, it may end up mobilising the metal so the metal actually accumulates in brain and fatty tissues.
Combination Therapy
Sometimes, a combination of two structurally different chelators is used when found to be more beneficial than using one chelator alone. The potential success of combination therapy is based on the possibility that two chelators will act through different mechanisms resulting in two distinct effects, or even support each other in a complementary manner leading to synergism. It is well known that hydrophobic vitamin E and hydrophilic vitamin C are synergetic partly due to being lipid soluble and water soluble, respectively. Likewise, hydrophobic and hydrophilic chelators could work well due to complementary properties, one removing metals from the extracellular regions and the other from the insides of cells and fatty tissues.
General Chemistry of Chelators
Metal ions can attach to ligands to form chelates. Ligands donate an electron pair to the metal ion to form a dative bond. Some ligands have just one donor pair of electrons and attach to the metal ion at one point. These are known as monodentate ligands (from Latin meaning ‘one-toothed’). Other ligands may donate several pairs of electrons and have several points of attachment to the metal ion. These are known as polydentate ligands. A ligand may have more than one different potential set of donor atoms. Which atom(s) donates might depend on factors such the metal ion present and also pH. Such ligands are known as ambidentate. Polydentate ligands can produce a chelate (from Greek meaning ‘claw’), a complex where the ligand forms a ring around the metal ion [21].
Since ligands donate electron pairs, it is useful to use the theory of Lewis acids and bases [22]. A Lewis acid is an electron pair acceptor and a Lewis base is an electron pair donator. With these definitions, the metal ion acts as a Lewis acid and ligands act as Lewis bases. A hard metal ion is loosely defined as one which retains valence electrons strongly. A hard metal ion usually has a small ion size and a high charge. Fe3+ is a good example of a hard metal ion. Conversely, a soft metal ion is one which is relatively large and does not retain electrons so firmly. Examples are heavy metal ions like mercury and lead but also the transition metal ion Cu+.
The above definitions are useful for predicting which parts of ambidentate ligands attach to which metal ions. As a rule, formation of stable complexes occurs when hard bases react with hard acids or soft bases react with soft acids. Hard bases (ligands) contain oxygen atoms as donor atoms. Also ligands which contain nitrogen atoms are usually described as hard to intermediate. This is consistent to the observed case of Fe(III) and Fe(II) interacting with O or N in heme or haemoglobin. The dative bonds formed are largely ionic (electrostatic) in character. On the other hand, ligands with electron donating groups containing sulphur or selenium are soft bases. In general, these form stable complexes with mercury, polonium, arsenic, lead and copper(I) which are soft metal ions. The dative bonds formed are covalent in character. In fact, mercury and arsenic also bond with the non-metal carbon to form organomercurials and organoarsenicals [21].
This chemistry can also be used to predict which atoms in amino groups bind to metals in metalloproteins. The following amino groups and their potential sites of metal ion attachment are given in Table 1.
The implications for copper biochemistry is that Cu(II) prefers oxygen found in aspartic and glutamic acid or imidazole nitrogen groups found in histidine while Cu(I) prefers thiol and thioether groups found in cysteine or methionine [21]. However, non-polar (hydrophobic) amino acids (cysteine and methionine) tend to be on inside of protein so metal chelation at these amino acid sites is protective. In contrast, uncharged polar and especially acidic and basic amino acids (all other amino acids considered above) tend to be on the outside of the protein [23]. Such proteins may chelate metals but the metal will be on the outside and exposed to the cytoplasm causing likely toxicity. This suggests, at first sight, that only soft ions like Cu(I) can be chelated by protein chelators. However, proteins are very versatile and could form an inner cavity to where the metal ion is somehow transported and safely positioned away from the cytoplasm. Nature seems to have succeeded in producing protein chelators (chaperones) but the detailed mechanisms of how the metal ion is placed in an unexposed part of the protein are complicated. The metal ions may attach to the part of the protein in the lipid membrane. Clearly, the design of synthetic protein metal ion chelators mimicking the natural system of regulation is not a simple task.
Furthermore, while the above is helpful in recognising biologically affinities, it is only based on the thermodynamics of chelation and does not give any information about the rates of chelation formation. Reaction rates are very relevant in biology. Porphin in haemoglobin is multidentate and thermodynamically stable under certain conditions. However, it is also important that the iron cannot be removed rapidly from binding sites by an exogenous competing chelator. This is a kinetic property and such a chelator is referred to as robust [21]. Chelation may involve several steps and may be impractically slow.
Chelators
Some Well-Known Examples of Chelators
Some of the problems of chelators used as pharmaceuticals are illustrated by some early well-known heavy metal chelators. These have well-known problems in clinical use and were mainly used to treat for heavy metals like arsenic, cadmium, mercury and lead poisoning (soft metal ions) rather than for iron and copper. While not directly relevant, they provide some insights into some of the problems of clinical chelators.
EDTA (=ethylendiamine-tetraacetic acid) is a polydentate chelator. Its donor atoms are O and N and so has high affinity for hard metal ions such as calcium, magnesium, iron, manganese and zinc. However, it has been used to treat lead poisoning. With Fe(III), it forms an open basket complex increasing catalytic activity (toxicity). For this reason and other reasons, it is unsuitable but important as a standard chelator in the study of chelation chemistry.
The two salts of EDTA which have been clinically used are Na2EDTA and CaNa2EDTA. Both had to be administered intravenously. Na2EDTA has high affinity for calcium and so was used for selected cases of hypercalcemia. On the other hand, the importance of calcium for metabolism also makes it potentially toxic causing severe electrolytic perturbations. It has caused serious side effects and deaths. Despite this, it is still sometimes used in the mistaken belief that it may remove calcium from atherosclerotic plaques and cause breakdown of plaques [24]. CaNa2EDTA was previously used in combination with British anti-Lewisite (BAL see below) for lead poisoning but treatment was found to be hazardous [25]. Without combination with other chelators, CaNa2EDTA has limitations and it has been replaced by other better chelators such as D meso-2,3-dimercaptosuccinic acid (MSA) (see below).
BAL (=British anti-Lewisite) (=2,3-dimercaprol) was developed during World War II and used clinically since 1949. It has a 3-carbon backbone with two –SH groups and a –OH group. It forms a five membered stable complex with arsenic and is most effective when administered immediately after exposure. It is oily and so not absorbed orally but by a deep painful intra-muscular injection [1••]. While it quickly mobilises arsenic and mercury, removing them from the body, it also causes these metals to be deposited in the brain. Furthermore, it causes many common adverse effects such as fever, eye inflammation, headaches and nausea. More serious complications such as liver damage and elevated blood pressure can also occur [1••].
DMSA (= meso-2,3-dimercaptosuccinic acid) was studied more than 20 years in China, Japan and Russia before it was recognised in the West. It is a dithiol compound and an analogue to BAL. In contrast to BAL, it can be administered orally. It is 95 % bound to plasma protein. This is probably due to the two –SH groups, one binding to cysteine residue on albumin and the other chelating the heavy metal [26].
It appears to be the least toxic of dithiol compounds and no sufficient loss of essential metals such as zinc, calcium and magnesium are observed. However, metabolism of copper may be altered [27]. Furthermore, the presence of dithiol groups may give it free-radical scavenging properties inhibiting lipid peroxidation.
Its main disadvantage is that it is unable to cross cell membranes and so can only reach its target heavy metal in the extracellular regions. This means that DMSA has to be administered shortly after metal poisoning before the heavy metals start to penetrate cells. Adverse reactions include gastrointestinal discomfort, skin reaction, mild neutropenia and elevated liver enzymes. Newer DMSA analogues (esters of DMSA) have been developed which improve its chelating and transport properties to the extent of overcoming the main disadvantage and crossing cell membranes.
Iron Chelators
Siderophores are small high-affinity iron chelating compounds secreted by microorganisms. For example, desferrioxamine or deferoxamine B, also known as deferral (Novartis), is produced by Streptomyces spp. [28•]. Siderophores compete with the body’s natural reserve of iron in ferritin. While deferral has been used in treatment, it is not surprising that siderophores cause problems by removing iron from the human’s natural iron proteins. This can weaken the system and promote infection by siderophore producing microorganism such as Yersinia enterocolitica and Candida albicans [29, 30].
Phytochemicals are another potential natural source of chelating compounds. Curcuminoids have been shown to bind in vitro and alleviate toxicity in thalassemic mice [31, 32]. The iron chelating activity of African walnut (Tetracarpidium conophorum) and wheat grass (Triticum aestivum) extracts have been studied and the results suggest this may have potential for iron overload in thalassaemia patients [33–35]. The African walnut contains a number of phenolic compounds which are believed to chelate to iron or copper [34]. The active chelators in wheat grass appear to be derivatives of mugineic acid, 3-hydroxy-mugineic acid and 2′-deoxy-mugineic acid [35]. More understanding of the role that chelation plays is needed as these natural compounds also affect gene control of foetal haemoglobin which helps thalassaemia patients [33].
Synthetically produced chelators also show promise. Ibuprofen has been shown to protect lipid peroxidation in vitro and phosgene-induced septic lung injury in rabbits due to chelation and suppression of ROS [36]. Some chelators, such as EDTA, do not fully cover the surface of Fe3+ (basket complex) [1••]. Thus, this exposes free ion to fluids of organism which may contain superoxide radicals and hydrogen peroxide. This may produce hydroxyl radicals (Fenton and Haber-Weiss reactions) which may be useful in killing cancer cells but harmful to proteins, lipids and DNA.
Kalinowski and Richardson suggest some potential new iron chelators which bind iron in stable (safe) manner [37•]. These are based on compounds like catechol, hydroxamate and hydroxypyridinone. The rationale is that they all provide hexadentate hydroxypyridinone functionality (like deferiprone) and so show promise as bacteriostatic agents in outcompeting siderophores of microbes. However, hydroxamate desferal and hydroxypyridinone deferiprone are actually assessable by various microorganisms [38]. The problem is that conventional chelators can end up providing microbes with an iron source.
Holbein and Mira reported that the new iron-chelating polymer containing hydroxypyridinone iron-ligand functionality developed by Chelation Partners Inc., known as DIBI, provided Fe-specific growth inhibition of C. albicans. [30]. DIBI is one example of a new approach to providing controlled molecular weight chelating-functional polymers. These may provide an additional advantage through providing compartmentalised sinks for iron thus reducing ROS or its bioavailability to pathogenic cancer or microbial cells.
Some of the most commonly used pharmaceutical chelators for iron are given in Table 2 and described in further detail below:
DFO = deferoxamine is a trihydroxamic acid secreted by Streptomyces pilosus. As it is a siderophore, it may compete with the normal system of iron control and may increase susceptibility to iron-loving pathogens. However, it has low affinity for other metals and is used to treat iron-related diseases such as thalassaemia major [39]. DFO needs to be administered mainly through intravenous injection or infusion (generally not more than 80 mg/kg/day). It is distributed mainly extracellularly and binding with proteins in the plasma is low (<10 %). It complexes with iron and is excreted rapidly as ferrioxamine mostly by the kidneys and one third into bile through faeces. Adequate urine output is thus important which can be facilitated by dialysis [1••].
Usually, DFO is well tolerated but the few cases of side effects include opthalamic and auditory toxicity, alterations in blood histology, allergic skin reactions and a few adverse effects on the lungs, kidneys and nervous system [40]. Also, as previously explained, patients may suffer some susceptibility to bacterial and fungal infections.
L1 = deferiprone = 1,2-dimethyl-3-hydroxypyrid-4-one is a suitable alternative to DFO in transfusional iron overload [41]. Clinical studies suggest a combination of DFO and L1 for 3 days a week to deplete iron overload and improve cardiac function in transfusion-dependent thalassaemia patients. DFO is known for reversing cardiac dysfunction. L1 has also shown cardio-protective effects but it is not clear whether this is a result of reducing iron overload or of some other mechanism.
L1 is administered orally rapidly being absorbed by the stomach and reaching circulation quickly. However, there may be a food-drug interaction or other factors delaying appearance of drug in the blood. It is mainly metabolised as glucuronide conjugates, excreted via the kidneys in the urine having a half-life of 47–134 min [1••]. However, a wide variation in metabolism and clearance has been observed among patients. This has been found to depend on iron overload and availability of chelatable iron [42]. An effective dose is believed to be 75–100 mg/kg/day in order to maintain a stable iron balance and reduce serum ferritin levels within 1 year of treatment in thalassaemia patients [1••].
Side effects during L1 therapy are arthropathy, gastrointestinal symptoms, headache, and moderate zinc deficiency. Adverse reactions can usually be reversed by reducing dose or discontinuing administration. Most patients have been able to continue with L1 therapy for a long time except, for a few patients, severe joint symptoms. The most severe complication is agranulocytosis or neutropenia although this is rare [43].
NTA = nitrilotriacetic acid is both an iron and copper chelator and is described below in copper chelators.
Copper Chelators
Brief descriptions of some copper chelators are given in Table 2 and below. The first three are in current pharmaceutical use. The fourth describes a group of chelators still undergoing investigation.
DPA or D-PEN = D-penicillamine = β-β-dimethylcysteine or 3-mercapto-D-valine is a sulfhydryl with amino acid and degradation product of penicillin. L-isomer causes optic neuritis and so not used. It was first used by Walshe to treat Wilson’s disease in 1957 [44]. It can be also be used for lead and mercury poisoning [45]. It has also been used as an anti-rheumatic drug [46••]. DPA can be administered orally or by intravenous route being 50 % absorbed orally but mainly extracellularly distributed. A small fraction is metabolised in the liver but most excreted unchanged in urine. Half-life elimination is 1 to 7 h or more.
DPA has side effects which disrupt the immune system, connective tissue and kidneys and may worsen neurological symptoms of patients with pre-existing neurological problems [47, 48]. Low adverse reactions have been reported but also more serious ones like thrombocytopenia and leukocytopenia (1–15 % incidence). In rare cases, aplastic anaemia may also occur. Prolonged DPA treatment can lead to anorexia, nausea and vomiting. Other toxic effects such as gastrointestinal disturbances (10–30 %), loss of taste (5–30 %), hair loss (1–2 %) and partly proteinuria (5–20 %) have also been reported. More severe adverse effects are autoimmune phenomena such as pemphigus, DPA-induced lupus erythematosus, polymyositis/dermatomyositis, membranous glomerulopathy and hypersensitivity pneumonitis [49]. Furthermore, DPA is a well-recognised teratogen and lathyrogen (causing skeletal, cutaneous and pulmonary abnormalities [50].
DPA is unsuitable (contraindication) for cases of renal insufficiency, patients allergic to penicillin, receiving gold therapy, antimalarial drugs, cytotoxic drugs, phenyl-butazone and oxyphenbutazone.
TETA = tetraethylenetetraamine or trientine is used to treat acute copper intoxication. It was introduced in 1982 as an alternative to DPA for patients intolerant to DPA [51] and believed to be safer [52•, 53]. Increased urinary excretion of copper is observed after its administration. Although it is usually administered orally, its absorption is relatively poor (5–18 % systemically absorbed). TETA is used to treat Wilson’s disease with a dosage of 0.75–2 g/day [1••].
Its two major metabolites are N 1 -acetyltriethylenetetramine (MAT) and N 1,N 10 -diacetyltriethylenetetramine (DAT). MAT appears to provide the role in the mechanism of extracting copper from the system [1••].
NTA = nitrilotriacetic acid is a polyamino carboxylic acid. It will chelate with Ca2+, Cu2+ and Fe3+. It is used similarly to EDTA but has the advantage of easy biogradability. NTA can be used in the form Na3NTA or FeNTA.
NTA also is able to mobilise nickel and manganese. While non-mutagenic in vitro high doses may produce epigenetic changes, perhaps related to sustained cytotoxicity of zinc ion transfer to urinary track. This causes damage to vacuolated cells of proximal tubules [54–56]. It is believed that both forms are potentially toxic, FeNTA causing iron overload, lipid peroxidation and genotoxicity [57, 58] and Na3NTA binding to zinc and calcium [59].
8-Hydroxyquinolines are a group of non-selective chelators which some studies show to have positive effects on Alzheimer’s patients and on in vivo tumours as well [60•, 61].
Clioquinol is a halogenated derivative of 8-hydroxyquinoline but was withdrawn from market in 1970s due to associations with subacute myelo-optic neuropathy. However, it remains a good prototype substance and 5,7-dichloro-2-[(dimethylamino)methyl]quinolin-8-ol (PBT2), a derivative of clioquinol, is currently under investigation as a marketable drug without the risk of neuropathy [62–64•].
Conclusion
Chelation therapy is a poor substitute to the natural polypeptides of the normally functioning copper and iron regulation systems. Furthermore, chelators may compete with the natural system and hoard copper, iron and perhaps other essential metals causing depleted levels for use in natural and essential metalloproteins. Therefore, there should be no surprise that chelators have side effects according to our current understanding. It is perhaps more surprising that they have been moderately successful in some cases.
Some alternatives to currently used chelators are being studied but require further research. Is it difficult to determine whether potential chelators are acting purely as chelators or are affecting the biochemistry in other ways for example some may affect gene expression.
Abbreviations
- ATOX:
-
Antioxidant protein
- ATP:
-
Adenosine triphosphate
- ATP7A:
-
ATPase copper transporting alpha protein/Menkes’ protein
- ATP7B:
-
ATPase copper transporting beta protein/Wilson disease protein
- BAL:
-
British anti-Lewisite/2,3-dimercaprol
- CCS:
-
Copper chaperone for Zn, Cu superoxide dismutase
- Cp:
-
Ceruloplasmin
- ctr1:
-
Copper transporter 1
- COX17:
-
Cytochrome c oxidase chaperone
- DAT:
-
N 1,N 10 -Diacetyltriethylenetetramine
- DFO:
-
Deferoxamine
- DIBI:
-
Iron-chelator developed by the company Chelation Partners Inc.
- DMSA:
-
Meso-2,3-dimercaptosuccinic acid
- DMT1:
-
Divalent metal transporter 1
- DNA:
-
Deoxyribonucleic acid
- DPA:
-
D-Penicillamine/β-β-dimethylcysteine/3-mercapto-D-valine
- D-PEN:
-
D-Penicillamine/β-β-dimethylcysteine/3-mercapto-D-valine
- EDTA:
-
Ethylendiamine-tetraacetic acid
- FPN:
-
Ferroportin
- HCP:
-
Heme carrier protein
- IRE:
-
Iron-responsive element
- IREG1:
-
Iron regulator transporter protein 1
- IRE-BP:
-
Iron-responsive element-binding protein
- IRP:
-
Iron regulatory protein
- L1:
-
Deferiprone/1,2-dimethyl-3-hydroxypyrid-4-one
- MAT:
-
N 1 -Acetyltriethylenetetramine
- MNK:
-
Menkes’protein/ATPase copper transporting alpha protein
- NADH:
-
Reduced form of nicotinamide adenine dinucleotide
- NTA:
-
Nitrilotriacetic acid
- PBT2:
-
5,7-Dichloro-2-[(dimethylamino)methyl]quinolin-8-ol
- SOD:
-
Superoxide dismutase
- SOD1:
-
Superoxide dismutase type 1
- SOD3:
-
Superoxide dismutase type 3
- spp.:
-
Plurality of species
- TETA:
-
Tetraethylenetetraamine/trientine
- Tf:
-
Transferrin
- Tfr:
-
Transferrin receptor
- TGN:
-
Trans golgi network
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Flora SJS, Vidhu P. Chelation in metal intoxication. Int J Environ Res Public Health. 2010;7:2745–88. doi:10.3390/ijerph7072745. Source of much of the basic information concerning chelators. This paper is quoted numerous times.
Rosenstein ED, Caldwell JR. Trace elements in the treatment of rheumatic conditions. Rheum Dis Clin N Am. 1999;25:929–35.
Dlouhy AC, Outten CE. The iron metallome in eukaryotic organisms. Met Ions Life Sci. 2013;12:241–78. doi:10.1007/978-94-007-5561-1_8.
Laufberger V. Sur la cristallisation de la ferritine. Bulletin de la Societe de chimie biologique. 1937;19:1575–82.
MacPherson IS, Murphy ME. Type-2 copper-containing enzymes. Cell Mol Life Sci. 2007;64:2887–99. doi:10.1007/s00018-007-7310-9.
Kosman DJ. Multi copper oxidases: a workshop on copper coordination chemistry, electron transfer, and metallophysiology. J Biol Inorg Chem. 2010;15:15–28. doi:10.1007/s00775-009-0590-9.
Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi:10.1016/j.tox.2011.03.001.
Cox DW. Disorders of copper transport. Br Med Bull. 1999;55:544–55.
Lane N. Oxygen the molecule that made the world. Oxford; 2002.
Brzóska K, Męczyńska S, Kruszewsk M. Iron-sulfur cluster proteins: electron transfer and beyond. Acta Biochim Pol. 2006;53:685–91.
Rouault TA. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech. 2012;5:155–64. doi:10.1242/dmm.009019.
Nairz M, Haschka D, Demetz E, Weiss G. Iron at the interface of immunity and infection. Front Pharmacol. 2014;5:1–10. doi:10.3389/fphar.2014.00152.
Cartwright GE, Lauritsen MA, Jones PJ, Merrill IM, Wintrobe MM. The anemia associated with chronic infection. Science. 1946;103:72–3. doi:10.1126/science.103.2664.72.
Waldvogel-Abramowskia S, Waeber G, Gassner C, Buser A, Frey BM, Favrat B, Tissot J-D. Transfus Med Hemother. 2014;41:165–9. doi:10.1159/000362888.
Vest KE, Hashemi HF, Cobine PA. Chapter 13 The copper metallome in eukaryotic cells. In Banci, Lucia Ed. Metallomics and the Cell. Metal Ions in Life Sciences 12. Springer; 2013.
Crichton RR, Charloteaux-Wauters M. Iron transport and storage. Eur J Biochem. 1987;164:485–506. doi:10.1111/j.1432-1033.1987.tb11155.x.
Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85. doi:10.1038/nchembio.72.
Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable free intracellular copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–8. doi:10.1126/science.284.5415.805. Paper reporting an important and surprising experimental finding.
Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482–93. doi:10.1038/ncpneuro0291.
Menkes JH, Alter M, Steigleder GK, Weakly DR, Sung JH. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–79.
Shriver DF, Atkins PW, Langford CH. Chapter 7 d-Metal complexes. In: Inorganic Chemistry, 5th ed., Oxford University Press; 2010.
Lewis G. Acids and bases. J Franklin Instit. 1938;226:293–313. doi:10.1016/S0016-0032(38)91691-6.
Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Chapter 2 Small molecules, energy, and biosynthesis. In: The Molecular Biology of the Cell, 2nd ed., Garland Publishing; 1989.
Seely DM, Wu P, Mills EJ. EDTA chelation therapy for cardiovascular disease: asystematic review. BMC Cardiovasc Disord. 2005;5:32.
Aaseth J. The effect of mercaptodextran on distribution and toxicity of mercuryin mice. Acta Pharmacol Tox. 1973;32:430–41. doi:10.1111/j.1600-0773.1973.tb01489.x.
Alan L, Miller ND. Dimercaptosuccinic acid (DMSA), a non-toxic, water-soluble treatment for heavy metal toxity. Altern Med Rev. 1998;3:199–207.
Aposian HV, Maiorino RM, Dart RC, Perry DF. Urinary excretion of meso-2,3-dimercaptosuccinic acid in human subjects. Clin Pharmacol Ther. 1989;45:520–6. doi:10.1038/clpt.1989.67.
Lehmann C, Islam S, Jarosch S, Zhou J, Hoskin D, Greenshields A, Al-Banna N, Sharawy N, Sczcesniak A, Kelly M, Wafa K, Cheliak W, Holbein B. The utility of iron chelators in the management of inflammatory disorders. Hindawi Publishing Corporation. Mediat Inflamm. 2015:1–12. doi:10.1155/2015/516740. A very recent paper.
Robins-Browne RM, Prpic JK. Effects of iron and desferrioxamine on infections with Yersina enterocolitica. Infect Immun. 1985;47:774–9.
Holbein BE, Mira de Orduna R. Effect of trace iron levels and iron withdrawal by chelation on the growth of Candida albicans and Candida vini. FEMS Microbiol Lett. 2010;307:19–24. doi:10.1111/j.1574-6968.2010.01956.x.
Srichairatanakool S, Thephinlap C, Phisalaphong C, Porter JB, Fucharoen S. Curcumin contributes to in vitro removal of non-transferrin bound iron by deferiprone and desferrioxamine in thalassemic plasma. Med Chem. 2007;3:469–74. doi:10.2174/157340607781745447.
Thephinlap C, Phisalaphong C, Fucharoen S, Porter JB, Srichairatanakool S. Efficacy of curcuminoids in alleviation of iron overload and lipid peroxidation in thalassemic mice. Med Chem. 2009;5:474–82. doi:10.2174/157340609789117912.
Aayush K, Wadhwa N, Tiwari A. Therapeutic role of natural agents in beta-thalassemia: a review. J Pharm Res. 2013;6:954–9. doi:10.1016/j.jopr.2013.10.002.
Olabinri BM, Eniyansoro OO, Okoronkwo CO, Olabinri PF, Olaleye MT. Evaluation of chelating ability of aqueous extract of Tetracarpidium conophorum (African walnut) in vitro. Int J Appl Res Nat Prod. 2010;3:13–8. doi:10.9734/EJMP/2012/1059.
Das P, Mukhopadhyay A, Mandal S, et al. In vitro studies of iron chelation activity of purified active ingredients extracted from Triticum aestivum Linn. (wheat grass). Eur J Med Plants. 2012;2:113–24.
Kennedy TP, Rao NV, Noah W, et al. Ibuprofen prevents oxidant lung injury and in vitro lipid peroxidation by chelating iron. J Clin Investig. 1990;86:1565–73. doi:10.1172/JCI114876.
Kalinowski DS, Richardson DR. Future of toxicology—iron chelators and differing modes of action and toxicity: the changing face of iron chelation therapy. Chem Res Toxicol. 2007;20:715–20. doi:10.1021/tx700039c. Paper reporting a possibly significant breakthrough in finding safe chelators.
Thompson MG, Corey BW, Si Y, Craft DW, Zurawski DV. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob Agents Chemother. 2012;56:5419–21. doi:10.1128/AAC.01197-12.
Giardina PJ, Grady RW. Chelation therapy in beta-thalassemia: an optimistic update. Semin Hematol. 2001;38:360–6. doi:10.1016/S0037-1963(01)90030-7.
Winship KA. Toxicity of aluminium: a historical review, part 2. Adverse Drug React Toxicol Rev. 1993;12:77–211.
Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood. 2003;102:17–24. doi:10.1182/blood-2002-06-1867.
Cappellini MD, Pattoneri P. Oral iron chelators. Annu Rev Med. 2009;60:25–38. doi:10.1146/annurev.med.60.041807.123243.
Kattamis A, Ladis V, Berdousi H, Kelekis NL, Alexopoulou E, Papasotiriou I, Drakaki K, Kaloumenou I, Galani A, Kattamis C. Iron chelation treatment with combination therapy with deferiprone and deferioxamine: a 12-month trial. Blood Cell Mol Dis. 2006;36:21–5. doi:10.1016/j.bcmd.2005.11.002.
Walshe JM. Penicillamine, a new oral therapy for Wilson’s disease. Am J Med. 1956;21:487–95. doi:10.1016/0002-9343(56)90066-3.
Roussaeux CG, MacNabb LG. Oral administration of D-pencillamine causes neonatal mortality without morphological defects in CD-1 mice. J Appl Toxicol. 1992;12:35–8. doi:10.1002/jat.2550120108.
Aaseth J, Skaug MA, Cao Y, Anderson O. Chelation in metal intoxication—principles and paradigms. J Trace Elem Med Biol. 2015;31:260–6. doi:10.1016/j.jtemb.2014.10.001. An excellent recent review and understanding of basic principles behind chelators.
Brewer GJ, Terry CA, Aisen AM, Hill GM. Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch Neurol. 1987;44:490–3. doi:10.1001/archneur.1987.00520170020016.
Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease. Gut. 2007;56:115–20. doi:10.1136/gut.2005.087262.
Grasedyck K. D-penicillamine-side effects, pathogenesis and decreasing the risks. Z Rheumatol. 1988;47:17–9.
Gupta B, Srivastava RK, Saxena KK, Prasad DN. A study on the penicillamine induced gastric ulceration in the rat. Ind J Pharmacol. 1980;12:247–52.
Walshe JM. Treatment of Wilson’s-disease with trientine (TriethyleneTetramine) dihydrochloride. Lancet. 1982;1:643–7. doi:10.1016/S0140-6736(82)92201-2.
Delangle P, Mintz E. Chelation therapy in Wilson’s disease: from D-penicillamine to the design of selective bioinspired intracellular Cu(I) chelators. Dalton Trans. 2012;41:6359–70. doi:10.1039/C2DT12188C. Good account of chelation therapy directed towards treating Wilson´s disease. Fairly recent paper.
Schilsky ML. Treatment of Wilson’s disease: what are the relative roles of penicillamine, trientine, and zinc supplementation? Curr Gastroenterol Rep. 2001;3:54–9. doi:10.1007/s11894-001-0041-4.
Alden CL, Kanerva RL, Anderson RL, Adkins AG. Short-term effects of dietary nitrilotriacetic acid in the male Charles River rat kidney. Vet Pathol. 1981;18:549–59. doi:10.1177/030098588101800413.
Hiasa Y, Kitahori Y, Konishi N, Shimoyama T. Dose-related effect of trisodium nitrilotnacetate monohydrate on renal tumorigenesis initiated with N-ethyl-N-hydroxyethylnitrosamine in rats. Carcinogenesis. 1985;6:907–10. doi:10.1093/carcin/6.6.907.
Miyashiro A. Promoting effect of trisodium nitrilotriacetate monohydrate on the development of tumors in kidneys and urinary bladders or rats treated with N-ethyl-N-hydroxyethylnitrosamine or N-butyl-N-(hydroxybutyl) nitrosamine. J Nara Med Assoc. 1984;35:550–65.
Hartwig A, Klyszcz-Nasko H, Schlepegrell R, Beyersmann D. Cellular damage by ferric nitrilitriacetate and ferric citrate in V79 cells: interrelationship between lipid peroxidation, DNA strand breaks and sister chromatid exchange. Carcinogenesis. 1993;14:107–12. doi:10.1093/carcin/14.1.107.
Umemura T, Hasegawa R, Sai-Kato K, Nishikawa A, Furukawa F, Toyokum S, Uchida K, Inouc T, Kurokawa Y. Prevention by 2-mercaptoethane sulfonate and N-acetylcysteine of renal oxidative damage in rats treated with ferric nitriltriacetate. Jpn J Cancer Res. 1996;87:882–6. doi:10.1111/j.1349-7006.1996.tb02115.x.
Anderson RL. The role of zinc in nitrilotriacetate (NTA)-associated renal tubular cell toxicity. Fd Cosmet Toxicol. 1981;19:639–50. doi:10.1016/0015-6264(81)90516-2.
Oliveri V, Giuffrida ML, Vecchio G, Aiello C, Viale M. Gluconjugates of 8-hydroxyquinolines as potential anti-cancer prodrugs. Dalton Trans. 2012;41:4530–5. doi:10.1039/C2DT12371A. Pertinent paper in showing how chelation therapy research has provided other important insights and possible applications, i.e. in cancer therapy.
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91.
Bareggi SR, Cornelli U. Clioquinoli review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci Ther. 2010;18:41–6. doi:10.1111/j.1755-5949.2010.00231.x.
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Sargum S, Bush AI, Murdoch R, Wilson J, Ritchie CW. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–86. doi:10.1016/S1474-4422(08)70167-4.
Crouch PJ, Barnham KJ. Therapeutic redistribution of metal ions to treat Alzheimer’s disease. Acc Chem Res. 2012;45:1604–11. doi:10.1021/ar300074t. Paper dealing with the highly popular topic of treating Alzheimer´s disease.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors M.K. Lawson, M. Valko, M.T.D. Cronin and K. Jomova declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Chemical and Molecular Toxicology
Rights and permissions
About this article

Cite this article
Lawson, M.K., Valko, M., Cronin, M.T.D. et al. Chelators in Iron and Copper Toxicity. Curr Pharmacol Rep 2, 271–280 (2016). https://doi.org/10.1007/s40495-016-0068-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-016-0068-8