
Abstract
The concomitant use of herbal products and synthetic drugs necessitates the assessment of their interaction potentials. The herbal hepatoprotective medicine, silybin A inhibits cytochrome P450 (CYP) 2C9 and 3A4 enzymes, thus, may interact with the drugs that are substrates of CYP2C9 and 3A4, such as losartan. The three most prominent genotypes, expressed by CYP2C9 are the CYP2C9*1/*1, CYP2C9*1/*2 and CYP2C9*1/*3. This study aimed to assess silybin A-losartan interaction in different CYP2C9 genotypes using physiological-based pharmacokinetic (PBPK) model approach. The individual PBPK models for silybin A and losartan were developed using PK-Sim®. Losartan pharmacokinetics was predicted with or without co-administration of silybin A in individuals of different CYP2C9 genotypes to find herbal-drug interaction. The predicted drug plasma curves and pharmacokinetic parameters were optimized using parameter identification tool and were compared with reported pharmacokinetic parameters from the published clinical studies for model validation. The silybin-losartan interactions were predicted by change in area under the curve (AUC) and peak systemic concentration (Cmax). The co-treatment of silybin A, 420 mg/24 h (140 mg/8 h) with losartan 50 mg/24 h, exhibited a genotype-dependent change in the losartan’s AUC and Cmax. In CYP 2C9*1/*1 genotype, AUC and Cmax of losartan were increased 1.16 and 1.37 folds, respectively falling in a range stipulated for negligible interaction. Increase in AUC and Cmax by 0.873 and 0.294 folds, respectively in CYP2C9*1/*3 after co-administration of silybin A exhibited a minor interaction with losartan. However, in individuals with CYP2C9*1/*2 genotype, the losartan’s AUC and Cmax were decreased by 0.01 folds, manifesting a moderate interaction. Hence, in CYP2C9*1/*1 and CYP2C9*1/*3 genotypes, silybin A is a weak CYP inhibitor for losartan while in CYP2C9*1/*2 genotype, the co-administration of silybin consequents into a moderate pharmacokinetic interaction with losartan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Occurrence of drug interactions is a global issue [1]. After co-administration of multiple drugs, e.g., over the counter products, foods or herbal medicines, most of the interactions may alter response due to changed systemic drug exposure [2]. Advances in scientific knowledge regarding the natural products and the perception of their lack of side effects, have persuaded the consumers to use herbal products [3]. Trend of the use of natural products is increasing in western pharmacotherapy because these are considered “natural”, which is linked with their safety [4]. However, the use of herbal medications increases the chances of drug-drug and drug-herbal interactions [5]. Silybum marianum L. Garnet., a medicinally important plant belongs to Asteraceae family and is used to treat liver disorders [6]. It contains 65–80% of flavonolignans silybin A, isosilybin B, silybin B, isosilybin A, silydianin, silychristin A, and silychristin B [7], yet silybin A is the major bioactive component. Silybin A, obtained from the Milk thistle or wild artichoke is among the top selling herbal product around the World [8]. Despite, the therapeutic effects of silybin, its safety cannot be overlooked [9]. Silybin A is reported to inhibit a variety of in-vitro CYP isozymes, e.g., CYP2C8, CYP2C9, CYP3A4, CYP2C19 and CYP2D6 [8], the most significant inhibition being observed for CYP2C9 and CYP3A4 [10]. It is also a P-glycoprotein (P-gp) inhibitor [11], though a consensus is awaited. Silybin A is 23–47% absorbed orally, an indication of its low bioavailability, secondary to its poor solubility [12] and poor permeability in gut epithelial cells. After absorption, silybin undergoes extensive enterohepatic circulation. Silymarin flavonolignans (silybin A and Silybin B) significantly inhibit organic anion transporters (OATP) in over-expressed cell lines and human hepatocytes [13]. Silybin A is metabolized by the intestinal and hepatic CYP2C9 and CYP3A4 enzymes. It is highly reactive with phase II conjugation and is rapidly eliminated [14], majorly through bile and 1.2% through urine with a half-life of 4 h [15, 16].
Silybin A is commonly prescribed and, self-administered along with various drugs such as losartan; an antihypertensive which is used for the treatment of heart failure [17]. When administered simultaneously with other drugs, silybin A may alter the effect of drugs because of its herb-drug interaction potential [18], as it inhibits P-gp, OATP, CYP3A4 and CYP2C9 [10, 11, 13]. Losartan, a prodrug is a non-peptide angiotensin II receptor antagonist, which inhibits the vasopressor effect of angiotensin II [19]. It is transported prominently by P-gp and to a lesser extent by other intestinal transporters [20]. Losartan assumes linear pharmacokinetics [21] and is bioavailable approximately 33% after oral administration [22]. It undergoes first pass metabolism [23] and is metabolized by the cytochrome P450 (CYP) 2C9 and 3A4 isozymes. OATPs are the influx transporters in hepatocytes and they play a role in losartan hepatic uptake [20]. Losartan is converted to an active metabolite, carboxylosartan, E-3174 approximately 14% by CYP2C9 through an aldehyde-natured intermediate metabolite, E-3179 [19, 21, 24], in known genotypes, except in CYP2C9*1/*3 [21]. Though the carboxylic acid metabolite of losartan is not a P-gp substrate, but displays considerably higher affinity for other transporters than the losartan [20]. The gene corresponding to CYP2C9 enzyme is highly polymorphic [25] and is prone to the single-nucleotide point mutations [26]. CYP2C9 has six polymorphic genes including: CYP2C9*1/*1, CYP2C9*1/*2, CYP2C9*1/*3, CYP2C9*2/*3, CYP2C9*2/*2 and CYP2C9*3/*3 [27]. Some polymorphisms are bland, such as CYP2C9*1/*1 which is a homozygous genotype and represents wild type activity [28]. A couple of genetic polymorphisms vary from the wild-type genotype by single point mutation [26], causing altered three-dimensional protein structure by changing amino acid sequence, which modifies the enzyme activity. For instance, the CYP2C9*1/*2 and CYP2C9*1/*3 genotypes show 12% and 5% lower enzyme activity for other drugs, respectively as compared to the wild type in different populations [29,30,31]. Furthermore, losartan is not bio-converted to EXP 3174, because later is absent in CYP2C9*3/*3 genotype [21]. The CYP2C9*1/*1 and CYP2C9*1/*3 variants of CYP2C9 are the major alleles while CYP2C9*1/*2 is rare in South-East and East-Asian populations [32]. The CYP genes’ polymorphism and its variability in genotype frequencies in different ethnic populations lead to altered drug response, therapeutic failure, toxicity or higher adverse drug reactions [33]. Losartan is secreted by OATP, similar to the classic p-aminohippurate (PAH) transporter [34]. Since losartan is a substrate for P-gp, OATP, CYP2C9 and CYP3A4 [35,36,37] and its gene coding for CYP2C9A are polymorphic [27], thus interactions of varying magnitude are expected in different genotypes with silybin A; an inhibitor of P-gp, OATP, CYP2C9 and CYP3A4 [10, 11, 13].
A real time study on silybin A-losartan interaction has already been carried out in human volunteers of two genotypes CYP2C9*1/*1 and CYP2C9*1/*3 in Chinese population [8]. To our best knowledge, the effect of silybin A on losartan pharmacokinetics has not been assessed in CYP2C9*1/*2 genotype. The recent Drug-Drug Interaction guidelines recommend using the modeling and simulation approaches to interpret drug-drug interactions [38]. Physiologically based pharmacokinetic (PBPK) modeling, a simulation approach is used to assess interaction involving herbs and drugs [39]. Hence, in this study, the herb-drug interactions between silybin A and losartan in different CYP2C9 genotypes, were assessed using the PBPK modeling.
Materials and methods
Software tools
PK-Sim®, Version 8.0.1 (Bayer Technology Services, Leverkusen, Germany) was used to develop PBPK models of silybin A and losartan. The blood level time profiles of silybin A and losartan from reference clinical studies were digitized using GetData Graph Digitizer, Version 2.25.0.32164. For making the graphs of senitivity analysis and goodness of fit plot, Microsoft Excel 2013 and statistical software R version 4.1.2 were used.
Development of PBPK model
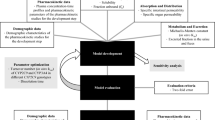
The scheme for the development of PBPK models is given in Fig. 1. Data from the reference clinical studies for silybin A and losartan [8, 21] were used to develop PBPK models in the virtual individual and then in the virtual population.

Workflow for the evaluation of drug interaction between silybin A and losartan through PBPK modelling
PBPK model of silybin A
The physicochemical properties of Silybin A including molecular weight, fraction unbound, lipophilicity, pKa, plasma protein binding and solubility, given in Table 1 were entered in the respective data nodes of PK-Sim. Demographics; including population ethnicity, population size, proportion of females, body mass index (BMI), height, age, weight, and the dose of silybin A (Table 2) were entered according to reference clinical study [8]. The formulation release was entered as Lint 80; the time interval added as an input parameter at which a dosage form releases 80% of the drug. For silybin A, 45 min was incorporated as an input parameter to release 80% of silybin A from its formulation [7]. The enterohepatic cycling of silybin A was taken into account in the model development. Hepatic Km and Vmax values of silybin A, respectively of 54 (µmol/l) and 138,000 (ml/l/min) were taken according to the reported study [4]. The PK-Sim-generated values for intestinal permeability (in cm/min), and biliary clearance were optimized, according to a procedure cited in literature [40] in order to accomplish the appropriate drug exposure according to the reference [4]. The value of CYP2C9 kinact was 0.221/min added in the inhibition parameters of PK-Sim, while reference values for CYP3A4 Ki and CYP3A4 kinact were 26.5 µmol/l and 10.22/min which were further optimized by PK-Sim, itself [40].
PBPK model of losartan
Table 1 shows the physicochemical properties (molecular weight, fraction unbound, lipophilicity, pKa, plasma protein binding and solubility) of losartan added in the respective data nodes of PK-Sim. Similarly, the demographics (population ethnicity, population size, proportion of females, BMI, height, age, weight) and dose of losartan (Table 2) were entered according to the reference [21, 47]. The values of losartan renal and hepatic clearance of 75 ml/min and 11.1 ml/min/kg, respectively, incorporated in the software were taken from the literature [41, 48]. Losartan release type was entered as Lint 80, whereby 15 min was entered for 80% losartan release. The reference values used for MDCK-MDR1 Km, Caco-2 km, MDCK-MDR1 Vmax and Caco-2 Vmax were 403.2 ± 15.2 µM, 232 ± 7.9 µM, 199.7 ± 8.8 nmol/cm2/min and 92.8 ± 2.9 nmol/cm2/min, respectively [49]. However, for losartan simulation, the values of intestinal permeability and renal clearance were optimized by PK-Sim [4]. Km and Vmax values for different genotypes were incorporated in the model development. The PK-Sim-default values for the small intestine transit and gastric emptying times for the drug were used. Individual models were combined through CYP2C9 and P-gp inhibition to determine the herb drug interaction between silybin A and losartan in CYP2C9*1/*1, CYP2C9*1/*2, and CYP2C9*1/*3 genotypes.
Parameter sensitivity analysis
The parameter sensitivity analysis of PBPK model was performed for silybin A, losartan and their interaction profile to assess the influence of already assessed model parameters on the simulated values of area under the curve (AUC). The sensitivity value of ± 1.0 indicates a change of 10% of the analysed parameters.
Validation of PBPK model
Model validation was performed by comparing the predicted and clinically acquired pharmacokinetic parameters obtained from literature [21, 47]. Furthermore, the simulated model of losartan in Korean population was designed only for the purpose of validation of losartan PBPK model and thus, no interaction was studied with the said simulation. The interaction model for CYP2C9*1/*1 and CYP2C9*1/*3 were validated using the reported clinical study in Chinese population [8]. For CYP2C9*1/*2 genotype, no interaction study was available in literature.
PBPK interaction model simulation
The model for silybin A (perpetrator drug) and losartan in Chinese (victim compound) were connected through the mechanism-based inhibition (irreversible) of hepatic CYP2C9 [4]. Simulations were conducted following an oral dose of 140 mg three times daily (420 mg/day) of silybin A to determine the clinical relevance of interactions, if any. For simulation of each genotype group, only the in-vitro metabolic rate was adjusted according to CYP2C9 genotype outcomes from the simulated profiles as given in Table 3.
Determination of magnitude of herbal-drug interaction
Among several criteria for inferring the magnitude of herb-drug interaction, the ratio of predicted AUC and Cmax after administration of losartan alone to the observed AUC after concomitant silybin A-losartan was considered. Where the above parameters were increased (i.e., where enzymes were inhibited), the interaction magnitude was interpreted as minor, if ratio was > 1.25 to < 2, moderate, i.e., ≥ 2 to 4.9 and severe, if the ratio was ≥ 5 [51]. When the above ratios of parameter were decreased after interaction (i.e., where the enzymes were induced), the ratios of the parameters were reversed and the same criteria was implemented for concluding the magnitude of interaction.
Results
Predicted pharmacokinetic parameters of silybin A and losartan in virtual individual and population
The predicted plasma concentration time profiles in comparison to the mean values of observed profile from reference clinical study of silybin A and losartan for their respective virtual individual and population are shown in Figs. 2 and 3. AUC after the administration of 140 mg TID silybin A tablet was 4.35 and 2.53 µmol.h/l in virtual individual and virtual population. The Cmax and tmax were 0.32 µmol/l, 3.2 h, for individual and 0.5 µmol/l, 2.75 h for population, respectively (Table 4). The AUC values after the administration of 50 mg losartan in Chinese virtual individual and population were 1082 ng.h/ml and 1125.65 ng.h/ml, respectively. The Cmax and tmax were 32.9 ng/ml and 1.1 h in individual and 289 ng/ml and 1.5 h, respectively in population. Furthermore, the AUC values after the administration of 50 mg losartan in Korean virtual individual and population were 1199.86 ng.h/ml and 1531 ng.h/ml, respectively. The Cmax and tmax were 440.76 ng/ml and 2.46 h in individual and 302 ng/ml and 2.8 h, respectively in population.

Predicted mean plasma concentration time profile of silybin A following 140 mg TID in comparison to clinical study [4] a virtual individual, b virtual population; shown as red dots: observed data, black solid line: predicted data

Predicted mean plasma concentration time profile of losartan following single oral administration of 50 mg compared to clinical studies [21, 47]: a Chinese virtual individual, b Chinese virtual population (blue and red dots): observed data (red and black solid lines): predicted data, shaded area shows 5th and 95th percentile, c Korean virtual individual and d Korean virtual population (green dots): observed data (black solid lines): predicted data, dotted lines shows 5th and 95th percentile
Predicted interaction of silybin A with losartan in different genotypes
The predicted interaction between silybin A 420 mg TID and losartan 50 mg OD in different CYP2C9 genotypes (Chinese) are given in Fig. 4 and Table 5. In virtual CYP2C9*1/*1 genotype, the AUC of losartan was increased from 1051 to1220 ng.h/ml (1.16 folds increase). The Cmax, was elevated from 250.88 to 346.07 ng/ml and tmax was decreased from 2.30 to 1.65 h (Table 6). In CYP2C9*1/*2 the AUC was decreased from 1211.85 to 22.57 ng.h/ml (− 4.6 folds), Cmax, decreased from 260.79 to 3.14 ng/ml (− 4.6 folds) and tmax was reduced from 2.30 to 1.35 h, respectively. In CYP2C9*1/*3 genotype the AUC and Cmax, were increased from 768.99 to 880.09 ng.h/ml (− 0.13 folds), and from 179.57 to 609.85 ng/ml (− 1.39 folds), respectively and tmax was reduced from 2.40 to 0.80 h (3 folds).

Predicted interaction of losartan 50 mg oral dosing with silybin A 420 mg multiple oral dosing in CYP2C9*1/*1 genotype: a individual, b population (blue line: silybin A concentration, red line: losartan concentration); CYP2C9*1/*2 genotype, c individual (blue line: silybin A concentration, red line: losartan concentration), d population (blue line: losartan concentration, red line: silybin A concentration), CYP2C9*1/*3 genotype, e individual, f population (Red line: losartan concentration, blue line: silybin A concentration) (color figure online)
Sensitivity analysis and goodness of fit plot
The results for sensitivity analysis and goodness of fit plot are shown in Fig. 5.

Sensitivity analysis for: a silybin A 140 mg, b losartan 50 mg, c interaction profile of silybin A 140 mg TID and losartan 50 mg OD and Goodness of fit plots (log scale) demonstrating the correlation of individual predicted (red) with observed (blue) for: (d) silybin A 140 mg, e losartan 50 mg, f interaction profile of silybin A 140 mg TID and losartan 50 mg OD).The solid line (black) represents the line of unity, the dashed line indicates 2 and 4-fold deviation (gray and black), respectively (color figure online)
Discussion
This study focused on prediction of impact of silybin A on the pharmacokinetics of losartan when co-administered in different CYP2C9 genotypes. PBPK models were developed, separately for the compound alone and in combination with herbal product. The drug physicochemical parameters, doses and the patient demographics were taken into consideration in the PBPK model development to obtain the same pharmacokinetic parameters as reported in the reference clinical studies for the respective drugs [4, 21]. The predicted models for both compounds were validated by comparing the observed pharmacokinetic parameters of both drugs to the in vivo studies. The sensitivity analysis and goodness of fit were employed to further optimize the predicted parameters for the developed models before and after interaction.
The oral PBPK model of silybin A was established by using a reference clinical study [4] at a dose of 140 mg TID. In the present study, the blood level time curve of silybin showed a rapid elimination; in line with the reported rapid conjugative metabolism along with excretion primarily through bile, resulting in shorter half-life and less systemic exposure [14]. A higher than the customary oral or intravenous doses are needed 2–3 times daily to overcome the extensive first pass metabolism of silybin A [52], that is a justification of TID dose of silybin A. The present predicted AUC of silybin in the virtual individual (4.35 µmol.h/l) was close to the reported AUC (5.92 µmol.h/l) [4] and in virtual population, it was lesser (2.53 µmol.h/l) due to interindividual variability. The predicted tmax of silybin A in virtual individual and population, 3.2 h and 2.75 h were close to the reported tmax (3.6 h) showing the validation of the developed PBPK model for the silybin A [4].
The model for losartan after 50 mg oral dosing was developed using reference study and the simulated pharmacokinetic parameters were reasonably well in accordance with the reference [21, 47, 48]. In model development, losartan metabolite was not considered as an individual compound, as metabolite concentration was dependent on the concentration of losartan. Therefore, entering losartan alone was deemed enough. Nevertheless, the influence on model without addition of metabolite could not be assessed. Furthermore, Silybin-A also inhibits losartan metabolism, leading to higher concentration of losartan’s active metabolite, E-3174 [53]. The predicted AUC of losartan in Chinese virtual individual (1082 ng.h/ml) and in population (1125.65 ng.h/ml) were close to the reported study [21]. The simulated tmax of losartan (1.1 h), was similar to the reported tmax of 1 h. However, the simulated tmax for the population (1.5 h) was slightly higher than the reported value possibly, due to interindividual variability. Moreover, the predicted AUC of losartan in Korean virtual individual (1199.86 ng.h/ml) and in population (1531 ng.h/ml) were close to the reference study [47]. The simulated tmax of losartan was 2.46 h and 2.8 h in individual and population, respectively. Both models were validated using the reported studies.
After co-administration of silybin A, AUC, Cmax and tmax were altered in a genotype-dependent manner. In the individuals of CYP2C9*1/*1 genotype, when, silybin A was co-administered, a 1.16 folds (ln ratio of 0.14) increase in AUC and 1.37 folds (ln ratio of 0.31) of losartan concentration was observed, which was comparable to the that of reported study in the same genotype [8]. Similarly, the inhibition of CYP2C9 in CYP2C9*1/*3 caused an increased in losartan AUC and Cmax, concentration to 0.873folds (ln ratio -0.13) and 0.294 folds (-1.39), respectively, also in accordance with the reported study in CYP2C9*1/*3 genotype [8]. Therefore, altered pharmacokinetic parameters in CYP2C9*1/*1 and CYP2C9*1/*3 genotypes could be ascribed to the silybin A-led inhibition of P-gp, and OATP but, primarily the inhibition of CYP2C9, an enzyme encoded by polymorphic gene and the major isoenzyme involved in oxidation [54]. An already reported 5% lower CYP2C9 activity [33] had not markedly effected the losartan systemic concentration in CYP2C9*1/*3 genotype. The genetic polymorphism of enzymes results in the interindividual variations in losartan oxidation and activation [50].
A strong inhibitor accounts for a > 5-fold (ln ratio of > 1.609) increase in AUC of substrate whereas a strong inducer causes a > 80% increase in clearance rate. Moderate inhibitors can produce a >2-fold (ln ratio of > 1.589) increase in AUC and a decrease clearance by 50–80%; while the weak inhibitors have capacity to cause a clinically relevant > 1.25– < 2-fold (ln ratio between 0.222 and 0.688) increase in AUC and a decrease in clearance rate up to 20–50% [55]. Based on the above criteria, silybin A in CYP2C9*1/*1 and CYP2C9*1/*3 genotype exhibited a minor interaction with losartan, since CYP2C9*1/*1 showed a wild type enzyme activity and CYP2C9*1/*3, exhibited only 5% lesser CYP2C9 activity [33]. Silybin A is a mechanism-based inhibitor of losartan metabolism that is why AUC was increased in these genotypes [56]. The reduced tmax indicated a faster absorption, resulting in prolongation of half-life and lessening of drug elimination due to lesser metabolism. All the above factors, albeit increased the systemic drug concentration, yet without a relevant interaction. Thus, silybin A in the above genotypes could be categorized as the weak inhibitor of losartan metabolism.
The enzyme coded with CYP2C9*1/*2 genotype manifests into a 12% decreased enzyme activity, thus in this genotype, the systemic losartan concentration has already been reported to be higher [33]. In this study, it was expected that the inhibition of P-gp, OATPs and CYP2C9 with co-administration of silybin A [10, 11, 13], would lead to an increase in systemic losartan concentration, thereby higher Cmax and AUC. Instead, in CYP2C9*1/*2 genotype, a tremendous decrease in losartan AUC and Cmax, to 0.01folds (ln ratio-4.6), was observed for both parameters, showing a moderate interaction between silybin A and losartan. A marked decrease in losartan absorption in the CYP2C9*1/*2 genotype was a surprise in this study. In an attempt to explain the above phenomenon, following reasons could be given.
The inhibition of the transporting function of P-gp, decreases the efflux (excretion) of drugs or their metabolites into urine, bile and intestinal lumen and increases their penetration and accumulation in tissues [57]. Nevertheless, most of the studies about silybin-led P-gp inhibition are pre-clinical, carried out in cells or animals, and indeed, the findings of the related clinical studies remained inconclusive. Milk thistle (containing 80% silymarin), at a daily dose of 900 mg does not affect P-gp substrate, digoxin [58], showing silybin has no effect on P-gp transporter. Contrarily, silymarin has been reported to significantly increase the plasma concentration of talinolol; a typical P-gp substrate [59], indicating inhibition of P-gp transporter. Silymarin has also been reported to increase the clearance of metronidazole with a concomitant decrease in half-life, Cmax, and AUC, which has been ascribed to the induction of both intestinal P-gp and CYP3A4 [60]. In line with the above, silybin A could be presumed to induce intestinal P-gp (and CYP3A4) in CYP2C9*1/*2 genotype, rather than inhibiting them. Furthermore, losartan itself is reported to be a P-gp inducer [57].
There may be a functional interaction between cytochrome P450 3A-dependent metabolism and P-gp transporter in a way, that losartan was converted to a metabolite [61]. An interplay exists between intestinal CYP enzymes and P-gp, causing a slow and steady concentration reaching to systemic circulation and facilitating a linear losartan pharmacokinetics [21]. If silybin was an inhibitor of the P-gp and CYP enzyme, this caused a lesser losartan metabolism, leading to a higher concentration that might initiate a saturation elimination kinetics where the half-life was usually prolonged while the systemic drug concentration was lesser. Furthermore, a higher level of protein binding of losartan, leaving less free drug in the blood could also be a reason for lower losartan concentration. Silybin A significantly inhibits OATP transports in hepatocytes which influx losartan into liver with an unestablished mechanism [20]. Thus, losartan’s hepatic uptake would be inhibited and lesser drug would be metabolised, leading to more concentration, a reason again for possible initiation of saturation kinetics. Losartan’s carboxylic acid metabolite which displays considerably higher affinity for the transporters, (other than P-gp) than the losartan, may compete for an unknown absorption-mediating transport of the losartan, leading to a lesser concentration. It can also be hypothesized, that the inhibitors or inducers of proteins, which are involved in the active transport of the losartan metabolite, may have an impact upon losartan absorption, although losartan itself might not be a substrate of such transporters [20]. A further reason for the decline in losartan’s AUC in CYP2C9*1/*2 genotype could probably be any other event(s) instead of the inhibition caused by silybin A. Nevertheless, to support the above phenomena, the literature is not available.
Furthermore, in genotype CYP2C9*1/*2, the lower predicted losartan concentration could be due to shortcomings in the PBPK model development. Nevertheless, the models were validated using the reported study for genotypes CYP2C9*1/*1 and CYP2C9*1/*3 [4, 8, 21] with in-built module of parameter identification as the predicted pharmacokinetic parameters were commensurable with the observed data. Therefore, the likelihood of potential perils in PBPK outcomes is very less. However, a real clinical and pharmacokinetics investigation is required because, hitherto no such studies are available for losartan with co-administered silybin A in the individuals of CYP2C9*1/*2 genotype. The above are important, not only to confirm the findings of this study but also because, any modulation of the CYP and P-gp activities could cause significant changes in the pharmacokinetics of losartan and its active metabolite, EXP3174 [62].
If a real time study confirms the present findings in CYP2C9*1/*2 genotype, there would be need for revisiting the assumption of inhibition of CYP2C9, when silybin A is concomitantly administered. The losartan concentration could be decreased in the genotype, to an extent where losartan might not produce its effect. Losartan, being a prodrug does not produce therapeutic effect till it is metabolized to active compounds. Administration of CYP450 inducer and inhibitor can lead to an exaggerated response or therapeutic failure, respectively. Administration of prodrug to the slower metabolisers can result in lower therapeutic response secondary to production of little or no active drug [63]. Therefore, the clinical strategy (dose, administration frequency of losartan) might be adjusted according to the genotype of the individuals. Furthermore, if the clinical studies confirm the modification of losartan in presence of silybin A, then its effect would also be expected for other commonly prescribed major CYP2C9-substrates. For instance, CYP2C9 is the major enzyme responsible for the metabolism of commonly prescribed drugs, e.g., S-warfarin, tolbutamide, glipizide, phenytoin and several non-steroidal anti-inflammatory drugs [25, 64]. Thus, such drugs are the good candidates for evaluation of drug-drug and herb drug interactions.
As stated, earlier, interaction magnitude could be inferred using a reported criterion [51]. Nevertheless, literature cites a couple of different criteria for different pharmacokinetic parameters, prominently AUC, Cmax, and clearance. The criterion in use requires reversing of ratio in case of the decrease in the AUC and Cmax, i.e., in enzyme-induction based interactions. To overcome the above limitations, the present study validated a literature-cited modified criterion obtained by converting the ratio of predicted AUC, Cmax and tmax to that of the observed respective parameters from the reference to log natural (ln). This ln-converted ratio, appropriately stated the effect-boundaries for interactions, with a side benefit of defining the nature of interactions, i.e., enzyme inhibition or induction, by the plus or negative signs, respectively, as shown in Table 6. The values of the proposed criteria, 0.222–0.688, 0.693–1.589 and greater than 1.609 reflect minor, moderate and higher interactions, respectively for enzyme inhibitors while the above values with negative signs demonstrate interactions magnitude for enzyme inducers.
Conclusion
Silybin A changed the AUC and Cmax of losartan in a genotype-dependent manner. The pharmacokinetic interactions between silybin A and losartan were minor when concomitantly administered at normal doses in genotypes CYP2C9*1/*1 and CYP2C9*1/*3. However, moderate interaction was found in CYP2C9*1/*2 genotype. Therefore, in clinical practice dose adjustment of losartan might not be required in CYP2C9*1/*1 and CYP2C9*1/*3 genotypes as silybin A weakly inhibited the losartan metabolism in the above genotypes while, dose adjustment might be considered in CYP2C9*1/*2 genotype. Nevertheless, real clinical studies are required to substantiate the above findings.
References
Huang SM, Lesko L (2004) Drug-drug, drug—dietary supplement, and drug—citrus fruit and other food interactions: what have we learned? J Clin Pharmacol 44(6):559–569
Arnold RJ, Tang J, Schrecker J, Hild C (2018) Impact of definitive drug–drug interaction testing on medication management and patient care. Drugs-Real World Outcomes 5(4):217–224
Vargesson N (2015) Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today 105(2):140–156
Brantley S, Gufford B, Dua R, Fediuk D, Graf TN, Scarlett Y, Frederick K, Fisher M, Oberlies N, Paine M (2014) Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb–drug interaction. Pharmacomet Syst Pharmacol 3(3):1–9
Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA (2002) Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA 287(3):337–344
Bijak M (2017) Silybin, a major bioactive component of milk thistle (Silybum marianum L. Gaernt.)—Chemistry, bioavailability, and metabolism. Molecules 22(11):1942
Monographs O United States Pharmacopeia-National Formulary (USP 25-NF 20). In: Rockville, Maryland: United States Pharmacopeial Convention Inc (2002)
Han Y, Guo D, Chen Y, Chen Y, Tan Z-R, Zhou H-H (2009) Effect of silymarin on the pharmacokinetics of losartan and its active metabolite E-3174 in healthy Chinese volunteers. Eur J Clin Pharmacol 65(6):585–591
Wesołowska O, Łania-Pietrzak B, Kużdżał M, Stańczak K, Mosiądz D, Dobryszycki P, Ożyhar A, Komorowska M, Hendrich AB, Michalak K (2007) Influence of silybin on biophysical properties of phospholipid bilayers 1. Acta Pharmacol Sin 28(2):296–306
Beckmann-Knopp S, Rietbrock S, Weyhenmeyer R, Böcker RH, Beckurts KT, Lang W, Hunz M, Fuhr U (2000) Inhibitory effects of silibinin on cytochrome P-450 enzymes in human liver microsomes. Pharmacol Toxicol 86(6):250–256
Chung SY, Sung MK, Kim NH, Jang JO, Go EJ, Lee HJ (2005) Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch Pharmacal Res 28(7):823–828
Gunaratna C, Zhang T (2003) Application of liquid chromatography–electrospray ionization-ion trap mass spectrometry to investigate the metabolism of silibinin in human liver microsomes. J Chromatogr B 794(2):303–310
Köck K, Xie Y, Hawke RL, Oberlies NH, Brouwer KLJDM, Disposition (2013) Interaction of silymarin flavonolignans with organic anion-transporting polypeptides. 41(5):958–965
Xie Y, Zhang D, Zhang J, Yuan J (2019) Metabolism, transport and drug–drug interactions of silymarin. Molecules 24(20):3693
Lorenz D, Lücker P, Mennicke W, Wetzelsberger N (1984) Pharmacokinetic studies with silymarin in human serum and bile. Methods Find Exp Clin Pharmacol 6(10):655–661
Barzaghi N, Crema F, Gatti G, Pifferi G, Perucca E (1990) Pharmacokinetic studies on IdB 1016, a silybin-phosphatidylcholine complex, in healthy human subjects. Eur J Drug Metab Pharmacokinet 15(4):333–338
Kjeldsen S, Stålhammar J, Hasvold P, Bodegard J, Olsson U, Russell D (2010) Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens 24(4):263–273
Venkataramanan R, Ramachandran V, Komoroski BJ, Zhang S, Schiff PL, Strom SC (2000) Milk thistle, a herbal supplement, decreases the activity of CYP3A4 and uridine diphosphoglucuronosyl transferase in human hepatocyte cultures. Drug Metab Dispos 28(11):1270–1273
Goa KL, Wagstaff AJ (1996) Losartan potassium. Drugs 51(5):820–845. https://doi.org/10.2165/00003495-199651050-00008
Soldner A, Benet LZ, Mutschler E, Christians, (2000) Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and Caco-2 cell monolayers. Br J Pharmacol 129(6):1235–1243
Lo MW, Goldberg MR, McCrea JB, Lu H, Furtek CI, Bjornsson TD (1995) Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin Pharmacol Ther 58(6):641–649
Munafo A, Christen Y, Nussberger J, Shum LY, Borland RM, Lee RJ, Waeber B, Biollaz J, Brunner HR (1992) Drug concentration response relationships in normal volunteers after oral administration of losartan, an angiotensin II receptor antagonist. Clin Pharmacol Ther 51(5):513–521
Gardiner P, Paine SW (2011) The impact of hepatic uptake on the pharmacokinetics of organic anions. Drug Metab Dispos 39(10):1930–1938
Sachinidis A, Ko Y, Weisser P, Zu Brickwedde Meyer M, Düsing R, Christian R, Wieczorek AJ, Vetter H (1993) EXP3174, a metabolite of losartan (MK 954, DuP 753) is more potent than losartan in blocking the angiotensin II-induced responses in vascular smooth muscle cells. J Hypertens 11(2):155–162
Van Booven D, Marsh S, McLeod H, Carrillo MW, Sangkuhl K, Klein TE, Altman RB (2010) Cytochrome P450 2C9-CYP2C9. Pharmacogenet Genom 20(4):277
Cabaleiro T, Román M, Ochoa D, Talegón M, Prieto-Pérez R, Wojnicz A, López-Rodríguez R, Novalbos J, Abad-Santos F (2013) Evaluation of the relationship between sex, polymorphisms in CYP2C8 and CYP2C9, and pharmacokinetics of angiotensin receptor blockers. Drug Metab Dispos 41(1):224–229
Kirchheiner J, Brockmöller J (2005) Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther 77(1):1–16
Miners JO, Birkett DJ (1998) Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol 45(6):525–538
Ingelman-Sundberg M, Rodriguez-Antona CJPTotRSBBS (2005) Pharmacogenetics of drug-metabolizing enzymes: implications for a safer and more effective drug therapy. 360(1460):1563–1570
D’ambrosio R, D’andrea G, Cafolla A, Faillace F, Margaglione M (2007) A new vitamin K epoxide reductase complex subunit-1 (VKORC1) mutation in a patient with decreased stability of CYP2C9 enzyme. J Thromb Haemost 5(1):191–193
Sosa-Macías M, Lazalde-Ramos B, Galaviz-Hernandez C, Rangel-Villalobos H, Salazar-Flores J, Martínez-Sevilla V, Martínez-Fierro M, Dorado P, Wong M-L, Licinio J (2013) Influence of admixture components on CYP2C9* 2 allele frequency in eight indigenous populations from Northwest Mexico. Pharmacogenomics J 13(6):567–572
Kurose K, Sugiyama E, Saito Y (2012) Population differences in major functional polymorphisms of pharmacokinetics/pharmacodynamics-related genes in Eastern Asians and Europeans: implications in the clinical trials for novel drug development. Drug Metab Pharmacokinet 27(1):9–54
Dorji PW, Tshering G, Na-Bangchang K (2019) CYP2C9, CYP2C19, CYP2D6 and CYP3A5 polymorphisms in South-East and East Asian populations: a systematic review. J Clin Pharm Therap 44(4):508–524
Sekine T, Cha SH, Endou H (2000) The multispecific organic anion transporter (OAT) family. Pflugers Arch 440(3):337–350
Zhang S, Morris ME (2003) Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. J Pharmacol Exp Ther 304(3):1258–1267
Zuber R, Modrianský M, Dvořák Z, Rohovský P, Ulrichová J, Šimánek V, Anzenbacher P (2002) Effect of silybin and its congeners on human liver microsomal cytochrome P450 activities. Phytotherap Res 16(7):632–638
Jančová P, Anzenbacherova E, Papoušková B, Lemr K, Lužná P, Veinlichová A, Anzenbacher P, Šimánek V (2007) Silybin is metabolized by cytochrome P450 2C8 in vitro. Drug Metab Dispos 35(11):2035–2039
Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, Dickins M, Phipps A, Darekar A, Hyland R (2009) Comparison of different algorithms for predicting clinical drug-drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos 37(8):1658–1666
Brantley S, Gufford B, Dua R, Fediuk D, Graf TN, Scarlett Y, Frederick K, Fisher M, Oberlies N, Paine M (2014) Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb–drug interaction. CPT 3(3):1–9
Rashid M, Sarfraz M, Arafat M, Hussain A, Abbas N, Sadiq MW, Rasool MF, Bukhari NI (2020) Prediction of lisinopril pediatric dose from the reference adult dose by employing a physiologically based pharmacokinetic model. BMC Pharmacology and Toxicity 21:56
Chem P (2020-02-08) Losartan https://pubchem.ncbi.nlm.nih.gov/compound/11751549.
Tronde A, Nordén B, Jeppsson A-B, Brunmark P, Nilsson E, Lennernäs H, Bengtsson UH (2003) Drug absorption from the isolated perfused rat lung–correlations with drug physicochemical properties and epithelial permeability. J Drug Target 11(1):61–74
Bolouri-Moghaddam MR, Le Roy K, Xiang L, Rolland F, Van den Ende W (2010) Sugar signalling and antioxidant network connections in plant cells. FEBS J 277(9):2022–2037
Ježko P, Žufková V, Remko M (2015) Modelling of absorption, distribution and physicochemical properties of AT1 receptor antagonists/Modelovanie absorpcie, distribúcie a fyzikálnochemických vlastnosti antagonistov AT1 receptorov. Europ Pharmac J 62(2):20–31
DrugBank (2015) Silybinin. https://www.drugbank.ca/drugs/DB09298. 2019
Yamasaki K, Sato H, Minagoshi S, Kyubun K, Anraku M, Miyamura S, Watanabe H, Taguchi K, Seo H, Maruyama T (2017) The binding of silibinin, the main constituent of silymarin, to site I on human serum albumin. Biol Pharmac Bull 40(3):310–317
Bae J-w, Choi C-i, Kim M-j, Oh D-h, Keum S-k, Park J-i, Kim B-h, Bang H-k, Oh S-g, Kang B-s (2011) Frequency of CYP2C9 alleles in Koreans and their effects on losartan pharmacokinetics. Acta Pharmacol Sin 32(10):1303–1308
Nguyen HQ, Lin J, Kimoto E, Callegari E, Tse S, Obach RS (2017) Prediction of losartan-active carboxylic acid metabolite exposure following losartan administration using static and physiologically based pharmacokinetic models. J Pharm Sci 106(9):2758–2770
Soldner A, Benet LZ, Mutschler E, Christians UJBjop (2000) Active transport of the angiotensin‐II antagonist losartan and its main metabolite EXP 3174 across MDCK‐MDR1 and Caco‐2 cell monolayers. 129(6):1235–1243
Yasar Ü, Tybring G, Hidestrand M, Oscarson M, Ingelman-Sundberg M, Dahl M-L, Eliasson E (2001) Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab Dispos 29(7):1051–1056
Wiggins BS, Saseen JJ, Page RL, Reed BN, Sneed K, Kostis JB, Lanfear D, Virani S, Morris PB (2016) Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation 134(21):e468–e495
Javed S, Kohli K, Ali M (2011) Reassessing bioavailability of silymarin. Altern Med Rev 16(3):239
Sica DA, Gehr TW, Ghosh S (2005) Clinical pharmacokinetics of losartan. Clin Pharmacokinet 44(8):797–814
Dorji PW, Tshering G, Na-Bangchang K (2019) CYP2C9, CYP2C19, CYP2D6 and CYP3A5 polymorphisms in South-East and East Asian populations: A systematic review. J Clin Pharm Ther 44(4):508–524
Palleria C, Di Paolo A, Giofrè C, Caglioti C, Leuzzi G, Siniscalchi A, De Sarro G, Gallelli L (2013) Pharmacokinetic drug-drug interaction and their implication in clinical management. J Res Med Sci 18(7):601
Hanley MJ, Masse G, Harmatz JS, Court MH, Greenblatt DJ (2012) Pomegranate juice and pomegranate extract do not impair oral clearance of flurbiprofen in human volunteers: divergence from in vitro results. Clin Pharmacol Ther 92(5):651–657
Tanigawara YJTdm (2000) Role of P-glycoprotein in drug disposition. 22(1):137–140
Gurley BJ, Barone GW, Williams DK, Carrier J, Breen P, Yates CR, Song P-f, Hubbard MA, Tong Y, Cheboyina SJDm, disposition (2006) Effect of milk thistle (Silybum marianum) and black cohosh (Cimicifuga racemosa) supplementation on digoxin pharmacokinetics in humans. 34(1):69–74
Han Y, Guo D, Chen Y, Tan Z-R, Zhou H-HJX (2009) Effect of continuous silymarin administration on oral talinolol pharmacokinetics in healthy volunteers. 39(9):694–699
Rajnarayana K, Reddy MS, Vidyasagar J, Krishna DRJA (2004) Study on the influence of silymarin pretreatment on metabolism and disposition of metronidazole. 54(02):109-113
Stieger B, Hagenbuch B (2014) Organic anion-transporting polypeptides. In: Current topics in membranes, vol 73. Elsevier, pp 205–232
Li H, Liu L, Xie L, Gan D, Jiang X (2016) Effects of berberine on the pharmacokinetics of losartan and its metabolite EXP3174 in rats and its mechanism. Pharm Biol 54(12):2886–2894
Lynch T, Price A (2007) The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician 76(3):391–396
Lee CR, Goldstein JA, Pieper JA (2002) Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenet Genom 12(3):251–263
Funding
Nil.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tanveer, A., Hussain, K., Tasneem, H. et al. Prediction of CYP-mediated silybin A-losartan pharmacokinetic interactions using physiological based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn 49, 311–323 (2022). https://doi.org/10.1007/s10928-022-09804-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-022-09804-0