
Abstract
Aromatic l-amino acid decarboxylase deficiency (AADC-DY) is caused by one or more mutations in the DDC gene, resulting in the deficit in catecholamines and serotonin neurotransmitters. The disease has limited therapeutic options with relatively poor clinical outcomes. Accumulated evidence suggests the involvement of neurodegenerative mechanisms in the etiology of AADC-DY. In the absence of neurotransmitters’ neuroprotective effects, the accumulation and the chronic presence of several neurotoxic metabolites including 4-dihydroxy-L-phenylalanine, 3-methyldopa, and homocysteine, in the brain of subjects with AADC-DY, promote oxidative stress and reduce the cellular antioxidant and methylation capacities, leading to glial activation and mitochondrial dysfunction, culminating to neuronal injury and death. These pathophysiological processes have the potential to hinder the clinical efficacy of treatments aimed at increasing neurotransmitters’ synthesis and or function. This review describes in detail the mechanisms involved in AADC-DY neurodegenerative etiology, highlighting the close similarities with those involved in other neurodegenerative diseases. We then offer novel strategies for the treatment of the disease with the objective to either reduce the level of the metabolites or counteract their prooxidant and neurotoxic effects. These treatment modalities used singly or in combination, early in the course of the disease, will minimize neuronal injury, preserving the functional integrity of neurons, hence improving the clinical outcomes of both conventional and unconventional interventions in AADC-DY. These modalities may not be limited to AADC-DY but also to other metabolic disorders where a specific mutation leads to the accumulation of prooxidant and neurotoxic metabolites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pathophysiology of AADC-DY
Aromatic l-amino acid decarboxylase deficiency (AADC-DY) is a rare autosomal recessive neurometabolic disorder caused by one or more mutations in the DDC gene, leading to a deficit in the AADC, the final enzyme in the biosynthesis of the monoamine neurotransmitters, known to catalyze the decarboxylation of l-3,4-dihydroxyphenylalanine (L-dopa) to dopamine and of L-5-hydroxytryptophan (5-HTP) to serotonin [1]. The deficit in dopamine synthesis can affect the three main brain dopaminergic pathways: the nigro-striatal pathway, the meso-limbic pathway, and the meso-cortical pathway, leading to motor and cognitive dysfunctions [2], whereas serotonin deficit has a significant impact on neurodevelopment with adverse effects on mood and behavior [3].
The affected individuals often present with high levels of metabolites upstream from the metabolic defect such as L-dopa, 3-O-methyldopa (3-OMD), and 5-HTP, and decreased levels of metabolites downstream from the metabolic defect, including homovanillic acid (HVA), 3-methoxy-4-hydroxyphenylglycol (MHPG), norepinephrine (NEP), epinephrine (EP), and 5-hydroxyindoleacetic acid (5-HIAA). As a result, AADC-deficient (DT) subjects experience neurodevelopmental delay, movement disorders; including oculogyric crises, hypotonia, dystonia, and hypokinesia; and autonomic dysregulations including sleep disorder and mood disturbances [4, 5]. These clinical symptoms are often manifested during the first months of life, although there is a considerable variability in the clinical presentation and severity of symptoms, depending in part to the degree of the impact of the enzyme deficiency on the synthesis of each of the 4 neurotransmitters, serotonin, dopamine, and its catecholamine derivatives, NEP and EP [4]. The disease has a high prevalence in Taiwan due to the Chinese DDC founder mutation [6].
Treatment of AADC-DY
The treatment modalities of AACD-DY, either the conventional or the unconventional, follow closely that of Parkinson’s disease (PD), a disease involving a selective loss of nigrostriatal dopaminergic neurons, where disease progression and cell death leads to the loss of brain AADC enzyme activity [7].
Dopamine agonists are the first line of treatment of AADC-DT patients, with the objective to increase brain monoamine neurotransmitter production, and more common are the ergot-derived dopamine agonists such as bromocriptine and pergolide [8]. Although these drugs have been shown to improve some of the clinical symptoms of the AADC-DY, they are known to cause fibrotic reactions at the level of heart, lung, and retroperitoneal space; also, such reactions are less likely with the non-ergot-derived dopamine agonists [9]. Bromocriptine, the most prescribed dopamine agonist, is an antidiabetic drug able to reduce plasma glucose and hepatic glucose productions [10]. Studies in rodents show that these effects of bromocriptine are accompanied by a concurrent significant reduction in the serotonergic and noradrenergic activities, indicated by reduced levels of the metabolites 5HIAA, MPG, and HVA in the ventromedial hypothalamus [11]. Therefore, the use of bromocriptine in AADC-DT subjects, who have neurotransmitters’ deficit and are often hypoglycemic, would likely be contraindicated. Similarly, the treatments with L-dopa and 5-HTP are contraindicated since AADC-DT subjects are unable to metabolize the high level of endogenous L-dopa and 5-HTP due to AADC enzyme deficiency, leading to poor clinical responses [4, 8].
An additional treatment modality for AADC-DY is the use of monoamine oxidase (MAO) inhibitors, with the objective to minimize dopamine and serotonin breakdowns. This regimen is often combined with dopamine agonists and or pyridoxine/pyridoxal phosphate, the AADC enzyme cofactor. Anticholinergics, melatonin, benzodiazepines, and α2-adrenergic agonists are among less common modalities to treat the AADC-DY clinical symptoms [4, 8, 12]. Although these different regimens either singly or combined have shown some clinical efficacy, most have unwanted side effects and the overall clinical outcomes of these limited treatment modalities remain poor [12, 13]. The non-pharmacological treatment of AADC-DT subjects includes ongoing physical, occupational, and speech therapies [8].
The newer treatment of AADC-DY has the objective to correct the AADC-related genetic defect, increasing brain dopamine production, using the recombinant adeno-associated viruses (AAV), the vector for the delivery of the functional DDC gene. Gene transfer in AADC-DY is based on earlier studies in PD patients receiving bilateral intraputamenal infusion of the non-pathogenic, low immunogenic AAV2 vector containing the human (h) DDC gene as a mean to enhance the AADC enzyme activity in the post-synaptic cells that express dopamine receptors, thus promoting the local metabolism of the administered L-dopa [14]. These studies reported improvements in the mean scores on the Unified Parkinson’s Disease Rating Scale [15, 16], as well as increases in the short- and long-term AADC enzyme activities [16].
The studies of AAV2 vector-mediated intraputamenal hDDC gene transfer in AADC-DY were conducted by Hwu and colleagues [17, 18] in Taiwan, reporting improvements in motor functions and increase in brain dopamine levels, indicated by enhanced putamen’s uptake of the AADC tracer 6-[(18) F]fluorodopa on PET imaging; these improvements were sustained at a 5-year follow-up [19]. A modification to the AAV2-mediated gene transfer in AADC-DT subjects was carried out by Pearson et al. [20], selecting the midbrain, instead of putamen, as the anatomical site, based on the notion that dopaminergic neurons in the midbrain and their axonal projections remain intact in the AADC-DT subjects [21]. The investigators reported increased dopamine in the site of gene delivery, in both putamen and caudate nucleus due to anterograde axonal transport via the intact nigrostriatal pathway, and improvements in both motor and non-motor functions. Based on these clinical outcomes, the AAV2-mediated functional hDDC gene transfer has been recently approved in Europe for the treatment of ≥ 18-month-old subjects with confirmed AADC-DY based on clinical, molecular, and genetic tests [22]. Nevertheless, the shape of the skull bones, if unsuitable for stereotactic surgery, and a pre-existing immunity to AAV2 limit the applicability of the AAV2-mediated gene transfer for AADC-DY treatment [18].
Accumulated Metabolites Induce Neuroinflammation
Among the underlying pathologies, which are overlooked in AADC-DY, are oxidative stress and inflammation, two processes that can lead to neuronal injury and death. In the following paragraphs, we describe the role of various metabolites and their potential to induce, either singly or in a synergistic manner, oxidative stress, glial activation, and neuroinflammation, thus promoting neuronal injury and death. We will highlight the similarities between those and mechanisms involved in other neurodegenerative diseases. This new concept of AAD-DY as a neurodegenerative disease would likely open a path to novel treatment strategies, that when used early in the course of the disease, would likely minimize neuronal injury, preserving the functional integrity of neurons, hence improving the clinical outcomes of both conventional and unconventional AADC-DY interventions. The effects of accumulated metabolites and the pathways to neuroinflammation are depicted in Fig. 1.

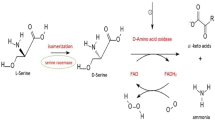
Adverse effects of upregulated metabolites in the brain of AADC-DT subjects. The deficit in dopamine to exert inhibitory effect on tyrosine hydroxylase enzyme leads to the accumulation of brain L-dopa and subsequent activation of the COMT enzyme [23], enhancing 3-OMD [4, 24], which in turn leads to the increase in the rate of o-methylation with the subsequent reduction in SAM and the accumulation of Hcy, SAH [25, 26], and HCA [27]. These processes enhance excitotoxity and reduce the level of the antioxidant precursor, cysteine [28]. In addition, metabolite-induced oxidative stress and excitotoxicity, singly or in synergy, reduce the activity of BCE [29], CSE [30], and MST [31] enzymes, as well as reduce the activity of the cofactor Trx in neurons [32], all of which are involved in the production of antioxidants and neuroprotective agents such as H2S, GSH, ALA, and SOD. The metabolite-induced prooxidant environment and the reduced cellular antioxidant capacity leads to glial activation [33,34,35,36], mitochondrial dysfunction [37], and neuroinflammation, promoting neuronal injury and death. Oxidative stress brought about L-dopa and Hcy has the potential to upregulate the KP pathway [38, 39], with the generation of QA which exerts neurotoxic effects via both increase in glutamate release and stimulation of NMDA receptors [40]. These many adverse effects of metabolites, combined with their ability to inhibit AADC enzyme activity [41], would have the potential to impede the clinical efficacy of treatments aiming to enhance neurotransmitters’ synthesis and or functions. The red highlights present the upregulated and the green highlights present the downregulated metabolites and products. The brown highlights present the enzymes, and the yellow highlights present the cofactors. Abbreviations: AADC, aromatic L amino acid decarboxylase; Ado, adenosine; ALA, α-lipoic acid; CAT, cysteine aminotransferase; CBS, cystathionine β-synthase; COMT, catechol-O-methyl-transferase; CSE, cystathionine γ-lyase; Cys, cysteine; Cyst, cystathionine; DA, dopamine; DHLA, dihydrolipoic acid; DOPAL, 3,4-dihydroxyphenylacetaldehyde; Glu, glutamine; GSH, glutathione; HCA, homocysteic acid; Hcy, homocysteine; H2S, hydrogen sulfide; 5-HT, serotonin; 5-HTP, 5-hydroxytryptophan; Ketog, ketoglutarate; KP, kynurenine pathway; L-dopa, 3,4-dihydroxy-L-phenylalanine; MAO, monoamine oxidase; MAT, methionine adenosyl transferase; Met, methionine; 3-MP, 3-mercaptopyruvate; MS, methionine synthase; MST, 3-mercaptopyruvate sulfurtransferase; MT, methyl transferase; MTHF, N-5-methyl tetrahydrofolate; 3-MT, 3-methoxytyramine; NMDAR, N-methyl-D-aspartate receptor; 3-OMD, 3-O-methyldopa; Os-Inf, oxidative stress-inflammation; QA, quinolinic acid; Qnes, quinones; SAH, s-adenosylhomocysteine; SAM, s-adenosylmethionine; SOD, superoxide dismutase; Tr, tryptophan; TrH, tryptophan hydroxylase; Trx, thioredoxin; Tyr, tyrosine; TyrH, tyrosine hydroxylase
Among metabolites known to be upregulated in AADC-DY are L-dopa and its major metabolite 3-OMD, which is produced by the enzymatic action of catecholamine-o-methyltransferase (COMT) enzyme. The increase in the levels of the two metabolites is the result of the lack of feedback inhibition, which is normally exerted by the end-product, dopamine, on tyrosine hydroxylase enzyme. Since dopamine is not produced, a high level of L-dopa accumulates, which is subsequently metabolized to 3-OMD, in a reaction that consumes the methyl donor, s-adenosylmethionine (SAM). The accelerated rate of o-methylation leads to the enhanced production of s-adenosylhomocysteine (SAH) which is rapidly converted to homocysteine (Hcy) and adenosine.
3-Methyldopa (3-OMD)
The major metabolite, 3-OMD, is produced from L-dopa in many organs including the brain, peripheral tissues, and the blood [25]. Comparing the level of 3-OMD and L-dopa, the CSF level of 3-OMD in AADC-DT subjects is many times higher than that of L-dopa [4, 24]. CSF levels of L-dopa in AADC-DT subjects range between 2 and 549 nm (reference range < 25 nm) whereas 3-OMD levels range between 54 and 4293 nm (reference range < 100 nm) [42]. There are several reasons for the higher CSF levels of 3-OMD compared to the levels of L-dopa.
The 1st reason is related to the differences in the biological half-life of the two metabolites. The half-life of L-dopa is about 1 h, that of 3-OMD is 15 h [43]. As a result, the ratio of 3-OMD to L-dopa concentration can be as high as 14:1 [44], leading to brain 3-OMD accumulation in AADC-DT subjects, with the potential to exert oxidative stress and cytotoxicity [45]. In PD patients, treated with L-dopa, the high concentration of plasma 3-OMD [46, 47] and the ratio of 3-OMD to L-dopa correlate with both dyskinesia [48] and poor response to L-dopa therapy [49]. In addition, the high CSF levels of 3-OMD correlate with the wearing-off phenomenon post-L-dopa therapy in PD [50], suggesting an association between 3-OMD levels and motor dysfunction. Although 3-OMD is a marker of AADC-DY diagnosis [51, 52], there are no studies investigating the relationship between 3-OMD or 3-OMD/L-dopa and the heterogeneity in the AADC-DY clinical presentation; or heterogeneity in response to treatment, whether conventional or post-AAV2-mediated gene transfer.
The 2nd reason for the higher CSF 3-OMD levels in AADC-DY is related to COMT enzyme characteristics, the activity of which is substrate-dependent. COMT activity is enhanced due to the excess L-dopa availability [23], leading to accelerated o-methylation rate of L-dopa to 3-OMD, through consumption of SAM. Although L-dopa initially increases SAM levels due to augmented expression and activity of methionine adenosyl transferase (MAT) [23], the enzyme that produces SAM, the significant increase in the rate of o-methylation subsequently decreases SAM levels and increases brain SAH [25, 26]. The SAM/SAH ratio is known to be an indicator of cellular methylation correlating with glutathione/oxidized glutathione (GSH/GSSG) ratio [53], an indicator of cellular antioxidant capacity and redox homeostasis, associated with neurodegeneration [54]. The increased rate of o-methylation can also accelerate dopamine methylation rate by COMT enzyme and the formation of the metabolite 3-methoxytyramine [23], which has low biological activity as a result of reduced binding affinity to dopamine D2 receptors [55]. Methylated dopamine can reduce the efficacy of AADC-DY treatments targeted to enhance dopamine function.
The 3rd reason for the higher 3-OMD levels may be related to the differential transport of L-dopa and 3-OMD across the blood–brain barrier (BBB). Like in the CSF, the level of 3-OMD in the plasma of AADC-DT subjects is high, approximating 2312% of the normal median range [24]. Both L-dopa and 3-OMD can cross the BBB into the CNS via a facilitated transport mechanism which is shared with other amino acids [56]. This bidirectional transport is inequal between L-dopa and 3-OMD, as 3-OMD is able to compete with the CNS uptake of L-dopa at the BBB [57].
In vivo studies show that a single intracerebroventricular (icv) injection of 3-OMD decreases dopamine turnover rate in the rat striatum and inhibits dopamine transporter and uptake in the brain striatal membrane, and 3-OMD administration for 5 days significantly reduces catecholamine levels and impairs locomotor activities [45]. These adverse effects have been attributed to the 3-OMD ability to exert oxidative stress and cytotoxicity [45]. 3-OMD is also able to enhance the L-dopa prooxidant and cytotoxic effects [45]. Therefore, the high 3-OMD level and accumulation in the AADC-DT brain have the potential to interfere with dopamine synthesis and/or function, and reduce the efficacy of treatment modalities aiming to enhance dopamine production and function. Therefore, 3-OMD has the potential to contribute neuronal injury and death, both directly and through enhancing L-dopa adverse effects, as described in the following paragraphs.
4-Dihydroxy-L-Phenylalanine (L-dopa)
The effects of L-dopa on dopaminergic neurons have been extensively studied in relation to PD. L-dopa, the precursor of dopamine, in combination with carbidopa, which prevents peripheral breakdown of L-dopa by AADC enzyme, is a common treatment modality in PD, aiming to restore brain dopamine levels. However, long-term L-dopa administration and the resulting high brain L-dopa levels were thought to play a role in neuronal injury in PD [58], and concerns have been raised on the contribution of chronic high levels of L-dopa to PD progression [59]. Studies in experimental models of PD report several protein expression profile differences and increased ratio of post-translational protein modifications in the dopamine depleted side of the brain when compared to the corresponding intact side [60]. In addition, proteomic studies in animal model of PD show the sensitivity of dopamine-depleted striatum to the first ever L-dopa exposure, causing irreversible rapid post-translational modification-based changes to several proteins involved in chronic L-dopa adverse effects [61].
L-dopa Autooxidation
L-dopa can exert neurotoxicity via several mechanisms. Undergoing autoxidation, L-dopa is able to generate hydroxyl and superoxide radicals, semiquinones, and quinones [62]. A recent rodent study shows that oral administration of 100 mg/kg L-dopa for 30 days significantly enhanced oxidative stress in rat striatum, indicated by a reduced GSH/GSSG ratio [63]. L-dopa also increased the level of malondialdehyde [63], a marker of lipid peroxidation, and led to a significant increase in glial fibrillary acidic protein (GFAP) immune expression. GFAP, a cytoskeletal intermediate filament, has long been used as an astrocyte-specific marker of cell activation and proliferation. However, astrocyte proliferation is not a universal response to all damages and can occur in the absence of disease and depends on the context of inflammation and neurodegeneration [64]. Histological examination of the striatal tissues showed L-dopa-induced microglial activation, neurons exhibiting degenerative changes and apoptotic cell death [63]. Similar results were reported in in vitro studies showing a relationship between L-dopa-induced oxidative stress and mitochondrial dysfunction, cell apoptosis, and reduction in neuronal cell survival [59, 65,66,67,68,69,70,71] in time- and dose-dependent manners [65, 72]. These adverse effects are enhanced in the presence of 3-OMD [45].
Several in vitro studies report protective effects of sub-toxic concentrations of L-dopa in neurons cocultured in the presence of glial cells [73, 74]. The neuroprotective effects of L-dopa in neurons-glial coculture were thought to be related to the L-dopa’s ability to induce the release of GSH, the glial major cellular antioxidant enzyme [74], an effect that seems to be independent of dopamine production [75]. The release of glial GSH is attributed to the ability of a compound to auto-oxidize [69]. As auto oxidizable compounds, subtoxic concentrations of both L-dopa [73] and dopamine [76] are able to induce GSH release from glial cells. However, L-dopa loses this ability in the absence of AADC enzyme [69] and in the presence of high levels of 3-OMD [74] and transition metals [77], suggesting that AADC-DT neurons are highly prone to injury. In young children, the under-developed excretory system can lead to accumulation of the transition metals, especially in under-developed countries due to higher daily exposure [78], a fact that could increase L-dopa neurotoxicity in AADC-DT children living in these regions. Furthermore, dopaminergic neurons and their projection targets are especially vulnerable to L-dopa cytotoxicity, since these neurons are known to possess relatively low glia cells’ density [79].
One could argue that the observed cytotoxicity may be attributed to 3-OMD and or dopamine rather than L-dopa. Although 3-OMD does not undergo autooxidation [69], it is still capable of inducing oxidative stress [45]. Similar to L-dopa, dopamine undergoes autooxidation, with the generation of reactive quinones [80]. L-dopa also undergoes enzymatic degradation by MAO with the generation of hydrogen peroxide and hydroxyl radicals [81]. Nevertheless, L-dopa cytotoxicity seems to be dopamine [75], 3-OMD- [67], and MAO- [65] independent, since it is apparent in the presence of the AADC inhibitor, NSD-1015, the COMT inhibitor, Ro 41–0960, and the MAO inhibitor, deprenyl, respectively. Therefore, despite the relatively short biological half-life, the chronic presence of L-dopa in the brain of AADC-DT subjects has the potential for significant neurotoxicity, independent of 3-OMD, but in synergy with it. In addition, L-dopa-induced prooxidant environment is enhanced in the absence of dopamine, as it is the case in AADC-DY, since dopamine is known to upregulate GSH synthesis, as well as enhance GSH trafficking from glial cells to neurons [82].
Mechanisms Involved in Neuroinflammation
Glial Activation
Chronic oxidative stress brought about by L-dopa and 3-OMD, and by homocysteine (Hcy) (discussed in the following paragraphs), can injure neurons, triggering the recruitment and activation of the microglia, the resident innate immune cells of the brain, which are known to normally regulate several physiological processes required for proper neuronal survival and brain function. Microglial activation has the potential for secreting a wide array of proinflammatory mediators including cytokines, chemokines, reactive oxygen–nitrogen species, and excitotoxins, such as glutamate, resulting in a vicious cycle of neuroinflammation, culminating to neuronal cell death, with dopaminergic neurons to be especially susceptible to neurotoxicity [33, 34]. These pathological processes play major roles in many pathophysiological processes that underly PD [83,84,85], Alzheimer’s disease (AD) [86, 87], and amyloid lateral sclerosis (ALS) [88, 89].
Microglial activation is sustained by a self-feedback loop involving the proinflammatory mediator, the high-mobility group protein box-1 (HMGB1), assisting with the transcription of proinflammatory genes [90]. In addition, there is a crosstalk between microglia and astrocytes, the other type of glial cells. Astrocytes proliferation and activation can be induced through proinflammatory cytokines secreted from the activated microglia [35, 36], further fueling the cascade of neuroinflammation. Therefore, microglia and astrocytes’ activation and the intimate crosstalk among the two are fundamental events in neuroinflammation. The damaged neurons can further activate glial cells by releasing ATP and other damage-associated molecular patterns (DAMPs) such as HMGB1 and nucleotides. Interacting with pattern recognition receptors, DAMPS can further perpetuate the cycle of inflammation [91].
Astrocytes are one of the most numerous glial cells in the CNS. Astrocytes’ reactivity is highly heterogenous and depends on the type of stimuli with the potential of two distinct functional phenotypes termed A2 as anti-inflammatory and neuroprotective, and A1 as proinflammatory [36, 64]. The A2 can upregulate neurotrophic factors and thrombospondins, able to promote neuronal growth and support synaptic repair [92]. A2 reactive astrocytes also produce antioxidative molecules in response to oxidative stress able to protect dopaminergic neurons [93], and have the potential for preventing glutamate excitotoxicity by taking up the glutamate released into the synaptic cleft [94].
IL-1α and TNFα, secreted from activated microglia, induce the cytotoxic A1 reactive astrocytes’ phenotype [35, 36]. A1 astrocytes lose the ability to promote neuronal survival, outgrowth, and synaptogenesis, and upregulate proinflammatory factors, IL-1α, IL-1β, and TNF-α [36], leading to the amplification of the microglia-driven inflammatory responses, which when combined with the astrocytes’ loss of glutamate-removing activity [95], would promote a neurotoxic environment. The reduced astrocytic glutamate uptake is the result of increased glutamate derived from activated microglia, which downregulates the expression of astrocytic glutamate transporters [96]. Therefore, the increase in inflammatory mediators and glutamate acts in concert to perpetuate neuronal cell death. Furthermore, astrocytes have the capacity to protect neurons against oxidative damage due to the preferential activation in astrocytes of the nuclear factor erythroid-2-related factor 2 (Nrf2), a transcription factor that governs an array of detoxifying and antioxidant enzymes [97]. However, reactive astrocytes may lose the ability to protect neurons due to Nrf2 downregulation [98].
The A1 phenotype is abundant in many neurodegenerative diseases such as PD, AD, and ALS, contributing to death of neurons and oligodendrocytes [36]. The role of astrocytes in the context of neuroinflammation in AADC-DY is specifically important since striatal astrocytes express dopaminergic receptors and D4-mediated signal transduction in response to dopamine [99]. The dysfunction of astrocytes combined with the absence of dopamine neuroprotective effects [82, 100] would have deleterious effects on dopaminergic neurons in AADC-DY. In addition, striatal astrocytes act as a reservoir, taking up the excess L-dopa [101]. However, the activated astrocytes may have an impaired ability to take up the excess L-dopa, similar to the loss of glutamate-removing activity [95].
Mitochondrial Dysfunction
As dynamic organelles, mitochondria adapt to physiological or environmental changes in cellular requirements, by processes of transport, biogenesis, fusion, fission, and mitophagy, the latter refers to the removal of damaged mitochondria through autophagy, which involves the two proteins, Parkin and Pink1 [102]. The reciprocal interactions between these processes are integral to mitochondrial homeostasis [103], and the increase in oxidative stress is able to adversely affect the different functions of mitochondria [37].
In microglia, oxidative stress–induced microglial activation leads to mitochondrial dysfunction. Fragmented mitochondria released from microglia are able to not only trigger A1-inflammatory-astrocytes’ activation [104], but also exacerbate the pro-inflammatory microglial M1 phenotype [105] and the release of neurotoxic cytokines, further enhancing the ROS formation [106].
Mitochondria are essential for ATP production through oxidative phosphorylation, involving a subset of the mitochondrial DNA (mtDNA). Compared to other cell types, neurons are more dependent on mitochondrial oxidative phosphorylation to fulfill their energy demands. This process generates reactive oxygen species (ROS). Neurons have limited capacity to upregulate glycolysis or to counteract oxidative damage. The generated ROS has the potential to damage mtDNA [107], resulting in mitochondrial dysfunction and dopaminergic neuronal cell loss [108]. A correlation between altered mitochondrial dynamics and neurodegeneration has been observed in PD and AD [109].
Functional astrocytes have the ability for reducing neuronal-derived ROS via enhancing the production of antioxidant molecules [93], as well as internalizing and degrading dysfunctional mitochondria [110], in order to protect neurons and maintain the quality of mitochondria for bioenergetic functions. However, reactive astrocytes may lose the capacity to reduce oxidative stress due to Nrf2 downregulation [98], and have impaired mitophagy capacity due to inflammation-induced Parkin downregulation [111].
Neuroinflammation in AADC-DY
Although the above pathological processes have not been examined in relation to AADC-DY, the neuronal injury and death may contribute to the neuroimaging abnormalities observed in AADC-DT subjects, such as cerebral [4] and cortical [112] atrophy, demyelination [113], and degenerative changes of white matter [4]. Furthermore, brain magnetic resonance imaging of 12 AADC-DT subjects showed hypomyelinations, reduced volume of caudate nucleus, and decreased density of the white matter fiber tracts [114]. Oxidative stress and microglial activation are known to also adversely affect BBB integrity [115, 116], especially in the presence of high Hcy levels [117].
Inflammation-induced loss of astrocytes’ neuroprotective functions can also adversely affect the BBB integrity, since astrocytes regulate brain microvascular permeability via astrocyte-endothelial communication and the release of regulatory factors, such as transforming growth factor (TGF)-α and glial-derived neurotrophic factor (GDNF); these factors are able to influence the permeability of tight junction in endothelial cells [118]. The increase in BBB permeability can result in the infiltration of systemic inflammatory mediators and immune cells into the CNS, further promoting microglial activation and neuronal cell death. The increase in striatal BBB permeability has been thought to underly PD disease progression [119], and could similarly be involved in the pathophysiology of AADC-DY.
It is noteworthy that the AAV2 vector used for the transfer of the functional hDDC gene has high degree of tropism to neurons, and low degree of tropism to microglia and astrocytes [120]. It remains an open question to whether the introduction of a functional copy of the hDDC gene to putamen and or midbrain of AADC-DT subjects would influence the activation state of glial cells and hence the cascade of neuroinflammatory processes.
Additional L-dopa Neurotoxic Effects
L-dopa-Induced Excitotoxicity
L-dopa can also induce glutamate excitotoxity in a Ca(2 +)-dependent, autooxidation-independent manner, although the timeline for neuronal injury and death due to L-dopa-induced glutamate excitotoxity is delayed when compared to the timeline of neuronal injury and death due to L-dopa autooxidation [100]. Studies show that ischemia-induced elevation of endogenous L-dopa increases glutamate release in rat striatal neurons, an effect that is Ca(2 +) dependent and dopamine independent [100], suggesting that elevation of endogenous L-dopa has the potential to exert neurotoxicity similar to the elevation due to L-dopa administration [121, 122]. The Ca(2 +)-induced enhanced glutamate release and the activation of the postsynaptic N-methyl-D-aspartate (NMDA) and non-NMDA receptors [123, 124] lead to caspase-3-activation [125] and neuronal cell death, a common pathway involved in the pathophysiology of many neurodegenerative disorders. Therefore, both L-dopa autooxidation and L-dopa excitotoxicity may work in concert to contribute to neurodegeneration.
The L-dopa-induced glutamate-related excitotoxity inhibits the AADC enzyme both in the striatum and in substantia nigra (SN) [41], and therefore, can adversely affect the clinical outcomes of AAV2-mediated hDDC gene therapy in AADC-DY. This notion is further supported by the results of a study showing that the central inhibition of the AADC enzyme by NSD-1015 amplifies the endogenous L-dopa-induced glutamate release and striatal neuronal cell death [100]. This is because dopamine negatively controls glutamate release and therefore dopamine depletion would have the potential to enhance glutamate release [100, 126]. The results collectively suggest that the L-dopa-induced glutamate excitotoxicity (as well the Hcy-induced glutamate excitotoxity described in the following paragraphs) is likely to be potentiated, playing a role in dopaminergic neuronal injury and death in AADC-DY.
L-dopa-Induced Protein Misfolding
An additional mechanism contributing to L-dopa cytotoxicity stems from structural similarities between L-dopa and the amino acid tyrosine. Due to these structural similarities, L-dopa can replace tyrosine in the polypeptide chain of proteins in the brain, promoting protein misfolding and protein aggregation with an adverse effect on mitochondrial function [127]. The glial dysfunction and the impaired mitophagy [111] can lead to the accumulation of misfolded proteins, enhancing neurodegeneration. The aggregation of misfolded brain proteins is the underlying cause of neuronal damage in several neurodegenerative disorders [128], and likely contribute to neurodegeneration associated with AADC-DY.
L-dopa-Induced Serotonergic Dysfunction
The dense serotonergic fibers projecting to the striatum are capable of a high affinity L-dopa uptake and conversion to dopamine [129]. Because of the dopaminergic lesions in PD, the serotonergic neurons are the main cells to use the exogenous L-dopa for dopamine synthesis in the striatum and SN [130]. Studies in a rodent model of PD show that high levels of brain L-dopa can also adversely affect the serotonergic neurons, with the reductions in both serotonin and dopamine in different parts of the brain, including striatum and SN [131], resulting in serotonin and 5-HIAA depletion and behavioral and cognitive dysfunctions [132]. Similarly, the chronic administration of L-dopa has been thought to exert toxicity toward serotoninergic neurons, hence contributing to physiological dysfunction in PD patients [133].
It is unknown whether the high levels of endogenous L-dopa found in AADC-DY undergo uptake into serotonergic neurons like the exogenous administrated L-dopa does, but if that is the case, then it can exert adverse effects on serotonergic neurons like it does in dopaminergic neurons, influencing their long-term survival. L-dopa adverse effects in serotonergic neurons would be enhanced due to serotonin depletion in AADC-DY since serotonin is known to exert antioxidant and neuroprotective effects [134, 135]. Furthermore, striatal astrocytes express serotonergic receptor 5-HT1A, a key mediator of serotonergic signaling in the CNS, and stimulation of these receptors activates Nrf2 in astrocytes, with the end results of upregulating the astrocytic antioxidant and neuroprotective capacities [136]. However, in AADC-DY, this neuroprotective pathway may be impaired due to glial dysfunction and serotonin deficit, negatively impacting neuronal survival.
L-dopa-Upregulation of Kynurenine Pathway
L-dopa oxidation has the potential to also upregulate the kynurenine pathway (KP), the major pathway in tryptophan catabolism, culminating in the generation of metabolites such as quinolinic acid [38, 39], known to exert neurotoxic effects via both increase in glutamate release and stimulation of NMDA receptors [40], further fueling oxidative stress and mitochondrial dysfunction [137]. Although high 5-HTP may contrast some of the L-dopa prooxidant effects [138], 5-HTP is known to dose-dependently inhibit AADC enzyme activity [139], and therefore could interfere with treatments aiming to increase dopamine and serotonin productions.
Quinones, the byproducts of L-dopa oxidation, can inactivate tryptophan hydroxylase, the rate-limiting enzyme in serotonin synthesis, adversely influencing serotonin production. The quinones exert these adverse effects through both modification of the cysteinyl residues within tryptophan hydroxylase, and via converting tryptophan hydroxylase to a redox-cycling quinoprotein; the latter is able to fulfill the role of a radical with neurotoxic properties [140]. Furthermore, quinones can covalently bind Parkin, inactivating ubiquitin ligase function of Parkin, thus impairing mitophagy in dopaminergic neurons [141], suggesting the vulnerability of these neurons to quinone toxicity. The data cumulatively demonstrate the many pathways through which the excess L-dopa is able to exert cytotoxicity, thus promoting neuronal injury and death in AADC-DY.
Homocysteine Neurotoxicity
Homocysteine (Hcy) is a non-protein, sulfur-containing amino acid that is generated during methionine transmethylation via several steps catalyzed sequentially by SAM synthetase, methyltransferase (MT), and SAH hydrolase enzymes [142]. Higher levels of Hcy have been regarded to be involved in the pathology of several disorders including neurodegeneration [143, 144]. PD patients show a positive correlation between Hcy plasma levels and L-dopa [145] and 3-OMD [146] concentrations. The effect of L-dopa on Hcy plasma levels in PD subjects is more pronounced under folate deficiency [147]. Neither plasma nor CSF levels of Hcy have been measured in AADC-DY. However, the excess brain L-dopa availability in AADC-DY, and the increase in the synthesis of 3-OMD by COMT enzyme, would result in the accelerated production of SAH, which would be readily converted to Hcy and adenosine by the action of SAH hydrolase enzyme, in a folate-dependent reaction, culminating to a reduced cerebral methyl donor, SAM, and reduced folate as the co-factor [148].
Homocysteine-Induced Oxidative Stress
Several mechanisms account for the adverse effects of Hcy. The subcutaneous administration of Hcy to rats increased both the plasma and brain levels of Hcy [149]. In the brain, Hcy exerted significant oxidative stress [149] and increased the level of inflammatory mediators resulting in mitochondrial dysfunction, DNA damage, and neuronal cell death, as measured in the hippocampus/cerebral cortex [150]. Similar adverse effects were observed after intracerebral injection of Hcy in mice, showing significant increase in lipid peroxidation and neuroinflammation which were accompanied with reduced GSH levels, culminating to cortical damage and cognitive deficit [151].
The oxidative stress, inflammation, and mitochondrial dysfunction in striatum and cerebellum have been also shown under chronic mild hyperhomocysteinemia [152], accompanied by a dose-dependent loss of striatal dopaminergic neurons, and motor dysfunction [153]. In PD subjects, Hcy plasma levels correlate with cognitive dysfunction [154] and Hcy-induced loss of dopaminergic neurons is thought to contribute to PD neurodegeneration [153].
It is assumed that Hcy promotes oxidative stress via reactive oxygen species generation upon disulfide bond formation. However, additional studies show that Hcy, unlike other thiol compounds, possess distinctive features enabling it to easily forms disulfide bonds with free thiol groups of cysteine residues in proteins forming thermodynamically stable Hcy-thyl-cysteine able to act as a radical after undergoing hydrogen atom transfer reaction [28]. This reaction can lead to the oxidation of DNA and proteins and limit the availability of the antioxidant precursor, cysteine.
Hcy reduces catecholamine and serotonin levels in the brain and increases MAOB activity [29]. The increase in MAOB will lead to accelerated dopamine catabolism to 3,4-dihydroxyphenylacetaldehyde (DOPAL), a reactive aldehyde derivative with cytotoxic effects [155]. The increase in MAOB can also reduce the activity of the antioxidant enzyme, superoxide dismutase (SOD) [156]. Furthermore, the BBB is especially sensitive to Hcy and even a mild increase in Hcy level can compromise BBB integrity, independent of Hcy-induced neuroinflammation [157]. Hcy neuronal damage is likely to occur in AADC-DY because of chronic presence and accumulation due a relatively long biological half-life (4 h) [158], with the potential for contributing to both motor and cognitive dysfunctions [29], the severity of which would be the function of both Hcy concentration and the length of exposure of neurons, especially dopaminergic neurons, to the insult.
Homocysteine-Induced Excitotoxicity
Similar to L-dopa, Hcy can exert excitotoxicity by interaction and activation of the NMDA receptors [159, 160], an adverse effect that is potentiated in the presence of high levels of glycine [159]. Glycine is known to be a NMDA receptor agonist [161]. AADC-DT subjects present with high CSF levels of glycine [8] with the potential to synergize with Hcy, further augmenting excitotoxicity and subsequent neuronal injury and death. Hcy activation of NMDA receptors is associated with the induction of seizures in young rats [162]. Whether cases of single seizures observed in some AADC-DT subjects [13] are related to Hcy excitotoxicity warrants further investigations.
Dopaminergic neurons extend their axons to the striatum, releasing dopamine, and striatal astrocytes express dopamine receptors, including D2 receptors [99]. A2 adenosine receptors and D2 dopaminergic receptors closely interact in an antagonistic manner to modulate glutamatergic transmission in striatal astrocytes through receptor heteromerization; D2 receptors’ activation inhibits glutamate release, while activation of A2A receptors abolishes the effect of D2 receptor–mediated inhibition of glutamate release from astrocytes [163]. Therefore, the intercellular communication between neurons and astrocytes via A2A-D2 heteromer can contribute to glutamatergic regulation.
Hcy has been shown to inhibit D2-mediated inhibition of glutamate release without affecting the functional interaction between A2A receptors and D2 receptors [164]. The absence of inhibition of glutamate release, due to dopamine depletion in AADC-DY, and the presence of high levels of Hcy and adenosine are factors leading to enhanced Hcy-induced excitotoxicity with adverse effects on dopaminergic neurons. Furthermore, similar to Hcy, high level of adenosine co-released with Hcy is able to increase SAH [165] and therefore can indirectly downregulate transmethylation processes. Such adenosine adverse effect is likely to be accentuated in the presence of high Hcy levels. Adenosine activation of A2 adenosine receptors is known to contribute to dopaminergic neurodegeneration, and antagonists to this receptor are considered for their beneficial effects in the treatment of PD [166]. The results collectively demonstrate the deleterious effects of chronic brain exposure to Hcy, a scenario likely to exist in AADC-DY.
Reduced Homocysteine Metabolism
Hcy levels are determined by the balance between biosynthesis and catabolism; the higher expected Hcy levels in AADC-DY can be the result of not only increase in the synthesis, but also reduced Hcy catabolism. Hcy is catabolized by two means: remethylation to methionine and transsulfuration to cysteine. Hcy remethylation occurs in both folate/vitamin B12-dependent and vitamin B12-independent mechanisms. The B12-dependent uses N-5-methyl tetrahydrofolate (MTHF) as methyl group donor catalyzed by methionine synthase (MS) with the aid of vitamin B12 as cofactor, whereas the remethylation pathway is independent of vitamin B12 and relies on betaine as methyl donor catalyzed by betaine-homocysteine methyltransferase (BHMT) enzyme.
Hcy transsulfuration is catalyzed by the two vitamin B6-dependent enzymes, the cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), as well a third enzyme, 3-mercaptopyruvate sulfurtransferase (MST). CBS is expressed mainly in glial/astrocyte lineage [167], whereas CSE [168] and 3-MST [169] catalyze Hcy metabolism mainly in neurons. The first step is the condensation between Hcy and serine by CBS produces cystathionine, which is further hydrolyzed by the CSE enzyme to produce cysteine and α-ketobutyrate [142, 170]. Cysteine is a precursor, critical for the synthesis of the antioxidant GSH and for the synthesis of H2S, a signaling molecule able to exert neuroprotective effects via multiple biological mechanisms including antioxidative, anti-inflammatory, and antiapoptotic [171].
The high Hcy levels, brought about by the accelerate rate of L-dopa-induced o-methylation, are able to diminish CBS enzyme activity [29] as well as reduce the level of the methyl donor, SAM, known to be the allosteric activator of the CBS enzyme [30], hence resulting in further Hcy accumulation. Hcy catabolism is also hampered due to dopamine and serotonin depletion since both neurotransmitters are known to upregulate CBS expression [172]. Therefore, factors such as depleted neurotransmitters, reduced SAM [8], high Hcy and adenosine levels, and depletion of cerebral folate due to L-dopa-induced oxidative stress [173] all have the potential for interfering with CBS enzyme activity and remethylation cycle, thus impairing Hcy catabolism in AADC-DY. This scenario is especially relevant to the brain due to the absence of BHMT enzyme [174], and brain dependency on folate/vitamin B12-pathway for Hcy remethylation, rendering the brain vulnerable to increased Hcy levels.
The physiological levels of CSE enzyme are especially low in the brain; in fact, CSE levels in the brain are > 100 folds lower than that in the liver [175] and more than 40-fold higher cystathionine levels are reported in the brain compared to other human tissues [176]. Low physiological levels of CSE enzyme in the brain combined with oxidative stress induced by L-dopa, 3-OMD [177], and Hcy [29] could significantly reduce CSE activity, resulting in Hcy accumulation, and subsequent reduction in the production of cysteine and its neuroprotective metabolites, GSH and H2S.
Homocysteine Downregulation of Cellular Antioxidant Status
AADC-DY-related deficits in dopamine and serotonin and the reduced activities of CBS and CSE enzymes [172], combined with reduced level of SAM [178], all are factors that could adversely affect H2S production and therefore, the antioxidant capacity of the brain, an organ known to be especially sensitive to oxidative stress [179]. CBS and CSE enzymes can both produce H2S directly from cysteine or through catalysis of the condensation of cysteine + cysteine and Hcy + cysteine. In addition, CSE can produce H2S directly from Hcy or through catalysis of the condensation of Hcy + Hcy [180]. The reactions catalyzed by both CBS and CSE are all vitamin B6-dependent [180]. In normal physiological state, CBS is the major enzyme in the production of H2S [181], which takes place mainly in astrocytes [182]. However, in the presence of high brain Hcy, as it is likely to be the case in AADC-DY, CSE becomes the principal enzyme for the H2S generation through CSE catalysis of Hcy + Hcy condensation [183]. Therefore, the physiologically low brain CSE levels and the reduced CSE activity in AADC-DY would interfere with H2S formation, promoting an environment of low antioxidant capacity, where the cytotoxicity of L-dopa is potentiated [68]. This prooxidant environment is further enhanced by the ability of Hcy to interact with H2S, forming Hcy-persulfide, thus reducing H2S bioavailability [184]. The disturbances in the H2S levels have been shown in many neurodegenerative diseases including PD and AD [185], and likely to be a part of AADC-DY pathophysiology.
H2S in the brain can be also generated by MST enzyme using 3-mercaptopyruvate (3-MP) as substrate, with thioredoxin (Trx) and dihydrolipoic acid (DHLA), the reduced form of α-lipoic acid (ALA) as cofactors [186]. Both Trx and DHLA act as sulfur acceptors for MST-bound cysteine persulfide (an intermediate generated by sulfur transfer from 3-MP to cysteine catalyzed by MST) to generate H2S [168, 187]. 3-MP is produced by the action of cysteine aminotransferase (CAT) enzyme on cysteine in the presence of α-ketoglutarate [180]. However, the physiological level of MST enzyme in the brain is 10 folds lower than the levels in the liver [188], and the oxidative stress exerted by the different metabolites could further reduce MST activity [31]. The chronic high level of L-dopa can also interfere with H2S production via reducing the level and the activity of the cofactor Trx in neurons [32]. Since H2S negatively regulate Hcy levels [189], the reduced production of H2S leads to further Hcy accumulation in the brain.
One of the many H2S biological effects in the brain is the regulation of the gastric juice, by reducing the gastric PH [190]. Most of AADC-DT subjects suffer from gastrointestinal problems, including digestion, diarrhea, constipation, reflux, nausea, and vomiting [191]. These clinical symptoms may be in part the result of hypochlorhydria due to reduced brain H2S production. Furthermore, H2S has been shown to regulate glucose homeostasis in a number of organs including hepatocytes, skeletal muscles, and the brain [192]. It is well known that neurotransmitters such as dopamine and serotonin regulate blood glucose [193]. The hypoglycemia observed in AADC-DT subjects may be the result of both neurotransmitters’ deficiency and H2S dysregulation.
Homocysteine Reduction in Cellular Methylation Status
Another consequence of elevated Hcy levels is the accumulation of SAH, since as a result of inefficient removal of Hcy through remethylation or degradation to cysteine, the equilibrium constant of SAH hydrolase favors SAH synthesis rather than hydrolysis (reverse catalysis) [175]. SAH elevation is known to disturb cell methylation capacity through the ability to competitively bind the catalytic region of most SAM-dependent methyltransferases, inhibiting transmethylation reactions [194], including the formation of SAM. The results of a combined genetic and dietary approach in rats shows an inverse relationship between SAM and SAH and a positive relationship between plasma Hcy and brain SAH [195]. The increase in SAH seems to be a better indicator of cellular methylation capacity than decrease in SAM [195]. Therefore, high levels of Hcy and SAH may play a role in reduced SAM levels observed in AADC-DY [148], suggesting suboptimal cellular antioxidant and methylation capacities.
Brain may be especially sensitive to SAH adverse effects since brain has a relatively low SAH hydrolase activity [175], and BHMT, the alternate Hcy remethylating enzyme, is not active in the brain [174]. Hypomethylation can adversely affect DNA and proteins, with adverse effects including neuronal susceptibility to injury and apoptosis [196]. SAM/SAH ratio also correlates with total GSH level and GSH/GSSG ratio [53]. The SAM/SAH ratio in plasma of healthy individuals is calculated to be 4.9 ± 1.7 (range, 1.6–9.5) [197], but this ratio is reduced in PD subjects, correlating with cognitive dysfunction [54]. Although this ratio is unknown, it is expected to be reduced in AADC-DT subjects as a result of L-dopa-induced reduction in SAM [198] and Hcy-induced increase in SAH.
Homocysteine Oxidation-HCA Generation
In a prooxidant environment, Hcy is prone to undergo oxidation generating 2-amino-4-sulfo-butanoic acid (homocysteic acid, HCA) [27], with enhanced production under folate deficiency [199]. HCA is an analogue of glutamic acid, able to bind and activate NMDA receptors. Even very low concentration of HCA is capable of over-activating the NMDA receptors leading to very high brain toxicity [200]. In fact, HCA is significantly more neurotoxic than Hcy [201, 202]. The icv infusion of HCA to immature rats caused seizures recorded in various parts of the brain, including striatum [203]. Brain levels of HCA are increased in the rodent model of AD, triggering memory deficit [204], and the administration of an anti-HCA antibody prevents cognitive impairment [204]. Compounds that reduce HCA to Hcy, such as H2S, have been shown to improve cognitive function in AD patients [205]. Similarly, ferulic acid, which inhibits HCA binding to NMDA receptors, improves HCA-induced cognitive impairment [205]. The data demonstrates the detrimental effects of HCA in the brain in the presence of high Hcy and a prooxidant environment that characterizes AADC-DY.
We propose that in the absence of AADC enzyme [69], and the absence of dopamine [82, 100] and serotonin neuroprotective effects [134, 135], the chronic presence of high levels of L-dopa, 3-OMD, and Hcy, would act in concert and synergy to generate a prooxidative and excitotoxic environment, resulting in glial activation, mitochondrial dysfunction, and neuroinflammation, hence marking the onset of neurodegeneration in AADC-DY. This hostile environment faced with the metabolites-induced suboptimal cellular antioxidant capacity would negatively impact neuronal survival in general and dopaminergic neurons in particular. The extent and severity of adverse effects would be in turn the function of the levels and duration of exposure to the toxic metabolites. This conclusion is supported by the results of the clinical outcomes of AAV2-mediated hDDC gene therapy to be more favorable in younger AADC-DT patients compared to older ones [18, 206], suggesting that the relatively shorter duration of the disease in younger subjects may be associated with a less opportunity for the toxic metabolites to cause an irreversible neuronal injury, compromising neurons’ functional integrity.
AADC-DY Novel Treatment Strategies
We have extensively discussed several upregulated metabolites in AADC-DY and their synergistic role in inducing oxidative stress and neuroinflammation, as well as interfering with the synthesis of cellular antioxidants. These effects of the metabolites combined with the absence of dopamine and serotonin antioxidant and neuroprotective effects would generate an unreceptive milieu impeding the success of AADC-DY treatments aiming to enhance neurotransmitters’ synthesis and or function. To improve treatment efficacy, either the levels of metabolites should be reduced or alternatively the cellular antioxidant capacity be increased by either supplementing with compounds that exert antioxidant effects or with precursors involved directly or indirectly in the generation of cellular antioxidants. These interventions, singly or combined, would be more effective if applied early in the course of the disease, before irreversible neuronal injury occurs. These novel strategies are depicted in Fig. 2.

Strategies to improve the treatment of AADC-DY. The new concept of AADC-DY as a neurodegenerative disease offers additional strategies to treat the disease. In addition to COMT inhibitors [207,208,209,210], H2S donors [29, 149, 151], sulfur-containing compounds (SAM, methionine, N-acetylcysteine, and 3-MP) [211,212,213], and the powerful antioxidant ALA [214,215,216] are strategies that could restore cellular methylation and cellular antioxidant capacities. Other strategies include the use of antioxidant enzymes (GSH, SOD, catalase) [217], vitamins such as ascorbate [132, 218, 219], α-tocopherol [45, 220, 221], and VD3 [222,223,224,225]. NMDA receptor antagonists are able to reduce glutamate excitotoxicity [41, 100], and erythropoietin is able to exert neuroprotective effects [226,227,228]. These strategies used singly or combined early in the course of the disease will minimize neuronal injury, preserving the functional integrity of the dopaminergic and serotonergic neurons. The red highlights present the upregulated metabolites. The purple highlights present the pathways to neuronal injury and death. The green highlights present the various treatment strategies. Abbreviations: ALA, α-lipoic acid; COMTI, catechol-O-methyl-transferase inhibitor; EPO, erythropoietin; GSH, glutathione; Hcy, homocysteine; H2S, hydrogen sulfide; L-dopa, 3,4-dihydroxy-L-phenylalanine; Met, methionine; 3-MP, 3-mercaptopyruvate; NAC, N-acetyl-cysteine; NMDAR-A, N-methyl-D-aspartate receptor-antagonist; 3-OMD, 3-O-methyldopa; SAM, s-adenosylmethionine; SOD, superoxide dismutase; VD3, vitamin D3; V-C, ascorbate; V-E, α-tocopherol
COMT Inhibitors
Among strategies for reducing the levels of metabolites, and restoring the cellular antioxidant and methylation capacities is the use of COMT inhibitors. Although not a part of treatment modality in AADC-DY, treatment of PD patients with the COMT inhibitor, tolcapone, results in a marked reduction in 3-OMD plasma level [207]. COMT inhibitors can also contrast the increase in Hcy levels, minimizing the related toxicity brought about L-dopa-induced accelerated o-methylation [208]. In addition, tea polyphenol, especially epigallocatechin-3-gallate (EGCG), and ( +)-catechin are effective COMT inhibitors, known to reduce 3-OMD levels in rat striatum [209, 210]. Having adequate bioavailability, these compounds cross BBB, exerting antioxidant, anti-inflammatory, and neuroprotective effects, even in low concentrations [209, 210]. The data suggests the possible benefits of treatment of AADC-DY with centrally acting COMT inhibitors as a mean for reducing 3-OMD and Hcy levels and improving both SAM/SAH and GSH/GSSG ratios.
H2S Donors
Alternatively, one can enhance the cellular antioxidant capacity to counteract metabolites-induced oxidative stress. Rodent studies show that intraperitoneal administration of the H2S donor, sodium hydrogen sulfide (NaHS), alleviated Hcy-induced toxicity by reducing oxidative damage, neuroinflammation, cells apoptosis, neurodegeneration, and cognitive deficit [29, 149, 151]. H2S administration also lowered the plasma and brain levels of Hcy, increased the level of the endogenous H2S, restored the activities of CBS and CSE enzymes, increased catecholamine levels, and improved cognitive and behavioral deficits in Hcy-treated animals [29, 151]. NaHS treatment of a 6-OHDA-induced PD model attenuated neuronal loss, lipid oxidation, and the accumulation of inflammatory markers, and improved movement dysfunction [229]. Similar effects have been shown in the MPTP-induced PD model, where H2S inhalation prevented movement disorder and the degeneration and apoptosis of TH-containing neurons [230]. In addition, the inhalation of low H2S levels has been shown to increase plasma glucose levels in postpartum rats [231], suggesting that such strategy may not only impact neurodegenerative processes, but also improve the hypoglycemia often reported in AADC-DT subjects.
Having a relatively high expression in the brain, the neuroprotective effects of H2S is attributed to the ability to prevent oxidative stress through Nrf2 upregulation [232] and preserve mitochondrial function, through upregulating Parkin activity [233]. H2S donors have been suggested as treatment modality for PD [234, 235], where the loss of Parkin activity has been documented [236]. These studies collectively suggest the beneficial effects of H2S as a part of AADC-DY treatment modality to enhance cellular antioxidant status, prevent mitochondrial dysfunction, and minimize neuronal injury.
Sulfur-Containing Compounds
The increase in H2S production can also be achieved by administration of several sulfur-containing compounds including SAM, methionine, N-acetylcysteine (NAC), and 3-MP. The administration of SAM, an activator of CBS, has the potential for restoring cellular methylation status, lowering Hcy levels, improving SAM/SAH ratio, and enhancing H2S production [211]. Although the direct effects of SAM supplementation on the brain are not well known, SAM is able to cross the BBB with slow accumulation in the CSF [237]. SAM administration to PD patients is well tolerated reducing the severity of depression [238]. Similarly, supplementation with methionine, the precursor of SAM, can improve the hypomethylation status, reducing Hcy and SAH levels [212], hence leading to increased H2S production. Supplementation with NAC can increase the levels of cysteine, the precursor for GSH, and enhance H2S production, while reducing Hcy via Hcy + cysteine condensation [212]. In addition, rodent studies show the potential of 3-MP to increase H2S; this effect is enhanced when 3-MP is administered in combination with α-lipoic acid (ALA) [213].
The enhancement in cellular antioxidant capacity can be also achieved by supplementation with antioxidant enzymes including SOD, catalase, and GSH [217], all of which fulfill important roles as antioxidants in the brain. PD patients, at advanced disease stages, show significantly lower SOD level in whole blood and in red blood cells, which correlates with disease duration [239]. Similarly, GSH level in the brain of PD patients is significantly reduced [240]. Studies show that SOD/catalase mimetics exert neuroprotective effects in a herbicide-mediated PD model [241]. Accumulated evidence supports the supplementation and enhancement of GSH and related antioxidants for improving PD pathology [242].
The ability of bromocriptine and pergolide to improve AADC-DY clinical symptoms [8] may stem in part from upregulating SOD [243]. In addition, studies in animal and cellular models of PD report that MAO inhibitors exert neuroprotective effects in part through increasing SOD and catalase activities in the brain regions containing dopaminergic neurons [244]. Furthermore, the clinical efficacy of vitamin B6 in AADC-DY treatment is attributed in part from B6 ability to induce GSH via Nrf2 activation [245]. The clinical efficacies of the dopamine-receptor agonists, MAO inhibitors, and vitamin B6 and their potential for increasing antioxidant levels, support the use of antioxidant enzymes and their mimetics for improving AADC-DY neuropathology.
Another powerful antioxidant, the organosulfur compound ALA, is an essential cofactor for several enzymes involved in mitochondrial energy metabolism. In vitro studies show the potential of ALA in protecting primary hippocampal and cortical neurons from HCA-mediated toxicity-induced neuronal cell death [214]. In addition, ALA reduced microglial activation and neuroinflammation in the MPTP-induced PD model [246]. In the 6-OHDA-lesioned rats, ALA reduced MDA levels and upregulated GSH activity in the striatum, thus preventing L-dopa activation of the proapoptotic caspase-3 signal in SN, resulting in improved neuronal survival [215]. The neuroprotective effects of ALA are attributed in part to the ability to downregulate inflammatory processes through targeting the proinflammatory transcription factor, the nuclear factor-KB pathway [216].
ALA is synthesized de novo in most cells in small amounts, using several intermediates including SAM [247] and therefore, reduced SAM is one of the many factors that may contribute to reduced levels of ALA in AADC-DY. In vivo, ALA could be reduced to its dithiol form, dihydrolipoic acid (DHLA), and in this form it functions as a sulfur acceptor during H2S production catalyzed by MST enzyme. Upon completion of this reaction, ALA is concomitantly released with H2S [187]. Therefore, the reduced MST activity and Trx levels are additional factors impeding not only the H2S production, but also that of ALA in AADC-DY. ALA is available as dietary supplement, and it is used as pharmaceutical drug in some European countries. Use of ALA in AADC-DT subjects, early in the course of the disease, will exert antioxidant and neuroprotective effects, preserving dopaminergic and serotonergic functions.
Vitamins
Metabolite-induced oxidative stress not only could deplete the level of cofactors such as folate [173], but it can also increase the rate of utilization of the two other antioxidant vitamins, ascorbate and α-tocopherol [248], thus adversely influencing cellular antioxidant capacity. In vitro studies show that ascorbic acid blocks L-dopa-induced reduction in the activity of the antioxidant enzyme catalase in mesencephalic cells [249], and prevents L-dopa-induced cell toxicity in striatal neurons [218]. These results were further corroborated in in vivo studies showing that ascorbate administration reduced Hcy, increased the activity of catalase and SOD enzymes in the hippocampus [219], and prevented serotonin depletion in the dorsal raphe nucleus, thus minimizing L-dopa-induced cognitive dysfunction in rats [132].
Similar results have been reported with α-tocopherol, able to reduce L-dopa- and 3-OMD-induced oxidative stress, preventing neuronal injury and motor dysfunction in rats [45]. The natural form of α-tocopherol, namely α-tocotrienol is multifold more potent than α-tocopherol, able to block neuronal injury resulting from not only oxidative stress, but also from Ca(2 +)-dependent glutamate-induced excitotoxicity [220]. This potency has been attributed to a faster cellular uptake of the former compared to the latter [250]. Treatment of PD patients with the combination of ascorbate and α-tocopherol reduced progression of the disease, indicated by the extension of the time to L-dopa treatment [221]. In addition, a randomized clinical study compared the benefit of α-tocopherol and NMDA receptor antagonist, memantine, in reducing the functional decline in patients with mild cognitive dysfunction, reporting encouraging results [251].
The other vitamin with the potential benefit in the treatment of AADC-DY is vitamin D3 (VD3), with the active metabolite being 1,25-dihydroxyvitamin D3. VD3 is a hormone with pleiotropic effects; in addition to the classical role in the regulation of intestinal, bone, and kidney calcium and phosphorus absorption, VD3 exerts antioxidant, anti-inflammatory, and neuroprotective effects [222]. Although there are no data on serum level of VD3 in AADC-DT subjects, PD patients show reduced level of this vitamin, and an inverse relationship between VD3 serum level and disease severity [252]. VD3 crosses the BBB [253], able to influence dopaminergic neurons through binding to and activation of the VD3 receptor/retinoic X receptor heterodimeric complex [254]. VD3 receptors act as a ligand-inducible transcription factors. VD3 belongs to the nuclear hormone receptor superfamily [255] with the highest concentrations reported to be located in the SN [256], indicating the important function of these receptors in dopaminergic neurons.
VD3 treatment protected rats’ nigrostriatal dopaminergic neurons against a partial lesion induced by the unilateral striatal injection of 6-OHDA, administered either before or after lesion induction [223]. The treatment prevented lipid peroxidation and the increase in the level of proinflammatory mediator TNF-α. VD3 also reversed the 6-OHDA-induced reduction in the level of dopamine transporter, an indicator of dopaminergic membrane integrity, and the level of tyrosine hydroxylase, the rate-limiting enzyme in dopamine synthesis, hence protecting dopaminergic neurons [223]. The neuroprotective effects are attributed in part to VD3’s ability to increase GDNF level in the striatum and SN, inhibiting cell apoptosis [224]. The results from rodents’ studies agree with those of a clinical trial showing that VD3 supplementation of PD patients prevented disease deterioration measured by modified Hoehn and Yahr stage and Unified Parkinson’s Disease Rating Scale [225]. The accumulated evidence support VD3 potential to downregulate pathophysiological processes that characterize neurodegeneration, and therefore may be of benefit in the treatment of AADC-DY.
NMDA Receptor Antagonists
The increase in the brain levels of metabolites also leads to an enhanced glutamate release via both oxidative- and Ca(2 +)-dependent pathways as described above. Rodent studies show that L-dopa-induced glutamate release and cell death are prevented by the administration of NMDA receptor antagonists CGP 40116, glycine/NMDA receptor antagonist HA-966, memantine, and the antiparkinsonian dugs budipine and amantadine, with a parallel increase in the level of the AADC enzyme [41]. Similar results were observed with the competitive DOPA antagonist, DOPA cyclohexyl ester [100], suggesting the potential beneficial effects of NMDA receptor antagonists in the treatment of AADC-DY.
Erythropoietin
An additional compound with possible beneficial effects in AADC-DY is the hypoxia-inducible growth factor erythropoietin (EPO). EPO has a role in hematopoiesis and the recombinant human EPO is used for the treatment of anemia. However, EPO and its receptor are also found in the brain, regulating cognition, neurogenesis, and neuroplasticity, while undergoing upregulation upon increase in oxygen demand and brain injury [257]. In vitro studies show that EPO reduced L-dopa-induced oxidative stress and caspase-3, and improved neuronal cell survival [226]. Furthermore, the systemic administration of the recombinant human EPO crosses the BBB, stimulating striatal dopamine release [227]. Similar results were observed in the PD model where EPO prevented neurodegeneration in the nigrostriatal dopaminergic system, improving cognitive function [228]. A clinical study of PD patients treated with intranasally administered EPO reports encouraging results [258]. Whether such an approach combined with other modalities could improve the neuropathology associated with AADC-DY warrants further investigations.
Concluding Remarks
We have extensively discussed oxidative stress in AADC-DY as an ignition for the downstream pathophysiological processes culminating to mitochondrial dysfunction, neuroinflammation, and neuronal injury and death. We believe that the heterogeneity in the clinical outcomes of established treatments and AAV2-mediated hDDC gene transfer in AADC-DT subjects may be related in part to the progression of the disease where the chronic presence of several upregulated metabolites leads to irreversible damage of neurons, compromising their functional integrity.
The new concept of AADC-DY as a neurodegenerative disease opens a path for additional treatment modalities to reduce the levels of the upregulated metabolites, and or minimize the downstream pathophysiological processes. This in turn would generate a more hospitable environment, where the clinical efficacy of AADC-DY treatments, aimed at restoring neurotransmitters’ synthesis and or function, would be enhanced. These additional treatment modalities include the use of COMT inhibitors, antioxidants, and neuroprotective agents such as H2S, ALA, EPO, and vitamins; sulfur-containing compounds such as SAM, methionine, NAC, and 3-MP, able to restore cellular methylation status and enhance cellular antioxidant capacity; antioxidant enzymes such as GSH, catalase, and SOD; and NMDA receptor antagonists with the potential to minimize metabolite-induced excitotoxicity. The use of these treatment modalities as single or in combination, early in the course of the disease, would minimize injury, hence preserving neuronal function. These therapies are not limited to AADC-DY but can be effective in other pathological states, where a mutation has the potential to lead to the accumulation of prooxidants and neurotoxic metabolites. Several disorders of vitamin B6 metabolism fall into this category.
Data Availability
NA.
Abbreviations
- AADC:
-
Aromatic L amino acid decarboxylase
- AAV:
-
Adeno-associated virus
- ALA:
-
α-Lipoic acid
- ALS:
-
Amyloid lateral sclerosis
- AD:
-
Alzheimer’s disease
- BBB:
-
Blood-brain barrier
- BHMT:
-
Betaine-homocysteine methyltransferase
- CAT:
-
Cysteine aminotransferase
- CBS:
-
Cystathionine β-synthase
- COMT:
-
Catechol-O-methyl-transferase
- CSE:
-
Cystathionine γ-lyase
- EP:
-
Epinephrine
- DAMPs:
-
Damage-associated molecular pattern
- DHLA:
-
Dihydrolipoic acid
- DOPAL:
-
3,4-Dihydroxyphenylacetaldehyde
- DT:
-
Deficient
- DY:
-
Deficiency
- EPO:
-
Erythropoietin
- GDNF:
-
Glial-derived neurotrophic factor
- GFAP:
-
Glial fibrillary acidic protein
- GSH:
-
Glutathione
- GSSG:
-
Oxidized glutathione
- HCA:
-
Homocysteic acid
- Hcy:
-
Homocysteine
- 5-HIAA:
-
5-Hydroxyindoleacetic acid
- H2S:
-
Hydrogen sulfide
- 5-HT:
-
Serotonin
- 5-HTP:
-
5-Hydroxytryptophan
- HMGB:
-
High mobility group protein box
- HVA:
-
Homovanillic acid
- icv:
-
Intracerebroventricular
- L-dopa:
-
3,4-Dihydroxy-L-phenylalanine
- MAO:
-
Monoamine oxidase
- MAT:
-
Methionine adenosyl transferase
- mDNA:
-
Mitochondrial DNA
- MHPG:
-
Methoxy-4-hydroxyphenylglycol
- 3-MP:
-
3-Mercaptopyruvate
- MS:
-
Methionine synthase
- MST:
-
3-Mercaptopyruvate sulfurtransferase
- MT:
-
Methyl transferase
- MTHF:
-
N-5-methyl tetrahydrofolate
- NAC:
-
N-acetyl-cysteine
- NaHS:
-
Sodium hydrogen sulfide
- NEP:
-
Norepinephrine
- NMDA:
-
N-Methyl-D-aspartate
- Nrf2:
-
Nuclear factor erythroid-2-related factor2
- 6-OHDA:
-
6-Hydroxydopamine
- 3-OMD:
-
3-O-methyldopa
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- SAH:
-
S-adenosylhomocysteine
- SAM:
-
S-adenosylmethionine
- SN:
-
Substantia nigra
- SOD:
-
Superoxide dismutase
- (TGF)-α:
-
Transforming Growth Factor
- Trx:
-
Thioredoxin
- VD3:
-
Vitamin D3
References
Hyland K, Clayton PT (1990) Aromatic amino acid decarboxylase deficiency in twins. J Inherit Metab Dis 13(3):301–304
Shih DF, Hsiao CD, Min MY, Lai WS, Yang CW, Lee WT, Lee SJ (2013) Aromatic L-amino acid decarboxylase (AADC) is crucial for brain development and motor functions. PLoS ONE 8(8):e71741
Gaspar P, Cases O, Maroteaux L (2003) The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci 4(12):1002–1012
Brun L, Ngu LH, Keng WT, Ch’ng GS, Choy YS, Hwu WL, Lee WT, Willemsen MA et al (2010) Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology 75(1):64–71
Pearson TS, Gilbert L, Opladen T, Garcia-Cazorla A, Mastrangelo M, Leuzzi V, Tay SKH, Sykut-Cegielska J et al (2020) AADC deficiency from infancy to adulthood: symptoms and developmental outcome in an international cohort of 63 patients. J Inherit Metab Dis 43(5):1121–1130
Hwu W-L (2023) Aromatic L-amino acid decarboxylase deficiency in Taiwan. JIMD Rep 64(5):387–392
Hitti FL, Yang AI, Gonzalez-Alegre P, Baltuch GH (2019) Human gene therapy approaches for the treatment of Parkinson’s disease: an overview of current and completed clinical trials. Parkinsonism Relat Disord 66:16–24
Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, Garcia-Cazorla A, Artuch R et al (2017) Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis 12(1):12
Andersohn F, Garbe E (2009) Cardiac and noncardiac fibrotic reactions caused by ergot-and nonergot-derived dopamine agonists. Mov Disord 24(1):129–133
Cincotta AH, Meier AH (1995) Bromocriptine inhibits in vivo free fatty acid oxidation and hepatic glucose output in seasonally obese hamsters (Mesocricetus auratus). Metabolism 44(10):1349–1355
Luo S, Meier AH, Cincotta AH (1998) Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology 68(1):1–10
Himmelreich N, Montioli R, Bertoldi M, Carducci C, Leuzzi V, Gemperle C, Berner T, Hyland K et al (2019) Aromatic amino acid decarboxylase deficiency: molecular and metabolic basis and therapeutic outlook. Mol Genet Metab 127(1):12–22
Manegold C, Hoffmann GF, Degen I, Ikonomidou H, Knust A, Laass MW, Pritsch M, Wilichowski E et al (2009) Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis 32(3):371–380
Bankiewicz KS, Eberling JL, Kohutnicka M, Jagust W, Pivirotto P, Bringas J, Cunningham J, Budinger TF et al (2000) Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol 164(1):2–14
Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, VanBrocklin HF, Wright JF et al (2009) Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73(20):1662–1669
Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K, Kawakami T, Urabe M et al (2010) A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther 18(9):1731–1735
Hwu WL, Muramatsu S, Tseng SH, Tzen KY, Lee NC, Chien YH, Snyder RO, Byrne BJ et al (2012) Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med 4(134):134ra61
Chien YH, Lee NC, Tseng SH, Tai CH, Muramatsu SI, Byrne BJ, Hwu WL (2017) Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, phase 1/2 trial. Lancet Child Adolesc Health 1(4):265–273
Tai CH, Lee NC, Chien YH, Byrne BJ, Muramatsu SI, Tseng SH, Hwu WL (2022) Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol Ther 30(2):509–518
Pearson TS, Gupta N, San Sebastian W, Imamura-Ching J, Viehoever A, Grijalvo-Perez A, Fay AJ, Seth N et al (2021) Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat Commun 12(1):4251
Lee WT, Weng WC, Peng SF, Tzen KY (2009) Neuroimaging findings in children with paediatric neurotransmitter diseases. J Inherit Metab Dis 32(3):361–370
Keam SJ (2022) Eladocagene exuparvovec: first approval. Drugs 82(13):1427–1432
Zhao WQ, Latinwo L, Liu XX, Lee ES, Lamango N, Charlton CG (2001) L-dopa upregulates the expression and activities of methionine adenosyl transferase and catechol-O-methyltransferase. Exp Neurol 171(1):127–138
Wassenberg T, Geurtz BPH, Monnens L, Wevers RA, Willemsen MA, Verbeek MM (2021) Blood, urine and cerebrospinal fluid analysis in TH and AADC deficiency and the effect of treatment. Mol Genet Metab Rep 27:100762
Miller JW, Shukitt-Hale B, Villalobos-Molina R, Nadeau MR, Selhub J, Joseph JA (1997) Effect of L-dopa and the catechol-O-methyltransferase inhibitor Ro 41–0960 on sulfur amino acid metabolites in rats. Clin Neuropharmacol 20(1):55–66
Liu XX, Wilson K, Charlton CG (2000) Effects of L-dopa treatment on methylation in mouse brain: implications for the side effects of L-dopa. Life Sci 66(23):2277–2288
Hasegawa T, Kosoku Y, Sano Y, Yoshida H, Kudoh C, Tabira T (2020) Homocysteic acid in blood can detect mild cognitive impairment: a preliminary study. J Alzheimers Dis 77(2):773–780
Sibrian-Vazquez M, Escobedo JO, Lim S, Samoei GK, Strongin RM (2010) Homocystamides promote free-radical and oxidative damage to proteins. Proc Natl Acad Sci U S A 107(2):551–554
Kumar M, Modi M, Sandhir R (2017) Hydrogen sulfide attenuates homocysteine-induced cognitive deficits and neurochemical alterations by improving endogenous hydrogen sulfide levels. BioFactors 43(3):434–450
Abe K, Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16(3):1066–1071
Modis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C (2013) Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun 433(4):401–407
Sabens EA, Distler AM, Mieyal JJ (2010) Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson’s disease. Biochemistry 49(12):2715–2724
Bachiller S, Jimenez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, Boza-Serrano A (2018) Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front Cell Neurosci 12:488
Muzio L, Viotti A, Martino G (2021) Microglia in neuroinflammation and neurodegeneration: from understanding to therapy. Front Neurosci 15:742065
Liddelow SA, Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity 46(6):957–967
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481–487
Guo C, Sun L, Chen X, Zhang D (2013) Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 8(21):2003–2014
Chen Y, Stankovic R, Cullen KM, Meininger V, Garner B, Coggan S, Grant R, Brew BJ et al (2010) The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox Res 18(2):132–142
Zhang Y, Wang L, Ren W (2022) Blast-related traumatic brain injury is mediated by the kynurenine pathway. NeuroReport 33(13):569–576
Popoli P, Pintor A, Domenici MR, Frank C, Tebano MT, Pezzola A, Scarchilli L, Quarta D et al (2002) Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J Neurosci 22(5):1967–1975
Fisher A, Starr MS (2000) Opposite effects of glutamate antagonists and antiparkinsonian drugs on the activities of DOPA decarboxylase and 5-HTP decarboxylase in the rat brain. Brain Res 868(2):268–274
Allen GFG (2010) The neurochemical consequences of aromatic L-amino acid decarboxylase deficiency. UCL Institute of Neurology, London
Kuruma I, Bartholini G, Tissot R, Pletscher A (1971) The metabolism of L-3-O-methyldopa, a precursor of dopa in man. Clin Pharmacol Ther 12(4):678–682
Marion MH, Stocchi F, Quinn NP, Jenner P, Marsden CD (1986) Repeated levodopa infusions in fluctuating Parkinson’s disease: clinical and pharmacokinetic data. Clin Neuropharmacol 9(2):165–181
Lee ES, Chen H, King J, Charlton C (2008) The role of 3-O-methyldopa in the side effects of L-dopa. Neurochem Res 33(3):401–411
Adamiak-Giera U, Jawien W, Pierzchlinska A, Bialecka M, Kobierski JD, Janus T, Gawronska-Szklarz B (2021) Pharmacokinetics of levodopa and 3-O-methyldopa in parkinsonian patients treated with levodopa and ropinirole and in patients with motor complications. Pharmaceutics 13(9):1395
Feuerstein C, Serre F, Gavend M, Pellat J, Perret J, Tanche M (1977) Plasma O-methyldopa in levodopa-induced dyskinesias. A bioclinical investigation. Acta Neurol Scand 56(6):508–524
Mena MA, Muradas V, Bazan E, Reiriz J, de Yebenes JG (1987) Pharmacokinetics of L-dopa in patients with Parkinson’s disease. Adv Neurol 45:481–486
Rivera-Calimlim L, Tandon D, Anderson F, Joynt R (1977) The clinical picture and plasma levodopa metabolite profile of parkinsonian nonresponders. Treatment with levodopa and decarboxylase inhibitor. Arch Neurol 34(4):228–232
Tohgi H, Abe T, Kikuchi T, Takahashi S, Nozaki Y (1991) The significance of 3-O-methyldopa concentrations in the cerebrospinal fluid in the pathogenesis of wearing-off phenomenon in Parkinson’s disease. Neurosci Lett 132(1):19–22
Kubaski F, Herbst ZM, Pereira DAA, Silva C, Chen C, Hwu PWL, van der Linden H, Lourenco CM et al (2021) Evaluation of 3-O-methyldopa as a biomarker for aromatic L-amino acid decarboxylase deficiency in 7 Brazilian cases. Mol Genet Metab Rep 27:100744
Burlina A, Giuliani A, Polo G, Gueraldi D, Gragnaniello V, Cazzorla C, Opladen T, Hoffmann G et al (2021) Detection of 3-O-methyldopa in dried blood spots for neonatal diagnosis of aromatic L-amino-acid decarboxylase deficiency: the northeastern Italian experience. Mol Genet Metab 133(1):56–62
Czarnecka AM, Hilgier W, Zielinska M (2020) S-adenosylmethionine deficiency and brain accumulation of S-adenosylhomocysteine in thioacetamide-induced acute liver failure. Nutrients 12(7):2135
Obeid R, Schadt A, Dillmann U, Kostopoulos P, Fassbender K, Herrmann W (2009) Methylation status and neurodegenerative markers in Parkinson disease. Clin Chem 55(10):1852–1860
Alachkar A, Brotchie JM, Jones OT (2010) Binding of dopamine and 3-methoxytyramine as l-DOPA metabolites to human alpha(2)-adrenergic and dopaminergic receptors. Neurosci Res 67(3):245–249
Deleu D, Northway MG, Hanssens Y (2002) Clinical pharmacokinetic and pharmacodynamic properties of drugs used in the treatment of Parkinson’s disease. Clin Pharmacokinet 41(4):261–309
Wade LA, Katzman R (1975) 3-O-methyldopa uptake and inhibition of L-dopa at the blood-brain barrier. Life Sci 17(1):131–136
Weiner WJ (2006) Levodopa–toxic or neuroprotective? Nat Clin Pract Neurol 2(10):518–519
Ziv I, Zilkha-Falb R, Offen D, Shirvan A, Barzilai A, Melamed E (1997) Levodopa induces apoptosis in cultured neuronal cells–a possible accelerator of nigrostriatal degeneration in Parkinson’s disease? Mov Disord 12(1):17–23
Pierson J, Norris JL, Aerni HR, Svenningsson P, Caprioli RM, Andren PE (2004) Molecular profiling of experimental Parkinson’s disease: direct analysis of peptides and proteins on brain tissue sections by MALDI mass spectrometry. J Proteome Res 3(2):289–295
Scholz B, Svensson M, Alm H, Skold K, Falth M, Kultima K, Guigoni C, Doudnikoff E et al (2008) Striatal proteomic analysis suggests that first L-dopa dose equates to chronic exposure. PLoS ONE 3(2):e1589
Pattison DI, Dean RT, Davies MJ (2002) Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology 177(1):23–37
Shehata AM, Ahmed-Farid OA, Rizk HA, Saber SM, Lashin FM, Re L (2020) Neurochemical, neurobehavioral and histochemical effects of therapeutic dose of l-dopa on striatal neurons in rats: protective effect of virgin coconut oil. Biomed Pharmacother 130:110473
Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J et al (2015) NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85(1):101–115
Mena MA, Pardo B, Casarejos MJ, Fahn S, Garcia de Yebenes J (1992) Neurotoxicity of levodopa on catecholamine-rich neurons. Mov Disord 7(1):23–31
Lai CT, Yu PH (1997) Dopamine- and L-beta-3,4-dihydroxyphenylalanine hydrochloride (L-Dopa)-induced cytotoxicity towards catecholaminergic neuroblastoma SH-SY5Y cells. Effects of oxidative stress and antioxidative factors. Biochem Pharmacol 53(3):363–372
Blessing H, Bareiss M, Zettlmeisl H, Schwarz J, Storch A (2003) Catechol-O-methyltransferase inhibition protects against 3,4-dihydroxyphenylalanine (DOPA) toxicity in primary mesencephalic cultures: new insights into levodopa toxicity. Neurochem Int 42(2):139–151
Mytilineou C, Walker RH, JnoBaptiste R, Olanow CW (2003) Levodopa is toxic to dopamine neurons in an in vitro but not an in vivo model of oxidative stress. J Pharmacol Exp Ther 304(2):792–800
Han SK, Mytilineou C, Cohen G (1996) L-DOPA up-regulates glutathione and protects mesencephalic cultures against oxidative stress. J Neurochem 66(2):501–510
Jang W, Park HH, Lee KY, Lee YJ, Kim HT, Koh SH (2015) 1,25-Dyhydroxyvitamin D3 attenuates L-DOPA-induced neurotoxicity in neural stem cells. Mol Neurobiol 51(2):558–570
Pardo B, Mena MA, de Yebenes JG (1995) L-dopa inhibits complex IV of the electron transport chain in catecholamine-rich human neuroblastoma NB69 cells. J Neurochem 64(2):576–582
Colamartino M, Padua L, Meneghini C, Leone S, Cornetta T, Testa A, Cozzi R (2012) Protective effects of L-dopa and carbidopa combined treatments on human catecholaminergic cells. DNA Cell Biol 31(11):1572–1579
Mena MA, Davila V, Sulzer D (1997) Neurotrophic effects of L-DOPA in postnatal midbrain dopamine neuron/cortical astrocyte cocultures. J Neurochem 69(4):1398–1408
Asanuma M, Miyazaki I (2016) 3-O-methyldopa inhibits astrocyte-mediated dopaminergic neuroprotective effects of L-DOPA. BMC Neurosci 17(1):52
Koshimura K, Tanaka J, Murakami Y, Kato Y (2000) Effects of dopamine and L-DOPA on survival of PC12 cells. J Neurosci Res 62(1):112–119
Jia Z, Zhu H, Misra BR, Li Y, Misra HP (2008) Dopamine as a potent inducer of cellular glutathione and NAD(P)H:quinone oxidoreductase 1 in PC12 neuronal cells: a potential adaptive mechanism for dopaminergic neuroprotection. Neurochem Res 33(11):2197–2205
Newcomer TA, Rosenberg PA, Aizenman E (1995) Iron-mediated oxidation of 3,4-dihydroxyphenylalanine to an excitotoxin. J Neurochem 64(4):1742–1748
Anyanwu BO, Ezejiofor AN, Igweze ZN, Orisakwe OE (2018) Heavy metal mixture exposure and effects in developing nations: an update. Toxics 6(4):65
Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52(1):1–6
Stokes AH, Hastings TG, Vrana KE (1999) Cytotoxic and genotoxic potential of dopamine. J Neurosci Res 55(6):659–665
Gotz ME, Kunig G, Riederer P, Youdim MB (1994) Oxidative stress: free radical production in neural degeneration. Pharmacol Ther 63(1):37–122
Allen GF, Ullah Y, Hargreaves IP, Land JM, Heales SJ (2013) Dopamine but not l-dopa stimulates neural glutathione metabolism. Potential implications for Parkinson’s and other dopamine deficiency states. Neurochem Int 62(5):684–694
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26-36 (discussion S36-8)
Badanjak K, Fixemer S, Smajic S, Skupin A, Grunewald A (2021) The contribution of microglia to neuroinflammation in Parkinson’s disease. Int J Mol Sci 22(9):4676
Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM (2020) Blood-brain barrier leakage is increased in Parkinson’s disease. Front Physiol 11:593026
Agostinho P, Cunha RA, Oliveira C (2010) Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des 16(25):2766–2778
Kurz C, Walker L, Rauchmann BS, Perneczky R (2022) Dysfunction of the blood-brain barrier in Alzheimer’s disease: evidence from human studies. Neuropathol Appl Neurobiol 48(3):e12782
Tam OH, Rozhkov NV, Shaw R, Kim D, Hubbard I, Fennessey S, Propp N, Consortium NA et al (2019) Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress, and activated glia. Cell Rep 29(5):1164-1177 e5
Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, Sallot S, Saporta S et al (2012) Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res 1469:114–128
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL et al (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26(24):6413–21
Hu X, Liou AK, Leak RK, Xu M, An C, Suenaga J, Shi Y, Gao Y et al (2014) Neurobiology of microglial action in CNS injuries: receptor-mediated signaling mechanisms and functional roles. Prog Neurobiol 119–120:60–84
Chan SJ, Niu W, Hayakawa K, Hamanaka G, Wang X, Cheah PS, Guo S, Yu Z et al (2019) Promoting neuro-supportive properties of astrocytes with epidermal growth factor hydrogels. Stem Cells Transl Med 8(12):1242–1248
Miyazaki I, Asanuma M, Kikkawa Y, Takeshima M, Murakami S, Miyoshi K, Sogawa N, Kita T (2011) Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 59(3):435–451
Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48(5):394–403
Tilleux S, Hermans E (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85(10):2059–2070
Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K (2012) L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation 9:275
Vargas MR, Johnson JA (2009) The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 11:e17
Li S, Zhou C, Zhu Y, Chao Z, Sheng Z, Zhang Y, Zhao Y (2021) Ferrostatin-1 alleviates angiotensin II (Ang II)-induced inflammation and ferroptosis in astrocytes. Int Immunopharmacol 90:107179
Miyazaki I, Asanuma M, Diaz-Corrales FJ, Miyoshi K, Ogawa N (2004) Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res 1029(1):120–123
Furukawa N, Arai N, Goshima Y, Miyamae T, Ohshima E, Suzuki F, Fujita K, Misu Y (2001) Endogenously released DOPA is a causal factor for glutamate release and resultant delayed neuronal cell death by transient ischemia in rat striata. J Neurochem 76(3):815–824
Asanuma M, Miyazaki I, Murakami S, Diaz-Corrales FJ, Ogawa N (2014) Striatal astrocytes act as a reservoir for L-DOPA. PLoS ONE 9(9):e106362
Springer W, Kahle PJ (2011) Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 7(3):266–278
Chen H, Chan DC (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet 18(R2):R169–R176
Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, Mochly-Rosen D (2019) Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci 22(10):1635–1648
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM (2000) M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 164(12):6166–6173
Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53(2):1181–1194
Nissanka N, Moraes CT (2018) Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett 592(5):728–742
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D et al (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis 3:30
Macdonald R, Barnes K, Hastings C, Mortiboys H (2018) Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem Soc Trans 46(4):891–909
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S et al (2014) Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A 111(26):9633–9638
Khasnavis S, Pahan K (2014) Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9(4):569–581
Pons R, Ford B, Chiriboga CA, Clayton PT, Hinton V, Hyland K, Sharma R, De Vivo DC (2004) Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology 62(7):1058–1065
Lee HF, Tsai CR, Chi CS, Chang TM, Lee HJ (2009) Aromatic L-amino acid decarboxylase deficiency in Taiwan. Eur J Paediatr Neurol 13(2):135–140
Lee WT, Lin JH, Weng WC, Peng SS (2017) Microstructural changes of brain in patients with aromatic L-amino acid decarboxylase deficiency. Hum Brain Mapp 38(3):1532–1540
Pun PB, Lu J, Moochhala S (2009) Involvement of ROS in BBB dysfunction. Free Radic Res 43(4):348–364
Walsh J, Tozer DJ, Sari H, Hong YT, Drazyk A, Williams G, Shah NJ, O’Brien JT et al (2021) Microglial activation and blood-brain barrier permeability in cerebral small vessel disease. Brain 144(5):1361–1371
Kamath AF, Chauhan AK, Kisucka J, Dole VS, Loscalzo J, Handy DE, Wagner DD (2006) Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 107(2):591–593
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7(1):41–53
Gray MT, Woulfe JM (2015) Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab 35(5):747–750
Aschauer DF, Kreuz S, Rumpel S (2013) Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS ONE 8(9):e76310
Maeda T, Cheng N, Kume T, Kaneko S, Kouchiyama H, Akaike A, Ueda M, Satoh M et al (1997) L-DOPA neurotoxicity is mediated by glutamate release in cultured rat striatal neurons. Brain Res 771(1):159–162
Goshima Y, Ohno K, Nakamura S, Miyamae T, Misu Y, Akaike A (1993) L-dopa induces Ca(2+)-dependent and tetrodotoxin-sensitive release of endogenous glutamate from rat striatal slices. Brain Res 617(1):167–170
Gill R, Foster AC, Woodruff GN (1987) Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J Neurosci 7(10):3343–3349
Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T (1990) 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247(4942):571–574
Pedrosa R, Soares-da-Silva P (2002) Oxidative and non-oxidative mechanisms of neuronal cell death and apoptosis by L-3,4-dihydroxyphenylalanine (L-DOPA) and dopamine. Br J Pharmacol 137(8):1305–1313
Golembiowska K, Dziubina A (2012) Effect of adenosine A(2A) receptor antagonists and L-DOPA on hydroxyl radical, glutamate and dopamine in the striatum of 6-OHDA-treated rats. Neurotox Res 21(2):222–230
Chan SW, Dunlop RA, Rowe A, Double KL, Rodgers KJ (2012) L-DOPA is incorporated into brain proteins of patients treated for Parkinson’s disease, inducing toxicity in human neuroblastoma cells in vitro. Exp Neurol 238(1):29–37
Davis AA, Leyns CEG, Holtzman DM (2018) Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol 34:545–568
Stansley BJ, Yamamoto BK (2013) L-dopa-induced dopamine synthesis and oxidative stress in serotonergic cells. Neuropharmacology 67:243–251
Kannari K, Yamato H, Shen H, Tomiyama M, Suda T, Matsunaga M (2001) Activation of 5-HT(1A) but not 5-HT(1B) receptors attenuates an increase in extracellular dopamine derived from exogenously administered L-DOPA in the striatum with nigrostriatal denervation. J Neurochem 76(5):1346–1353
Navailles S, Bioulac B, Gross C, De Deurwaerdere P (2011) Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis 41(2):585–590
Stansley BJ, Yamamoto BK (2015) Behavioral impairments and serotonin reductions in rats after chronic L-dopa. Psychopharmacology 232(17):3203–3213
Stansley BJ, Yamamoto BK (2015) L-dopa and brain serotonin system dysfunction. Toxics 3(1):75–88
Kalogiannis M, Delikatny EJ, Jeitner TM (2016) Serotonin as a putative scavenger of hypohalous acid in the brain. Biochim Biophys Acta 1862(4):651–661
Xia C, Tong X (2018) Moschamine-related indole alkaloids, Alkaloids. Chem Biol 79:139–189
Miyazaki I, Asanuma M, Murakami S, Takeshima M, Torigoe N, Kitamura Y, Miyoshi K (2013) Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol Dis 59:244–256
Mor A, Tankiewicz-Kwedlo A, Krupa A, Pawlak D (2021) Role of kynurenine pathway in oxidative stress during neurodegenerative disorders. Cells 10(7):1603
Keithahn C, Lerchl A (2005) 5-Hydroxytryptophan is a more potent in vitro hydroxyl radical scavenger than melatonin or vitamin C. J Pineal Res 38(1):62–66
Soares-da-Silva P, Parada A, Serrao P (2000) The O-methylated derivative of L-DOPA, 3-O-methyl-L-DOPA, fails to inhibit neuronal and non-neuronal aromatic L-amino acid decarboxylase. Brain Res 863(1–2):293–297
Kuhn DM, Arthur RE Jr (1999) L-DOPA-quinone inactivates tryptophan hydroxylase and converts the enzyme to a redox-cycling quinoprotein. Brain Res Mol Brain Res 73(1–2):78–84
LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ (2005) Dopamine covalently modifies and functionally inactivates parkin. Nat Med 11(11):1214–1221
Skovierova H, Vidomanova E, Mahmood S, Sopkova J, Drgova A, Cervenova T, Halasova E, Lehotsky J (2016) The molecular and cellular effect of homocysteine metabolism imbalance on human health. Int J Mol Sci 17(10):1733
Moretti R, Caruso P (2019) The controversial role of homocysteine in neurology: from labs to clinical practice. Int J Mol Sci 20(1):231
Obeid R, McCaddon A, Herrmann W (2007) The role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric diseases. Clin Chem Lab Med 45(12):1590–1606
Muller T, Woitalla D, Muhlack S (2011) Inhibition of catechol-O-methyltransferase modifies acute homocysteine rise during repeated levodopa application in patients with Parkinson’s disease. Naunyn Schmiedebergs Arch Pharmacol 383(6):627–633
Muller T, Woitalla D, Fowler B, Kuhn W (2002) 3-OMD and homocysteine plasma levels in parkinsonian patients. J Neural Transm (Vienna) 109(2):175–179
Miller JW, Selhub J, Nadeau MR, Thomas CA, Feldman RG, Wolf PA (2003) Effect of L-dopa on plasma homocysteine in PD patients: relationship to B-vitamin status. Neurology 60(7):1125–1129
Obeid R, Herrmann W (2006) Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett 580(13):2994–3005
Kumar M, Ray RS, Sandhir R (2018) Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: in vitro and in vivo studies. Neurochem Int 120:87–98
da Cunha AA, Ferreira AG, Wyse AT (2010) Increased inflammatory markers in brain and blood of rats subjected to acute homocysteine administration. Metab Brain Dis 25(2):199–206
Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, Tyagi N (2013) Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 252:302–319
Dos Santos TM, Ramires Junior OV, Alves VS, Coutinho-Silva R, Savio LEB, Wyse ATS (2021) Hyperhomocysteinemia alters cytokine gene expression, cytochrome c oxidase activity and oxidative stress in striatum and cerebellum of rodents. Life Sci 277:119386
Bhattacharjee N, Paul R, Giri A, Borah A (2016) Chronic exposure of homocysteine in mice contributes to dopamine loss by enhancing oxidative stress in nigrostriatum and produces behavioral phenotypes of Parkinson’s disease. Biochem Biophys Rep 6:47–53
Christine CW, Auinger P, Joslin A, Yelpaala Y, Green R, D.I. Parkinson Study Group (2018) Vitamin B12 and homocysteine levels predict different outcomes in early Parkinson’s disease. Mov Disord 33(5):762–770
Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ et al (2013) Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem 126(5):591–603
Gsell W, Reichert N, Youdim MB, Riederer P (1995) Interaction of neuroprotective substances with human brain superoxide dismutase. An in vitro study. J Neural Transm Suppl 45:271–279
Chu M, Teng J, Guo L, Wang Y, Zhang L, Gao J, Liu L (2021) Mild hyperhomocysteinemia induces blood-brain barrier dysfunction but not neuroinflammation in the cerebral cortex and hippocampus of wild-type mice. Can J Physiol Pharmacol 99(9):847–856
Guttormsen AB, Mansoor AM, Fiskerstrand T, Ueland PM, Refsum H (1993) Kinetics of plasma homocysteine in healthy subjects after peroral homocysteine loading. Clin Chem 39(7):1390–1397
Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS (1997) Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A 94(11):5923–5928
Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A (2003) Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res 5(4):237–243
Zafra F, Ibanez I, Bartolome-Martin D, Piniella D, Arribas-Blazquez M, Gimenez C (2017) Glycine transporters and its coupling with NMDA receptors. Adv Neurobiol 16:55–83
Folbergrova J (1994) NMDA and not non-NMDA receptor antagonists are protective against seizures induced by homocysteine in neonatal rats. Exp Neurol 130(2):344–350
Cervetto C, Venturini A, Passalacqua M, Guidolin D, Genedani S, Fuxe K, Borroto-Esquela DO, Cortelli P et al (2017) A2A–D2 receptor-receptor interaction modulates gliotransmitter release from striatal astrocyte processes. J Neurochem 140(2):268–279
Cervetto C, Venturini A, Guidolin D, Maura G, Passalacqua M, Tacchetti C, Cortelli P, Genedani S et al (2018) Homocysteine and A2A–D2 receptor-receptor interaction at striatal astrocyte processes. J Mol Neurosci 65(4):456–466
Kredich NM, Martin DV Jr (1977) Role of S-adenosylhomocysteine in adenosinemediated toxicity in cultured mouse T lymphoma cells. Cell 12(4):931–938
Hauser RA, Schwarzschild MA (2005) Adenosine A2A receptor antagonists for Parkinson’s disease: rationale, therapeutic potential and clinical experience. Drugs Aging 22(6):471–482
Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H (2005) Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J 19(13):1854–1856
Kimura H, Shibuya N, Kimura Y (2012) Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal 17(1):45–57
Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H (2009) 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal 11(4):703–714
Schalinske KL, Smazal AL (2012) Homocysteine imbalance: a pathological metabolic marker. Adv Nutr 3(6):755–762
Nagpure BV, Bian JS (2015) Brain, learning, and memory: role of H2S in neurodegenerative diseases. Handb Exp Pharmacol 230:193–215
Talaei F, Bouma HR, Van der Graaf AC, Strijkstra AM, Schmidt M, Henning RH (2011) Serotonin and dopamine protect from hypothermia/rewarming damage through the CBS/H2S pathway. PLoS ONE 6(7):e22568
Fuchs D, Jaeger M, Widner B, Wirleitner B, Artner-Dworzak E, Leblhuber F (2001) Is hyperhomocysteinemia due to the oxidative depletion of folate rather than to insufficient dietary intake? Clin Chem Lab Med 39(8):691–694
Sunden SL, Renduchintala MS, Park EI, Miklasz SD, Garrow TA (1997) Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch Biochem Biophys 345(1):171–174
Finkelstein JD (1990) Methionine metabolism in mammals. J Nutr Biochem 1(5):228–237
Tallan HH, Moore S, Stein WH (1958) L-cystathionine in human brain. J Biol Chem 230(2):707–716
Diwakar L, Ravindranath V (2007) Inhibition of cystathionine-gamma-lyase leads to loss of glutathione and aggravation of mitochondrial dysfunction mediated by excitatory amino acid in the CNS. Neurochem Int 50(2):418–426
Eto K, Kimura H (2002) The production of hydrogen sulfide is regulated by testosterone and S-adenosyl-L-methionine in mouse brain. J Neurochem 83(1):80–86
Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R (2006) A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem 281(47):35785–35793
Yang Q, He GW (2019) Imbalance of homocysteine and H2S: significance, mechanisms, and therapeutic promise in vascular injury. Oxid Med Cell Longev 2019:7629673
Kabil O, Vitvitsky V, Xie P, Banerjee R (2011) The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 15(2):363–372
Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP (2010) Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13(11):1763–1811
Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R (2009) H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J Biol Chem 284(17):11601–11612
Nakano S, Ishii I, Shinmura K, Tamaki K, Hishiki T, Akahoshi N, Ida T, Nakanishi T et al (2015) Hyperhomocysteinemia abrogates fasting-induced cardioprotection against ischemia/reperfusion by limiting bioavailability of hydrogen sulfide anions. J Mol Med (Berl) 93(8):879–889
Kumar M, Sandhir R (2018) Hydrogen sulfide in physiological and pathological mechanisms in brain. CNS Neurol Disord Drug Targets 17(9):654–670
Mikami Y, Shibuya N, Kimura Y, Nagahara N, Ogasawara Y, Kimura H (2011) Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem J 439(3):479–485
Bilska-Wilkosz A, Iciek M, Kowalczyk-Pachel D, Gorny M, Sokolowska-Jezewicz M, Wlodek L (2017) Lipoic acid as a possible pharmacological source of hydrogen sulfide/sulfane sulfur. Molecules 22(3):388
Yadav PK, Vitvitsky V, Carballal S, Seravalli J, Banerjee R (2020) Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J Biol Chem 295(19):6299–6311
Tyagi N, Moshal KS, Sen U, Vacek TP, Kumar M, Hughes WM Jr, Kundu S, Tyagi SC (2009) H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid Redox Signal 11(1):25–33
Sun H, Yang J, Shi Y, Wang Y, Li C, Zhu M (2020) Hydrogen sulfide in the nucleus tractus solitarii regulates gastric acid secretion in rats. J Physiol Pharmacol 71(4). https://doi.org/10.26402/jpp.2020.4.05
Saberian S, Rowan P, Hammes F, Patel P, Fernandez-Cortes F, Buesch K, Beitia Ortiz de Zarate I (2022) Burden of illness of aromatic L-amino acid decarboxylase deficiency: a survey of physicians in Southern Europe. Curr Med Res Opin 38(7):1115–1123
Untereiner A, Wu L (2018) Hydrogen sulfide and glucose homeostasis: a tale of sweet and the stink. Antioxid Redox Signal 28(16):1463–1482
Umhau JC, Petrulis SG, Diaz R, Rawlings R, George DT (2003) Blood glucose is correlated with cerebrospinal fluid neurotransmitter metabolites. Neuroendocrinology 78(6):339–343
Chen NC, Yang F, Capecci LM, Gu Z, Schafer AI, Durante W, Yang XF, Wang H (2010) Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J 24(8):2804–2817
Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME et al (2001) Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J Nutr 131(11):2811–2818
Miller AL (2003) The methionine-homocysteine cycle and its effects on cognitive diseases. Altern Med Rev 8(1):7–19
Ivanov AV, Dubchenko EA, Kruglova MP, Virus ED, Bulgakova PO, Alexandrin VV, Fedoseev AN, Boyko AN et al (2019) Determination of S-adenosylmethionine and S-adenosylhomocysteine in blood plasma by UPLC with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci 1124:366–374
Surtees R, Hyland K (1990) L-3,4-dihydroxyphenylalanine (levodopa) lowers central nervous system S-adenosylmethionine concentrations in humans. J Neurol Neurosurg Psychiatry 53(7):569–572
Nieraad H, de Bruin N, Arne O, Hofmann MCJ, Gurke R, Schmidt D, Ritter M, Parnham MJ et al (2021) Effects of Alzheimer-like pathology on homocysteine and homocysteic acid levels-an exploratory in vivo kinetic study. Int J Mol Sci 22(2):927
Olney JW, Price MT, Salles KS, Labruyere J, Ryerson R, Mahan K, Frierdich G, Samson L (1987) L-homocysteic acid: an endogenous excitotoxic ligand of the NMDA receptor. Brain Res Bull 19(5):597–602
Sommer S, Hunzinger C, Schillo S, Klemm M, Biefang-Arndt K, Schwall G, Putter S, Hoelzer K et al (2004) Molecular analysis of homocysteic acid-induced neuronal stress. J Proteome Res 3(3):572–581
Gortz P, Hoinkes A, Fleischer W, Otto F, Schwahn B, Wendel U, Siebler M (2004) Implications for hyperhomocysteinemia: not homocysteine but its oxidized forms strongly inhibit neuronal network activity. J Neurol Sci 218(1–2):109–114
Folbergrova J, Haugvicova R, Mares P (2000) Behavioral and metabolic changes in immature rats during seizures induced by homocysteic acid: the protective effect of NMDA and non-NMDA receptor antagonists. Exp Neurol 161(1):336–345
Hasegawa T, Mikoda N, Kitazawa M, LaFerla FM (2010) Treatment of Alzheimer’s disease with anti-homocysteic acid antibody in 3xTg-AD male mice. PLoS ONE 5(1):e8593
Hasegawa T, Ukai W (2016) Targeting therapy for homocysteic acid in the blood represents a potential recovery treatment for cognition in Alzheimer’s disease patients. Aging (Albany NY) 8(9):1838–1843
Kojima K, Nakajima T, Taga N, Miyauchi A, Kato M, Matsumoto A, Ikeda T, Nakamura K et al (2019) Gene therapy improves motor and mental function of aromatic l-amino acid decarboxylase deficiency. Brain 142(2):322–333
Blandini F, Nappi G, Fancellu R, Mangiagalli A, Samuele A, Riboldazzi G, Calandrella D, Pacchetti C et al (2003) Modifications of plasma and platelet levels of L-DOPA and its direct metabolites during treatment with tolcapone or entacapone in patients with Parkinson’s disease. J Neural Transm (Vienna) 110(8):911–922
Muller T, Muhlack S (2009) Peripheral COMT inhibition prevents levodopa associated homocysteine increase. J Neural Transm (Vienna) 116(10):1253–1256
Kang KS, Wen Y, Yamabe N, Fukui M, Bishop SC, Zhu BT (2010) Dual beneficial effects of (-)-epigallocatechin-3-gallate on levodopa methylation and hippocampal neurodegeneration: in vitro and in vivo studies. PLoS ONE 5(8):e11951
Kang KS, Yamabe N, Wen Y, Fukui M, Zhu BT (2013) Beneficial effects of natural phenolics on levodopa methylation and oxidative neurodegeneration. Brain Res 1497:1–14
Tang XQ, Shen XT, Huang YE, Chen RQ, Ren YK, Fang HR, Zhuang YY, Wang CY (2011) Inhibition of endogenous hydrogen sulfide generation is associated with homocysteine-induced neurotoxicity: role of ERK1/2 activation. J Mol Neurosci 45(1):60–67
Chen X, Jhee KH, Kruger WD (2004) Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem 279(50):52082–52086
Coletta C, Modis K, Szczesny B, Brunyanszki A, Olah G, Rios EC, Yanagi K, Ahmad A et al (2015) Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/H2S pathway: functional impairment by hyperglycemia and restoration by DL-alpha-lipoic acid. Mol Med 21(1):1–14
Lockhart B, Jones C, Cuisinier C, Villain N, Peyroulan D, Lestage P (2000) Inhibition of L-homocysteic acid and buthionine sulphoximine-mediated neurotoxicity in rat embryonic neuronal cultures with alpha-lipoic acid enantiomers. Brain Res 855(2):292–297
Zhang SF, Xie CL, Lin JY, Wang MH, Wang XJ, Liu ZG (2018) Lipoic acid alleviates LDOPAinduced dyskinesia in 6OHDA parkinsonian rats via antioxidative stress. Mol Med Rep 17(1):1118–1124
Mao J, Gao H, Bai W, Zeng H, Ren Y, Liu Y, Yang X (2021) Lipoic acid alleviates LPS-evoked PC12 cell damage by targeting p53 and inactivating the NF-kappaB pathway. Acta Neurobiol Exp (Wars) 81(4):375–385
VonVoigtlander PF, Fici GJ, Althaus JS (1998) Pharmacological approaches to counter the toxicity of Dopa. Amino Acids 14(1–3):189–196
Cheng N, Maeda T, Kume T, Kaneko S, Kochiyama H, Akaike A, Goshima Y, Misu Y (1996) Differential neurotoxicity induced by L-DOPA and dopamine in cultured striatal neurons. Brain Res 743(1–2):278–283
Machado FR, Ferreira AG, da Cunha AA, Tagliari B, Mussulini BH, Wofchuk S, Wyse AT (2011) Homocysteine alters glutamate uptake and Na+, K+-ATPase activity and oxidative status in rats hippocampus: protection by vitamin C. Metab Brain Dis 26(1):61–67
Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK (2006) Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem 98(5):1474–1486
Fahn S (1992) A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Ann Neurol 32(Suppl):S128–S132
Kubis AM, Piwowar A (2015) The new insight on the regulatory role of the vitamin D3 in metabolic pathways characteristic for cancerogenesis and neurodegenerative diseases. Ageing Res Rev 24(Pt B):126–137
Lima LAR, Lopes MJP, Costa RO, Lima FAV, Neves KRT, Calou IBF, Andrade GM, Viana GSB (2018) Vitamin D protects dopaminergic neurons against neuroinflammation and oxidative stress in hemiparkinsonian rats. J Neuroinflammation 15(1):249
Orme RP, Middleditch C, Waite L, Fricker RA (2016) The role of vitamin D(3) in the development and neuroprotection of midbrain dopamine neurons. Vitam Horm 100:273–297
Suzuki M, Yoshioka M, Hashimoto M, Murakami M, Noya M, Takahashi D, Urashima M (2013) Randomized, double-blind, placebo-controlled trial of vitamin D supplementation in Parkinson disease. Am J Clin Nutr 97(5):1004–1013
Park KH, Choi NY, Koh SH, Park HH, Kim YS, Kim MJ, Lee SJ, Yu HJ et al (2011) L-DOPA neurotoxicity is prevented by neuroprotective effects of erythropoietin. Neurotoxicology 32(6):879–887
Yamamoto M, Koshimura K, Kawaguchi M, Sohmiya M, Murakami Y, Kato Y (2000) Stimulating effect of erythropoietin on the release of dopamine and acetylcholine from the rat brain slice. Neurosci Lett 292(2):131–133
Huang CK, Chang YT, Amstislavskaya TG, Tikhonova MA, Lin CL, Hung CS, Lai TJ, Ho YJ (2015) Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Behav Brain Res 294:198–207
Hu LF, Lu M, Tiong CX, Dawe GS, Hu G, Bian JS (2010) Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 9(2):135–146
Kida K, Yamada M, Tokuda K, Marutani E, Kakinohana M, Kaneki M, Ichinose F (2011) Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid Redox Signal 15(2):343–352
Hayden LJ, Goeden H, Roth SH (1990) Exposure to low levels of hydrogen sulfide elevates circulating glucose in maternal rats. J Toxicol Environ Health 31(1):45–52
Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L et al (2013) Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18(15):1906–1919
Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI et al (2013) Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 4:1626
Zhou L, Wang Q (2023) Advances of H(2)S in regulating neurodegenerative diseases by preserving mitochondria function. Antioxidants (Basel) 12(3):652
Kimura H (2013) Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int 63(5):492–497
Martin I, Dawson VL, Dawson TM (2011) Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet 12:301–325
Lu SC, Mato JM (2012) S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev 92(4):1515–1542
Di Rocco A, Rogers JD, Brown R, Werner P, Bottiglieri T (2000) S-adenosyl-methionine improves depression in patients with Parkinson’s disease in an open-label clinical trial. Mov Disord 15(6):1225–1229
Bostantjopoulou S, Kyriazis G, Katsarou Z, Kiosseoglou G, Kazis A, Mentenopoulos G (1997) Superoxide dismutase activity in early and advanced Parkinson’s disease. Funct Neurol 12(2):63–68
Perry TL, Yong VW (1986) Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett 67(3):269–274
Peng J, Stevenson FF, Doctrow SR, Andersen JK (2005) Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. J Biol Chem 280(32):29194–29198
Asanuma M, Miyazaki I (2021) Glutathione and related molecules in parkinsonism. Int J Mol Sci 22(16):8689
Ihara Y, Chuda M, Kuroda S, Hayabara T (1999) Hydroxyl radical and superoxide dismutase in blood of patients with Parkinson’s disease: relationship to clinical data. J Neurol Sci 170(2):90–95
Maruyama W, Akao Y, Carrillo MC, Kitani K, Youdium MB, Naoi M (2002) Neuroprotection by propargylamines in Parkinson’s disease: suppression of apoptosis and induction of prosurvival genes. Neurotoxicol Teratol 24(5):675–682
Wei Y, Lu M, Mei M, Wang H, Han Z, Chen M, Yao H, Song N et al (2020) Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat Commun 11(1):941
Zhang J, Wang M, Zhao Y, Zhang Y, Gao Y, Zhang X, Yang G (2022) Alpha-lipoic acid improved motor function in MPTP-induced Parkinsonian mice by reducing neuroinflammation in the nigral and spinal cord. Neurosci Lett 781:136669
Solmonson A, DeBerardinis RJ (2018) Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem 293(20):7522–7530
Vatassery GT, Smith WE, Quach HT (2006) Effect of oxidative stress induced by L-dopa on endogenous antioxidants in PC-12 cells. Ann N Y Acad Sci 1074:330–336
Han SK, Cohen G (1996) Inhibition of catalase in mesencephalic cultures by L-DOPA and dopamine. Neurochem Int 29(6):645–649
Saito Y, Nishio K, Akazawa YO, Yamanaka K, Miyama A, Yoshida Y, Noguchi N, Niki E (2010) Cytoprotective effects of vitamin E homologues against glutamate-induced cell death in immature primary cortical neuron cultures: tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic Biol Med 49(10):1542–1549
Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD et al (2014) Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311(1):33–44
Peterson AL, Mancini M, Horak FB (2013) The relationship between balance control and vitamin D in Parkinson’s disease-a pilot study. Mov Disord 28(8):1133–1137
Gascon-Barre M, Huet PM (1983) Apparent [3H]1,25-dihydroxyvitamin D3 uptake by canine and rodent brain. Am J Physiol 244(3):E266–E271
Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G (2016) Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev 96(1):365–408
Kato S (2000) The function of vitamin D receptor in vitamin D action. J Biochem 127(5):717–722
Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ (2005) Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat 29(1):21–30
Wakhloo D, Scharkowski F, Curto Y, Javed Butt U, Bansal V, Steixner-Kumar AA, Wustefeld L, Rajput A et al (2020) Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat Commun 11(1):1313
Garcia-Llano M, Pedroso-Ibanez I, Morales-Chacon L, Rodriguez-Obaya T, Perez-Ruiz L, Sosa-Teste I, Amaro-Gonzalez D, Bringas-Vega ML (2021) Short-term tolerance of nasally-administered NeuroEPO in patients with Parkinson disease. MEDICC Rev 23(1):49–54
Acknowledgements
The author thanks Benjamin Lazar for help in the generation of figures.
Author information
Authors and Affiliations
Contributions
ZS contributed to the conception, design, analysis, writing, and the revision of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This is an observational study. The University at Buffalo Research Ethics Committee has confirmed that no ethical approval is required.
Consent to Participate
NA.
Consent for Publication
NA.
Competing Interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sternberg, Z. Neurodegenerative Etiology of Aromatic L-Amino Acid Decarboxylase Deficiency: a Novel Concept for Expanding Treatment Strategies. Mol Neurobiol 61, 2996–3018 (2024). https://doi.org/10.1007/s12035-023-03684-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03684-2