
Abstract
Neurological diseases place a substantial burden on public health and have a serious impact on the quality of life of patients. Despite the multifaceted pathological process involved in the occurrence and development of these neurological diseases, each disease has its own unique pathological characteristics and underlying molecular mechanisms which trigger their onset. Thus, it is unlikely to achieve effective treatment of neurological diseases by means of a single approach. To this end, we reason that it is pivotal to seek an efficient strategy that implements multitherapeutic targeting and addresses the multifaceted pathological process to overcome the complex issues related to neural dysfunction. In recent years, natural medicinal plant–derived monomers have received extensive attention as new neuroprotective agents for treatment of neurological disorders. Fisetin, a flavonoid, has emerged as a novel potential molecule that enhances neural protection and reverses cognitive abnormalities. The neuroprotective effects of fisetin are attributed to its multifaceted biological activity and multiple therapeutic mechanisms associated with different neurological disorders. In this review article, we summarize recent research progression regarding the pharmacological effects of fisetin in treating several neurological diseases and the potential mechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neurological disorders, especially those in the central nervous system (CNS), are still one of the leading causes of mortality and disability worldwide. There is a substantial increase in the prevalence of neurological diseases with age and an acceleration of the aging population globally, which poses a serious challenge to global community health. There is growing evidence that a number of toxic insults, including oxidative stress, trophic support loss, accumulation of protein aggregates, dysfunction of the neurovascular system, and immune system activation [1], have been identified as the main contributions to most neurological diseases. This multiplicity of insults and the possibility that each of these factors will have varying relative importance mainly depend on the individual [1]. In addition, most neurological diseases involve complex signaling pathways, making effective treatment more difficult. To effectively curtail and alleviate these neurodegenerative diseases, it is vital to comprehensively understand multiple molecular pathways and unveil detailed molecular mechanisms involved in these diseases [2]. In general, most neurological disorders do not share a common mechanism that elicits their onset. Several molecular pathways contribute to their pathogenesis in addition to the primary mechanism [2, 3]. Therefore, the exhaustive investigation of compounds that direct at different targets and modulate multiple molecular mechanisms simultaneously is very necessary and urgent. Moreover, these compounds must target multiple pathological process. Thus, compelling research has focused on naturally derived, nontoxic biomolecules from plants, such as flavonoids, for the treatment of neurological diseases.
Flavonoids are a broad category the dietary plant-derived compounds that are consumed by the human beings as part of their diet. Importantly, these plants are easily accessible [4]. Due to the extensive and immense health benefits of flavonoids, people are becoming increasingly interested in the long-term effects of consuming flavonoid-rich plants. Until now, numerous studies have showed that flavonoids possess the broad potential therapeutic effects, including antioxidants, antiviral, anti-inflammatory, anticancer, anti-bacterial, neurotrophic, neuroprotective, and immunostimulatory effects. Thus, flavonoids are also regarded as promising neuroprotective compounds that can aid in the treatment of prevalent neurodegenerative diseases [4, 5]. The flavonoid fisetin belongs to the class of flavonoids known as polyphenolic compounds, which has been extensively studied by researchers as a result of its potential to target multiple pathways. In this review, we will focus mainly on the therapeutic efficacy of fisetin in multiple animal neurological disease models, including Alzheimer’s disease (AD) [6], Parkinson’s disease (PD) [7], Huntington’s disease (HD) [8], stroke [9], epilepsy [10], depression [11], glioma [12], amyotrophic lateral sclerosis (ALS) [13] and the neurological complications. Additionally, the detailed cellular mechanisms of fisetin in the treatment of neurological diseases are discussed. Finally, the neuroprotective potential of fisetin in treating neurological diseases is systemically summarized to better understand its therapeutic effects.
Fisetin Pharmacodynamics, Pharmacokinetics, and Toxicity
Fisetin, a bioactive natural hydrophobic flavonol, is broadly found in vegetables and fruits such as cucumber, onion, strawberry, apple, lotus root, kiwifruit, peach, tomato, and persimmon, as seen in the food-plant chemical composition table [14]. Based on some recent studies and numerous previous reports, increasing research attention has been given to fisetin’s pronounced pharmaceutical properties. To date, a growing body of evidence indicates that fisetin shares distinct biological and functional properties with a plethora of other plant polyphenols, exhibiting a wide range of pharmacological activities, including anticancer [15], antioxidant [16], anti-inflammatory [17], and neuroprotective properties [18], which are tightly associated with the pharmacotherapectic strategies for multiple diseases, including neurodegenerative disorders.
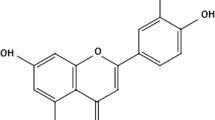
In general, the protective ability of the compounds is related to their chemical structures and substituents. As shown in Fig. 1, there are two aromatic rings in fisetin, which are connected by a 3-carbon-oxygenated heterocyclic ring, which has four hydroxyl substitutions and one oxo substitution. The structure consists of 6-hydrogen bond acceptors, 4-hydrogen bond donors, and one rotatable bond with one covalently bonded unit. The benzene structure without 5-hydroxy group indicates they can protect DNA from singlet molecular oxygen-induced single-strand breaks [19]. The hydroxylated C3, an unsaturated C ring, and hydrophobicity structure of fisetin can protect against exogenous glutamate [20]. The physiological effects of laccasein are closely related to the loss of electrons in the aromatic ring of its structure, preventing lipid peroxidation, scavenging oxygen radicals, binding glucuronides, and inhibiting arachidonic acid. Due to these unique pleiotropic pharmacological properties, fisetin exhibits activities against various diseases, including cancer and neurological and inflammatory diseases. Despite having the above pharmacological properties, the in vivo administration of fisetin remains challenging due to its poor aqueous solubility, high lipophilicity, extensive first-pass metabolism, and low oral bioavailability. Emerging studies indicate that oral bioavailability of fisetin is only 44% [21,22,23], resulting in low bioavailability and therapeutic effect in humans and animals. As a result, an effective, controlled release and safe fisetin-delivery systems for clinical use is urgently needed. Over a period of time, a variety of approaches, such as lipidosomes [24], cochleates, solid lipid nanoparticles, nano micelles, polymeric micelles, nano-emulsion, silica nanoparticles, co-crystals, cyclodextrins, and cyclosophoroase complexation, have been used to overcome solubility and bioavailability challenges [22]. These novel drug delivery options showed more evenly distributed fisetin in targeted tissues [10]. Regarding the treatment of CNS diseases, the delivery of drug molecules to the brain is often precluded by a variety of physiological, metabolic, and biochemical obstacles, such as the blood-brain barrier (BBB), blood cerebrospinal fluid barrier, and blood tumor barrier. Through a transfected Madin Darby canine kidney cell assay, Lapchak PA found that the BBB permeability rate of fisetin was low [25]. Therefore, the rational design of polymer-based drug delivery systems is essential for the drug to enter the brain interstitium. Using these drug delivery techniques, fisetin can be rapidly diffused to the brain’s blood vessels, followed by a slower dispersion to the parenchyma [26]. This indicates that using the drug delivery techniques the BBB penetration rate of fisetin can be effectively enhanced. During pharmacokinetic studies, the Cmax and area under the curve are higher, and the average residence time of drugs in plasma is also increased [27, 28]. In addition, there was no pathological evidence of renal, hepatic, or other organs dysfunction after the administration of drug delivery system of fisetin [12, 28].

Chemical structure of fisetin
Pharmacological Effects of Fisetin in Treating Neurological Diseases
Fisetin and Alzheimer’s Disease
AD is the most common type of dementia. The neuropathologic hallmarks of AD are extracellular neuroinflammatory plaques containing amyloid beta(Aβ)peptide [6] and intracellular neurofibrillary tangles containing tau [29]. The clinical manifestation of AD is the progressive loss of cognitive abilities, memory, and personality and visual-spatial confusion, which eventually leads to inability to perform daily functional activities. The current treatment for AD is only symptomatic and provides short-term improvements in cognitive function without halting the pathological progression of the disease.
Fisetin has been identified as a disease-related active small molecule [30] and plays a possible pharmacological role in neurodegeneration. The therapeutic effect of fisetin was investigated in APPswe/PS1dE9 double transgenic AD model mice aged 9 to 12 months [31]. The study showed that oral administration of fisetin can cause AD-related behavioral and pathophysiological changes in an age-dependent manner, as demonstrated by the Morris water maze test. At 9 months of age, fisetin had no significant effect on the behavior of the wild-type mice in the acquisition task. In contrast, oral administration of fisetin to AD mice improved their learning ability and memory, and animal’s task performance was almost the same as that of wild-type mice. To substantiate this hypothesis, mice were tested again at 12 months to determine whether fisetin continues to reduce memory deficits. As expected, the test result showed that it takes longer time for AD mice to find the destination than for wild-type mice. In contrast, there was no significant difference between AD mice fed with fisetin and wild-type mice. The findings imply that fisetin exerts a potential preventive or therapeutic role in AD. The deposition of Aβ aggregates can cause synaptic dysfunction, tau hyperphosphorylation, and neurodegeneration, leading to cognitive impairment. To identify the effect of fisetin on the accumulation of Aβ and tau protein, Ahmad et al. injected Aβ1-42 (3μl/5min/mouse) into the lateral ventricle of mice and concomitantly conducted intraperitoneal injection of fisetin (20 mg/kg/day). Two weeks later, Aβ aggregation and tau hyperphosphorylation were significantly reduced [6], suggesting that fisetin exerts neuroprotection by suppressing Aβ and tau aggregation. This finding is consistent with the previous experimental results showing that fisetin can regulate aluminum chloride-induced Aβ aggregationand reduce the level of phosphorylated tau [32, 33].
Apart from the neuroprotection, several research groups have shown therapeutic effect of fisetin on AD through electrophysiological changes [34]. Electrophysiology and multi-unit activity (MUA) revealed that the relative spectral power of α and β declined along with the MUA count in aged rats compared to young. However, supplementing fisetin for 4 weeks elevated the relative α-power, β-power, and MUA counts in aged rats. The findings demonstrated that fisetin prevents the aging-associated decline in relative spectral power of α, β and linked MUA in the cortex and behavioral alterations. These results provide further support for the idea that fisetin could be useful for the treatment of AD.
Fisetin and Parkinson’s Disease
PD is the second most prevalent progressive neurodegenerative disorder after AD [29] and is characterized by long-lasting depletion of striatal dopamine caused by the loss or degeneration of DA neurons in the substantia nigra (SN) of the midbrain. The clinical characteristics of PD include resting tremor, bradykinesia (slowness of movement), rigidity, and postural instability. Until now, no curative treatment for PD is available although existing treatments such as deep brain stimulation surgery and pharmacotherapies including levodopa monoamine oxidase B can alleviate some symptoms. Based on the molecular pathology of PD, it is crucial to seek an efficient multitherapeutic strategy to treat this complex disease. Since flavonoids have a wide range of biological activities, some of these have been identified specifically in the context of PD [35] using in vitro and in vivo PD models. As for the potential therapeutic effects of flavonoid fisetin in vitro models of PD, primary mid-brain neurons or cell line, such as PC12 cells, have been used for in vitro study of fisetin activities. Intriguingly, fisetin can significantly suppress neuronal cell degeneration and death by PD-relevant insults such as rotenone [7, 27, 34] and MPTP/MPP+ [36,37,38]. These studies suggest that fisetin likely has a potential protective effect on PD by attenuating cytotoxic insults. In addition, Kumar et al. [27] used a self-nanoemulsifying drug delivery system (SNEDDS) to identify the neuroprotective activity of fisetin in rotenone-treated rat PD model. Fascinatingly, they found that fisetin can effectively ameliorate the behavioral alterations. In light of current data, we speculated that fisetin may prevent neurodegeneration and can be utilized as neuroprotective agent against PD. Nevertheless, it is necessary to further investigate the underlying mechanism by which fisetin exerts its neuroprotective activities of in the complex physiological context of PD.
Fisetin and Huntington’s Disease
HD, a late-onset, progressive, and inherited neurodegenerative disorder, is caused by an expansion of a trinucleotide repeat that encodes an abnormally long polyglutamine tract in the huntingtin protein. Hitherto, no effective therapy has been identified. Since HD is an inherited neurodegenerative disorder, gene-based therapies such as a reduction in the expression of the abnormal huntingtin gene by antisense oligonucleotide and small interfering RNA (siRNA) may be an effective therapy. Nevertheless, there are still several issues to be resolved for their use in the treatment of HD, including their epigenetic memory, mismatch gene mutation, and tumor-forming risk. Thus, complex pharmacotherapies might be a promising option for treating HD. At the present, fisetin biological activity has been identified in several different models of HD including PC12 cells expressing mutant Httex1, Drosophila expressing Httex1, and R6/2 mice [8]. In an in vitro experiment, fisetin was added to PC12 cells pretreated with 5uM ponasterone (PA) to express the entire Htt exon 1 fused to EGFP. The results showed that fisetin significantly increased cell survival in a dosedependent manner, and the maximal effective doses were 5 to 10 μM. Interestingly, fisetin at 10 μM for 24 h provided equal or greater protection. In contrast, the pre-treated with 5uM PA resulted in ~45% cell death within 72 h. The data suggest that fisetin could reduce or eliminate the effect of Httex1-103QP already present in cells. In addition, fluorescence microscopy also confirmed this conclusion, and a similar number of EGFP-labeled Httex1-103QP aggregates could be seen in PA-treated PC12 cells with or without fisetin [8].
Given the positive results with fisetin in the cell-based assay, it was subsequently tested in mammalian R6/2 mouse model of HD. Oral administration of fisetin significantly ameliorated neurological deficits of R6/2 mice, improving animal performance and prolongating lifespan from 104 to 139 days [8]. These results indicate that fisetin can attenuate the effect of mutant Huntington protein in variety of HD models, highlighting that the potential therapeutic effect of fisetin in treating HD is likely attributed to its antioxidation, neurotrophic, and neuroprotection. These interesting findings have an important implication for utilizing flavonoid-based treatments for HD patients. Although the molecular mechanism needed to be investigated, these data provided important insights for future clinical applications.
Fisetin and Stroke
Stroke, including ischemic stroke and hemorrhagic stroke, is an acute neuro-deficit disease with high disability rate. Ischemic stroke, a common type of stroke, occurs when the normal blood supply to an area of the brain is disrupted. Hemorrhagic stroke is mainly due to bleeding into the brain by a sudden rupture of vessel. Hematoma usually causes oligemia, neuro-transmitter release, mitochondrial dysfunction, and cellular swelling. In addition, thrombin released from blood activates microglia and causes inflammation and edema. In ischemic stroke, high levels of low-density lipoprotein (LDL) and cholesterol constitute a major risk factor for atherosclerotic plaques that can easily form embolisms and block blood vessels. Thus, it is critical to reduce LDL and cholesterol levels to prevent stroke occurrence. An early study found that fisetin, as a natural anti-atherosclerosis component in the diet, can effectively prevent LDL oxidation and reduce oxLDL [39]. This finding has attracted increasing attentions to the application of fisetin for the treatment of ischemic stroke.
Emerging data from in vitro and in vivo studies indicate that fisetin possesses neuroprotection and considerably reduces the infarct area following ischemia stroke. For the study of neuroprotective capacity of fisetin, Dajas et al. used PC12 cells and a permanent focal ischemia (permanent middle cerebral artery occlusion-p MCAO) model [40] and treated PC12 cells with hydrogen peroxide and flavonoid fisetin of different concentration. Their result showed that fisetin significantly attenuate the decrease of cell viability caused by hydrogen peroxide. In addition, they administered fisetin to rat model of focal ischemia-induced brain injury through the intraperitoneal route [41]. Consistent with the in vitro studies, fisetin promotes the recovery of focal ischemia-induced brain injury in rats. This finding shows that fisetin has multiple beneficial actions that reduce ischemia-induced brain damage. Notably, compared with aqueous formulation of fisetin, liposomal preparation of fisetin was more beneficial and neuroprotective in focal ischemia experiments.
Moreover, fisetin was also found to exert neuroprotective effects in the temporary middle cerebral artery occlusion stroke model [42]. The researchers first developed a mouse model of temporary middle cerebral artery occlusion by the intraluminal filament method and intraperitoneally injected fisetin 20 min before the onset of ischemia. Inspiringly, a significant, dose-dependent protective effect of fisetin on stroke infarct size was observed, and animals immediately injected with fisetin before the onset of ischemia showed a trend towards smaller infarcts than those of the control placebo animals. In comparison, the delivery of higher dose of fisetin (50 mg/kg) resulted in a significantly smaller infarct infarct size. More importantly, fisetin still retained its protective capabilities even when injected 3 h after the onset of ischemia [42]. On the basis of the findings, the use of flavonoid-rich plant is capable of preventing ischemic stroke, mitigating the symptoms, and promoting early recovery.
In general, the resulting outcomes of severe stroke are deterioration of consciousness and neurological dysfunction. The development of brain tissue injury in stroke is accompanied by inflammatory response, disruption of the BBB, edema, overproduction of free radicals such as reactive oxygen species (ROS), glutamate-induced excitotoxicity, and release of hemoglobin and iron [9]. Among them, a number of clinical and animal studies have highlighted that the inflammatory response greatly contributes to the severity of brain damage and numerous sequelae after stroke [43]. Therefore, effectively antagonizing inflammatory responses may be a promising treatment strategy to facilitate functional recovery after stroke. Regarding the anti-inflammation of fisetin following stroke, one study focused on the effect of fisetin on early brain injury(EBI)after subarachnoid hemorrhage (SAH), a fatal subtype of stroke in rats [43]. The main findings of this study are as follows: (1) intraperitoneal delivery of high dose (50 mg/kg) of fisetin significantly attenuated EBI and improved neurological function after SAH; (2) fisetin significantly reduced the production of pro-inflammatory cytokines; and (3) the neuroprotective role of fisetin might be associated with the suppression of TLR 4/nuclear factor-κB (NF-κB)-mediated inflammatory signaling pathway. Due to a multifaced pathological process after SAH, cerebral hemorrhage might cause neuroinflammation in aged mice [44], and the neuroprotection of fisetin against cerebral ischemia-reperfusion injury is mediated by inhibiting oxidative stress and inflammatory factors [9]. Although the precise mechanism(s) by which fisetin exert neuroprotective role during ischemia injury remains to be elucidated, the anti-inflammatory properties of fisetin are responsible, at least in part, for the mechanisms underlying the observed cellular events.
Fisetin and Epilepsy
Epilepsy is characterized by abnormal and frequent electrical activity in the brain. Epilepsy occurrence is intimately associated with severe brain injury. In addition, stroke and central nervous system infections cause seizures and epilepsy. In general, administration of antiepileptogenic drugs following acute brain insults has certain curative effect on late epilepsy. Nevertheless, the proper choice of disease models and target populations is extremely difficult in screening desirable drugs for treatment of epilepsy. Currently, an iron-induced experimental model of traumatic epilepsy is widely used to study the effect of antiepileptogenic agents [45]. To date, it has been shown that fisetin prevents the development of iron-induced epileptic electrophysiological activity, and corresponding MUA records also showed that the pretreatment with fisetin resulted in significantly decreased MUA counts [45]. The data highlights the fisetin as a useful antiepileptogenic agent for treatment of epilepsy. Inflammation and apoptosis cascade activation are serious neurological sequelae during seizures. Fisetin, as a flavonoid molecule, is considered for its effective anti-inflammatory and anti-apoptotic properties. Thus, these properties are indicative of probable antiseizure activity of fisetin. As a result, an increasing number of studies have been performed to investigate the antiseizure activity of fisetin. The study of experimental epilepsy experiment is usually divided into two stages (acute and chronic) [10]. For the acute study, increasing current electroshock (ICES) and pentylenetetrazole (PTZ)-induced epilepsy tests were conducted. For the chronic study, the kindling model was established by the administration of PTZ at subconvulsive dose. Concomitantly, animals were treated with fisetin at different dose. The results showed that fisetin administration increased the epilepsy threshold current (STC) in the ICES test, implying fisetin neuroprotective effect [10, 45]. Likewise, in PTZ-induced epileptic model mice, administration of fisetin increased the latency for myoclonic jerks and generalized seizures, and in an induced kindling model, fisetin dose-dependently suppressed the development of kindling, downregulated mRNA expressions of the inflammatory molecules NF-κB, and COX-2, and reduced the neuronal damage in the experimental animals, which supports the potential of fisetin to prevent electrically induced seizures [10]. The data suggest that fisetin plays a critical role in seizure events and epileptogenesis by inhibiting neuroinflammatory cascade. Additionally, these studies offer a rationale that the fisetin inhibition of the NF-κB signaling pathway could be an important target for inflammation-linked disorders.
Fisetin and Depression
Depression is the leading cause of disease-related disability, which impairs quality of life in patients. Depression is usually associated with both neuropsychiatric SLE [46] and inflammation [47]. Depression commonly occurs due to the insufficient concentration of neurotransmitter (NT) in monoaminergic synapses. The imbalance of monoamine NTs might cause depressive illness. Currently, the treatments for depression and anxiety conditions are based on pharmacotherapy and cognitive behavioral therapy. A vast body of evidence supports the antidepressant effect of fisetin. Yu et al. evaluated the effect of fisetin against lipopolysaccharide and restraint stress-induced behavioral deficits and found that pre-treatment with fisetin (orally) at different dose for 7 days could reverse lipopolysaccharide (LPS)-induced increase in immobility time in forced swimming and tail suspension tests [47]. Moreover, at a higher dose, fisetin decreased the overexpression of pro-inflammatory cytokine mRNA and nitrite levels through modulation of NF-κB. Further studies showed that the antidepressant effects of fisetin involved modulation of the serotonergic system [48] by reducing serotonin metabolism, possibly through mild inhibition of monoamine oxidase A, which metabolizes serotonin, leading to amelioration of the comorbidly behavioral symptoms of depression.
In addition, fisetin has been shown to produce the antihyperalgesic effect after repeated treatment and prevent chronic neuropathic pain-induced depressive-like behavior in a dose-dependent manner [46, 48]. Mechanistically, it was shown that fistein is likely to activate serotoninergic 5-HT1A receptors and exert both antihyperalgesic and antidepressant-like effect [48]. Fisetin also appeared to have an impact on the number of senescent nerve cells in lupus model mice with depression-like behavior [46]. Neurochemical observation showed that doses of 5 and 10 μM fisetin decreased the fraction of SPiDER-b-Gal–positive senescent Neuro-2a cells in the CA3 region of the hippocampus [46], which is associated with depression.
Fisetin and Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is an age-related neurodegenerative disorder characterized by progressive degeneration of upper and lower motor neurons in the brain and spinal cord that supply voluntary muscles. Although ALS has genetic and environmental triggers, the exact cause of ALS is still unknown. It has been assumed that glutamate-induced excitotoxicity is one of the possible causes of complex pathogenesis since excess glutamate in the synapse or extracellular space is transported into neuronal and glial cells via glutamate transporter 1 (GLT-1) [13]. ROS are another factor that enhances the occurrence of ALS. Due to mutations in the gene encoding superoxide dismutase (SOD1), the ability of neuronal cells to scavenge ROS is impaired, leading to oxidative stress [45]. It has been shown that Copper/zinc SOD1 gene mutation is linked with ALS pathogenesis. Regarding this point, researchers have used three different hSOD1-related mutant models drosophila expressing mutant hSOD1-G85R, hSOD1-G93A in NSC34 cells to investigate whether mutant hSOD1 actively contributes to ALS pathogenesis [13]. The results are in accordance with what was expected. It is thus acknowledged that the pathological mechanism of ALS might correlate with the glutamate and oxidative stress–induced cell death. The antioxidant, anti-inflammatory, and antiapoptotic properties of fisetin may aid the survival of motor neurons. Oral administration of fisetin (9 mg/kg/day) to 2-month-old SOD1-G93A transgenic mice effectively improve motor functions by delaying motor deficits [13]. In addition, the delivery of fisetin significantly promotes the survival of motor neurons in the spinal cord [13]. This flavonoid also suppressed the progression of ALS and increased patient survival. The neuroprotective effect of fisetin against ALS is likely mediated at least partially through activation of ERK phosphorylation and upregulation of HO-1, GPx, and catalase to reduce oxidative stress [1, 35]. In addition, this flavonoid preserves mitochondrial SOD1 and counteracts mitochondrial DNA breaks. As a result, fisetin functions as an ideal agent to prevent the development of ALS.
The Mechanisms Underlying of Neuroprotective Effect of Fisetin in Neurological Diseases
Several lines of research literature elucidate multiple molecular signaling pathways by which fisetin participates in beneficial health effects in humans and animal models of neurological disorders. The critical mechanisms underlying fisetin’s role in neurological diseases are discussed below.
Antioxidant and Chelating Activity
Oxidative stress (OS) disrupts the chemical integrity of macromolecules and increases the risk of neurological damage [49, 50]. Neural cells’ death from oxidative stress has been studied with a variety of inducers, including exogenous glutamate, d-galactose, LPS, and rotenone [51,52,53,54]. Fisetin is a relatively suitable flavonoid that exhibits potent antioxidant properties and protects nerve cells from OS [16, 18, 20, 55]. It decreases the level of oxidative stress markers LPO and protein carbonyl [56, 57]. Pro-oxidant effects of fisetin at high concentrations have been reported. In contrast, in the tunicamycin (Tm)-mediated PC12 cell model, fisetin (<20μM) restored cell viability and inhibited apoptosis, autophagy, and ROS production [58]. In addition, fisetin reduced iron-induced lipid peroxidation in epileptic animals [45]. This activity might be related to the ability of fisetin to efficiently chelate iron [59]. Iron exhibits multiple oxidation states and is therefore redox active. This oxidation state of iron property is critical for biological activity including generation of hydroxyl radicals (–OH), ROS such as highly reactive hydroxyl radical, via the Fenton reaction. By chelating iron, fisetin could effectively reduce the harmful effects of oxidative stress and deleterious effects of iron accumulation in the brain, implying that fisetin chelating activity might play a pivotal role in its beneficial neuroprotective effects.
Regulation of Inflammatory Cytokines
Neuroinflammation is one of the important mechanisms involved in the progression of neurodegenerative diseases such as AD, PD, HD, stroke, and ALS. Numerous scientific studies have showed that inflammation may promote the progression of these neurological disorders. Microglial cells, once activated, are involved in the inflammatory response, promoting the release of pro-inflammatory cytokines and chemokines and NF-κB protein [60]. They modulate the activity of inducers, sensors, transducers, and effectors, which trigger neurodegenerative mechanisms, resulting in neuronal dysfunction and death. Several studies suggested a potential neuroprotective and anti-inflammatory role of flavonoid-rich extract. Fisetin attenuated neuroinflammatory responses by suppressing the activation of microglial cells and the production of TNF-α and IL-1β [60, 61]. Other research groups have demonstrated a fisetin-mediated inhibitory effect against pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-12) in macrophages and dendritic cells [17, 62]. The production of inflammation mediators, including TNF-α, IL-1β, and iNOS, in the brains of mice was significantly reduced after fisetin treatment [63]. Furthermore, fisetin is also known to reduce the neurological parameters and inflammatory cytokines TNF-α, IL-1β, IL-6 IL-1, iNOS, COX-2, and PGE2 [9, 44] in the brains of stroke patients. In particular, in a flavonoid mixture, it was reported that fisetin blocked NLRP3 inflammasome activation by promoting mitophagy in cerebral microvascular endothelial cells and possessed the greatest inhibitory effects against the secretion of IL-1β into the CNS, reducing neuroinflammation and contributing to the amelioration of cognitive impairment [64]. From these data, the inhibition of neuroinflammation emerges as the main protective mechanism of fisetin against neurodegenerative disease processes.
Maintenance of GSH
Glutathione (GSH) is one of the most powerful intracellular antioxidant [23]. GSH and GSH-associated enzymes provide the major defense against oxidative and other forms of toxic stress to protect cells from damage [51]. In addition, as part of the major redox couple in cells, GSH plays a central role in maintaining cellular redox homeostasis [65]. Fisetin is a flavonoid that exhibits potent antioxidant properties and protects the cells against oxidative stress. The antioxidant activities of fisetin protect the neural cells from apoptotic degeneration. In general, oxidative damage directly or indirectly accelerates development of AD, PD, HD, and ALS. GSH is a reducing factor that prevents the excessive oxidation of sensitive cellular components. More importantly, GSH also influences protein structure and activity through changes in thiol-disulfide balance. Therefore, GSH levels are associated with cell survival. Several data have shown that fisetin antioxidant effects are due to enhancement of GSH synthesis to inhibit the production of ROS and reduce oxidative damage, eventually leading to rat cortical neurons survive and neurite extension [66]. Moreover, fisetin can regulate the level of GSH. A study on the ability of fisetin to protect mice from aluminum chloride toxicity showed that oral administration of fisetin at 15 mg/kg could greatly reduce the loss of total GSH from both the cortex and hippocampus [67]. It has also been proposed that fisetin can increase GSH levels either by enhancing the influx of cysteine and/or by boosting the activity of GCL (glutamate cysteine ligase) [23, 51]. Accordingly, the maintenance and generation of GSH by fisetin are likely to be among the mechanisms underlying its neuroprotective effect of fisetin.
Restoration of Synaptic Proteins
Synapse dysfunction has been shown to adversely affect neuronal connections and nervous system functions. Direct evidence for an age-related decline in synaptic transmission is attributable to a shift in the balance between synaptic plasticity. This reduction in plasticity precedes neural death in several neurodegenerative diseases, contributing to cognitive decline. In addition, exposure to the neurotoxic environmental contaminant methylmercury (MeHg) during pregnancy severely affects synaptic transmission and plasticity in the brain of developing offspring [67]. For example, iron homeostasis is continuously kept under the physiological condition. Once the balance of certain iron of release and uptake is dysregulated, cell degeneration and apoptosis occur. Several evidence have also shown that fisetin can control the homeostasis of Zn2+ release from synapses and uptake by adjacent neurons. More uptake over release will progressively lead to toxic concentration, causing neuronal death [68]. Likewise, the aggregation of α-synuclein is also linked to neurological disorders [36]. For example, α-synuclein aggregation leads to neuronal death and affects the course of PD and other related diseases. However, fisetin can attenuate the expression level of α-synuclein and promote neuroprotection [36]. The dysregulation of synaptic proteins at both pre-synaptic and post-synaptic junctions elicits pathological changes in several neurodegenerative diseases. Fisetin can facilitate the maintenance of synaptic function and resist cognitive decline and neurological disorders. Several studies showed that fisetin treatment significantly reversed synaptic dysfunction by increasing the levels of presynaptic (SYN and SNAP-25) and postsynaptic proteins (PSD-95, SNAP-23, p-GluR1 (Ser 845), p-CREB (Ser 133), and pCAMKII (Thr 286)) and ultimately improved mouse memory [6, 69]. In another study, dizocilpine (MK-801) caused disruption of N-methyl d-aspartate receptors in rat model, leading to cognitive and memory decline [69]. Fisetin treatment can reverse these deficits of hippocampal synaptic plasticity and improve memory [70]. In addition, fisetin has ability to inhibit α-Syn fibrillation [71]. On the basis of the above data, it can be inferred that fisetin can improve synaptic dysfunction and memory by regulating synaptic proteins, synaptic transmission and plasticity, and α-synuclein aggregation.
Modulation of CREB
Learning and memory are controlled by the transcription factor cAMP-response element-binding protein (CREB), which interacts with the cAMP-response element in the promoter region of genes that encodes the corresponding proteins. Activation of CREB by phosphorylation is dependent on ERK activation. Fisetin treatment activates ERK and induces CREB phosphorylation, promoting long-term potentiation [72, 73]. In addition, fisetin upregulates gene expressions of CREB, and improves cell viability in the main brain areas related to recognition and memory [73], thus improving learning and memory abilities.
Regulation of PI3K/Akt Signaling Pathway
The PI3K/Akt pathway plays a pivotal role in apoptosis, survival, and angiogenesis [74, 75]. Fisetin upregulates the expression of the endogenous antioxidant HO-1 by activating of the PI3K/AKT signaling pathways in microglia [61], attenuating oxidative stress and inflammatory responses. Similarly, fisetin upregulated PI3K expression and phosphorylation of Akt [76, 77]. In a detailed study, pretreatment with fisetin obviously upregulated the high glucose-inhibited expression of BDNF, GDNF, Syp, and Gria1; attenuated LDH release and malondialdehyde (MDA) overproduction; and increased SOD activity in HT22 cells [51, 78]. In addition, the decreased phosphorylation of PI3K, Akt, and CREB was rescued by fisetin treatment. These findings indicate that fisetin has potent neuroprotective effect and prevents HG-induced neurotoxicity by activating the PI3K/Akt/CREB pathway [78]. Consistently, in SH-SY5Y cells, fisetin treatment increased cell viability by upregulating Bcl-2 and p38/JNK-MAPK and activating PI3K, Akt, and GSK-3β signaling pathways [79]. Collectively, these results suggest that PI3K/Akt signaling pathways contribute, at least partially, to fisetin-induced neuroprotection against various insults.
Regulation of NF-κB Signaling Pathway
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway has a remarkable role in pathologic conditions. The effects of fisetin on this pathway have been investigated in several cancer cell lines, including human lung adenocarcinoma H1299 cells, human Burkitt lymphoma Daudi cells, and human embryonic kidney A293 cells [17]. The data showed that fisetin inhibits TNF-induced IκBα degradation, NF-dependent IκBα phosphorylation and ubiquitination, TNF-induced IκBα kinase activation, p65 phosphorylation and its nuclear translocation, NF-κBdependent anti-apoptotic gene expression (cIAP1/2, survivin, Bcl-2, XIAP, Bcl-xL, and TRAF-1), and expression of cyclin D1, c-Myc, COX-2, MMP-9, VEGF, and intercellular adhesion molecule-1 (ICAM-1), implying the modulatory effect of fisetin on this pathway [80]. In another study, it was shown that fisetin can counteract human brain microvascular endothelial cells apoptosis caused by MMP-9 treatment or overexpression of COX-2 through the inhibition of the NF-kB pathway [81]. In addition, fisetin treatment significantly ameliorated lipopolysaccharide and restraint stress-induced behavioral deficits through suppression of NF-κB and IDO-1 (indoleamine 2,3-dioxygenase) activation, indicating fisetin as a nutraceutical for the management of inflammation-associated neurological disorders [11]. In a recent study, fisetin was found to decrease the phosphorylation of IκBα, p65, JNK, p38, and MEK and reduce the nuclear translocation of NF-κB p65 [17]. The regulation of NF-κB signaling by fisetin is likely to be involved in its neuroprotection against inflammation-associated neurological disorders.
Regulation of p38MAPK Pathway
The NF-κB and p38 mitogen-activated protein kinase (MAPK) signaling pathways have been shown to play a critical role in the expression of proinflammatory cytokines and iNOS in glia cells [60]. Using LPS-stimulated BV-2 microglia cells, fisetin inhibits neuroinflammation by suppressing IκB degradation, nuclear translocation of NF-κB, and phosphorylation of p38 MAPK [60]. Fisetin inhibits microglia activation through modulating p38 phosphorylation levels, resulting in the activation of its downstream signaling molecules, which subsequently attenuates inflammation-associated microglia activation and coordination defects in mice [61]. Experimentally, fistein treatment of RAW264.7 mouse macrophage cells decreased the activation of the NF-κB and MAPK signaling pathways [6]. This suggests that fisetin contributes to abrogation of inflammatory responses and progressive neuronal cell death.
Regulation of ERK Signaling Pathway
The ERK signaling pathway controls diverse cellular processes such as proliferation, differentiation [82], and survival. Fisetin is one of the flavonoids that has been found to activate ERK signaling in rat hippocampal slices, and enhance object recognition in animals [72]. Also, fisetin has been documented to prevent the development of learning and memory deficits by modulating ERK phosphorylation and reducing protein carbonylation [31]. Similarly, fisetin restored ERK1/2 in MK-801-treated rats [70]. In addition, in three different models of HD, fisetin reduced the impact of mutant huntingtin and activated ERK [8], causing improvement of psychiatric, cognitive, and motor functions. Via the Akt/Erk signaling pathway, fisetin inhibits aggressive astrocyte phenotype, which result in abortion of astrocyte migration and proliferation [77]. Furthermore, in mutant hSOD1 ALS models, fisetin upregulates the expression of antioxidant factors to inhibit oxidative damage and DNA break damage by enhancing phosphorylated ERK [13].
Regulation of Nrf2 Signaling Pathways
Nuclear erythroid 2-related factor 2 (Nrf2) is a key regulator to defense and survival against endogenous and exogenous stress. It has been found that fisetin reduces the levels of phosphorylated tau through the autophagy pathway activated by TFEB and Nrf2 [33]. Treatment of diabetic neuropathy rats with fisetin caused a progressive improvement of motor nerve conduction velocity and improved sciatic nerve blood flow deficits. Molecularly, it also reduced the levels of IL-6 and TNF-α, which are closely associated with suppression of the Nrf2 and NF-κB signaling pathways [83]. Additionally, fisetin induced the expression of HO-1, GCL, and cystine/glutamate transporter (xCT/SLC7A11) through activation of the Nrf2, MAPK, and SIRT1 cellular stress response pathways, which restored cell viability and represses cell apoptosis, autophagy, and ROS production to protect neuronal-like catecholaminergic PC12 cells from Tm-induced cytotoxicity [58]. Apart from these, fisetin decreased MDA, reduced the activity of glutathione peroxidase (GPx), and activated the Nrf2-antioxidant response element pathway following traumatic brain injury [84]. Recently, Hassan and Zhang et al. [85] found that fisetin translocated Nrf2 into the nucleus and dramatically increased the expression of downstream HO-1 gene by inhibiting the degradation of Nrf2 at the post-transcriptional level. Although these data provide a molecular mechanism for understanding the cellular antioxidant activity of fisetin [16, 85, 86], the details mechanism of action of fisetin still needs to be further investigated.
Prospects
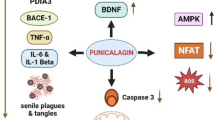
Fisetin and quercetin are two of the most common plant flavonoids that are present in many fruits and vegetables. The anti-cancer, anti-inflammatory, and antioxidant effects of these flavonols are related to their ability to inhibit apoptosis and mediate cell cycle arrest. A growing number of in vivo and in vitro experiments have shown that fisetin has the potential to maintain brain function and protects neuronal cells from various damages. The unique features of fisetin appear to orchestrate the molecular signaling for many processes related to neuroprotection in a coordinated fashion through the modulation of related signaling pathways. A profound understanding of the different molecular biological characteristics of fisetin is very important for utilization at the appropriate stage for specific clinical neurological diseases. Moreover, this review also provides new insights for elucidating the mechanism of these therapeutically active flavonols. Although this review highlights the modulatory potential of fisetin in different signaling pathways, such as Akt, Nrf2, ERK, p38MAPK, and NF-κB pathway, a detailed and dedicated outline of the therapeutic potential of fisetin in treating neurological health complications is still needed. In short, the underlying mechanisms by which fisetin exerts its neuroprotective role in the treatment of neurological diseases are illustrated in Fig. 2. Based on the aforementioned, fisetin, as novel therapeutic agent for neurodegenerative disease, may possess excellent clinical prospect in the future.

Schematic diagram of the potential mechanism underlying the neuroprotective effects of fisetin against neurological disease. The bioactive potential of fisetin has been highlighted in the modulation of different neuroprotection related signaling pathways such as PI3K/Akt, CREB, p38MAPK, NF-κB, and Nrf2, which are associated with a variety of cell events. All these events are crucial in preventing or inhibiting the initiation and progression of neurological diseases
Data Availability
Not applicable
References
Prior M, Chiruta C, Currais A, Goldberg J, Ramsey J, Dargusch R, Maher PA, Schubert D (2014) Back to the future with phenotypic screening. ACS Chem Neurosci 5(7):503–513
Kashyap D, Garg VK, Tuli HS, Yerer MB, Sak K, Sharma AK, Kumar M, Aggarwal V et al (2019) Fisetin and quercetin: promising flavonoids with chemopreventive potential. Biomolecules 9(5):E174
Maher P (2015) How fisetin reduces the impact of age and disease on CNS function. Front Biosci (Schol Ed) 7(1):58–82
Khan N, Syed DN, Ahmad N, Mukhtar H (2013) Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal 19(2):151–162
Rice-Evans CA, Miller NJ, Paganga G (1996) Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med 20(7):933–956
Ahmad A, Ali T, Park HY, Badshah H, Rehman SU, Kim MO (2017) Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice. Mol Neurobiol 54(3):2269–2285
Kumar R, Kumar R, Khurana N, Singh SK, Khurana S, Verma S, Sharma N, Vyas M et al (2022) Improved neuroprotective activity of fisetin through SNEDDS in ameliorating the behavioral alterations produced in rotenone-induced Parkinson’s model. Environ Sci Pollut Res Int 29(33):50488–50499
Maher P, Dargusch R, Bodai L, Gerard PE, Purcell JM, Marsh JL (2011) ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Genet 20(2):261–270
Zhang P, Cui J (2021) Neuroprotective effect of fisetin against the cerebral ischemia-reperfusion damage via suppression of oxidative stress and inflammatory parameters. Inflammation 44(4):1490–1506
Khatoon S, Agarwal NB, Samim M, Alam O (2021) Neuroprotective effect of fisetin through suppression of IL-1R/TLR axis and apoptosis in pentylenetetrazole-induced kindling in mice. Front Neurol 12:689069
Choubey P, Kwatra M, Pandey SN, Kumar D, Dwivedi DK, Rajput P, Mishra A, Lahkar M (2019) Ameliorative effect of fisetin against lipopolysaccharide and restraint stress-induced behavioral deficits via modulation of NF-κB and IDO-1. Psychopharmacol (Berl) 236(2):741–752
Wang G, Wang J, Guan R (2020) Novel Phospholipid-based labrasol nanomicelles loaded flavonoids for oral delivery with enhanced penetration and anti-brain tumor efficiency. Curr Drug Deliv 17(3):229–245
Wang TH, Wang SY, Wang XD, Jiang HQ, Yang YQ, Wang Y, Cheng JL, Zhang CT et al (2018) Fisetin exerts antioxidant and neuroprotective effects in multiple mutant hSOD1 models of amyotrophic lateral sclerosis by activating ERK. Neurosci 379:152–166
Arai Y, Watanabe S, Kimira M, Shimoi K, Mochizuki R, Kinae N (2000) Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J Nutr 130(9):2243–2250
Kumar RM, Kumar H, Bhatt T, Jain R, Panchal K, Chaurasiya A, Jain V (2023) Fisetin in cancer: attributes, developmental aspects, and nanotherapeutics. Pharmaceuticals (Basel) 16(2):196
Hassan SSU, Samanta S, Dash R, Karpiński TM, Habibi E, Sadiq A, Ahmadi A, Bunagu S (2022) The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: Focus on the role of oxidative stress. Front Pharmacol 13:1015835
Zhong R, Miao L, Zhang H, Tan L, Zhao Y, Tu Y, Angel Prieto M, Simal-Gandara J et al (2022) Anti-inflammatory activity of flavonols via inhibiting MAPK and NF-κB signaling pathways in RAW264.7 macrophages. Curr Res Food Sci 5:1176–1184
Elsallabi O, Patruno A, Pesce M, Cataldi A, Carradori S, Gallorini M (2022) Fisetin as a senotherapeutic agent: biopharmaceutical properties and crosstalk between cell senescence and neuroprotection. Mol Basel Switz 27(3):738
Devasagayam TP, Subramanian M, Singh BB, Ramanathan R, Das NP (1995) Protection of plasmid pBR322 DNA by flavonoids against single-stranded breaks induced by singlet molecular oxygen. J Photochem Photobiol B 30(2-3):97–103
Ishige K, Schubert D, Sagara Y (2001) Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med 30(4):433–446
Bothiraja C, Yojana BD, Pawar AP, Shaikh KS, Thorat UH (2014) Fisetin-loaded nanocochleates: formulation, characterisation, in vitro anticancer testing, bioavailability and biodistribution study. Expert Opin Drug Deliv 11(1):17–29
Mehta P, Pawar A, Mahadik K, Bothiraja C (2018) Emerging novel drug delivery strategies for bioactive flavonol fisetin in biomedicine. Biomed Pharmacother Biomedecine Pharmacother. 106:1282–1291
Chiruta C, Schubert D, Dargusch R, Maher P (2012) Chemical modification of the multitarget neuroprotective compound fisetin. J Med Chem 55(1):378–389
Renault-Mahieux M, Vieillard V, Seguin J, Espeau P, Le DT, Lai-Kuen R, Mignet N, Paul M et al (2021) Co-encapsulation of fisetin and cisplatin into liposomes for glioma therapy: from formulation to cell evaluation. Pharmaceutics 13(7):970
Lapchak PA (2013) Drug-like property profiling of novel neuroprotective compounds to treat acute ischemic stroke: guidelines to develop pleiotropic molecules. Transl Stroke Res 4(3):328–342
Krasieva TB, Ehren J, O’Sullivan T, Tromberg BJ, Maher P (2015) Cell and brain tissue imaging of the flavonoid fisetin using label-free two-photon microscopy. Neurochem Int 89:243–248
Kumar R, Kumar R, Khurana N, Singh SK, Khurana S, Verma S, Sharma N, Kapoor B et al (2020) Enhanced oral bioavailability and neuroprotective effect of fisetin through its SNEDDS against rotenone-induced Parkinson’s disease rat model. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc 144:111590
Krishnakumar IM, Jaja-Chimedza A, Joseph A, Balakrishnan A, Maliakel B, Swick A (2022) Enhanced bioavailability and pharmacokinetics of a novel hybrid-hydrogel formulation of fisetin orally administered in healthy individuals: a randomised double-blinded comparative crossover study. J Nutr Sci 11:e74
Xiao S, Lu Y, Wu Q, Yang J, Chen J, Zhong S, Eliezer D, Tan Q et al (2021) Fisetin inhibits tau aggregation by interacting with the protein and preventing the formation of β-strands. Int J Biol Macromol 178:381–393
Dash R, Emran TB, Uddin MMN, Islam A, Junaid M (2014) Molecular docking of fisetin with AD associated AChE, ABAD and BACE1 proteins. Bioinformation 10(9):562–568
Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P (2014) Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 13(2):379–390
Prakash D, Sudhandiran G (2015) Dietary flavonoid fisetin regulates aluminium chlorideinduced neuronal apoptosis in cortex and hippocampus of mice brain. J Nutr Biochem 26(12):1527–1539
Kim S, Choi KJ, Cho SJ, Yun SM, Jeon JP, Koh YH, Song J, Johnson GV et al (2016) Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci Rep 6:24933
Das J, Singh R, Ladol S, Nayak SK, Sharma D (2020) Fisetin prevents the aging-associated decline in relative spectral power of α, β and linked MUA in the cortex and behavioral alterations. Exp Gerontol 138:111006
Maher P (2017) Protective effects of fisetin and other berry flavonoids in Parkinson’s disease. Food Funct 8(9):3033–3042
Patel MY, Panchal HV, Ghribi O, Benzeroual KE (2012) The neuroprotective effect of fisetin in the MPTP model of Parkinson’s disease. J Park Dis 2(4):287–302
Chen TJ, Feng Y, Liu T, Wu TT, Chen YJ, Li X, Li Q, Wu YC (2020) Fisetin regulates gut microbiota and exerts neuroprotective effect on mouse model of Parkinson’s disease. Front Neurosci 14:549037
Rosado-Ramos R, Godinho-Pereira J, Marques D, Figueira I, Fleming Outeiro T, Menezes R, Nunes Dos Santos C (2021) Small molecule fisetin modulates alpha–synuclein aggregation. Molecules 26(11):3353
de Whalley CV, Rankin SM, Hoult JR, Jessup W, Leake DS (1990) Flavonoids inhibit the oxidative modification of low density lipoproteins by macrophages. Biochem Pharmacol 39(11):1743–1750
Dajas F, Rivera F, Blasina F, Arredondo F, Echeverry C, Lafon L, Morquio A, Heinzen H (2003) Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox Res 5(6):425–432
Rivera F, Urbanavicius J, Gervaz E, Morquio A, Dajas F (2004) Some aspects of the in vivo neuroprotective capacity of flavonoids: bioavailability and structure-activity relationship. Neurotox Res 6(7-8):543–553
Gelderblom M, Leypoldt F, Lewerenz J, Birkenmayer G, Orozco D, Ludewig P, Thundyil J, Arumugam TV et al (2012) The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 32(5):835–843
Zhou CH, Wang CX, Xie GB, Wu LY, Wei YX, Wang Q, Zhang HS, Hang CH et al (2015) Fisetin alleviates early brain injury following experimental subarachnoid hemorrhage in rats possibly by suppressing TLR 4/NF-κB signaling pathway. Brain Res. 1629:250–259
Chen C, Yao L, Cui J, Liu B (2018) Fisetin protects against intracerebral hemorrhage-induced neuroinflammation in aged mice. Cerebrovasc Dis Basel Switz 45(3-4):154–161
Das J, Singh R, Sharma D (2017) Antiepileptic effect of fisetin in iron-induced experimental model of traumatic epilepsy in rats in the light of electrophysiological, biochemical, and behavioral observations. Nutr Neurosci 20(4):255–264
Saito Y, Miyajima M, Yamamoto S, Sato T, Miura N, Fujimiya M, Chikenji TS (2021) Accumulation of senescent neural cells in murine lupus with depression-like behavior. Front Immunol 12:692321
Yu X, Jiang X, Zhang X, Chen Z, Xu L, Chen L, Wang G, Pan J (2016) The effects of fisetin on lipopolysaccharide-induced depressive-like behavior in mice. Metab Brain Dis 31(5):1011–1021
Zhao X, Wang C, Cui WG, Ma Q, Zhou WH (2015) Fisetin exerts antihyperalgesic effect in a mouse model of neuropathic pain: engagement of spinal serotonergic system. Sci Rep 5(1):9043
Radi E, Formichi P, Battisti C, Federico A (2014) Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 42(Suppl 3):S125–S152
Zhao X, Li XL, Liu X, Wang C, Zhou DS, Ma Q, Zhou WH, Hu ZY (2015) Antinociceptive effects of fisetin against diabetic neuropathic pain in mice: engagement of antioxidant mechanisms and spinal GABAA receptors. Pharmacol Res 102:286–297
Cho N, Choi JH, Yang H, Jeong EJ, Lee KY, Kim YC, Sung SH (2012) Neuroprotective and anti-inflammatory effects of flavonoids isolated from Rhus verniciflua in neuronal HT22 and microglial BV2 cell lines. Food Chem Toxicol 50(6):1940–1945
Ahmad S, Khan A, Ali W, Jo MH, Park J, Ikram M, Kim MO (2021) Fisetin rescues the mice brains against d-galactose-induced oxidative stress, neuroinflammation and memory impairment. Front Pharmacol 12:612078
Singh S, Singh AK, Garg G, Rizvi SI (2018) Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci 193:171–179
Ahmad A, Ali T, Rehman SU, Kim MO (2019) Phytomedicine-based potent antioxidant, fisetin protects CNS-Insult LPS-induced oxidative stress-mediated neurodegeneration and memory impairment. J Clin Med 8(6):E850
Maher P (2009) Modulation of multiple pathways involved in the maintenance of neuronal function during aging by fisetin. Genes Nutr 4(4):297–307
Alikatte K, Palle S, Rajendra Kumar J, Pathakala N (2021) Fisetin improved rotenone-induced behavioral deficits, oxidative changes, and mitochondrial dysfunctions in rat model of Parkinson’s disease. J Diet Suppl 18(1):57–71
Jacob S, Thangarajan S (2017) Effect of gestational intake of fisetin (3,3’,4’,7- Tetrahydroxyflavone) on developmental methyl mercury neurotoxicity in F1 generation rats. Biol Trace Elem Res 177(2):297–315
Yen JH, Wu PS, Chen SF, Wu MJ (2017) Fisetin Protects PC12 Cells from TunicamycinMediated Cell death via reactive oxygen species scavenging and modulation of Nrf2-driven gene expression, SIRT1 and MAPK signaling in PC12 Cells. Int J Mol Sci 18(4):E852
Maher P (2020) Modulation of the neuroprotective and anti-inflammatory activities of the flavonol fisetin by the transition metals iron and copper. Antioxid Basel Switz 9(11):E1113
Zheng LT, Ock J, Kwon BM, Suk K (2008) Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol 8(3):484–494
Chuang JY, Chang PC, Shen YC, Lin C, Tsai CF, Chen JH, Yeh WL, Wu LH (2014) Regulatory effects of fisetin on microglial activation. Mol Basel Switz 19(7):8820–8839
Liu SH, Lin CH, Hung SK, Chou JH, Chi CW, Fu SL (2010) Fisetin inhibits lipopolysaccharideinduced macrophage activation and dendritic cell maturation. J Agric Food Chem 58(20):10831–10839
Prakash D, Gopinath K, Sudhandiran G (2013) Fisetin enhances behavioral performances and attenuates reactive gliosis and inflammation during aluminum chloride-induced neurotoxicity. Neuromolecular Med 15(1):192–208
Ding H, Li Y, Chen S, Wen Y, Zhang S, Luo E, Li X, Zhong W (2022) Fisetin ameliorates cognitive impairment by activating mitophagy and suppressing neuroinflammation in rats with sepsis-associated encephalopathy. CNS Neurosci Ther 28(2):247–258
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30(11):1191–1212
Maher P (2008) The flavonoid fisetin promotes nerve cell survival from trophic factor withdrawal by enhancement of proteasome activity. Arch Biochem Biophys 476(2):139–144
Jacob S, Sumathi T (2019) Extenuation of in utero toxic effects of MeHg in the developing neurons by fisetin via modulating the expression of synaptic transmission and plasticity regulators in hippocampus of the rat offspring. Chem Biol Interact 305:3–10
Cai AL, Zipfel GJ, Sheline CT (2006) Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur J Neurosci 24(8):2169–2176
Yang W, Tian ZK, Yang HX, Feng ZJ, Sun JM, Jiang H, Cheng C, Ming QL et al (2019) Fisetin improves lead-induced neuroinflammation, apoptosis and synaptic dysfunction in mice associated with the AMPK/SIRT1 and autophagy pathway. Food Chem Toxicol 134:110824
Zhan JQ, Chen CN, Wu SX, Wu HJ, Zou K, Xiong JW, Wei B, Yang YJ (2021) Flavonoid fisetin reverses impaired hippocampal synaptic plasticity and cognitive function by regulating the function of AMPARs in a male rat model of schizophrenia. J Neurochem 158(2):413–428
Rane AR, Paithankar H, Hosur RV, Choudhary S (2021) Modulation of α-synuclein fibrillation by plant metabolites, daidzein, fisetin and scopoletin under physiological conditions. Int J Biol Macromol 182:1278–1291
Maher P, Akaishi T, Abe K (2006) Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci USA. 103(44):16568–16573
Khatoon S, Samim M, Dahalia M (2023) Fisetin provides neuroprotection in pentylenetetrazole-induced cognition impairment by upregulating CREB/BDNF. Eur J Pharmacol 944:175583
Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E (2014) PI3K/AKT signaling pathway and cancer: an updated review. Ann Med 46(6):372–383
Mayer IA, Arteaga CL (2016) The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med 67:11–28
Watanabe R, Kurose T, Morishige Y, Fujimori K (2018) Protective effects of fisetin against 6- OHDA-induced apoptosis by activation of PI3K-Akt signaling in human neuroblastoma SHSY5Y cells. Neurochem Res 43(2):488–499
Wang N, Yao F, Li K, Zhang L, Yin G, Du M, Wu B (2017) Fisetin regulates astrocyte migration and proliferation in vitro. Int J Mol Med 39(4):783–790
Zhang S, Xue R, Geng Y, Wang H, Li W (2020) Fisetin prevents HT22 cells from high glucoseinduced neurotoxicity via pi3k/akt/creb signaling pathway. Front Neurosci 14:241
Rajendran M, Ramachandran R (2019) Fisetin protects against rotenone-induced neurotoxicity through signaling pathway. Front Biosci Elite Ed 11(1):20–28
Sung B, Pandey MK, Aggarwal BB (2007) Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kappaB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkappaBalpha kinase activation. Mol Pharmacol 71(6):1703–1714
Tahanian E, Sanchez LA, Shiao TC, Roy R, Annabi B (2011) Flavonoids targeting of IκB phosphorylation abrogates carcinogen-induced MMP-9 and COX-2 expression in human brain endothelial cells. Drug Des Devel Ther 5:299–309
Sagara Y, Vanhnasy J, Maher P (2004) Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J Neurochem 90(5):1144–1155
Sandireddy R, Yerra VG, Komirishetti P, Areti A, Kumar A (2016) Fisetin imparts neuroprotection in experimental diabetic neuropathy by modulating Nrf2 and NF-κB pathways. Cell Mol Neurobiol 36(6):883–892
Zhang L, Wang H, Zhou Y, Zhu Y, Fei M (2018) Fisetin alleviates oxidative stress after traumatic brain injury via the Nrf2-ARE pathway. Neurochem Int 118:304–313
Zhang H, Zheng W, Feng X, Yang F, Qin H, Wu S, Hou DX, Chen J (2019) Nrf2−ARE signaling acts as master pathway for the cellular antioxidant activity of fisetin. Mol Basel Switz 24(4):E708
Jazvinšćak Jembrek M, Oršolić N, Mandić L, Sadžak A, Šegota S (2021) Anti-oxidative, anti-inflammatory and anti-apoptotic effects of flavonols: targeting Nrf2, NF-κB and p53 pathways in neurodegeneration. Antioxid Basel Switz 10(10):1628
Acknowledgements
The authors would like to Dr. Huiming Xu for the critical reading of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 82071551 and 81830077), and the key research and development program in Ningxia Hui Autonomous Region (grant nos. 2022BEG02032)
Author information
Authors and Affiliations
Contributions
HY conceived the review and supervised the project. YZJ and XWT wrote the manuscript. PD and CJ contributed to the literature review and editing. YQH contributed to the literature review and drew the figure. YH and DJH contributed to the content and editing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical Approval and Consent to Participate
Not applicable
Research Involving Human Participants and/or Animals
Not applicable
Informed Consent
Not applicable
Consent for Publication
Not applicable
Conflicts of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jiang, Y., Tang, X., Deng, P. et al. The Neuroprotective Role of Fisetin in Different Neurological Diseases: a Systematic Review. Mol Neurobiol 60, 6383–6394 (2023). https://doi.org/10.1007/s12035-023-03469-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03469-7