
Abstract
The current study is aimed to assess the therapeutic potential of fisetin, a phytoflavonoid in streptozotocin (STZ)-induced experimental diabetic neuropathy (DN) in rats. Fisetin was administered (5 and 10 mg/kg) for 2 weeks (7th and 8th week) post STZ administration. Thermal and mechanical hyperalgesia were assessed by measuring tactile sensitivity to thermal and mechanical stimuli, respectively. Motor nerve conduction velocity (MNCV) was determined using power lab system and sciatic nerve blood flow (NBF) was determined using laser Doppler system. Nerve sections were processed for TUNEL assay and NF-κB, COX-2 immunohistochemical staining. Sciatic nerve homogenate was used for biochemical and Western blotting analysis. MNCV and sciatic NBF deficits associated with DN were ameliorated in fisetin administered rats. Fisetin treatment reduced the interleukin-6 and tumour necrosis factor-alpha in sciatic nerves of diabetic rats (p < 0.001). Protein expression studies have identified that the therapeutic benefit of fisetin might be through regulation of redox sensitive transcription factors such as nuclear erythroid 2-related factor 2 (Nrf2) and nuclear factor kappa B (NF-κB). Our study provides an evidence for the therapeutic potential of fisetin in DN through simultaneous targeting of NF-κB and Nrf2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic neuropathy (DN) is one of the most common crippling complications affecting peripheral nerves of patients suffering from diabetes with an overall prevalence of 50–60 %. The most common symptoms include hyperalgesia, allodynia, intractable and burning pain in the extremities in a stocking–glove pattern (Huizinga and Peltier 2007). Loads of research done in the field of DN have identified wide array of pathogenetic mechanisms contributing to altered functional, behavioural, biochemical deficits in DN (Winkler and Kempler 2010). These mechanisms include classical pathways of hyperglycaemia such as advanced glycation end products (AGE) formation, polyol pathway, protein kinase c (PKC) pathway in addition to newer mechanisms like poly ADP ribose polymerase (PARP) activation, mitogen-activated protein kinase (MAPK) activation and mitochondrial dysfunction (Singh et al. 2014). However, the drug discovery in this area is limited to agents providing symptomatic relief from neuropathic pain (Jensen et al. 2006). The lack of any treatment option in DN emphasizes the necessity of research in this area.
In recent times, diabetes-associated neuronal dysfunction is being realized to be a result of maladaptive cellular signalling rather than the mere result of classical hyperglycaemic flux. Indeed research has been progressed to identify the organellar pathology in hyperglycaemic neuronal injury especially that of mitochondria and endoplasmic reticulum (Leinninger et al. 2006; Cameron 2013). Nonetheless, most of these neuronal disturbances employ oxidative stress and neuroinflammation as principal channels of their pathological implication (Sandireddy et al. 2014). Several clinical and experimental evidences of inflammatory and oxidative stress biomarkers identified in DN subjects are in line with this observation and thus targeting oxidative stress, and neuroinflammation-associated neuronal signalling provides new avenue for the treatment of diabetes-associated neuronal dysfunction (Kumar et al. 2013).
Nuclear erythroid 2-related factor 2 (Nrf2) is a redox sensitive cellular transcription factor involved in cytoprotection (Wakabayashi et al. 2010). Kelch-like ECH-associated protein 1 (Keap1) is a cytosolic repressor of Nrf2, acts by promoting the degradation of Nrf2 through ubiquitin proteasome system. The cysteine rich structural configuration of Keap1 makes it a target of several anti oxidants and electrophiles. When Nrf2 is released from Keap1, it gets translocated to nucleus, where along with small musculoaponeurotic fibrosarcoma (Maf) proteins, it binds to antioxidant response element (ARE) of genome and enhances the expression of several cytoprotective, antioxidant enzymes (Wakabayashi et al. 2010). Stimulation of Nrf2 signalling has been reported to ameliorate ageing and other chronic diseases whose pathogenesis is driven by oxidative stress (Lewis et al. 2010).
Nuclear factor kappa B (NF-κB) is considered to be a prime mediator of inflammatory signalling and is present in inactive state as a triad with inhibitory kappa B (IκB) (Tak and Firestein 2001). On its activation, it gets translocated to nucleus and results in elevated expression of several inflammatory mediators, cytokines etc. It has also been identified that these mediators in turn produce a positive feedback on NF-κB signalling and thus amplifies the inflammatory process (Cameron and Cotter 2008). Elevated levels of proinflammatory mediators like tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) has been reported to contribute to nerve damage in peripheral neuropathy (Kumar et al. 2012; Kumar and Sharma 2010).
Previous studies have identified the therapeutic potential of several phyto flavonoids such as resveratrol, sesamol and melatonin in experimental DN (Kumar et al. 2007; Chopra et al. 2010; Negi et al. 2011b). The therapeutic basis of natural flavonoids in most of the experimental DN cases has not been elucidated out in detail although anti oxidant and anti inflammatory properties may account for it (Sandireddy et al. 2014). There are very few studies which have exemplified the therapeutic potential of simultaneous targeting of Nrf2, NF-κB pathways in DN (Yerra et al. 2013). The current study assessed the potential of dual targeting of Nrf2, NF-κB pathways in DN. We have used fisetin for this study which is a naturally occurring flavonoid found in many fruits and vegetables such as strawberries, apples, onions and cucumbers. This study is aimed to evaluate potential of fisetin against neurobehavioural, functional and biochemical deficits in experimental neuropathy induced by streptozotocin (STZ).
Materials and Methods
Animals
Healthy male Sprague–Dawley rats (250–270 g) were fed on standard diet and water ad libitum. They were housed in plastic cages (two in each) at a temperature of 24 ± 1 °C and humidity of 55 ± 5 %, with 12 h light dark cycle. All experiments were conducted in accordance with the regulations of Institutional animal ethics committee (IAEC)-NIPER-Hyderabad.
Drugs and Chemicals
Unless otherwise specified all the chemicals used in the study were purchased from Sigma-Aldrich Ltd. Isoflurane was purchased from Raman & Weil Pvt. Ltd. (Mumbai, India). Glucose oxidase–peroxidase kit was purchased from Accurex (Mumbai, India).
Induction of Diabetes and Experimental Design
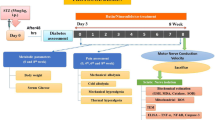
Diabetes was induced by STZ at a dose of 55 mg/kg (i.p.). Blood samples were collected from the tail vein after 48 h of STZ administration. The rats with blood glucose levels more than 250 mg/dL are considered as diabetic and were included in this study. The experimental groups comprised normal control, diabetic control and diabetic rats treated with fisetin at 5 and 10 mg/kg (p.o). A group of 6 normal animals were administered with fisetin 10 mg/kg dose for 2 weeks to assess the effect of fisetin on neurobehavioural and functional changes in normal healthy rats. Treatment was started 6 weeks after diabetes induction and continued for 2 weeks. The functional, behavioural and biochemical parameters were measured 24 h after administration of last dose (Kumar and Sharma 2010).
Functional Parameters
Motor Nerve Conduction Velocity
Motor nerve conduction velocity (MNCV) was calculated in posterior sciatic tibial system using power lab 4sp. Animals were anaesthetized with mixture of isoflurane and oxygen gaseous anaesthesia system. Needle electrodes were used for stimulation of sciatic nerve both at sciatic notch and ankle with 3V stimulus. The bipolar surface receiving electrodes were kept in foot muscle, and the latencies of sciatic and tibial stimulation were measured. Latencies were calculated by measuring the time from starting of the stimulation point to the onset of negative M-wave deflection. MNCV was calculated using the following formula and results were expressed as m/s (Kumar et al. 2012):
Measurement of Sciatic Nerve Blood Flow
Animals were anaesthetized and their left flank was exposed to visualize the sciatic nerve. Probe of the LASER Doppler system was placed slightly over the sciatic nerve in an area free from epineurial and perineurial blood vessels. Normal saline was applied at the exposing point to avoid tissue dehydration and the probe was left latent for 10–15 min for stabilization of the recording. Flux measurements were made from same part of the nerve and for similar period (10 min) among different animals and reported as arbitrary perfusion units (Kumar and Sharma 2010).
Behavioural Parameters
Thermal Hyperalgesia
Thermal hyperalgesia to both hot (45 °C) and cold (10 °C) stimuli was performed. Animals were acclimatized prior to the experiment. The tail flick response latency or any signs of struggle was observed as endpoint response. The cut-off time 15 s was kept in both tests. Six consecutive readings were taken at an interval of 10 min (Narenjkar et al. 2011).
Mechanical Hyperalgesia
Mechanical Sensitivity to noxious stimuli was assessed by quantifying the withdrawal threshold of the hind paw using a von Frey anaesthesiometer and Randall–Sellitto callipers (IITC Life Sciences, USA). Pressure was applied on both paws and time difference of 5 min was kept in between two consecutive readings (Kumar et al. 2007).
Biochemical Parameters
Measurement of Glucose Levels
Blood collected from the tail was centrifuged at 4000 rpm for 5 min to obtain plasma. The estimation of plasma glucose levels was done using GOD-POD kit according to the manufacturer’s instructions (Kumar et al. 2007).
Measurement of Lipid Peroxidation
Malondialdehyde (MDA) levels were measured by adding 0.2 ml of plasma with 0.2 ml of 8.1 % SDS, 1.5 ml of 20 % glacial acetic acid solution (pH 3.4) and 1.5 ml of 0.8 % thiobarbituric acid. The mixture was made up to 4 ml with distilled water, and it was heated on water bath at 95 °C for 60 min. After that, mixture was cooled under tap water, and it was centrifuged at 10,000 rpm. Absorbance of supernatant was measured at 532 nm. Results are expressed as nM/ml of plasma (Kumar et al. 2007).
Estimation of Glutathione
Briefly sciatic nerve was homogenized with 10 times 0.1 % (w/v) sodium phosphate buffer (pH 7.4). This homogenate was then centrifuged with 5 % Trichloroacetic acid (TCA) to precipitate out the proteins. To this, 100 μl of Ellman’s reagent [dithiobis-2-nitrobenzoic acid (DTNB) in PBS (pH 8)] solution was added (Arora et al. 2008). The mixture was then incubated for 10 min at 37 °C, and the absorbance was recorded at 412 nm.
Estimation of IL-6 and TNF-α
ELISA estimation was done as described by Kumar et al. (Kumar and Sharma 2010). Sciatic nerves were homogenized in lysis buffer containing 1 % Triton X-100, 150 mM Nacl, 1 mM EDTA, 2.5 mM Sodium pyrophosphate, 1 mM β-glycerophosphate and 20 mM Tris buffer (pH 7.5) with protease inhibitor cocktail. Homogenate was kept in ice-cold water for 30 min and then sonicated. The resultant homogenate was centrifuged at 12,000×g for 20 min in centrifuge at 4 °C, and the supernatant was used for estimations. The protein concentrations for the homogenates were calculated by Bradford method. ELISA estimations were then performed using commercial IL-6, TNF-α assay kits (ebiosciences, USA) as per manufacturer instructions. The colour developed in the assay plates was read at 450 nm using spectrophotometer (Spectramax M4). The values obtained were normalized to the mg protein present in homogenate, and the final values were expressed as pg/mg protein.
DNA Fragmentation and Protein Expression Studies
TUNEL Assay
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) assay was performed to study DNA fragmentation. Sciatic nerves were dehydrated and paraffin embedded, and 4–5 μm sections were made using microtome (Leica). The naked ends of fragmented DNA were labelled with fluorescein isothiocyanate (FITC) using TdT-FragEL kit as per manufacturer’s instructions (Calbiochem, USA). Mounting media containing 4′,6-diamidino-2-phenylindole (DAPI) was added to sections. Total cell population and TUNEL-positive cells were counted under fluorescent microscope (Nikon). At least 25 different frames were counted for total cell population and number of positive cells. The sections were blinded for avoiding any bias during counting the number of positive cells. TUNEL-positive cells were expressed as percentage of total cells (Negi et al. 2011b).
Immunohistochemistry
Sciatic nerves were fixed in 10 % buffered formaldehyde and dehydrated through graded concentrations of alcohol, paraffin embedded and sectioned. The sections were mounted on slides, deparaffinized and hydrated followed by blocking with bovine serum albumin (BSA) for 15 min. Then sections were incubated with primary antibodies for NF-κB P65 subunit and COX-2 (rabbit polyclonal; Cell Signalling Technology) at a dilution of 1:400 at 4 °C for overnight. Sections were washed with tris buffered saline with tween 20 (TBST) and incubated with secondary antibody, at room temperature for 1 h. Thereafter, sections were washed and then incubated with a 3,3-o-diaminobenzidine solution followed by counterstaining with haematoxylin. Sections were then dehydrated and mounted with DPX mountant for observation under light microscope. Nerves from 4 to 6 rats of each group were collected and at least five different regions of each section were studied. Samples were blinded for eliminating any bias during scoring/counting (Joshi et al. 2013).
Western Blotting Analysis
Protein lysate were obtained by homogenizing sciatic nerves with lysis buffer containing 1 % Triton X-100, 150 mM Nacl, 1 mM EDTA, 2.5 mM Sodium pyrophosphate, 1 mM β-glycerophosphate and 20 mM Tris buffer (pH 7.5) with protease inhibitor cocktail. Volumes of sample containing equal quantity of protein were loaded and separated by SDS-PAGE and transferred to a polyvinyl difluoride (PVDF) membrane. Membrane was blocked using 3 % BSA followed by incubation with primary rabbit polyclonal antibodies NF-κB, IκBα, P-IκB, β-actin (Cell Signaling Technology, USA) (1:1000),Nrf2 and HO-1 (Santhacruz, USA) for 12 h at 4 °C. After washing, membranes were incubated with an alkaline phosphatase-conjugated secondary antibody (1:20,000), and bound antibody was visualized by a coloured reaction with solutions of nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate (BCIP-NBT). The relative band densities were quantified by densitometry using image J software (NIH, USA). Equal loading was confirmed by β-actin expression (Negi et al. 2011b).
Statistical Analysis
The data obtained in this study were expressed as the mean of six replicate determinations plus or minus the standard error of mean (SEM). Statistical comparisons were made with “Bonferroni’s Multiple Comparison Test”. The intergroup variations was measured by one-way analysis of variance (ANOVA) using the Graph Pad Prism, version 5.0. Results with p value <0.05 were considered to be statistically significant.
Results
Effect of Fisetin on Plasma Glucose
Plasma glucose level of the diabetic animals was found to be significantly high when compared non diabetic animals (STZ-D 483.7 ± 5.2 and ND 100.6 ± 2.0). Fisetin treatment has not shown any significant effect on plasma glucose values and is comparable with that of diabetic control rats (STZ-D + F5 474.9 ± 3.1 and STZ-D + F10 458.3 ± 4.0).
Effect of Fisetin on Functional Parameters
The MNCV of diabetic rats was significantly decreased (p < 0.001) when compared to the normal control rats (STZ-D 44.1 ± 1.6 vs. ND 69.8 ± 3.1). Treatment with fisetin 10 mg/kg for 2 weeks significantly reversed the conduction impairment at high dose in diabetic rats (STZ-D+F10 58.5 ± 2.3) (Fig. 1a). A significant reduction in the endoneurial blood flow in diabetic rats was observed in comparison to vehicle treated rats (STZ-D 48.5 ± 1.8 and ND 109.9 ± 4.4). Two-week treatment of fisetin showed dose-dependent improvement of sciatic nerve blood flow (NBF) as compared to the disease control rats (STZ-D + F5 79.2 ± 8.6 and STZ-D + F10 94.9 ± 6.3) (Fig. 1b). Normal rats treated with fisetin at 10 mg/kg have not shown any significant changes in nerve conduction velocity and NBF when compared to normal control animals.

Effect of fisetin treatment on nerve functions: a motor nerve conduction velocity (MNCV) and b Nerve blood flow (NBF) of diabetic animals and the effect of 2-week treatment with fisetin (5 and 10 mg/kg). Results are expressed as mean ± SEM (n = 6). ND non diabetic, STZ-D diabetic, STZ-D+F5 and STZ-D+F10 are diabetic rats treated with fisetin 5 and 10 mg/kg, respectively, ND+F10 normal control rats treated with fisetin 10 mg/kg. ^^^ p < 0.001 versus ND, ** p < 0.01, ***p < 0.001 versus STZ-D
Effect of Fisetin on Behavioural Parameters
Tail flick latency was decreased significantly (p < 0.001) at the end of eighth week in diabetic animals in both cold as well as hot immersion test as compared to normal animals. Increased tail withdrawal latencies were observed in fisetin-treated rats. Paw withdrawal pressure in diabetic rats was significantly (p < 0.001) diminished in both Randall–Selitto method and von Frey method as compared to normal control rats. Paw withdrawal pressure was observed to be increased by fisetin treatment in von Frey as well as Randall–Selitto tests (Table 1).
Effect of Fisetin on Biochemical Parameters
MDA level in the plasma of diabetic rats was increased by twofold to threefold as that of normal rats (p < 0.001). Two-week treatment with fisetin reduced the MDA levels in diabetic rats (p < 0.01 and p < 0.001 at 5 and 10 mg/kg of fisetin). In order to assess the antioxidant capacity of peripheral neuronal tissue, GSH was assessed in sciatic nerve homogenate. A profound reduction in GSH level was observed in diabetic rats (p < 0.001). Oral administration of fisetin to diabetic rats improved neuronal GSH level significantly (p < 0.001 at both the doses) (Table 1).
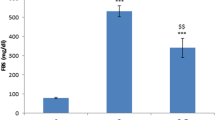
IL-6 and TNF-α are important inflammatory mediators generated through the activation of NF-κB signalling. We found a clear elevation of IL-6 (STZ-D 23.1 ± 1.2 and ND 6.6 ± 0.6) (Fig. 2a) and TNF-α (STZ-D 20.7 ± 0.9 and ND 5.4 ± 0.8) (Fig. 2b) in the sciatic nerve homogenates of diabetic rats (p < 0.001). Fisetin-treated rats have shown a dose-dependent reduction in IL-6 and TNF-α profile when compared to the diabetic control animals which indicates the anti inflammatory action of the compound.

Effect of fisetin on Pro inflammatory cytokine levels: a interleukin-6 (IL-6) and b tumour necrosis factor-α (TNF-α) levels in sciatic nerve homogenates of normal, diabetic animals and the fisetin-treated (5 and 10 mg/kg) animals. Results are expressed as mean ± SEM (n = 6). ND non diabetic, STZ-D diabetic, STZ-D+F5 and STZ-D+F10 are diabetic rats treated with fisetin 5 and 10 mg/kg, respectively. ^^^ p < 0.001 versus ND, ** p < 0.01, *** p < 0.001 versus STZ-D
Effect of Fisetin on DNA Fragmentation
To assess the oxidative damage at molecular level, we have carried out TUNEL assay in sciatic nerve microsections. As observed in results (Fig. 3), an enhanced number of TUNEL-positive cells were found in the sciatic nerves of diabetic rats (p < 0.001). A total number of nuclei were counted by DAPI staining. Administration of fisetin to diabetic rats for 2 weeks reduced the oxidative damage in peripheral nervous tissue as evident by reduced TUNEL-positive nuclei in the sciatic microsections (p < 0.001 at both the doses).

Effect of fisetin treatment on oxidative DNA damage, NF-κB and COX-2 immunochemistry: pictorial representations of DNA fragmentation [TUNEL images (1st row) and DAPI images (2nd row)] and expression of NF-κB (3rd row) and COX-2 (4th row) in sciatic nerve microsections. Bar graphs represent the average percentage of positive cells among the total number of nuclei. Values are expressed as mean ± SEM (n = 3). ND non diabetic, STZ-D diabetic, STZ-D+F5 and STZ-D+F10 are diabetic rats treated with fisetin 5 and 10 mg/kg, respectively. ^^^ p < 0.001 versus ND, **p < 0.01, ***p < 0.001 versus STZ-D
Effect of Fisetin on NF-ΚB and COX-2 Immunochemistry
Immunolocalization of inflammatory markers was visualized by performing immunohistochemistry to sciatic nerve microsections. In line with the previous observations, enhanced NF-κB and COX-2 positive nuclei (p < 0.001) were found in diabetic nerve sections when compared to healthy rats. Oral administration of fisetin to diabetic rats significantly reduced sciatic expression of NF-κB (p < 0.01 and p < 0.001 at 5 and 10 mg/kg of fisetin) and COX-2 (p < 0.001 at both doses) proteins (Fig. 3). This observation points towards NF-κB inhibitory action of fisetin in DN.
Effect of Fisetin on NF-κB, Nrf2, HO-1, IκBα and P-IκBα Levels
Chronic diabetes caused an enhanced NF-κB expression (p < 0.001) with increased phosphorylation of IκBα, a cytosolic inhibitor of the former protein. Treatment with fisetin hampered the NF-κB over expression (p < 0.001 at both the doses) and also significantly reduced the phosphorylation of IκBα (significantly at 10 mg/kg) and hence found to reduce the NF-κB mediated neuroinflammation in sciatic nerves of diabetic rats (Fig. 4). Similarly, it has been also observed that hyperglycaemia impaired Nrf2 and its downstream enzyme HO-1 signalling (p < 0.001) in diabetic rats. Fisetin supplementation to diabetic rats boosted the Nrf2 as well as HO-1 protein expression significantly at both the doses (p < 0.001) (Fig. 4).

Effect of fisetin treatment on NF-κB, IκBα, phospho-IκBα, Nrf2 and HO-1 expression levels: Results are expressed as mean ± SEM (n = 3). ND non diabetic, STZ-D diabetic, STZ-D+F5 and STZ-D+F10 are diabetic rats treated with fisetin 5 and 10 mg/kg, respectively. ^^^ p < 0.001 versus ND, ***p < 0.001 versus STZ-D and ### p < 0.001 versus IκB
Discussion
In the current study, we demonstrate that fisetin protects against oxidative stress and neuroinflammation-mediated functional, behavioural and biochemical deficits in DN through NF-κB inhibition and Nrf2-positive modulation as depicted in Fig. 5. This study further supports the therapeutic benefits of simultaneous targeting NF-κB-Nrf2 axis in diabetic complications including DN (Yerra et al. 2013). Moreover, the neuroprotective role of fisetin in oxidative stress may also involve the strengthening of antioxidant defence by enhancing Nrf2 activity. Nrf2 modulation in turn augments the expression of several cytoprotective, antioxidant enzymes such as hemeoxygenase-1 (HO-1), NADPH quinone oxidoreductase 1(NQO-1), glutamate cysteine ligase (GCL), glutathione peroxidase (GPx), thioredoxin (Trx), thioredoxin reductase (TrxR), peroxiredoxin (Prx), glutathione S-transferase (GST) and multidrug resistance-associated proteins (MRPs) (Zhang et al. 2013). Inhibition of NF-κB signalling pathways appears to mediate the protective role against inflammation led damage in diabetic nerves by controlling the production of proinflammatory mediators like TNF-α, IL-6, iNOS and COX-2 as observed in the peripheral nerves of STZ induced diabetic rats. Thus, the current results provides a substantial support to those previously observed reports of neuroprotection by targeting oxidative-nitrosative stress and neuroinflammation in diabetic complications including peripheral neuropathy (Negi et al. 2011b; Kumar and Sharma 2010).

Probable mechanism of neuroprotection shown by fisetin in experimental diabetic neuropathy. Fisetin, being a phytoflavonoid, may inhibit the ROS mediated IKK activation and corresponding phosphorylation of IκB. Inhibition of IκB phosphorylation thus halts NF-κB nuclear translocation and its activation. Keap1 is a cytosolic repressor of Nrf2 which directs the later for ubiquitin mediated proteasomal degradation in the absence of any antioxidant stimulus. Fisetin might also interfere with Keap1 association with Nrf2 by modifying the cysteine residues on Keap1 and thus facilitates the Nrf2 nuclear translocation and promotes antioxidant response element of genome. (HO-1 hemeoxygenase-1, IκB inhibitory kappa B, IL-6 interleukin-6, Keap1 kelch-like ECH-associated protein, NQO1 NAD(P)H dehydrogenase [quinone] 1, Nrf2 nuclear erythroid 2-related factor 2, TNF-α tumour necrosis factor-α)
Compromised nerve functionality is a cardinal feature observed in DN in both animal models as well as in diabetic subjects suffering from neuropathy and manifested as decline in nerve conduction (MNCV and SNCV) and nutritive NBF (Coppey et al. 2000). Slowing of nerve conduction results from demyelination of large nerve fibres, alteration of action potential, defect in axonal transport, etc. (Obrosova 2009). The myelin fragments and sulphatide released as a result of demyelination were also found to activate NF-κB signalling in neurons which further creates a vicious cycle of inflammation locally (Sun et al. 2010). The inflammation generated can cause endothelial damage which may cause decline in nutritive NBF, malnourished nerve and eventual nerve damage in diabetic animals (Cameron et al. 1991). Several natural phyto flavonoids have been found to have the ability to modulate the cav1.2 channels of the vascular smooth muscle cells and thereby regulate vascular tone (Saponara et al. 2011). The present observed effect of improved NBF in fisetin administered animals may be due to the fisetin’s ability to modulate voltage-dependent L-type Ca2+ current (I ca,L) current and calcium-regulated K+ current in the vascular cells (Wu et al. 2003; Wu et al. 2008). Although a relation between the transcriptional regulation and ion current has not been studied in detail, the evidences of NF-κB mediated transcription of Cav1.2 channel in the arterial smooth muscle cells may partially explain the fisetin’s ability to produce vasodilatation indirectly through NF-κB inhibition (Narayanan et al. 2010). Fisetin corrected motor NCV and also improved the NBF in treated diabetic rats. These effect forms a major basis of our claims of protective effect of fisetin against functional deficits in DN.
Changes in sensorimotor perceptions are the symptomatic endpoints in any form of neuropathy. Thermal and mechanical perception is altered in the diabetic animals which may be due to nerve sensitization due to local inflammation and nerve fibre damage (Tavee and Zhou 2009). In this regard, the observed decreased activity of COX-2 and NF-κB may have directly contributed to improvement in behavioural deficits.
Glutathione depletion may exacerbate free radical generation, culminating in neuronal death. Additionally increased lipid peroxidation is also considered as a picture of declined antioxidant defence and raised oxidative-nitrosative stress in diabetes (Tiwari et al. 2013). Fisetin decreased malondialdehyde, marker for lipid peroxidation, and contributed to the replenishment of neuronal GSH levels along with improvement in Nrf2 expression in sciatic nerves. This may be due to its capability of positively modulating Nrf2 signalling which controls expression of various cytoprotective enzymes which may act against oxidant induced nerve damage (Negi et al. 2011a). Fisetin also decreased the DNA fragmentation as seen in TUNEL assay outcomes in nerve microsection of treated diabetic animals. Increased DNA fragmentation has been reported to be due to elevated superoxide and peroxynitrite levels. Fisetin may limit the DNA fragmentation in fisetin-treated groups due to its antioxidant effect and Nrf2 modulatory potential. The enhanced sciatic expression of HO-1 in fisetin-treated rats also warrants its anti oxidant potential in DN.
Fisetin decreased the NF-κB expression as well as its activation as evident from the decrease p-IκB levels in treated groups. If there is decline in IκB phosphorylation, it will also limit the nuclear translocation of NF-κB, and hence, results in decreased expression of proinflammatory proteins and enzymes which were seen in fisetin-treated groups (Kumar et al. 2013). Decreased levels of IL-6 and TNF-alpha are in line with this outcome. We have also observed decrease in COX-2 immunopositivity in nerve microsections of treated rats, which further fortify our claim of NF-κB inhibitory activity of fisetin.
In this study, we clearly show that fisetin prevents oxidative stress and neuroinflammation evoked by diabetes-associated peripheral nerve damage. Improvement in nerve functional and behavioural parameters along with improved biochemical deficits points towards attenuation of peripheral nerve damage in animal model of diabetic peripheral neuropathy. We can speculate that the use of nutraceuticals like fisetin may provide an additive regimen for management of DN.
References
Arora M, Kumar A, Kaundal RK, Sharma SS (2008) Amelioration of neurological and biochemical deficits by peroxynitrite decomposition catalysts in experimental diabetic neuropathy. Eur J Pharmacol 596(1):77–83
Cameron NE (2013) Role of endoplasmic reticulum stress in diabetic neuropathy. Diabetes 62(3):696–697
Cameron NE, Cotter MA (2008) Pro-inflammatory mechanisms in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Curr Drug Targets 9(1):60–67
Cameron NE, Cotter MA, Low PA (1991) Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am J Physiol 261(1):E1–E8
Chopra K, Tiwari V, Arora V, Kuhad A (2010) Sesamol suppresses neuro-inflammatory cascade in experimental model of diabetic neuropathy. J Pain 11(10):950–957
Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA (2000) Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. J Diabetes Res 1(2):131–143
Huizinga MM, Peltier A (2007) Painful diabetic neuropathy: a management-centered review. Clin Diabetes 25(1):6–15
Jensen TS, Backonja M-M, Jimenez SH, Tesfaye S, Valensi P, Ziegler D (2006) New perspectives on the management of diabetic peripheral neuropathic pain. Diab Vasc Dis Res 3(2):108–119
Joshi RP, Negi G, Kumar A, Pawar YB, Munjal B, Bansal AK, Sharma SS (2013) SNEDDS curcumin formulation leads to enhanced protection from pain and functional deficits associated with diabetic neuropathy: an insight into its mechanism for neuroprotection. Nanomedicine 9(6):776–785
Kumar A, Kaundal RK, Iyer S, Sharma SS (2007) Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life Sci 80(13):1236–1244
Kumar A, Negi G, Sharma SS (2012) Suppression of NF-kB and NF-kB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IkB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie 94(5):1158–1165
Kumar A, Negi G, Sharma SS (2013) Neuroprotection by resveratrol in diabetic neuropathy: concepts & mechanisms. Curr Med Chem 20(36):4640–4645
Kumar A, Sharma SS (2010) NF-kappaB inhibitory action of resveratrol: a probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem Biophys Res Commun 394(2):360–365. doi:10.1016/j.bbrc.2010.03.014
Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL (2006) Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol 2(11):620–628
Lewis KN, Mele J, Hayes JD, Buffenstein R (2010) Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr Comp Biol 50(5):829–843
Narayanan D, Xi Q, Pfeffer LM, Jaggar JH (2010) Mitochondria control functional CaV1. 2 expression in smooth muscle cells of cerebral arteries. Circ Res 107(5):631–641
Narenjkar J, Roghani M, Alambeygi H, Sedaghati F (2011) The effect of the flavonoid quercetin on pain sensation in diabetic rats. Basic Clin Neurosci 2(3):51–57
Negi G, Kumar A, Joshi RP, Sharma SS (2011a) Oxidative stress and Nrf2 in the pathophysiology of diabetic neuropathy: old perspective with a new angle. Biochem Biophys Res Commun 408(1):1–5
Negi G, Kumar A, Sharma SS (2011b) Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kB and Nrf2 cascades. J Pineal Res 50(2):124–131
Obrosova IG (2009) Diabetes and the peripheral nerve. Biochim Biophys Acta 1792(10):931–940
Sandireddy R, Yerra VG, Areti A, Komirishetty P, Kumar A (2014) Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets. Int J Endocrinol 2014:1–10
Saponara S, Carosati E, Mugnai P, Sgaragli G, Fusi F (2011) The flavonoid scaffold as a template for the design of modulators of the vascular Cav1. 2 channels. Br J Pharmacol 164(6):1684–1697
Singh R, Kishore L, Kaur N (2014) Diabetic peripheral neuropathy: current perspective and future directions. Pharmacol Res 80:21–35
Sun X, Wang X, Chen T, Li T, Cao K, Lu A, Chen Y, Sun D, Luo J, Fan J (2010) Myelin activates FAK/Akt/NF-kB pathways and provokes CR3-dependent inflammatory response in murine system. PLoS One. doi:10.1371/journal.pone.0009380
Tak PP, Firestein GS (2001) NF-kB: a key role in inflammatory diseases. J Clin Invest 107(1):7–11
Tavee J, Zhou L (2009) Small fiber neuropathy: a burning problem. Clevel Clin J Med 76(5):297–305
Tiwari BK, Pandey KB, Abidi AB, Rizvi SI (2013) Markers of oxidative stress during diabetes mellitus. J Biomark. doi:10.1155/2013/378790
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW (2010) When NRF2 talks, who’s listening? Antioxid Redox Signal 13(11):1649–1663
Winkler G, Kempler P (2010) Pathomechanism of diabetic neuropathy: background of the pathogenesis-oriented therapy. Orv Hetil 151(24):971–981
Wu S-N, Chen B-S, Hsu C-L, Peng H (2008) The large-conductance Ca 2-activated K+ channel: A target for the modulators of estrogen receptors. Curr Top Biochem Res 10(2):93–101
Wu SN, Chiang HT, Shen AY, Lo YK (2003) Differential effects of quercetin, a natural polyphenolic flavonoid, on L-Type calcium current in pituitary tumor (GH3) cells and neuronal NG108-15 cells. J Cell Physiol 195(2):298–308
Yerra VG, Negi G, Sharma SS, Kumar A (2013) Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kB pathways in diabetic neuropathy. Redox Biol 1(1):394–397
Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47
Acknowledgments
The authors would like to acknowledge the financial support provided by the Department of Pharmaceuticals, Ministry of Chemical and fertilizers, Government of India for carrying out this work. The authors would also like to acknowledge the support of NIPER-Hyderabad for preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Sandireddy, R., Yerra, V.G., Komirishetti, P. et al. Fisetin Imparts Neuroprotection in Experimental Diabetic Neuropathy by Modulating Nrf2 and NF-κB Pathways. Cell Mol Neurobiol 36, 883–892 (2016). https://doi.org/10.1007/s10571-015-0272-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0272-9