
Abstract
Status epilepticus (SE) is defined as continuous and self-sustaining seizures, which trigger hippocampal neurodegeneration, mitochondrial dysfunction, oxidative stress, and energy failure. During SE, the neurons become overexcited, increasing energy consumption. Glucose uptake is increased via the sodium glucose cotransporter 1 (SGLT1) in the hippocampus under epileptic conditions. In addition, modulation of glucose can prevent neuronal damage caused by SE. Here, we evaluated the effect of increased glucose availability in behavior of limbic seizures, memory dysfunction, neurodegeneration process, neuronal activity, and SGLT1 expression. Vehicle (VEH, saline 0.9%, 1 μL) or glucose (GLU; 1, 2 or 3 mM, 1 μL) were administered into hippocampus of male Wistar rats (Rattus norvegicus) before or after pilocarpine to induce SE. Behavioral analysis of seizures was performed for 90 min during SE. The memory and learning processes were analyzed by the inhibitory avoidance test. After 24 h of SE, neurodegeneration process, neuronal activity, and SGLT1 expression were evaluated in hippocampal and extrahippocampal regions. Modulation of hippocampal glucose did not protect memory dysfunction followed by SE. Our results showed that the administration of glucose after pilocarpine reduced the severity of seizures, as well as the number of limbic seizures. Similarly, glucose after SE reduced cell death and neuronal activity in hippocampus, subiculum, thalamus, amygdala, and cortical areas. Finally, glucose infusion elevated the SGLT1 expression in hippocampus. Taken together our data suggest that possibly the administration of intrahippocampal glucose protects brain in the earlier stage of epileptogenic processes via an important support of SGLT1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Status epilepticus (SE) is defined as continuous and self-sustaining seizures lasting > 30 min, reaching a significant number of patients [1,2,3,4]. Because it is an epileptogenic insult, SE is capable of leading to temporal lobe epilepsy (TLE) and promoting severe damage to the central nervous system (CNS), such as activation of a recurrent excitatory circuit, neurodegeneration, aberrant neurogenesis, and mossy fiber sprouting [5,6,7]. Despite treatment with AEDs, about 30% of patients may be refractory to standard drugs, experiencing frequent and lasting seizures capable of promoting brain damage [8,9,10].
Putative neuroprotective substances have been increasingly identified using animal models of seizures. Intrahippocampal administration of pilocarpine has typically been used to induce TLE in rodents, mimicking epileptic seizures in humans, which initiate as focal and then evolve to generalized [11,12,13]. After the infusion of H-PILO, the animal behavior is altered, presenting wet dog shake (WDS), forelimb myoclonus, rearing, and falling [14,15,16]. As a consequence of PILO-induced SE, especially after 24 h, selective cell death occurs in DG hilus, CA3 and CA1 hippocampal subareas, as well as in extrahippocampal regions, including subiculum, thalamus, amygdala, substantia nigra, and cortical areas [17,18,19,20,21]. Furthermore, pilocarpine-induced SE increases reactive oxygen species (ROS) production, directly associated with neuronal degeneration, increasing malondialdehyde (MAD) levels, and decreasing catalase (CAT) and superoxide dismutase (SOD) antioxidant enzymes’ activity [22,23,24,25]. In addition, cell death in the hippocampus and adjacent limbic areas can lead to memory and learning impairments [26,27,28].
Glucose is the main source of energy for the mammalian brain, and energy deficit can lead to neuronal dysfunction [29]. During epileptic seizures, glucose uptake increases in hyperexcited neurons above the body’s supply capacity [30,31,32,33]. Furthermore, oxidative stress, mitochondrial dysfunction, energy failure, and tricarboxylic acid cycle (TCA) failure typically occur in the hippocampus and nearby area after SE, working together contribute to neuronal damage [31, 34, 35]. Interestingly, both hypo- and hyperglycemia lead to aggravated epileptic seizures and, consequently, compromised the physiology of CNS [36,37,38,39,40]. Therefore, cerebral glucose control may be an interesting approach to protect against damage following pilocarpine-induced SE, but its underlying mechanisms of action remain uncertain. Since the neuronal membranes are impermeable to glucose, the transport of glucose into the neuron is mediated by facilitated diffusion and secondary active transport, via glucose transporters (GLUTs) and sodium/glucose cotransporters (SGLTs), respectively [41, 42]. SGLT1 isoform is expressed in the hippocampus and other brain regions, including amygdala, hypothalamus, basal ganglia, and cortical areas [30, 43, 44]. We have previously observed that SGLTs play a crucial role in protecting against pilocarpine-induced SE damage. When we inhibited hippocampal SGLTs with phlorizin, a nonspecific inhibitor, there was an increase in WDS number, seizure severity, and neuronal death pattern after SE [13]. In addition, other authors have shown that glycemic index control was able to suppress neuronal death following kainate-induced SE, indicating that hippocampal glucose modulation may be a critical therapeutic target [36].
Starting from this, we tested the hypothesis that increased glucose availability upregulate SGLTs translocation, which is correlated to the neuroprotective effect in the acute phase of epileptogenesis.
Methods
Animals
This study was conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals of the Brazilian Society of Laboratory Animals Science (SBCAL). All experimental procedures were approved by the Ethical Committee of the Federal University of Alagoas (Protocol # 04/2016), according to Ethical Principles adopted by the Brazilian College of Animal Experimentation (COBEA). Animal studies are reported in compliance with the approved guidelines. Experiments were conducted in male Wistar rats (Rattus norvegicus [n = 81, 240–340 g, 2–3 months]) from the main breeding stock of the Federal University of Alagoas. Animals were maintained on a 12-h/12-h light/dark cycle at 21 ± 2 °C, with lights on at 07:00 AM and lights off at 07:00 PM. They were individually housed in plastic cages with food and water ad libitum. All experiments were designed to minimize animal suffering and to limit the number of animals used.
All animals were monitored by research staff at least 2 times per day, in order to observe signs of illness or impairment by observing the general body condition, respiration rate, dehydration, posture, immobility, social interaction, and response to manipulation. For the animals submitted to SE, monitoring the health was carried out for 2 h/day until the complete post-ictal recovery (about 2 days after SE; note that intrahippocampal pilocarpine model allows rapid recovery and a high rate of survival) [12, 13]. During this period, animals were treated with electrolyte and nutrient replacement (ip injection of saline 0.9%, and by feeding animals with pasty food). None of the animals presented clinical/behavior signal of pain or unexpected distress, used as humane endpoint criteria for euthanasia.
Surgical Procedure
Animals were anesthetized with ketamine (100 mg/kg, ip) and xylazine (10 mg/kg, ip) and received 0.1 mL/100 g veterinary pentabiotic (Fort Dodge®, subcutaneous) before the surgery. After fixing on stereotaxic, animals received local anesthetic (lidocaine with epinephrine, subcutaneous [Astra®]). Posteriorly, a cannula was implanted stereotaxically in the hilus of the dentate gyrus (DG) of the left hippocampus, according to following coordinates: − 6.30 mm anterior-posterior (AP, reference: bregma); 4.50 mm medial-lateral (ML, reference: sagittal sinus); − 4.50 mm dorsal-ventral (DV, reference: dura mater) [7, 12, 13, 45]. After the surgery, animals were 7 days in recovery.
Intrahippocampal Microinjections
Animals received either glucose, pilocarpine, or vehicle by microinjections (1 μL) of glucose in the left hilus of the DG of hippocampus, as described below. The rats were divided into 8 experimental groups: VEH (n = 10), H-PILO (n = 10), G+P 1 mM (n = 10), G+P 2 mM (n = 10), G+P 3 mM (n = 10), P+G 1 mM (n = 10), P+G 2 mM (n = 10), and P+G 3 mM (n = 11).
Animals were gently immobilized, and the drug microinjection was performed. VEH animals received only one administration of intrahippocampal saline 0.9%. H-PILO animals received microinjections of vehicle (saline 0.9%), and after 30 min, pilocarpine (1.2 mg) was administrated to evoke limbic seizures. G+P animals received microinjections of 1, 2, or 3 mM glucose [diluted in saline 0.9%]), and after 30 min, pilocarpine (1.2 mg) was administrated to evoke limbic seizures. In addition, P+G animals received microinjections of pilocarpine (1.2 mg), and after 5 min, 1, 2, or 3 mM glucose [diluted in saline 0.9%]) were administrated. We used a 5-μL syringe (Hamilton Company, Reno, NV, USA) connected to a microinjection pump (Harvard Apparatus PHD 2000, Holliston, MA, USA) at a speed of 0.5 μl/min.
All animals that develop SE were rescued with diazepam (5 mg/kg; ip) after 90 min of SE onset. Furthermore, animals that did not develop SE received the injection of diazepam under the same conditions.
Behavioral Analysis
SE Seizures
After microinjection of intrahippocampal pilocarpine, behavioral activity was recorded by video camera (Full HD Digital Camcorder Sony DCR-PJ6) for a period of 90 min, which is enough time to observe neurodegeneration [12, 13]. Racine’s scale [46] was used to categorize the behavioral analysis into the following classes observed: (0) immobility, (1) facial movements, (2) head nodding, (3) forelimb clonus, (4) rearing, and (5) rearing and falling.
Furthermore, the latency period for the SE was analyzed. Number of wet dog shake (WDS) was quantified before and along SE. During the SE, the 90-min observation time was split into 18 windows of 5 min and the most severe seizure with more frequency in each interval was used to represent the window [12]. In addition, the number and total time of class 3–5 seizures were analyzed to better understand the severity and evolution of seizures along the SE among different experimental groups. Finally, to determine the severity of seizures, the representative scales of each window were summed, and the result was divided by the total number of windows.
Inhibitory Avoidance Test (IAT)
In the inhibitory avoidance test (IAT), all animals were placed in an automatically operated box (40 × 25 × 25 cm) with a wall glass front, being the floor constituted by a steel grid coupled to an energy generating box. The test was divided into (1) learning/training session, before SE, animal was kept on the platform and received a shock (2.0 s of 0.2 mA) after getting off with its four paws on the grid, and (2) test session, 24 h after training and SE, animal was placed in the same apparatus, under the same environmental conditions, without the aversive stimulus.
Biochemical Assessments
Animals (n = 14) that survived within 24 h after intrahippocampal pilocarpine-induced SE were guillotined, and the brains were directly placed on an ice-plate and dissected in order to remove the hippocampus. Hippocampi were immediately frozen in liquid nitrogen for further future use and stored at − 80 °C.
Total Thiol Content (Sulfhydryl Groups)
Sulfhydryl content was determined from reaction with compound DTNB (5,5′-Dithiobis (2-nitrobenzoic acid). Aliquot of homogenate (100 μg protein) was incubated in the dark with 25 μL of DTNB (20 mM) and the final volume of 1 mL with extraction buffer was completed. Absorbance reading was taken on spectrophotometer (AJX-6100PC) at 412 nm [47]. Results were expressed in mmol/mg protein.
Lipid Peroxidation
The colorimetric technique was used for the determination of thiobarbituric acid reactive substances (TBARS) [48]. A total of 0.3 mg/mL of hippocampi homogenate were added to 200 μL of 30% (w/v) trichloroacetic acid and stirred for 1 min. Then, 200 μL of 10 mM TRIS HCl, pH 7.4, was added to the material, stirred for 1 min, and then centrifuged at 2500 rpm for 10 min at 4 °C. The collected supernatant (450 μl) was mixed with 0.73% (w/v) thiobarbituric acid (450 μL) which reacted with the lipoperoxidation products to form a pink-colored compound. The mixture was incubated for 15 min at 100 °C and then cooled. The absorbance reading was performed in a spectrofluorimeter (Flex Station 3, Molecular Devices) at 535 nm. The results were expressed as mmol/mg protein.
Superoxide Dismutase (SOD) Activity
Determination of SOD activity was carried out accordingly to Misra and Fridovich [49]. The hippocampi homogenate (100 μg protein) was incubated in sodium carbonate buffer (50 mM, pH 10.2, + 0.1 mM EDTA) in a water bath at 37 °C. The reaction was initiated by the addition of 30 μL of epinephrine (150 mM) in acetic acid (0.05%) in a final volume of 1 mL. The absorbance was read at 480 nm for 1 min on the spectrophotometer (AJX-6100PC). One unit of SOD was defined as the amount of protein required to inhibit the autoxidation of 1 μmol of epinephrine per minute. The results were expressed in U/mg protein.
Catalase Activity (CAT)
Catalase activity was monitored accordingly to Aebi [50]. The test is based on the determination of the decomposition of H2O2. Eighty micrograms of proteins was added to a 50 mM phosphate buffer (sodium phosphate monobasic monohydrate + dibasic sodium phosphate), pH 7.0, 35 °C. The reaction was then started with 0.3 mM H2O2 in a final volume of 1 mL. The decrease in absorbance was monitored at 240 nm on a spectrophotometer (AJX-6100PC) for 1 min. One unit of CAT was defined as the amount of protein required to convert 1 μmol de H2O2 per minute to H2O. The results were expressed in U/mg protein.
Histological Processing
In order to perform the histological procedures, animals were injected with an overdose of xylazine and ketamine at 24 h after SE induction and were transcardially perfused with 0.1 M phosphate-buffered saline, pH 7.4 (PBS), followed paraformaldehyde solution (4%, diluted in PBS). Afterwards, the brains were removed, cryoprotected with sucrose 20%, frozen at − 20 °C for 3 h and stored at − 80 °C. Sections were then cut (30 μm thickness) using a cryostat (Leica CM 1850) at a temperature ranging from − 18 to − 22 °C and were processed for FJ-C staining and immunofluorescence techniques.
FJ-C Staining Procedure
Brain sections were placed onto slides and then subjected to successive washes of 100% ethanol for 3 min, 70% ethanol for 1 min, distilled water for 1 min. Afterwards, slides were transferred to a solution of 0.06% potassium permanganate for 15 min on a rotating platform. Slides were rinsed three times for 1 min in distilled water and then transferred to the FJ staining solution (0.0001%) for 30 min. After, slides were rinsed three times for 1 min in distilled water [51]. Finally, slides were coverslipped using fluoromount (EMS). The sections were examined, and images captured using a fluorescence microscope (Nikon DS RI1).
SGLT1 and c-Fos Immunofluorescences
We used an antibody that binds to SGLT1 or nuclear protein c-fos. After intrahippocampal pilocarpine-induced SE, there is an increased glucose uptake via SGLT and c-Fos overexpression, which indicates a neuronal hyperexcitability. Immunofluorescence was used to analyze the expression of both proteins primarily in the hippocampus and other areas of the brain.
The immunohistochemistry assays were done in histological slide, and the protocol used for detection of SGLT1 and c-Fos antigens was the same, with only alteration of primary antibodies. The following primary antibodies were used: rabbit polyclonal IgG to SGLT1 (Catalog Number - Orb11364, Biorbyt®, 1:100) and rabbit polyclonal IgG to c-Fos (Lot # C1010, Santa Cruz Biotechnology®, 1:50). Secondary antibody was used: Alexa Fluor 594 Donkey secondary antibody (anti-rabbit IgG, Biolegend®, San Diego, CA; 1: 2000). Briefly, the immunohistochemistry protocol begins with brain tissue slices submerged in methanol (10 min), followed by two baths of 10 min in PBS 1x. An antigenic rescue was then performed with citrate buffer (pH 6) for 10 min (output 6) and, after a cooling period (30 min, room temperature), immersed in the same solution. Sections were then incubated in an autofluorescence blocking solution with PBS/glycine 3% (1 h, room temperature), followed by a second blocking solution for nonspecific sites using fish skin gelatin in 0.05% in PBS 1X and equine serum 1.5% (1 h, room temperature, Product code – 11540636, Gibco™® Horse Serum, heat inactivated). Shortly thereafter, it was incubated with the anti-SGLT1 or anti-cfos primary antibody diluted in fish skin gelatin in 0.05% in PBS 1X (overnight, 4 °C). In the second stage, the slices were washed with PBS 1x (2 times in 10 min), followed by incubation with Alexa 594 diluted in fish skin gelatin 0.05% in PBS 1X (1 h, room temperature). Sections were washed with PBS 1x (two baths of 5 min), and DNA was counterstained with fluorescent dye 4′,6-diamidino-2-phenylindole (DAPI, ab104139, Abcam®, USA; 1:1000, diluted in PBS 1x, 15 min, room temperature). Finally, the sections were washed (PBS 1x, five baths of 2 min) and used as mounting medium PBS/glycerol. To control for binding specificity, sections were subjected to the same protocol with omission of anti-SGLT1 and anti-cfos primary antibodies. Sections were examined and images captured using a fluorescence microscope (Nikon DS RI1).
Cell Counting and Densitometry
SGLT1 expression was quantified by densitometry, while fluoro-Jade-positive (FJ+) and c-Fos-positive (c-Fos+) cells were quantified by using the ImageJ software (Wayne Rasband; Research Services Branch, National Institute of Mental Health, Bethesda, MD, USA). In order to quantify the FJ+ and c-Fos+ neurons in the hippocampus and extrahippocampal areas, different coordinates were used [45]. All cells were counted on the contralateral side because animals that received microinjection of intrahippocampal pilocarpine developed a scar around the microinjection site [12].
In the hippocampus, we sampled in three different coordinates: CA1, CA3, and hilus of dentate gyrus, (AP − 2.56 mm, AP − 3.30 mm, and AP −6.30 mm), as showed by Castro et al. [12]. These regions were selected because of the high sensitivity to the neurodegenerative process. In addition, for the mapping of cortical areas, we used three different coordinates: motor primary and secondary (M1 and 2), somatosensory primary (S1), retrosplenial granular (RSGc), agranular insular (AIP), ectorhinal (Ect), perirhinal (PRh), and piriform (Pir) (AP − 2.64 mm, AP − 3.36 mm, AP − 4.80 mm). Additionally, the mapping of the dorsal subiculum (DS) and the substantia nigra (reticular part, SNR) were made based on three other coordinates: DS (AP − 4.92 mm, AP − 5.04 mm, AP − 5.20 mm) and SNR (AP − 4.80 mm, AP − 4.92 mm, AP − 5.04 mm). Finally, the thalamic (lateral posterior, mediorostral part [LPMR], and centrolateral [CL]), hypothalamic (paraventricular, posterior part [PVP]), and amygdaloid (lateral, dorsolateral part [LaDL]) nuclei were mapped according to the following coordinates: thalamus and hypothalamus (AP − 2.92 mm, AP − 3.36 mm, AP − 3.72 mm) and amygdala (AP − 2.92 mm; AP − 3.48 mm; AP − 3.84 mm).
Statistical Analysis
All experimental values are presented as mean ± SEM, and a significance level of 5% (described as p < 0.05) was adopted for all statistical tests. Comparisons of most of the results were performed by unpaired t test or one-way analysis of variance (ANOVA), followed by Dunnett’s post-test (GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego, CA, USA). Only in the inhibitory avoidance test, the data were expressed as median with interquartile range and compared by the Kruskal-Wallis test. The number of animals is cited in the figure legends.
Results
Intrahippocampal Glucose Supply Does Not Change Latency for Seizures but Increases the Number of WDS
Typically, as expected, after administration of intrahippocampal pilocarpine, the animals had a change in behavior, including immobility, facial movements, head nodding, and myoclonic movements of the limbs that evolved to continuous tonic clonic convulsive seizures, indicating the onset of SE.
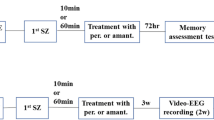
In the initial periods after intrahippocampal pilocarpine microinjection, animals had a latency interval without manifesting epileptic seizures. In order to verify if the increase in glucose supply at different concentrations (1, 2 or 3 mM) before and after intrahippocampal pilocarpine interferes with the generation time of SE, the animal behavior was carefully evaluated before SE (Fig. 1A and B). The latency for SE was similar in all groups that received the different concentrations of glucose in relation to the control (one-way ANOVA, F (6, 64) = 0.4881, P = 0.8149; Fig. 1C).

Glucose control does not alter latency for seizures and memory dysfunction but increases the number of WDS after pilocarpine-induced SE. The experimental scheme (A, B). Rats received glucose microinjections 30 min before (A) or 5 min after intrahippocampal pilocarpine (B). Glucose infusion prior and after intrahippocampal pilocarpine do not change the latency to SE (one-way ANOVA, F (6, 64) = 0.5160, P = 0.7941; C). The supply of hippocampal glucose (3 mM) increased the number of total WDS (one-way ANOVA, F (6, 64) = 0.8919, P = 0.5063; D). After 24h of SE, long-term memory consolidation was analyzed by inhibitory avoidance test in both rats receiving glucose before (A) or after (B) intrahippocampal pilocarpine. Initially, all animals were submitted to an aversive stimulus (training session) prior to SE induction (E). Memory consolidation was not protected from increased glucose (1, 2, or 3 mM) administration before (gray, blue, and red bar) and after (gray, blue and red bar outline) intrahippocampal pilocarpine (Kruskal-Wallis test, p = 0.0006 vs VEH; F). Latency and WDS data represent the mean ± S.E.M. of 10–11 rats. *P < 0.05; one-way ANOVA with Dunnett’s post hoc test or unpaired t test. Memory dysfunction data represent the median with interquartile range. *P < 0.05, ***P < 0.001, and ****P < 0.0001 compared with VEH; one-way ANOVA with Kruskal-Wallis test with Dunn’s post hoc test. VEH, vehicle; H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, status epilepticus; SEM, standard error of the mean
In intrahippocampal pilocarpine models, it is common to observe the presence of repetitive movements in the head and neck of rodents, motor pattern that stereotypes the shaking of a wet dog. As expected, wet dog shake (WDS) was quite common and frequent during the latency period in all animals, reducing drastically after the beginning of the SE. Increased glucose supply (3 mM) after intrahippocampal pilocarpine increased the number of total WDS when compared to the control (t test, t16 = 2.252, P = 0.0387; Fig. 1D).
The Impairment in Memory Consolidation Is Maintained After Increased Glucose Availability
In order to evaluate the effect of increased hippocampal glucose availability on memory consolidation, the inhibitory avoidance test was performed (Fig. 1A and B). During the training period, all animals spent little time on the platform (Fig. 1E). In contrast, in the test period, the group that did not have SE consolidated a long-term memory. Typically, the group that received only H-PILO had impaired memory consolidation, as expected (Kruskal-Wallis test, p = 0.0006 vs VEH; Fig. 1F). Increased glucose availability (1, 2 or 3 mM) before and after intrahippocampal pilocarpine was not able to reverse memory dysfunction following pilocarpine-induced SE (Kruskal-Wallis test, p = 0.0006 vs VEH; Fig. 1F).
Intrahippocampal Glucose Supply Attenuates the Severity of Seizures During SE
In order to assess the impact of glucose modulation on the severity of seizures, epileptic seizures were analyzed during 90 min of SE according to Racine’s scale [46] (Fig. 2A and B). The number and total time of class 3, 4, and 5 seizures remained unchanged after administration of the different glucose concentrations (1, 2 and 3 mM) before and (1 or 2 mM) after intrahippocampal pilocarpine (p > 0.05). However, the higher glucose concentration (3 mM) after intrahippocampal pilocarpine was able to reduce the frequency and total time of class 3, 4, and 5 seizures compared to control (Fig. 2C, D, F, G, I, J; p < 0.05).

Increased glucose availability reduces seizure severity following intrahippocampal pilocarpine-induced SE. The experimental scheme (A, B). Rats received glucose microinjections 30 min before (A) or 5 min after intrahippocampal pilocarpine (B). The epileptic seizures were analyzed during the 90 min of SE, according to the Racine’s scale. Over the 90 min of SE, the total number (one-way ANOVA, F (6, 64) = 1.716, P = 0.04; C) and time (one-way ANOVA, F (6, 64) = 2.035, P = 0.01; D) of class 3 seizures (head and neck myoclonus) were decreased when administered glucose (3 mM; red bar outline) into the hippocampus. Class 3 was reduced at the beginning and at the end of SE (E). Glucose administration (3 mM; red bar outline) reduced the total number (one-way ANOVA, F (6, 64) = 2.714, P = 0.01; F) and time (one-way ANOVA, F (6, 63) = 2.328, P = 0.02; G) of class 4 seizures, decreasing significantly in the 30–50 time range (H). Similarly, the total number (one-way ANOVA, F (6, 63) = 1.742, P = 0.03; I) and time (one-way ANOVA, F (6, 63) = 1.744, P = 0.02; J) of class 5 was attenuated by increased glucose availability (3 mM; red bar outline). During the evolution of class 5, glucose interfered strongly between times 30–60 (K). Increased glucose supply (3 mM; red bar outline) was able to decrease the severity of seizures (one-way ANOVA, F (6, 64) = 1.65, P = 0.04; L). Error bars indicate the SEM. Data represent the mean ± S.E.M. of 10–11 rats. *P < 0.05, **P < 0.01, and ***P < 0.001; one-way ANOVA with Dunnett’s post hoc test or unpaired t test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, status epilepticus; SEM, standard error of the mean
When analyzed in detail the effect of increased glucose concentration (3 mM) in the evolution of the seizures over the SE, we observed that in the intervals of 20–40 and 30–50 min, class 3 and 4 seizures remained reduced, respectively (Fig. 2 E and H, p < 0.05). In all other final times, class 3 and 4 seizures of the P+G (3 mM) group remained like those of the control, except class 4 seizure that decreases again at 90 min (Fig. 2H, p < 0.05). In addition, class 5 seizure is decreased in the range of 30–60 min (p < 0.05), remaining the same to the control until the end of the SE (Fig. 2K).
Using the average of the most severe seizures along the 18 windows of SE, the seizure severity was evaluated. Corroborating previous behavioral findings, only the high availability of glucose (3 mM) after intrahippocampal pilocarpine was able to reduce the severity of seizures compared to the H-PILO group (one-way ANOVA, F (6, 64) = 1.65, P = 0.04; Fig. 2L). In other words, glucose availability may exert an anticonvulsive effect on the SE.
Brain Glucose Supply Reduces Neuronal Death in the Hippocampus and Other Brain Areas
Classically, pilocarpine-induced SE triggers neuronal death in several brain areas, especially in the hippocampus [12, 13]. In order to evaluate whether brain glucose modulation can prevent neuronal damage caused by SE, the neurodegeneration process was evaluated by histochemistry of FJ. When glucose was administered prior to intrahippocampal pilocarpine at all concentrations (1–3 mM), the number of FJ + neurons was reduced in the DG hilus (one-way ANOVA, F (6, 33) = 4.985, P = 0.001), CA3 (one-way ANOVA, F (6, 33) = 4.976, P = 0.001), and CA1 (one-way ANOVA, F (6, 33) = 9.216, P < 0.0001) subfields of hippocampus compared to the control (Fig. 4 A1–4, B1–4, and C1–4). Similarly, all concentrations of glucose after intrahippocampal pilocarpine were able to attenuate neuronal death in the same areas of the hippocampus (P < 0.001, Fig. 3 A5–8, B5–8, and C5–8). In other words, besides promoting an anticonvulsive role, hippocampal glucose modulation prevents the neuronal damage characteristic of SE. The highest concentration of glucose (3 mM) administered after intrahippocampal pilocarpine showed better efficiency in the behavior of seizures and in the neurodegenerative process; therefore, it was chosen for the analysis of the other methodological approaches.

Increased glucose availability attenuates neurodegeneration process in the hippocampus following intrahippocampal pilocarpine-induced SE. After 24 h of SE, the neurodegeneration process was evaluated by Fluoro-Jade C (FJ-C) histochemistry in both rats receiving glucose 30 min before or 5 min after intrahippocampal pilocarpine. Hilar interneurons (A1–7) and pyramidal neurons of the CA3 (B1–7) and CA1 (C1–7) regions were labeled with FJ (FJ+, green). Increased glucose administration at all concentrations before and after intrahippocampal pilocarpine was able to reduce the number of FJ + neurons in the DG hilus (one-way ANOVA, F (6, 33) = 4.985, P = 0.001; A8), as well as in the CA3 (one-way ANOVA, F (6, 33) = 4.976, P = 0.001; B8) and CA1 (one-way ANOVA, F (6, 33) = 9.216, P < 0.0001; C8) regions. Representative digital zoom was done on the photomicrographs of the control (A1, B1 ,and C1; see squares) Arrows represent the DG hilus, CA3, or CA1 regions. Magnification, 100×; scale bar, 100 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 5–7 rats. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with H-PILO; one-way ANOVA with Dunnett’s post hoc test or unpaired t test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, Status epilepticus; SEM, standard error of the mean
Some infusion of glucose administered before and after intrahippocampal pilocarpine were also able to attenuate the number of FJ+ neurons in motor (one-way ANOVA, F (6, 30) = 4.81, P = 0.0015), somatosensory (one-way ANOVA, F (6, 19) = 2.17, P = 0.09), insular (one-way ANOVA, F (6, 26) = 6.16, P = 0.0004), ectorhinal (one-way ANOVA, F (6, 29) = 3.79, P = 0.006), perirhinal (one-way ANOVA, F (6, 26) = 3.73, P = 0.008), and piriform (one-way ANOVA, F (6, 23) = 4.36, P = 0.004) cortices compared to control (Fig. 4 A–F 1–8). In addition, thalamic [lateral posterior (one-way ANOVA, F (6, 27) = 8.66, P < 0.0001) and centrolateral (one-way ANOVA, F (6, 26) = 15.06, P < 0.0001)], hypothalamic [posterior paraventricular (one-way ANOVA, F (6, 27) = 10.96, P < 0.0001)], and amygdaloid lateral (one-way ANOVA, F (6, 27) = 4.27, P = 0.004) nuclei were protected due to high glucose administration (Fig. 5 A–D 1–8). Similarly, the supply of cerebral glucose was able to reduce neuronal damage in the subiculum (one-way ANOVA, F (6, 33) = 6.38, P = 0.0002) and the substantia nigra (one-way ANOVA, F (6, 30) = 4.48, P = 0.002) (Fig. 5 E, F 1–8). Besides, presenting an anticonvulsive effect, modulation of hippocampal glucose plays a neuroprotective role in several brain regions.

Increased glucose supply decreases the neurodegeneration process in cortical areas after intrahippocampal pilocarpine-induced SE. After 24 h of SE, the neurodegeneration process was evaluated by Fluoro-Jade C (FJ-C) histochemistry in both rats receiving glucose 30 min before or 5 min after intrahippocampal pilocarpine. The concentration of 3 mM glucose had a more significant potential in protecting neuronal damage following SE. Some concentrations of hippocampal glucose (1, 2, or 3 mM) before and after intrahippocampal pilocarpine reduced FJ+ neurons in M1 (one-way ANOVA, F (6, 30) = 4.81, P = 0.0015; A1–8; dark blue rectangle), S1 (one-way ANOVA, F (6, 19) = 2.17, P = 0.09; B1–8; light blue rectangle), AIP (one-way ANOVA, F (6, 26) = 6.16, P = 0.0004; C1–8; green rectangle), Ect (one-way ANOVA, F (6, 29) = 3.79, P = 0.006; D1–8; yellow rectangle), PRh (one-way ANOVA, F (6, 26) = 3.73, P = 0.008; E1–8; orange rectangle) and Pir (one-way ANOVA, F (6, 23) = 4.36, P = 0.004; F1–8; pink rectangle) areas compared with control (H-PILO, black bar). Arrows represent the FJ+ neurons in each cortical area. A digital zoom was performed with the same proportions in all groups of animals to better show the FJ + neurons in the different brain areas. Magnification, 100×; scale bar, 100 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 5–7 rats. *P < 0.05 and **P < 0.01 compared with H-PILO; one-way ANOVA with Dunnett’s post hoc test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; M1, motor; S1, somatosensory; AIP, insular; Ect, ectorhinal; PRh, perirhinal; Pir, piriform; SEM, standard error of the mean

Glucose control reduces the neurodegeneration process in the thalamus, amygdala, subiculum, and substantia nigra after intrahippocampal pilocarpine-induced SE. The concentration of 3 mM glucose had a more significant potential in protecting neuronal damage following SE. Most hippocampal glucose concentrations (1, 2, or 3 mM) before and after intrahippocampal pilocarpine decreased FJ+ neurons in PVP (one-way ANOVA, F (6, 27) = 10.96, P < 0.0001; A1–8; red rectangle), CL (one-way ANOVA, F (6, 26) = 15.06, P < 0.0001; B1–8; pink rectangle), LPMR (one-way ANOVA, F (6, 27) = 8.66, P < 0.0001; C1–8; dark red rectangle), LaDL (one-way ANOVA, F (6, 27) = 4.27, P = 0.004; D1–8; blue rectangle), DS (one-way ANOVA, F (6, 33) = 6.38, P = 0.0002; E1–8; light blue rectangle) and SNR (one-way ANOVA, F (6, 30) = 4.48, P = 0.002; F1–8; green rectangle) areas compared with control (H-PILO, black bar). Arrows represent the FJ+ neurons in each brain area. A digital zoom was performed with the same proportions in all groups of animals to better show the FJ + neurons in the different brain areas. Magnification, 100×; scale bar, 100 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 5–7 rats. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with H-PILO; one-way ANOVA with Dunnett’s post hoc test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; PVP, posterior paraventricular th ncl; CL, centrolateral th ncl; LPMR, lateral posterior th ncl; LaDL, lateral amygdaloid ncl; DS, subiculum; SNR, substantia nigra; ncl, nucleus; th, thalamic; SEM, standard error of the mean
Effects of Brain Glucose Supply on Oxidative Stress Markers and Antioxidants Enzymes Activity in the Hippocampus
Typically, pilocarpine-induced SE exacerbates reactive oxygen species (ROS) levels in hippocampus. In order to analyze whether hippocampal glucose control interferes with oxidative stress promoted by SE, oxidative stress markers and antioxidants enzymes activity were assessed (Online Resource 1A). As the higher concentration of glucose (3 mM) administered after intrahippocampal pilocarpine had a more significant result, it was chosen for all subsequent analyzes. MDA formation was significantly increased in hippocampus of H-PILO animals when compared with VEH, but increased glucose (3 mM) supply after intrahippocampal pilocarpine was not able to prevent elevated MDA levels (one-way ANOVA, F (2, 8) = 5.182, P = 0.036) (Online Resource 1B). In addition, as a result of intrahippocampal pilocarpine-induced SE, the total thiol number was markedly reduced in the hippocampus of H-PILO animals and hippocampal glucose infusion did not change this condition (one-way ANOVA, F (2, 10) = 6.048, P = 0.019) (Online Resource 1C). Concordant with the elevated MDA level, we found significant decreases in the activity of antioxidant enzymes CAT (one-way ANOVA, F (2, 8) = 24.57, P = 0.0004) and SOD (one-way ANOVA, F (2, 9) = 4.947, P = 0.0355) in P+G 3 mM and H-PILO animals, compared with VEH (Online Resource 1D and E). Specifically, hippocampal glucose modulation did not reverse decreased antioxidant enzymatic activity. Taken together, these data indicate that increased glucose availability was not able to interfere with oxidative stress caused by intrahippocampal pilocarpine-induced SE.
Increased Glucose Availability Attenuates Neuronal Activity in the Hippocampus and Other Brain Areas
Neuronal hyperexcitability is a typical characteristic of pilocarpine-induced SE [12, 52, 53]. In order to identify whether modulation of hippocampal glucose interferes with neuronal hyperexcitability, cellular activity was evaluated by cFOS immunofluorescence. In hippocampus, increased glucose (3 mM) after intrahippocampal pilocarpine reduced the total number of cFOS+ neurons in the DG hilus (t test, t8 = 2.481, P = 0.0380), CA3 (t test, t10 = 5.651, P = 0.0002), and CA1 (t test, t8 = 2.735, P = 0.0257) subfield compared to control (Fig. 6 A–F 1–4).

Glucose modulation decreases neuronal activity in the hippocampus after intrahippocampal pilocarpine-induced SE. After 24 h of SE, the neuronal activity was analyzed by cFOS immunofluorescence in both rats receiving glucose 5 min after intrahippocampal pilocarpine. The nuclei were labeled with DAPI (blue, middle panels). Fluorescent labeling of the hippocampus shows strong cFOS immunoreactivity (red, left panels) in hilar interneurons (DG hilus, A1) and pyramidal neurons (CA3, C1; and CA1, E1) in H-PILO rats. Merge of cFOS and DAPI is shown in right panels. Quantitative analysis of cFOS immunofluorescent labeling of pyramidal neurons and interneurons of the H-PILO (black bars) and P+G (red bar outline) rats shown in B4, D4 and F4. Glucose control (3 mM) after intrahippocampal pilocarpine reduced the number of cFOS+ neurons in the DG hilus (t test, t10 = 2.521, P = 0.0303; B4), as well as in the CA3 (t test, t10 = 5.750, P = 0.0002; D4) and CA1 (t test, t9 = 2.581, P = 0.0296; F4) regions. Representative digital zoom was done on the photomicrographs of the control (A1–3, C1–3, and E1–3; see squares) Arrows represent the DG hilus, CA3, or CA1 regions. All images were digitally adjusted to the same proportions in all groups of animals to better highlight and specify the different hippocampal areas (hilus, CA3 and CA1). Magnification, 200×; scale bar, 50 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 3–6 rats. *P < 0.05 and ***P < 0.001 compared with H-PILO; unpaired t test. H-PILO, pilocarpine and saline; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, status epilepticus; SEM, standard error of the mean
As in neurodegenerative processes, a hippocampal-like result was observed in neuronal activity in other brain areas. Elevated hippocampal glucose supply (3 mM) after intrahippocampal pilocarpine decreased neuronal activity in retrosplenial (one-way ANOVA, F (6, 22) = 3.59, P = 0.012), perirhinal (one-way ANOVA, F (6, 22) = 3.59, P = 0.012), and piriform (one-way ANOVA, F (6, 16) = 3.70, P = 0.017) cortices in relation to control (Online Resource 2 A-F 1–4). In addition, the total number of cFOS+ neurons was lower in the subiculum of the group that received the glucose infusion when compared to control (one-way ANOVA, F (6, 20) = 6.51, P = 0.0006; Online Resource 2 G-H 1–4). Similarly, intrahippocampal administration of glucose was able to attenuate neuronal activity in thalamic [lateral posterior (one-way ANOVA, F (6, 22) = 10.94, P < 0.0001) and centrolateral (one-way ANOVA, F (6, 22) = 3.54, P = 0.01); Online Resource 3 A-D 1–4], hypothalamic [posterior paraventricular (one-way ANOVA, F (6, 20) = 5.21, P = 0.0023); Online Resource 3 E-F 1–4], and amygdaloid (lateral) nuclei (one-way ANOVA, F (3, 9) = 9.65, P = 0.0036; Online Resource 3 G-H 1–4).
Translocation of the Sodium/Glucose Cotransporter 1 (SGLT1) Increases After High Hippocampal Glucose Availability Followed by Intrahippocampal Pilocarpine
During a metabolic deficit, such as SE, the uptake of glucose is increased by greater SGLT function, which is essential during the epileptic seizures [13, 30, 43, 44]. We analyzed SGLT1 expression by immunofluorescence to test whether increased glucose availability alters SGLT1 translocation to the neuronal membrane. The higher glucose concentration (3 mM) administered after intrahippocampal pilocarpine was able to exacerbate the SGLT1 translocation in the DG hilus (t test, t7 = 2.407, P = 0.0470), as well as in CA3 (t test, t8 = 2.926, P = 0.0191) and CA1 (t test, t8 = 3.467, P = 0.0085) subareas of hippocampus compared to the control (Fig. 7 A–F 1–4). Thus, modulation of hippocampal glucose plays an anticonvulsive and neuroprotective role possibly involving SGLT1.

Increased glucose supply increases the SGLT1 translocation in hippocampal neurons after intrahippocampal pilocarpine-induced SE. After 24 h of SE, the SGLT1 expression was analyzed by immunofluorescence and quantified by densitometry in rats receiving glucose 5 min after intrahippocampal pilocarpine. The nuclei were labeled with DAPI (blue, middle panels). Fluorescent labeling of the hippocampus shows strong SGLT1 immunoreactivity (red, left panels) in hilar interneurons (DG hilus, A1) and pyramidal neurons (CA3, C1; and CA1, E1) in P+G rats. Merge of SGLT1 and DAPI shown in right panels. Quantitative analysis of SGLT1 immunofluorescent labeling of pyramidal neurons and interneurons of the H-PILO (black bars) and P+G (red bar outline) rats shown in B4, D4, and F4. Glucose modulation (3 mM) after intrahippocampal pilocarpine intensified the SGLT1 expression in neurons of DG hilus (t test, t7 = 2.407, P = 0.0470; B4), CA3 (t test, t8 = 2.926, P = 0.0191; D4) and CA1 (t test, t8 = 3.467, P = 0.0085; F4) regions. Representative digital zoom was done on the photomicrographs of the control (B1–3, D1–3, and F1–3; see squares). Arrows represent the DG hilus, CA3, or CA1 regions. All images were digitally adjusted to the same proportions in all groups of animals to better highlight and specify the different hippocampal areas (hilus, CA3 and CA1). Magnification, 400×; scale bar, 25 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 4–6 rats. *P < 0.05 and **P < 0.01 compared with H-PILO; unpaired t test. H-PILO, pilocarpine and saline; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, status epilepticus; SEM, standard error of the mean
Discussion
Glucose consumption is acutely accentuated during epileptic seizures [30, 54], and the effects of its modulation are vast and very complex in the epileptic brain. Along the SE, characterized by continuous and self-sustained seizures [2, 55,56,57,58], there is a marked increase in cerebral blood flow and oxygen consumption, and consequently, in glucose utilization [59]. Most patients with SE present an intense secondary hypometabolism, which is capable of compromising several brain areas [60,61,62,63,64,65]. Severe hypometabolism coupled with limited glucose availability [66,67,68] may be directly associated with the typical process of neuronal death in the hippocampus and nearby regions. In view of this, the control of glycemic status can be a determining factor during SE. In the present study, we evaluated the effects of high availability of hippocampal glucose on the severity of epileptic seizures and histological changes following intrahippocampal pilocarpine-induced SE. Overall, our main finding was that modulation of the hippocampal glucose index is able to protect the brain from damage resulting from intrahippocampal pilocarpine-induced SE.
After intrahippocampal pilocarpine injection, a latency period preceding SE is common in animal models [7, 12, 13]. The alteration in latency may be an important factor to be considered for the genesis of epileptic seizures; however, we observed that the latency interval for SE was not altered after glucose administration. In addition, WDS is typically observed throughout this latency phase prior to kainic acid- or H-PILO-induced SE [14, 69,70,71]. Our findings demonstrated that glucose infusion after intrahippocampal pilocarpine was able to increase the number of WDS during latency. Previous studies have argued that WDS elevation can act as an endogenous anticonvulsant mechanism [14]. In other words, the pronounced motor manifestation of the WDS after cerebral glucose supply may be indicative of the reduction in seizures severity, considering that WDS and epileptic seizures may possibly be propagated through different pathways [69, 72].
Classically, epileptic seizures worsen throughout SE [11], starting with milder seizures, such as chewing behavior and head nodding, that intensify for forelimb myoclonus, rearing, and falling [12, 13]. In order to observe the effect of hippocampal glucose control on the severity of seizures, epileptic seizures followed by glucose infusion were evaluated according to Racine’s scale [46]. We demonstrated that intrahippocampal increased glucose availability after pilocarpine was able to attenuate the number and total time of classes 3–5, indicating the decrease of the severity of the seizures.
Decreased score and duration of seizures have been associated with the anticonvulsant effect of several substances with antiepileptic potential [73, 74]. In contrast to our findings, the intraperitoneal infusion of pyruvate, a natural metabolite of glucose, is not able to alter the time or severity of seizures [75]. On the other hand, it has been previously described that the glycolytic inhibitor 2-deoxy-D-glucose (2-DG) is capable of attenuating susceptibility to seizures [76], similarly to our data. The glycolytic inhibitor 2-DG has shown an efficient seizures suppressor effect in several animal models [77, 78], reducing epileptiform burst, severity and duration of seizures, as well as increasing the latency for intrahippocampal pilocarpine-induced seizures in rats [79, 80]. Because it is a glucose analog, 2-DG enters the cell via glucose transporters and is converted to 2-DG-6-phosphatase by hexokinase [81,82,83], which impedes its metabolism by glycolysis [84]. Interestingly, 2-DG attenuates the levels of ATP that activate the non-voltage dependent potassium channel (KATP) regulated by intracellular ATP/ADP status [85, 86], as well as upregulates the protein and mRNA expression of Kir6.1 and Kir6.2 subunits of this channel [76], which together indicate an antiepileptic role [87].
Hyperglycemia and hypoglycemia have also influenced seizure susceptibility [36,37,38, 40, 88]. Although no interference was observed in seizure sensitivity of hypoglycemic B6 mice, an interesting study showed that hyperglycemia was able to modulate seizure susceptibility [36]. As previous studies [89, 90], these authors found that streptozotocin (STZ)-induced hyperglycemia (experimental diabetes—chronic or sustained hyperglycemia model) was able to accentuate the duration of seizures during KA-induced SE in B6 mice, indicating a higher susceptibility to seizures. In contrast, using an acute hyperglycemia model, they showed that the severity of seizures was reduced due to the increase in latency time to onset of severe seizures. Based on this, the glycolytic pathway has been shown to be a determinant factor for the modulation of seizure propagation, but the underlying mechanism for difference in seizures sensitivity in both models is uncertain. Thus, hippocampal glucose control may act as a potential anticonvulsant agent.
Glucose transporter activity is associated with epilepsy [13, 91]; however, the effect of glucose in additional pathways with anticonvulsant effects activated by N-methyl-d-aspartate (NMDA), 5-HT1B/1D, and GABA receptors remain unclear. Interestingly, it was showed that chronic administration of high doses of morphine can promote an anticonvulsant effect in mice with pentylenetetrazole-induced seizures [92]. In addition, it was suggested that the N-methyl-d-aspartate receptor (NMDA-R)/nitric oxide (NO) pathway is involved with the anticonvulsant role of morphine. Sumatriptan promotes reduction in the severity of seizures in lithium-PILO-induced SE model, suggesting a mechanism mediated through 5-HT1B/1D receptors and associated with anti-inflammatory properties [93]. Furthermore, protection against motor seizures can also be mediated through GABA receptors [94]. Although our data indicates that glucose modulation plays a crucial role in reducing the severity of the seizures, the intracellular mechanism of action underlying the anticonvulsant effect of glucose control remains uncertain. All these data discussed support the hypothesis that the activation of the glycolytic pathway can be multifactorial, since in our results the improvement observed in the behavior of the seizures was associated with greater local bioavailability of glucose. Thus, we believe that present study opens new perspectives for prevention and developing novel epilepsy therapies.
Typically, pilocarpine-induced SE causes hyperexcitability neuronal and neuronal death in DG hilus, CA3, and CA1 subfields of the hippocampus [12, 13, 55]. The role of glucose control in the neurodegeneration process is complex and uncertain. It is known that during the SE there is an exacerbated release of glutamate and excessive intracellular calcium that may lead also to ROS increase [95,96,97,98], promoting cell death by excitotoxicity [96, 99, 100]. In addition, limited energy availability [66, 68], impaired ATP production [101], and the release of ROS [102,103,104] may contribute to SE-induced excitotoxic cell death. Therefore, excitotoxic cell death may be sensitive to energy availability.
We report here that the increased infusion of intrahippocampal glucose before and after intrahippocampal pilocarpine decreased neuronal activity and cell loss in areas of the hippocampus. Our findings corroborate previous studies that have shown that infusion of glucose or its metabolites exert a neuroprotective effect against SE-induced neuronal death. A relevant study demonstrated that administration of exogenous glucose (20%, ip) for 3 consecutive days following KA-induced SE significantly reduced cell loss in hilus DG, and CA3 and CA1 subfields [36]. Similarly, another study showed that pyruvate (500 mg/kg, ip) was able to attenuate neuronal death caused by KA-induced SE in the same regions of the hippocampus [75]. These authors have interestingly correlated the lower neuronal loss promoted by pyruvate to the reduced accumulation of zinc in hippocampal neuronal cell bodies, suggesting a putative mechanism of action of the glycolytic pathway that justifies the potential neuroprotective effect. Many studies indicate zinc as an endogenous mediator of KA-induced neuronal death [105,106,107], which has been shown to occur through oxidative necrosis [108, 109] associated with mitochondrial damage and energy failure [110, 111], as well as NADPH oxidase induction and poly (ADP-ribose) polymerase (PARP) activation [112, 113]. These findings support the idea that controlled infusion of glucose or its metabolites may play a neuroprotective role, reducing neuronal activity and, consequently, the degeneration process caused by SE.
SE-induced seizures preferentially affect one or more extrahippocampal areas, including thalamus, amygdala, substance nigra, and neocortical regions, increasing glucose utilization, excitability, and neuronal death [18,19,20,21, 52, 114]. Thalamus is a strategically located region that communicates through afferent and efferent pathways with motor areas of the cerebral cortex, amygdaloid nuclei, and motor-related subcortical structures, such as the basal ganglia [115, 116]. Since the basal ganglia-thalamo-cortical loops are responsible for controlling voluntary movement, involving events such as muscular contraction, motor planning, and execution [12, 117, 118], its dysfunction is associated with the development of generalized motor seizures [70], neuronal damage, and cognitive deficit [18, 21, 119]. We demonstrated that intrahippocampal glucose supply was able to reduce neuronal activity and death following intrahippocampal pilocarpine-induced SE in the amygdaloid and thalamic nuclei, subiculum, substantia nigra, and cortical areas. Similar to our data, as in the hippocampus, pyruvate protected the cortex and thalamus from neuronal death, which was correlated with decreased zinc in these regions [75]. Since there is mitochondrial dysfunction, tricarboxylic acid cycle (TCA) damage, and oxidative stress in hippocampal formation and other brain areas, such as the cortex and thalamus [31, 34, 35], cerebral glucose control may be a putative therapeutic approach to protect against neuronal damage caused by SE.
Hippocampal and extrahippocampal regions are responsible for memory consolidation and learning processes. Pilocarpine-induced SE can cause damage to the hippocampus and several adjacent brain areas, triggering neuronal death and, consequently, learning deficits and memory dysfunction [26,27,28, 119,120,121,122]. In order to evaluate the effect of hippocampal glucose modulation on memory consolidation, cognitive deficit followed by intrahippocampal pilocarpine-induced SE was analyzed by the inhibitory avoidance test. Our findings showed that glucose supply after intrahippocampal pilocarpine was not able to prevent memory dysfunction. According to our findings, it has been established that STZ-induced diabetic hyperglycemia worsens the memory and learning performances followed by pilocarpine-induced SE [89]. These authors demonstrated that chronic and progressive glycemic exacerbation plays an excitotoxic role that leads to neuronal death, which justifies cognitive damage. Although our data showed that controlled glucose increase in hippocampal region can protect from neuronal loss, the deficit in memory consolidation and learning processes remained unchanged.
Additionally, pilocarpine-induced SE is able to raise ROS generation by mitochondria, which are their major target [22,23,24,25]. Mitochondrial dysfunction includes deficits in mitochondrial oxygen consumption ratios to form ATP [123], decrease of respiratory chain complex I activity from 20 h of SE [31, 34, 124], mitochondrial ultrastructural damage, and reduction of cytochrome oxidase III (complex IV of respiratory chain) mRNA and protein levels in chronic epileptic rat tissue [125]. Although attenuated neurodegeneration, our results showed that increased glucose availability did not interfere with oxidative stress followed 24 h after SE, both in MDA (product of lipid peroxidation) levels and in CAT and SOD oxidant enzyme activity. The effect of hippocampal glucose modulation on oxidative stress in pilocarpine-induced SE is poorly described, but a study indicated that the use of a synthetic antioxidant prevented oxidative stress, deficits in mitochondrial oxygen consumption rates, hippocampal neuronal death, and cognitive damage [123]. Therefore, the neuroprotective effect of hippocampal glucose control is not associated with oxidative stress.
We observed an additional benefit to the 3 mM glucose infusion after pilocarpine administration. Both G+P 3 mM and P+G 3 mM promoted similar effects to reduce neurodegeneration process in hilus, CA3 and CA1 of hippocampus, M1, ECT, thalamus, amygdala, subiculum, and substantia nigra regions. P+G 3 mM promoted additional benefits as increase WDS; reduces the number, time, and severity of seizures; and reduced the neurodegeneration process in S1, AIP, PRH, and PIR areas. Therefore, our findings suggest that these additional benefits of glucose administration after intrahippocampal pilocarpine are related with the higher glucose availability when the neuronal excitability increases during epileptic seizures. In contrast, when glucose is infused 30 min before intrahippocampal pilocarpine administration may occur an intracellular glucose flux to neurons and dispersion, which reduces the local supply of glucose at the onset of seizures. Thus, the bioavailability of hippocampal glucose can influence the different results observed.
During SE, a metabolic deficit, it has been established that SGLTs play a crucial compensatory role in glucose uptake by hippocampal neurons [30, 43, 44]. Previously, we demonstrated that nonspecific inhibition of SGLTs with phlorizin was able to enhance severity seizures and neuronal death, indicating the importance of their expression during SE [13]. Here, we also showed for the first time that intrahippocampal infusion of glucose possibly via glucose sensors T1R2/T1R3 [126] increased SGLT1 expression in hippocampal subfields that have been associated with neuroprotection process similar to presented in our results (Online Resource 4).
Conclusion
In summary, we showed that hippocampal glucose availability increases the amount of WDS, as an anticonvulsant attempt, and reduces the severity of seizures during intrahippocampal pilocarpine-induced SE. In addition, the ectopic intrahippocampal supply of glucose attenuates neuronal activity and the process of neurodegeneration in hippocampal and extrahippocampal regions, without preventing memory deficit and oxidative stress. Finally, we observed that these findings can be sustained by increased SGLT1 expression. Although hypo- and hyperglycemia have been reported as the reason for increased seizure susceptibility and neuronal damage following SE, our results support the hypothesis that local glucose control exerts a neuroprotective profile via SGLT1 translocation, during the acute phase of epileptogenesis, indicating a putative therapeutic strategy of epilepsy.
References
Sloviter RS (1999) Status epilepticus-induced neuronal injury and network reorganization. Epilepsia 40:34–39. https://doi.org/10.1111/j.1528-1157.1999.tb00876.x
Sánchez S, Rincon F (2016) Status epilepticus: epidemiology and public health needs. J Clin Med 5. https://doi.org/10.3390/jcm5080071
Lowenstein DH, Bleck T, Macdonald RL (1999) It’s time to revise the definition of status epilepticus. Epilepsia 40:120–122
Santos VR, Melo IS, Pacheco ALD, Castro OW (2019) Life and death in the hippocampus : what’ s bad ? Epilepsy Behav 106595:106595. https://doi.org/10.1016/j.yebeh.2019.106595
Upadhya D, Castro OW, Upadhya R, Shetty AK (2018) Prospects of cannabidiol for easing status epilepticus-induced epileptogenesis and related comorbidities. Mol Neurobiol 55:6956–6964. https://doi.org/10.1007/s12035-018-0898-y
Castro OW, Upadhya D, Kodali M, Shetty AK (2017) Resveratrol for easing status epilepticus induced brain injury, inflammation, epileptogenesis, and cognitive and memory dysfunction—are we there yet? Front Neurol 8:603. https://doi.org/10.3389/fneur.2017.00603
De Furtado MA, Braga GK, Oliveira JAC et al (2002) Behavioral, morphologic, and electroencephalographic evaluation of seizures induced by intrahippocampal microinjection of pilocarpine. Epilepsia 43:37–39. https://doi.org/10.1046/j.1528-1157.2002.043s2037.x
Kälviäinen R, Reinikainen M (2019) Management of prolonged epileptic seizures and status epilepticus in palliative care patients. Epilepsy Behav 101:106288. https://doi.org/10.1016/j.yebeh.2019.04.041
Ho C-J, Lin C-H, Lu Y-T, Shih FY, Hsu CW, Tsai WC, Tsai MH (2019) Perampanel treatment for refractory status epilepticus in a neurological intensive care unit. Neurocrit Care 31:24–29. https://doi.org/10.1007/s12028-019-00704-9
Trinka E, Kälviäinen R (2017) 25 years of advances in the definition, classification and treatment of status epilepticus. Seizure 44:65–73. https://doi.org/10.1016/j.seizure.2016.11.001
Furtado MA, Castro OW, Del Vecchio F et al (2011) Study of spontaneous recurrent seizures and morphological alterations after status epilepticus induced by intrahippocampal injection of pilocarpine. Epilepsy Behav 20:257–266. https://doi.org/10.1016/j.yebeh.2010.11.024
Castro OW, Furtado MA, Tilelli CQ et al (2011) Comparative neuroanatomical and temporal characterization of FluoroJade-positive neurodegeneration after status epilepticus induced by systemic and intrahippocampal pilocarpine in Wistar rats. Brain Res 1374:43–55. https://doi.org/10.1016/j.brainres.2010.12.012
Melo IS, Santos YMO, Costa MA, Pacheco ALD, Silva NKGT, Cardoso-Sousa L, Pereira UP, Goulart LR et al (2016) Inhibition of sodium glucose cotransporters following status epilepticus induced by intrahippocampal pilocarpine affects neurodegeneration process in hippocampus. Epilepsy Behav 61:258–268. https://doi.org/10.1016/j.yebeh.2016.05.026
Rodrigues MCA, Rossetti F, Foresti ML, Arisi GM, Furtado MA, Dal-Cól MLC, Bertti P, Fernandes A et al (2005) Correlation between shaking behaviors and seizure severity in five animal models of convulsive seizures. Epilepsy Behav 6:328–336. https://doi.org/10.1016/j.yebeh.2005.02.005
Lai M-C, Lin K-M, Yeh P-S, Wu SN, Huang CW (2018) The novel effect of immunomodulator-glatiramer acetate on epileptogenesis and epileptic seizures. Cell Physiol Biochem 50:150–168. https://doi.org/10.1159/000493965
Wu Q, Wang H (2018) The spatiotemporal expression changes of CB2R in the hippocampus of rats following pilocarpine-induced status epilepticus. Epilepsy Res 148:8–16. https://doi.org/10.1016/j.eplepsyres.2018.10.002
Oliveira TL, Candeia-Medeiros N, Cavalcante-Araújo PM, Melo IS, Fávaro-Pípi E, Fátima LA, Rocha AA, Goulart LR et al (2016) SGLT1 activity in lung alveolar cells of diabetic rats modulates airway surface liquid glucose concentration and bacterial proliferation. Sci Rep 6:21752. https://doi.org/10.1038/srep21752
Jung K-H, Chu K, Lee S-T, Kim JH, Kang KM, Song EC, Kim SJ, Park HK et al (2009) Region-specific plasticity in the epileptic rat brain: a hippocampal and extrahippocampal analysis. Epilepsia 50:537–549. https://doi.org/10.1111/j.1528-1167.2008.01718.x
Zenki KC, Kalinine E, Zimmer ER, dos Santos TG, Mussulini BHM, Portela LVC, de Oliveira DL (2018) Memantine decreases neuronal degeneration in young rats submitted to LiCl-pilocarpine-induced status epilepticus. Neurotoxicology 66:45–52. https://doi.org/10.1016/j.neuro.2018.03.005
Scholl EA, Dudek FE, Ekstrand JJ (2013) Neuronal degeneration is observed in multiple regions outside the hippocampus after lithium pilocarpine-induced status epilepticus in the immature rat. Neuroscience 252:45–59. https://doi.org/10.1016/j.neuroscience.2013.07.045
Loss CM, Córdova SD, de Oliveira DL (2012) Ketamine reduces neuronal degeneration and anxiety levels when administered during early life-induced status epilepticus in rats. Brain Res 1474:110–117. https://doi.org/10.1016/j.brainres.2012.07.046
Santos LFL, Freitas RLM, Xavier SML, Saldanha GB, Freitas RM (2008) Neuroprotective actions of vitamin C related to decreased lipid peroxidation and increased catalase activity in adult rats after pilocarpine-induced seizures. Pharmacol Biochem Behav 89:1–5. https://doi.org/10.1016/j.pbb.2007.10.007
dos Santos PS, Costa JP, Tomé ADR et al (2011) Oxidative stress in rat striatum after pilocarpine-induced seizures is diminished by alpha-tocopherol. Eur J Pharmacol 668:65–71. https://doi.org/10.1016/j.ejphar.2011.06.035
Shakeel S, Rehman MU, Tabassum N et al (2017) Effect of naringenin (A naturally occurring flavanone) against pilocarpine-induced status epilepticus and oxidative stress in mice. Pharmacogn Mag 13:S154–S160. https://doi.org/10.4103/0973-1296.203977
Xue Y, Xie N, Cao L, Zhao X, Jiang H, Chi Z (2011) Diazoxide preconditioning against seizure-induced oxidative injury is via the PI3K/Akt pathway in epileptic rat. Neurosci Lett 495:130–134. https://doi.org/10.1016/j.neulet.2011.03.054
Shetty AK (2014) Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection? Epilepsy Behav 38:117–124. https://doi.org/10.1016/j.yebeh.2013.12.001
Khalil A, Kovac S, Morris G, Walker MC (2017) Carvacrol after status epilepticus (SE) prevents recurrent SE, early seizures, cell death, and cognitive decline. Epilepsia 58:263–273. https://doi.org/10.1111/epi.13645
Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, Prockop DJ, Shetty AK (2017) Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc Natl Acad Sci 114:E3536–E3545. https://doi.org/10.1073/pnas.1703920114
Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27:1766–1791. https://doi.org/10.1038/sj.jcbfm.9600521
Poppe R, Karbach U, Gambaryan S, Wiesinger H, Lutzenburg M, Kraemer M, Witte OW, Koepsell H (1997) Expression of the Na+-D-glucose cotransporter SGLT1 in neurons. J Neurochem 69:84–94
McDonald TS, Carrasco-Pozo C, Hodson MP, Borges K (2017) Alterations in cytosolic and mitochondrial [U- 13 C]-glucose metabolism in a chronic epilepsy mouse model. Eneuro 4:ENEURO.0341-16.2017. https://doi.org/10.1523/ENEURO.0341-16.2017
Vielhaber S, Von Oertzen JH, Kudin AF et al (2003) Correlation of hippocampal glucose oxidation capacity and interictal FDG-PET in temporal lobe epilepsy. Epilepsia 44:193–199
Nehlig A, Rudolf G, Leroy C, Rigoulot MA, Simpson IA, Vannucci SJ (2006) Pentylenetetrazol-induced status epilepticus up-regulates the expression of glucose transporter mRNAs but not proteins in the immature rat brain. Brain Res 1082:32–42. https://doi.org/10.1016/j.brainres.2006.01.078
Folbergrová J, Ješina P, Kubová H, Druga R, Otáhal J (2016) Status epilepticus in immature rats is associated with oxidative stress and mitochondrial dysfunction. Front Cell Neurosci 10:136. https://doi.org/10.3389/fncel.2016.00136
Smeland OB, Hadera MG, McDonald TS et al (2013) Brain mitochondrial metabolic dysfunction and glutamate level reduction in the pilocarpine model of temporal lobe epilepsy in mice. J Cereb Blood Flow Metab 33:1090–1097. https://doi.org/10.1038/jcbfm.2013.54
Schauwecker PE (2012) The effects of glycemic control on seizures and seizure-induced excitotoxic cell death. BMC Neurosci 13:94. https://doi.org/10.1186/1471-2202-13-94
Xia L, Lei Z, Shi Z, Guo D, Su H, Ruan Y, Xu ZC (2016) Enhanced autophagy signaling in diabetic rats with ischemia-induced seizures. Brain Res 1643:18–26. https://doi.org/10.1016/j.brainres.2016.04.054
Chou I-C, Wang C-H, Lin W-D, Tsai FJ, Lin CC, Kao CH (2016) Risk of epilepsy in type 1 diabetes mellitus: a population-based cohort study. Diabetologia 59:1196–1203. https://doi.org/10.1007/s00125-016-3929-0
Maheandiran M, Mylvaganam S, Wu C, el-Hayek Y, Sugumar S, Hazrati L, Campo M, Giacca A et al (2013) Severe hypoglycemia in a juvenile diabetic rat model: presence and severity of seizures are associated with mortality. PLoS One 8:e83168. https://doi.org/10.1371/journal.pone.0083168
Moseley B, Bateman L, Millichap JJ, Wirrell E, Panayiotopoulos CP (2013) Autonomic epileptic seizures, autonomic effects of seizures, and SUDEP. Epilepsy Behav 26:375–385. https://doi.org/10.1016/j.yebeh.2012.08.020
Sabino-Silva R, Mori RC, David-Silva A et al (2010) The Na +-/glucose cotransporters: from genes to therapy. Braz J Med Biol Res 43:1019–1026. https://doi.org/10.1590/S0100-879X2010007500115
Wright EM, Loo DDF, Hirayama BA (2011) Biology of human sodium glucose transporters. Physiol Rev 91:733–794. https://doi.org/10.1152/physrev.00055.2009
Yu AS, Hirayama BA, Timbol G et al (2010) Functional expression of SGLTs in rat brain. Am J Phys Cell Phys 299:C1277–C1284. https://doi.org/10.1152/ajpcell.00296.2010
Yu AS, Hirayama BA, Timbol G et al (2013) Regional distribution of SGLT activity in rat brain in vivo. Am J Phys Cell Phys 304:C240–C247. https://doi.org/10.1152/ajpcell.00317.2012
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates, 6th Editio. Academic Press, San Diego
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77. https://doi.org/10.1016/0003-9861(59)90090-6
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–310. https://doi.org/10.1016/S0076-6879(78)52032-6
Misra HP, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247:3170–3175
Aebi H (1984) [13] catalase in vitro. Methods Enzymol 105:121–126. https://doi.org/10.1016/S0076-6879(84)05016-3
Schmued LC, Albertson C, Slikker W (1997) Fluoro-jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res 751:37–46. https://doi.org/10.1016/S0006-8993(96)01387-X
Sanabria ERG, da Silva AV, Spreafico R, Cavalheiro EA (2002) Damage, reorganization, and abnormal neocortical hyperexcitability in the pilocarpine model of temporal lobe epilepsy. Epilepsia 43(Suppl 5):96–106
Tejada J, Garcia-Cairasco N, Roque AC (2014) Combined role of seizure-induced dendritic morphology alterations and spine loss in newborn granule cells with mossy fiber sprouting on the hyperexcitability of a computer model of the dentate gyrus. PLoS Comput Biol 10:e1003601. https://doi.org/10.1371/journal.pcbi.1003601
Yamada A, Momosaki S, Hosoi R, Abe K, Yamaguchi M, Inoue O (2009) Glucose utilization in the brain during acute seizure is a useful biomarker for the evaluation of anticonvulsants: effect of methyl ethyl ketone in lithium-pilocarpine status epilepticus rats. Nucl Med Biol 36:949–954. https://doi.org/10.1016/j.nucmedbio.2009.06.008
Cameron S, Lopez A, Glabman R, Abrams E, Johnson S, Field C, Gulland FMD, Buckmaster PS (2019) Proportional loss of parvalbumin-immunoreactive synaptic boutons and granule cells from the hippocampus of sea lions with temporal lobe epilepsy. J Comp Neurol. https://doi.org/10.1002/cne.24680
Mohapel P, Ekdahl CT, Lindvall O (2004) Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol Dis 15:196–205. https://doi.org/10.1016/j.nbd.2003.11.010
Meldrum BS, Brierley JB (1973) Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 28:10–17
Meldrum BS, Horton RW (1973) Physiology of status epilepticus in primates. Arch Neurol 28:1–9
Franck G, Sadzot B, Salmon E, Depresseux JC, Grisar T, Peters JM, Guillaume M, Quaglia L et al (1986) Regional cerebral blood flow and metabolic rates in human focal epilepsy and status epilepticus. Adv Neurol 44:935–948
Farooque P, Hirsch L, Levy S, Testa F, Mattson R, Spencer D (2017) Surgical outcome in adolescents with mesial temporal sclerosis: is it different? Epilepsy Behav 69:24–27. https://doi.org/10.1016/j.yebeh.2016.10.028
Fernández-Torre JL, Pascual J, Quirce R, Gutiérrez A, Martínez-Martínez M, Rebollo M (2006) Permanent dysphasia after status epilepticus: long-term follow-up in an elderly patient. Epilepsy Behav 8:677–680. https://doi.org/10.1016/j.yebeh.2006.01.014
Kim HY, Kim JY, Un KG et al (2012) Alien hand syndrome after epilepsia partialis continua: FDG PET and MRI studies. Epilepsy Behav 23:71–73. https://doi.org/10.1016/j.yebeh.2011.08.043
Sakakibara E, Takahashi Y, Murata Y et al (2014) Chronic periodic lateralised epileptic discharges and anti-N-methyl-D-aspartate receptor antibodies. 16:218–222. https://doi.org/10.1684/epd.2014.0655
Van Bogaert P, Goldman S, Rodesch G et al (1994) Cerebral lesions following convulsive partial status epilepticus. Clinical, neuroradiologic and PET study of a case. J Neuroradiol 21:176–180
García-García L, Shiha AA, Fernández de la Rosa R, Delgado M, Silván Á, Bascuñana P, Bankstahl JP, Gomez F et al (2017) Metyrapone prevents brain damage induced by status epilepticus in the rat lithium-pilocarpine model. Neuropharmacology 123:261–273. https://doi.org/10.1016/j.neuropharm.2017.05.007
Sapolsky RM, Stein BA (1989) Status epilepticus-induced hippocampal damage is modulated by glucose availability. Neurosci Lett 97:157–162
Meldrum BS (1983) Metabolic factors during prolonged seizures and their relation to nerve cell death. Adv Neurol 34:261–275
Johansen FF, Diemer NH (1986) Influence of the plasma glucose level on brain damage after systemic kainic acid injection in the rat. Acta Neuropathol 71:46–54
Grimes L, McGinty J, McLain P, Mitchell C, Tilson H, Hong J (1988) Dentate granule cells are essential for kainic acid-induced wet dog shakes but not for seizures. J Neurosci 8:256–264
Lothman EW, Collins RC (1981) Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res 218:299–318. https://doi.org/10.1016/0006-8993(81)91308-1
Shin E-J, Jeong JH, Chung YH, Kim TW, Shin CY, Kim WK, Ko KH, Kim HC (2009) Decrease in the kainate-induced wet dog shake behavior in genetically epilepsy-prone rats: possible involvement of an impaired synaptic transmission to the 5-HT(2A) receptor. J Pharmacol Sci 110:401–404
Lee PH, Hong JS (1990) Ventral hippocampal dentate granule cell lesions enhance motor seizures but reduce wet dog shakes induced by mu opioid receptor agonist. Neuroscience 35:71–77. https://doi.org/10.1016/0306-4522(90)90121-J
Citraro R, Scicchitano F, De Fazio S et al (2011) Preclinical activity profile of α-lactoalbumin, a whey protein rich in tryptophan, in rodent models of seizures and epilepsy. Epilepsy Res 95:60–69. https://doi.org/10.1016/j.eplepsyres.2011.02.013
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y (2004) Anticonvulsant action of hippocampal dopamine and serotonin is independently mediated by D2 and 5-HT1A receptors. J Neurochem 89:834–843. https://doi.org/10.1111/j.1471-4159.2004.02355.x
Kim T-Y, Yi J-S, Chung S-J, Kim DK, Byun HR, Lee JY, Koh JY (2007) Pyruvate protects against kainate-induced epileptic brain damage in rats. Exp Neurol 208:159–167. https://doi.org/10.1016/j.expneurol.2007.08.013
Yang H, Guo R, Wu J, Peng Y, Xie D, Zheng W, Huang X, Liu D et al (2013) The antiepileptic effect of the glycolytic inhibitor 2-deoxy-d-glucose is mediated by upregulation of KATP channel subunits Kir6.1 and Kir6.2. Neurochem Res 38:677–685. https://doi.org/10.1007/s11064-012-0958-z
Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP (2009) Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann Neurol 65:435–447. https://doi.org/10.1002/ana.21603
Gasior M, Yankura J, Hartman AL, French A, Rogawski MA (2010) Anticonvulsant and proconvulsant actions of 2-deoxy-d-glucose. Epilepsia 51:1385–1394. https://doi.org/10.1111/j.1528-1167.2010.02593.x
Lian X-Y, Khan FA, Stringer JL (2007) Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci 27:12007–12011. https://doi.org/10.1523/JNEUROSCI.3163-07.2007
Stafstrom CE, Roopra A, Sutula TP (2008) Seizure suppression via glycolysis inhibition with 2-deoxy-D-glucose (2DG). Epilepsia 49:97–100. https://doi.org/10.1111/j.1528-1167.2008.01848.x
Bissonnette P, Gagne H, Blais A, Berteloot A (1996) 2-Deoxyglucose transport and metabolism in Caco-2 cells. Am J Physiol Liver Physiol 270:G153–G162. https://doi.org/10.1152/ajpgi.1996.270.1.G153
Sols A, Crane RK (1954) Substrate specificity of brain hexokinase. J Biol Chem 210:581–595
Kimmich GA, Randles J (1976) 2-Deoxyglucose transport by intestinal epithelial cells isolated from the chick. J Membr Biol 27:363–379
Chen W, Guéron M (1992) The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie 74:867–873
Yamada K, Ji JJ, Yuan H et al (2001) Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science (80- ) 292:1543–1546. https://doi.org/10.1126/science.1059829
Hernandez-Sanchez C, Basile AS, Fedorova I, Arima H, Stannard B, Fernandez AM, Ito Y, LeRoith D (2001) Mice transgenically overexpressing sulfonylurea receptor 1 in forebrain resist seizure induction and excitotoxic neuron death. Proc Natl Acad Sci 98:3549–3554. https://doi.org/10.1073/pnas.051012898
Ma W, Berg J, Yellen G (2007) Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci 27:3618–3625. https://doi.org/10.1523/JNEUROSCI.0132-07.2007
Falip M, Miró J, Carreño M, Jaraba S, Becerra JL, Cayuela N, Perez Maraver M, Graus F (2014) Hypoglycemic seizures and epilepsy in type I diabetes mellitus. J Neurol Sci 346:307–309. https://doi.org/10.1016/j.jns.2014.08.024
Huang CW, Cheng JT, Tsai JJ, Wu SN, Huang CC (2009) Diabetic hyperglycemia aggravates seizures and status epilepticus-induced hippocampal damage. Neurotox Res 15:71–81. https://doi.org/10.1007/s12640-009-9008-2
Stafstrom CE (2003) Hyperglycemia lowers seizure threshold. Epilepsy Curr 3:148–149. https://doi.org/10.1046/j.1535-7597.2003.03415.x
de Melo IS, Pacheco ALD, dos Santos YMO, Figueiredo LM, Nicacio DCSP, Cardoso-Sousa L, Duzzioni M, Gitaí DLG et al (2020) Modulation of glucose availability and effects of hypo- and hyperglycemia on status epilepticus: what we do not know yet? Mol Neurobiol:1–15. https://doi.org/10.1007/s12035-020-02133-8
Zamanian G, Shayan M, Rahimi N, Bahremand T, Shafaroodi H, Ejtemaei-Mehr S, Aghaei I, Dehpour AR (2020) Interaction of morphine tolerance with pentylenetetrazole-induced seizure threshold in mice: the role of NMDA-receptor/NO pathway. Epilepsy Behav 112:112. https://doi.org/10.1016/j.yebeh.2020.107343
Eslami F, Rahimi N, Ostovaneh A et al (2020) Sumatriptan reduces severity of status epilepticus induced by lithium–pilocarpine through nitrergic transmission and 5-HT 1B/D receptors in rats: a pharmacological-based evidence. Fundam Clin Pharmacol:fcp.12590. https://doi.org/10.1111/fcp.12590
Taiwe GS, Kouamou ALN, Ambassa ARM, Menanga JR, Tchoya TB, Dzeufiet PDD (2017) Evidence for the involvement of the GABA-ergic pathway in the anticonvulsant activity of the roots bark aqueous extract of Anthocleista djalonensis A. Chev. (Loganiaceae). J Basic Clin Physiol Pharmacol 28:425–435. https://doi.org/10.1515/jbcpp-2017-0048
Vishnoi S, Raisuddin S, Parvez S (2016) Glutamate excitotoxicity and oxidative stress in epilepsy: modulatory role of melatonin. J Environ Pathol Toxicol Oncol 35:365–374. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2016016399
During MJ, Spencer DD (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet (London, England) 341:1607–1610
Ambrogini P, Torquato P, Bartolini D, Albertini MC, Lattanzi D, di Palma M, Marinelli R, Betti M et al (2019) Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: the role of vitamin E. Biochim Biophys Acta Mol basis Dis 1865:1098–1112. https://doi.org/10.1016/j.bbadis.2019.01.026
Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60:1215–1226. https://doi.org/10.1002/glia.22341
Rossi AR, Angelo MF, Villarreal A, Lukin J, Ramos AJ (2013) Gabapentin administration reduces reactive gliosis and neurodegeneration after pilocarpine-induced status epilepticus. PLoS One 8:e78516. https://doi.org/10.1371/journal.pone.0078516
Chiu KM, Wu CC, Wang MJ, Lee MY, Wang SJ (2015) Protective effects of bupivacaine against kainic acid-induced seizure and neuronal cell death in the rat hippocampus. Biol Pharm Bull 38:522–530. https://doi.org/10.1248/bpb.b14-00633
Wang LY, Dudek EM, Browning MD, MacDonald JF (1994) Modulation of AMPA/kainate receptors in cultured murine hippocampal neurones by protein kinase C. J Physiol 475:431–437. https://doi.org/10.1113/jphysiol.1994.sp020083
Mishra V, Shuai B, Kodali M, Shetty GA, Hattiangady B, Rao X, Shetty AK (2015) Resveratrol treatment after status epilepticus restrains neurodegeneration and abnormal neurogenesis with suppression of oxidative stress and inflammation. Sci Rep 5. https://doi.org/10.1038/srep17807
Dariani S, Baluchnejadmojarad T, Roghani M (2013) Thymoquinone attenuates astrogliosis, neurodegeneration, mossy fiber sprouting, and oxidative stress in a model of temporal lobe epilepsy. J Mol Neurosci 51:679–686. https://doi.org/10.1007/s12031-013-0043-3
Pestana RRF, Kinjo ER, Hernandes MS, Britto LRG (2010) Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci Lett 484:187–191. https://doi.org/10.1016/j.neulet.2010.08.049
Yi J-S, Lee S-K, Sato T-A, Koh J-Y (2003) Co-induction of p75(NTR) and the associated death executor NADE in degenerating hippocampal neurons after kainate-induced seizures in the rat. Neurosci Lett 347:126–130
Lee JY, Cole TB, Palmiter RD, Koh JY (2000) Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci 20:RC79
Lee J-Y, Kim J-H, Palmiter RD, Koh J-Y (2003) Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol 184:337–347
Kim EY, Koh JY, Kim YH, Sohn S, Joe E, Gwag BJ (1999) Zn2+ entry produces oxidative neuronal necrosis in cortical cell cultures. Eur J Neurosci 11:327–334
Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY (1999) Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience 89:175–182
Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH (2001) Zn 2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem 276:47524–47529. https://doi.org/10.1074/jbc.M108834200
Sensi SL, Yin HZ, Weiss JH (2000) AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur J Neurosci 12:3813–3818
Kim Y-H, Koh J-Y (2002) The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol 177:407–418
Noh KM, Koh JY (2000) Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 20:RC111
Sakurai M, Kurokawa H, Shimada A, Nakamura K, Miyata H, Morita T (2015) Excitatory amino acid transporter 2 downregulation correlates with thalamic neuronal death following kainic acid-induced status epilepticus in rat. Neuropathology 35:1–9. https://doi.org/10.1111/neup.12141
Young CB, Sonne J (2019) Neuroanatomy. StatPearls Publishing, Basal Ganglia
Bosch-Bouju C, Hyland BI, Parr-Brownlie LC (2013) Motor thalamus integration of cortical, cerebellar and basal ganglia information: implications for normal and parkinsonian conditions. Front Comput Neurosci 7:163. https://doi.org/10.3389/fncom.2013.00163
Iseki K, Hanakawa T (2010) The functional significance of the basal ganglia-thalamo-cortical loop in gait control in humans: a neuroimaging approach. Brain Nerve 62:1157–1164
Hooks BM (2017) Sensorimotor convergence in circuitry of the motor cortex. Neurosci 23:251–263. https://doi.org/10.1177/1073858416645088
Ma DL, Qu JQ, Goh ELK, Tang FR (2016) Reorganization of basolateral amygdala-subiculum circuitry in mouse epilepsy model. Front Neuroanat 9:167. https://doi.org/10.3389/fnana.2015.00167
Gröticke I, Hoffmann K, Löscher W (2007) Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp Neurol 207:329–349. https://doi.org/10.1016/j.expneurol.2007.06.021
Lenck-Santini P-P, Holmes GL (2008) Altered phase precession and compression of temporal sequences by place cells in epileptic rats. J Neurosci 28:5053–5062. https://doi.org/10.1523/JNEUROSCI.5024-07.2008
Peixoto-Santos JE, Velasco TR, Galvis-Alonso OY, Araujo D, Kandratavicius L, Assirati JA, Carlotti CG, Scandiuzzi RC et al (2015) Temporal lobe epilepsy patients with severe hippocampal neuron loss but normal hippocampal volume: Extracellular matrix molecules are important for the maintenance of hippocampal volume. Epilepsia 56:1562–1570. https://doi.org/10.1111/epi.13082
Pearson JN, Rowley S, Liang LP, White AM, Day BJ, Patel M (2015) Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol Dis 82:289–297. https://doi.org/10.1016/j.nbd.2015.07.005
Folbergrová J, Ješina P, Kubová H, Otáhal J (2018) Effect of resveratrol on oxidative stress and mitochondrial dysfunction in immature brain during epileptogenesis. Mol Neurobiol 55:7512–7522. https://doi.org/10.1007/s12035-018-0924-0
Gao J, Yao H, Pan XD, Xie AM, Zhang L, Song JH, Ma AJ, Liu ZC (2014) Alteration of mitochondrial function and ultrastructure in the hippocampus of pilocarpine-treated rat. Epilepsy Res 108:162–170. https://doi.org/10.1016/j.eplepsyres.2013.11.016
Ren X, Zhou L, Terwilliger R et al (2009) Sweet taste signaling functions as a hypothalamic glucose sensor. Front Integr Neurosci 3:12. https://doi.org/10.3389/neuro.07.012.2009
Acknowledgments
DLGG was supported by the Research Productivity Scholarship Program in Brazilian National Council for Scientific and Technological Development (CNPq). We thank CAPES-Brazil for PhD Research Fellowship to I.S.M., Y.M.O.S., A.L.D.P., M.A.C., and J.F.S.
Funding
This project was supported by FAPEAL, CNPq, and CAPES.
Author information
Authors and Affiliations
Contributions
Conceptualization, I.S.M., R.S.S., and O.W.C.; methodology, I.S.M., Y.M.O.S., A.L.D.P., M.A.C., V.O.S., J.F.S., C.M.B.C., R.C.S.F, A.C.R.L., R.S.S., and O.W.C.; investigation, I.S.M., A.C.R.L., A.U.B., R.S.S., and O.W.C.; formal analysis, I.S.M., R.C.S.F, A.C.R.L., R.S.S., and O.W.C.; supervision and fund acquisition, O.W.C.; writing—review and editing, I.S.M., D.G.L.G., M.D., A.U.B., R.S.S., and O.W.C.; resources, M.D., A.U.B., R.S.S., and O.W.C.
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interest.
Ethics Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Experiments were performed in accordance with the NIH guidelines for the care and use of laboratory animals, and with approval of the Federal University of Alagoas Animal Use Ethics Committee.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Online Resource 1
Effects of increased glucose availability on status of oxidative stress levels in rat hippocampus after 24 h of intrahippocampal pilocarpine-induced SE. Modulated levels of malondialdehyde (MDA), total thiol, catalase (CAT), and superoxide dismutase (SOD) were assessed (A). Glucose-(3 mM)-treated rats showed a significant increase in MDA levels (B) and a significant decrease in total thiol (C), CAT (D) and SOD (E) levels compared with saline-treated rats. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 4–6 rats. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with VEH; one-way ANOVA with Dunnett’s post-hoc test. VEH, saline-treated vehicle; H-PILO, pilocarpine and saline; P+G, pilocarpine followed by glucose infusion; DZP, diazepam; SE, Status epilepticus; SEM, standard error of the mean. (PNG 1620 kb)
Online Resource 2
Glucose modulation decreases the cFOS expression in cortical and subiculum areas after intrahippocampal pilocarpine-induced SE. The nuclei were labeled with DAPI (blue, middle panels, A-H2). Fluorescent labeling strong cFOS immunoreactivity (red, left panels) in RSGr (A1), Prh (C1), Pir (E1) and DS (G1) in H-PILO rats. Merge of cFOS and DAPI are shown in right panels (A-H3). Quantitative analysis of cFOS+ neurons in H-PILO (black bars), G+P (gray, blue and red bar) and P+G (gray, blue and red bar outline) rats are shown in B4, D4, F4 and H4. Some concentrations of hippocampal glucose (1, 2 or 3 mM) before and after intrahippocampal pilocarpine attenuated the number of cFOS+ neurons in RSGr (one-way ANOVA, F (6, 22) = 3.59, P = 0.012; B4), Prh (t-test, t5 = 3.79, P = 0.013; D4), Pir (one-way ANOVA, F (6, 16) = 3.70, P = 0.017; F4) and DS (one-way ANOVA, F (6, 20) = 6.51, P = 0.0006; H4) areas. The 3 mM glucose concentration was used as a representative image. Arrows represent the brain areas. Magnification, 200x; scale bar, 50 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 3–6 rats. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with H-PILO; one-way ANOVA with Dunnett’s post-hoc test and unpaired t-test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; RSGr, retrosplenial; PRh, perirhinal; Pir, piriform; DS, subiculum; SEM, standard error of the mean. (PNG 4306 kb)
Online Resource 3
Increased glucose supply decreases the cFOS expression in thalamic and amygdaloid areas after intrahippocampal pilocarpine-induced SE. The nuclei were labeled with DAPI (blue, middle panels). Fluorescent labeling of the hippocampus shows strong cFOS immunoreactivity (red, left panels) in LPMR (A1), CL (C1), PVP (E1) and LaDL (G1) in H-PILO rats. Merge of cFOS and DAPI shown in right panels. Quantitative analysis of cFOS+ neurons in H-PILO (black bars), G+P (gray, blue and red bar) and P+G (gray, blue and red bar outline) rats are shown in B4, D4, F4 and H4. Some concentrations of hippocampal glucose (1, 2 or 3 mM) before and after intrahippocampal pilocarpine decreased the number of cFOS+ neurons in LPMR (one-way ANOVA, F (6, 22) = 10.94, P < 0.0001; B4), CL (one-way ANOVA, F (6, 22) = 3.54, P = 0.01; D4), PVP (one-way ANOVA, F (6, 20) = 5.21, P = 0.0023; F4) and LaDL (one-way ANOVA, F (3, 9) = 9.65, P = 0.0036; H4) areas. The 3 mM glucose concentration was used as a representative image. Arrows represent the brain areas. Magnification, 200x; scale bar, 50 μm. Error bars indicate the SEM. Data represent the mean ± S.E.M. of 5–6 rats. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with H-PILO; one-way ANOVA with Dunnett’s post-hoc test. H-PILO, pilocarpine and saline; G+P, glucose followed by pilocarpine infusion; P+G, pilocarpine followed by glucose infusion; PVP, posterior paraventricular th ncl; CL, centrolateral th ncl; LPMR, lateral posterior th ncl; LaDL, lateral amygdaloid ncl; SEM, standard error of the mean. (PNG 4591 kb)
Online Resource 4
Glucose supply reduces the seizures severity but does not change memory dysfunction and oxidative stress. Cell death and neuronal activity are attenuated in the hippocampus and extrahippocampal areas because of this glucose control, via elevation of sodium-glucose cotransporter translocation in hippocampus. (PNG 3670 kb)
Rights and permissions
About this article

Cite this article
de Melo, I.S., dos Santos, Y.M.O., Pacheco, A.L.D. et al. Role of Modulation of Hippocampal Glucose Following Pilocarpine-Induced Status Epilepticus. Mol Neurobiol 58, 1217–1236 (2021). https://doi.org/10.1007/s12035-020-02173-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02173-0