
Abstract
Cadmium (Cd), a ubiquitous environmental pollutant, has neurotoxicity to humans and animals. Quercetin (QE), the main component of flavonoids, has strong antioxidant and anti-inflammatory effects. However, little is reported about the influence of Cd exposure on necroptosis in the chicken brain and the antagonistic impacts of QE against Cd-induced brain necroptosis. The aim of this study was to ascertain the alleviative mechanism of QE on Cd-induced necroptosis in the chicken brain. Two hundred 3.5-month-old Isa hens were randomly divided into four groups, control group, QE group, Cd group, and Cd + QE co-administration group. The histopathological analysis indicated that necrosis features were observed in the Cd-intoxicated chicken brains. Meanwhile, the expression levels of RIPK1, RIPK3, and MLKL were elevated and the level of Caspase 8 was reduced in the Cd group, which further testified Cd triggered the occurrence of necroptosis in the chicken brain. Cd exposure obviously increased Cd accumulation, ROS generation, and MDA level; weakened the activities of antioxidase (SOD, GPx, and CAT); enhanced iNOS activity and NO production; promoted the expression of inflammatory factors (NF-κB, TNFα, COX-2, iNOS, PTGEs, and IL-1β); and activated HSPs (HSP27, HSP40, HSP60, HSP70, and HSP90). But, these Cd-caused variations were obviously attenuated in the Cd + QE group. This study indicated that QE had an alleviative effect on Cd-induced necroptosis in the chicken brain through inhibition ROS/iNOS/NF-κB pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd), as a poisonous heavy metal, is a universal environmental contaminant. Cd exposure poses severe health risks to animals and humans through the food chain associated with Cd pollutant. Cd intoxication associates with serious poisoning effects of the heart, the kidney, the liver, the lung, and the immune organs [1, 2]. It has been reported that Cd has the capability to cross the blood-brain barrier and consequently accumulate in the brain tissue leading to neurotoxicity [3], such as Parkinson’s symptoms, Alzheimer’s disease, schizophrenia, anxiety, depression, learning disabilities, and attention deficit disorder [4, 5]. In addition, early exposure to Cd can damage neurogenesis to result in failure of neuronal differentiation and axon formation and eventually lead to neuron death [6, 7]. Even though the mechanism of Cd-induced neurotoxicity is not fully understood, growing researches indicated that excessive reactive oxygen species (ROS) triggered by Cd is considered to be a key pathogenic factor [8]. This is well known that excessive production of ROS can result in oxidative stress, which further damage tissues and cells via changing lipid peroxidation and the structure and function of protein or nucleic acid. In recent years, a variety of medicinal plant extracts and natural products have been reported as therapeutic agents to reduce Cd-induced neurotoxicity. Protocatechuic acid can counteract neurotoxicity caused by Cd through lessening oxidative stress and restraining inflammation and apoptosis [9]. Elkhadragy et al. [10] discovered that the methanolic extract of Fragaria ananassa prevents the brain tissue against Cd-induced neuronal toxicity via enhancing the antioxidant system and boosting antiapoptotic and anti-inflammatory activities of the rat brain.
Necroptosis is a special cell death form differed from apoptosis and necrosis. Disequilibrium between the oxidative system and the oxidation defense system is related to triggering necroptosis [11]. Necroptosis is regulated by a particular molecular pathway, which involves the proteins receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like (MLKL) [12]. In addition, aspartate-specific cysteine protease (Caspase 8) is certified as an extremely important signal molecule for blocking necroptosis via separating RIPK1 and RIPK3 [13]. Previous researches showed that cell death mediated by RIPK1/RIPK3 pathway probably relies on the induction of ROS, which can promote necrosome formation [14, 15]. Han et al. [16] found that oxidative stress led to necroptosis in the rat lung after hyperoxia exposure. Lots of ROS can also induce an inflammatory response and promote the secretion of a variety of inflammatory cytokines, for instance, nuclear factor-kappa B (NF-κB), inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2), interleukin 1-beta (IL-1β), and prostaglandin E synthase (PTGEs), which in turn further maintain the induction between oxidative stress and inflammation [17, 18]. The NF-κB pathway exerts a key role in the inflammatory reaction via regulating the expression of inflammatory cytokines [19]. TNF-α is an important downstream cytokine in NF-κB inflammatory pathway, and NF-κB activation can promote the expression and release of TNF-α. It was reported that TNF-α also has been considered to be a vital initiator of necroptosis [20]. TNF-α mediated the occurrence of necroptosis in bronchial epithelial cells [21]. The high expression of NF-κB and TNF-α resulted from lead exposure participated in inducing the necroptosis in the chicken spleen [22]. iNOS, as another downstream inflammatory cytokine of NF-κB, can synthesize nitric oxide (NO) and excessive NO helps to trigger the infiltration of inflammatory cells which release numerous proinflammatory mediators [23]. A high level of NO also can participate in multiple pathological processes and evoke various inflammatory diseases [24]. Cd can incur oxidative stress and enhance the expression of inflammation-related genes including iNOS, TNF-α, NF-κB, COX-2, and PTGEs in the breast muscles of the chicken [25]. Guo et al. [26] indicated that Cd exposure increased iNOS activities, induced cardiac inflammation, and upregulated inflammation factor expression in the cardiac tissue. Cd addition is reported to motivate ROS overproduction triggering oxidative stress and ultimately cause necroptosis in chicken peripheral blood lymphocytes [27]. As a highly conserved protein family, heat shock proteins (HSPs), could respond to environmental stress [28]. Wang et al. [29] found that the expression levels of HSP60, HSP70, and HSP90 were dramatically enhanced during selenium deficiency-mediated necroptosis in tracheal epithelial cells, which showed that HSPs participated in the emergence and developing process of necroptosis. Zhang et al. [30] also indicated that HSP27, HSP40, HSP60, HSP70, and HSP90 mRNA and protein levels were drastically enhanced in chicken lymphocytes undergoing lead-induced necroptosis.
Quercetin (QE), one of natural flavonoid compounds, is widely distributed in vegetables and fruits. It was reported that QE possesses antioxidative, anti-inflammatory, antimicrobial, antiviral, antitumor, antihypertensive, and hypolipidemic properties [31]. QE can play protective roles against many exogenetic toxicity substances, such as alcohol, hydrogen peroxide, bisphenol A, acrylamide, and heavy metal. Yang et al. [32] indicated that QE administration lessened oxidative stress and inflammatory reaction, thereby ameliorating renal function. QE exerts its antioxidant potential to relieve neurotoxicity triggered by Cd via declining ROS production and regulating mitochondrial integrity and mitogen-activated protein kinase (MAPK) signaling [33]. Nna et al. [34] reported that QE antagonized Cd-triggered oxidative stress in the uterus and ovary of a female rat. QE also was demonstrated to obviously alleviate oligodendrocytes necroptosis after spinal cord injury in rats [35]. Although many studies have researched the attenuation effect of QE against hazardous substances, but whether QE can protect the chicken brain against the necroptosis induced by Cd and the alleviative mechanism of QE on Cd-caused necroptosis in the brain are unknown. In the current study, we administrated to add Cd or/and QE in the diet of chicken for 60 days, then detected Cd concentration, histopathological changes, ROS production, and the status of oxidative stress, NO level, and iNOS activity. We also evaluated the expression levels of necroptosis markers, inflammatory cytokines, and HSPs in the chicken brain. The objective of this study was to elucidate the mechanism about the alleviative effect of QE on Cd-induced necroptosis in the chicken brain.
Material and Methods
Animals and Experimental Design
All procedures used in our experiment were approved by the Institutional Animal Care and Use Committee of Northeast Agricultural University. Two hundred 3.5-month-old Isa hens were randomly divided into four groups (n = 50 per group). Group I (control) was fed a basal diet, group II (QE-administrated group) was fed with the basal diet added 500 mg/kg QE, group III (Cd-exposed group) was fed the basal diet added with 150 mg/kg CdCl2, and group IV (Cd/QE co-treated group) was fed with the basal diet added with 150 mg/kg CdCl2 + 500 mg/kg QE. All animals lived in a normal diet condition. On the 60th day of this experiment, the chickens were euthanized. The brain tissues were separated and partially fixated in 10% formalin. The remaining was stored at − 80 °C for other analysis.
Estimation of Cd Concentrations in the Brain
The Cd content in the brains of chicken was examined by using the inductively coupled plasma mass spectrometry (ICP-MS, iCAPQ, Thermo, USA). The standard parameters of an instrument in our experiment are shown in Table 1. Each sample (1 g) was dissolved in 5 ml nitric acid (65%) and 2 ml hydrogen peroxide (30%) and then diluted to 10 ml final volume with deionized water. All sample solutions were limpid. The samples were dissolved in a microwave system according to the following conditions: 1 800 W and 100 °C for 3 min, 1 800 W and 150 °C for 10 min, and 1 800 W and 180 °C for 45 min. The same method was used for blank digestion. Before ICP-MS determination, all digested samples were diluted to a final volume of 50 ml with ultrapure water and fully mixed.
Histological Observation of Brain Tissue
Some brain tissues were fixed for at least 24 h with 10% neutral formalin. Then, the brain tissues were dehydrated with an increasing concentration of alcohol following embedded by paraffin. Sections about 5-μm thickness were obtained, stained with hematoxylin and eosin (H&E), and then observed using a light microscope (XDS-1B, Olympus, Japan).
Determination of Oxidative Stress Indexes
A ROS assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) was used to measure the production of ROS in the chicken brain. A single-cell suspension of the chicken brain was prepared by enzymatic digestion. The isolated cells were suspended with PBS and inoculated in the black 96-well plate. ROS were determined by the conversion of nonfluorescent dye chloromethyl-2,7-dichloro fluorescindiacetate (DCF-DA). DCF-DA was added into the cells and incubated for 30 min. Then, the fluorescence was quantified as RFU at 485 nm excitation wavelength and 525 nm emission wavelength by using a Cytation 5 fluorescence spectrophotometer.
The homogenates of the chicken brain tissue were prepared in ice bath circumstances. The content of malondialdehyde (MDA), the activities of superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT) were measured in the light of the instructions of the determination kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Assay of NO Production and iNOS Activity
NO production and iNOS activity were detected by using NO and iNOS determination kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Real-time Quantitative PCR Analysis of Genes on mRNA Levels
The total RNA from the chicken brain tissues was extracted with Trizol reagent, and then, cDNA was synthesized by reverse transcription reaction on the basis of the manufacturer’s instructions (Roche, Shanghai, China). The sequences of all the primers used in this analysis were shown in Table 2. qRT-PCR was carried using the Light Cycler® 480 System (Roche, Basel, Switzerland) and Fast Universal SYBR Green Master Mix (Roche, Basel, Switzerland) to assess the mRNA expression levels of genes. The internal reference gene is β-actin. The mRNA relative abundance of the target genes was calculated using the 2-ΔΔCT method.
Western Blot Analysis
Western blot was used to measure the protein expression. Total proteins were extracted from the chicken brains, and under reducing conditions were separated using SDS-polyacrylamide gel electrophoresis on 12% gel. Then, we transferred the protein bands to the nitrocellulose membrane by using a pot transfer (200 mA) fulfilling Tris-glycine buffer containing 20% methanol at 4 °C for 2 h. Afterwards, the membranes blocking were performed with 5% skim milk at 37 °C for 2 h, followed by incubating at 4 °C with diluted primary antibodies including NF-κB (1:500), COX-2 (1:500), TNF-α (1:500), iNOS (1:1000), MLKL (1:1000), RIPK1 (1:1500), RIPK3 (1:1500), HSP70 (1:500), HSP90 (1:500), and β-actin (1:1000) for a night. Next day, we incubated the membrane at room temperature using peroxidase-conjugated secondary antibodies against rabbit IgG (1:5000, Santa Cruz, USA) for 1 h. The protein signal was measured using X-ray films (TransGen Biotech Co., Beijing, China) by using an ECL kit (Kangweishiji Biotechnology, Beijing, China). In our experiment, β-actin was used as the internal reference protein, the ratios of the optical density of each protein to that of β-actin denoted the relative expression levels of above target proteins.
Statistical Analysis
All datas in our study were expressed as the mean ± standard deviation. Statistical analysis of obtained datas was performed utilizing SPSS for Windows (version21; SPSS Inc., Chicago, IL, USA). One-way ANOVA with Tukey’s correction was used where appropriate. Samples with different superscripted letters represent statistically significant differences (P < 0.05).
Results
Cd Concentrations
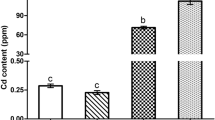
To evaluate the effect of QE on Cd accumulation in the brain tissue of chickens, we detected the Cd content of the brains. The results showed that after adding Cd or/and QE to the diet for 60 days, the content of Cd in the brain of each group was significantly different (Fig. 1). Compared with the control and QE group, Cd content in the Cd group rose markedly (P < 0.05). But, the change was alleviated by the co-treatment of QE and Cd. Cd content in the Cd/QE combined treatment group was evidently lower than that in the Cd group (P < 0.05), and significantly higher than that in the control group and QE group (P < 0.05).

Effect of QE and Cd on Cd contents in the chicken brains. Each value represents the mean ± SD of 10 individuals. The different lowercase letters at the top of the bars represent significant statistical differences (P < 0.05) between different groups
Histological Analysis
To ascertain whether QE can attenuate the brain injury induced by Cd, we assessed the pathological variations of the brain tissue with HE staining. The results showed that the control and QE group displayed normal structure and regular morphology of the brain tissue. The cellularity of neurons and glial cells was complete and healthy (Fig. 2a, b). Compared with the control group, the brain tissue of the Cd group showed obvious morphological damages. The morphology of neurons and glial cells was irregular. The necrocytosis and vacuolization of cells were frequently observed. The cell nucleus was dissolved and fragmented. Moreover, there was serious inflammatory cell infiltration in Cd-induced brain tissue (Fig. 2c). QE can significantly alleviate the pathological changes of the brain caused by Cd. The Cd/QE combined treatment group displayed the majority of neurons and glial cells were healthy. The necrotic neurons were infrequently discovered. In addition, only a small number of neurons remained damaged with inflammatory cell infiltration (Fig. 2d).

Effect of QE and Cd on the histological changes in the chicken brains. These pictures are at ×400 magnification. a The brains of the control group. b The brains of QE group. c The brains of the Cd group. d The brains of the Cd/QE co-treated group. The red arrows show necrosis, and the black arrows show inflammatory cell infiltration
The Levels of Oxidative Stress Biomarkers
To ascertain the effect of QE on Cd-induced oxidative stress in the chicken brain, we measured ROS production, MDA content, and antioxidase activities. The production of ROS and MDA content in the chicken brain induced by Cd or/and QE treatment were shown in Fig. 3a, b. Compared to the control group and QE group, the levels of ROS and MDA in the brain tissue of the Cd exposure group were dramatically increased (P < 0.05). However, the levels of ROS and MDA in the Cd + QE group were evidently lower than those in the Cd group (P < 0.05), but they did not restore to the levels of the control group and QE group (P < 0.05).

Effect of QE and Cd on oxidative stress indicators in the chicken brains. a Effect of QE and Cd on ROS production in the brain. b Effect of QE and Cd on MDA content in the brain. c Effect of QE and Cd on SOD activity in the brain. d Effect of QE and Cd on GPx activity in the brain. e Effect of QE and Cd on CAT activity in the brain. Each value represents the mean ± SD of 10 individuals. The different lowercase letters at the top of the bars represent significant statistical differences (P < 0.05) between different groups
The activities of SOD, GPx, and CAT in the brain from chicken treated by Cd or/and QE were shown in Fig. 3c–e. SOD, GPx, and CAT activities in the brain of the Cd-induced chickens were dramatically lower than the levels in the control group and QE group (P < 0.05). Compared to the Cd group, QE addition in the diet of Cd-induced chickens evidently elevated the activities of SOD, GPx, and CAT (P < 0.05), but QE addition did not recover these antioxidant activities to the levels in the control and QE group (P < 0.05). Compared with other groups, QE administration alone could remarkably increase the activities of SOD, GPx, and CAT in the brain tissue (P < 0.05).
The Levels of Inflammatory Relevant Factors
To elucidate the influence of QE on the iNOS/NF-κB signal pathway induced by Cd exposure, we measured NO contents, iNOS activities, and the expression of major inflammatory cytokines in iNOS/NF-κB pathway in the brains. As presented in Fig. 4a, b, NO levels and iNOS activities in the Cd-poisoned brain tissue were prominently higher than those levels in the control group and QE group (P < 0.05). Compared with the Cd group, QE/Cd combined treatment could alleviate the above changes (P < 0.05), but could not recover to the levels of the control group and QE group (P < 0.05).

Effect of QE and Cd on NO content, iNOS activity, and the expression of genes related to iNOS/NF-κB pathway in chicken brains. a Effect of QE and Cd on NO content in the brains. b Effect of QE and Cd on iNOS activity in the brains. c Effect of QE and Cd on the mRNA expression of IL-1β, NF-κB, TNF-α, COX-2, PTGEs, and iNOS in the chicken brains. d Effect of QE and Cd on the protein expression of NF-κB, TNF-α, COX-2, and iNOS in the chicken brains. Each value represents the mean ± SD of 10 individuals. The different lowercase letters at the top of the bars represent significant statistical differences (P < 0.05) between different groups
The mRNA and protein expression of the major inflammatory cytokines were exhibited in Fig. 4c, d. Compared to the control group and QE group, Cd significantly increased the mRNA expression of NF-κB, TNF-α, COX-2, PTGEs, iNOS, IL-1β, and the protein levels of NF-κB, TNF-α, COX-2, and iNOS in the Cd-induced chicken brains (P < 0.05). The expression levels of those inflammatory cytokines in Cd and QE co-administrated chicken brains were evidently lower than the levels of the Cd group and apparently higher than the levels of the control and QE group (P < 0.05).
The Expression of HSPs
To discover the impact of QE on HSPs in the chicken brains after Cd exposure, we examined the mRNA and protein levels of HSPs. As shown in Fig. 5, the mRNA levels of HSP27, HSP40, HSP60, HSP70, and HSP90 and the protein levels of HSP70 and HSP90 in the Cd group were dramatically improved compared with the control group and QE group (P < 0.05). Cd/QE co-administration significantly inhibited the mRNA expression of HSP27, Hsp40, HSP60, HSP70, and Hsp90, and the protein expression of HSP70 and HSP90 compared with the Cd group (P < 0.05), but did not recover to the mRNA and protein levels of the control group and QE group (P < 0.05).

Effect of QE and Cd on the expression of HSPs in the chicken brains. a Effect of QE and Cd on the mRNA expression of HSP27, HSP40, HSP60, HSP70, and HSP90 in the chicken brains. b Effect of QE and Cd on the protein expression of HSP70 and HSP90 in the chicken brains. Each value represents the mean ± SD of 10 individuals. The different lowercase letters at the top of the bars represent significant statistical differences (P < 0.05) between different groups
The Expression of Necroptosis Markers
To illustrate the effect of QE on the necroptosis signaling pathway in the chicken brain after Cd exposure, the mRNA and protein expressions of brain necroptosis-related factors in chickens were detected. As shown in Fig. 6. The mRNA level and protein expression of MLKL, RIPK1, and RIPK3 in the chicken brain exposed to Cd were distinctly higher than those levels in the control group and QE group (P < 0.05). However, compared with the Cd group, Cd and QE co-administration could obviously decrease the mRNA and protein levels of MLKL, RIPK1, and RIPK3 (P < 0.05). Conversely, Cd markedly reduced the mRNA expression of Caspase 8 compared with the control group and QE group (P < 0.05), whereas QE and Cd co-administration could dramatically elevate the expression of Caspase 8 mRNA compared to the Cd group (P < 0.05).

Effect of QE and Cd on the expression of necroptosis-related factors in the chicken brains. a Effect of QE and Cd on the mRNA levels of RIPK1, RIPK3, MLKL, and Caspase 8 in the chicken brains. b Effect of QE and Cd on the protein levels of RIPK1, RIPK3, and MLKL in the chicken brains. Each value represents the mean ± SD of 10 individuals. The different lowercase letters at the top of the bars represent significant statistical differences (P < 0.05) between different groups
Discussion
Cd is a toxic heavy metal that widely distributes in environment. The brain is one of the target organs influenced by Cd [25]. The neurotoxicity resulted from Cd exposure is closely related with disturbances in brain redox state, neurotransmitters, neurodegeneration, neuroinflammation, and neuronal death [36]. QE has long been used as antioxidant and antiinflammatory agent to protect the liver and kidney from exogenous stimuli-induced damages [37, 38]. However, the neuroprotective effects of QE are complicated and still remain unclear. In our study, we found that dietary exposure to Cd can cause necrotic injury in the chicken brain tissue. Cd induced ROS overproduction and targeted iNOS/NF-κB pathway to activate RIPK3/MLKL, thereby leading to necroptosis, and the HSPs involved in this pathological process. Furthermore, we also firstly found that QE can exert ameliorative roles on Cd-triggered necroptosis in the chicken brain by inhibition ROS/iNOS/NF-κB pathway.
Cd can pass the blood-brain barrier, assemble in the brain tissue, and then induce toxicity in the nervous tissue. A few studies manifested the poisonous effects and probable toxic mechanisms of Cd exposure on the brain [7, 39]. In our study, we discovered that the Cd-exposed chicken brains have higher Cd content than normal chickens. But QE and Cd co-fed chickens markedly decreased brain Cd level. It indicated that the residual Cd in the chicken brain likely causes the neurotoxicity and QE is a potentially antidotal substance against Cd-induced toxicity on the chicken brain. Moreover, our histopathological results displayed that Cd administration induced serious morphological alterations of the brain tissue, including irregular cell morphology, unnormal cell nucleus, and neuronal cell necrosis. Necroptosis is a special manner of cell death, which frequently occurs in multiple pathophysiological processes and commonly exhibits morphological features of necrosis [40]. It was well known that necroptosis is mainly regulated by MLKL, RIPK1, and RIPK3 when Caspase 8 is inhibited. Once necroptosis was triggered, RIPK1 and RIPK3 interact via the RIPK homotypic interaction motifs (RHIMs) to activate RIPK3, which in turn could recruit and phosphorylate MLKL to play corresponding necroptosis effects. Inhibition of Caspase 8 induces RIPK1 and RIPK3 to form necrosomes, which lead to the autophosphorylation of RIPK1 and RIPK3, and finally result in the emergence of necroptosis [41]. Cd was testified to induce the necroptosis of the chicken liver in which Cd evidently promoted the expression of RIPK1, RIPK3, and MLKL and dramatically reduced the expression of Caspase 8 [42]. Cd exposure triggered the necroptosis of rainbow trout cell lines [43]. In the current study, we also found a notable decrease in the mRNA level of caspase 8 and obvious increases in the mRNA and protein expression levels of the key signal moleculars (RIPK1, RIPK3, MLKL) from Cd-intoxicated chicken brain, which indicated that dietary exposure to Cd can induce brain necroptosis in chickens by activating RIPK3/MLKL pathway and inhibiting the expression of Caspase 8. QE was reported to alleviate RIPK3/MLKL-mediated necroptosis of oligodendrocytes after spinal cord injury in rats [35]. Our histopathological results showed that the majority of nerve cells in brain tissues from Cd and QE co-treated chickens were normal and healthy. QE administration markedly decreased the incidence of necrocytosis in the chicken brain. Meanwhile, QE treatment evidently reduced the mRNA and protein expression levels of RIPK1, RIPK3, and MLKL, but Caspase 8 evidently elevated in the brain of Cd-exposed chicken. It manifested QE had an antagonistic effect against Cd-evoked necroptosis in the chicken brain.
ROS play a vital role in cell signal and regulation of cell physiological function. Oxidative stress triggered by excessive ROS can injure tissues and cells. ROS participate in the development of oxidative stress induced by Cd, which can cause lipid peroxidation and inhibit antioxidase activities [44]. Zhang et al. [45] reported that ROS contributes to elicit paraquat-caused necroptosis in the mice heart, and the increased expressions of RIPK1, RIPK3, and MLKL were accompanied by overgeneration of ROS in cardiac tissues. Oxidative stress is one of the mechanisms of neurotoxicity caused by Cd [46]. It has been reported that Cd can cause the decrease of total anti-oxidation capacity and the upregulation of MDA in the rat brain [36]. Cd exposure elevated the levels of MDA in the cortical tissue of the rat brain, but the activity and mRNA expression of antioxidant enzymes showed remarkable decreases [9]. Oxidative stress is also believed as the promoter for necroptosis. Han et al. [16] showed that oxidative stress can induce the activation of necroptosis in hyperoxic acute lung damage. Wang et al. [47] manifested that Cd-triggered liver necroptosis is closely related to oxidative stress and MAPK pathway activation. Cd administration elevated ROS level and reduced antioxidase activities, which cause the death of hepatocytes mainly through the necrosis pathway [48]. In the present study, Cd evidently also increased the levels of ROS and MDA, and inhibited the activities of antioxidant enzymes in the chicken brain. We presumed that Cd exposure led to oxidative stress induced by a large amount of ROS in the chicken brain, which is helpful to trigger the necroptosis of the brain tissue. iNOS/NF-κB pathway implements an important effect in regulating the inflammatory response [49]. Zhang et al. [50] showed that selenium deficiency induced inflammatory response in the pig brain via iNOS/NF-κB pathway. iNOS is an important downstream mediator of inflammation, which is induced by a variety of cytokines or exogenous stimuli to synthesize excessive NO [51]. NO is a vital signal molecule and its levels could be regarded as a biomarker to estimate the influence of toxins and also reflect the inflammation state [52]. Excessive NO also contributes to pathological processes of many diseases. Cd was reported that it could trigger inflammation in chicken splenic lymphocytes and increase iNOS activity and NO production [53]. Wang et al. [54] revealed that Cd evidently enhanced iNOS activity, NO production, inflammatory factors, and HSP expression in the chicken liver. NF-κB is an important nuclear transcription factor in inflammatory signaling pathway. As a downstream cytokine of NF-κB pathway, iNOS expression could be motivated by NF-κB in the choroid plexus cells following meningitis [55]. When exogenous substances stimulate or injure the cells, activated NF-κB could promote and regulate the expression of inflammatory cytokines (such as iNOS and TNF-α), which further contribute to pro-inflammatory responses [52]. It has been reported that excessive ROS can activate pro-inflammatory signaling pathway and prompt the release of inflammatory mediators [56]. Dietary exposure to Cd obviously increased the mRNA and protein expression of inflammation-related genes (NF-κB, TNF-α, COX-2, PTGEs, and IL-1β) in the hen liver [57]. Cd caused rat pancreatitis coupled with overexpression of iNOS, NF-κB, IL-6, and TNF-α and pathological changes [58]. Interestingly, inflammation also can promote the occurrence of necroptosis. Yang et al. [59] indicated that inflammation caused cardiomyocyte necroptosis. Atrazine, a common pesticide pollutant, promoted the expression of TNF-α and further motivated NF-κB inflammatory pathway, thereby causing lymphocyte necroptosis [60]. Gallic acid-induced TNF-α signaling-mediated necroptosis in activated hepatic stellate cells [61]. In our study, histopathological observation results showed that inflammatory intracellular infiltration existed in the brain tissue suffering Cd exposure. Moreover, the content of NO, iNOS activities, and the expression of inflammatory cytokines (NF-κB, iNOS, TNF-α, COX-2, IL-1β, and PTGE) in NF-κB pathway were all increased in Cd-intoxicated brains, which illustrated that the upregulation of NO-iNOS system may take part in the process of brain necroptosis and Cd could activate iNOS/NF-κB pathway to promote the brain necroptosis in chicken.
QE is usually used as an effective antioxidant and inflammatory inhibitor. In the recent reports, QE can protect both goat sperm and preimplantation embryos against Cd-induced oxidative stress [62]. QE relieves Cd-induced hepatotoxicity and nephrotoxicity via enhancing the antioxidant defense system [63]. Ma et al. [37] showed that QE administration prominently restrained the inflammatory response in the mice livers caused by carbon tetrachloride through evidently reducing the protein expression of pro-inflammatory markers (NF-κBp65, iNOS, IL-1β, COX-2) and NO production in the livers. QE inhibited lipopolysaccharide-induced NO release and the mRNA and protein expressions of iNOS, COX-2, TNF-α, IL-1, and IL-6 [64]. QE antagonized manganese-triggered oxidative stress and neuroinflammation [65]. In the current study, QE administration was found to significantly reduce ROS and MDA levels and dramatically elevate antioxidant enzyme activities in the chick brains exposed to Cd. QE also could reduce iNOS activity and succedent NO generation in Cd-exposed chicken brain. Furthermore, the mRNA or protein expression levels of inflammatory cytokines associated with NF-κB signal pathway in the brain of chickens fed with Cd were decreased by QE administration. So we speculated QE maybe attenuated Cd-induced necroptosis in the chicken brain by inhibiting ROS/iNOS/NF-κB pathway, through which QE restrained ROS overproduction triggered by Cd and oxidative stress, and alleviated Cd-caused inflammatory response. HSPs can induct stimuli and activate necroptosis and inflammatory reaction. HSP70 can significantly irritate the secretion of TNF-α and IL-1β in head kidney leukocytes of grass carp [66]. HSP 90 regulates the stability of MLKL and RIPK3 and is also requisite for TNF-induced necroptosis [67]. It has been reported that lead exposure-induced necroptosis in the chicken spleen was coupled with high expression levels of HSP27, HSP40, HSP60, HSP70, and HSP90 [22]. Wang et al. [29] found that selenium deficiency-mediated necroptosis gave rise to the upregulation of HSP expression. QE, as a HSP repressor, could efficiently restrain the expression of HSP27, HSP70, and HSP90 in human breast cancer cells [68]. QE played an obvious inhibiting effect on the whole expression of HSPs in human hepatocellular carcinoma [69]. Our results displayed that Cd could remarkably promote the mRNA expression of HSP27, HSP40, HSP60, HSP70, and HSP90 and the protein expression of HSP70 and HSP90 in the chicken brain, while QE/Cd combined treatment could reduce the expression levels. These results elucidated that HSPs maybe participated in Cd-caused necroptosis; meanwhile, we inferred that QE may inhibit HSPs’ activation to help attenuate Cd-induced necroptosis.
In conclusion, our present investigation demonstrated that Cd exposure could promote ROS overgeneration to trigger oxidative stress, activate iNOS and NF-κB pathway, and increase the expression HSPs, which led to the necroptosis of the brain tissue in chicken. Furthermore, QE exerted alleviative roles against Cd-induced necroptosis through inhibiting ROS/iNOS/NF-κB pathway in the chicken brain. Our discovery will offer new ideas for further elucidating the detoxification effect of QE on neurotoxicity in birds induced by heavy metals.
References
Zhang Y, Yin H, Shao B, Xue H, Huang B, Liu H, Li S (2020) Antagonistic effect of VDR/CREB1 pathway on cadmium-induced apoptosis in porcine spleen. Ecotoxicol Environ Saf 209:111819. https://doi.org/10.1016/j.ecoenv.2020.111819
Qu K, Wang Z, Tang K, Zhu Y, Fan R (2019) Trehalose suppresses cadmium-activated Nrf2 signaling pathway to protect against spleen injury. Ecotoxicol Environ Saf 181:224–230
Batool Z, Agha F, Tabassum S, Batool T, Siddiqui R, Haider S (2019) Prevention of cadmium-induced neurotoxicity in rats by essential nutrients present in nuts. Acta Neurobiol Exp 79(2):169–183
Ashok A, Rai N, Tripathi S, Bandyopadhyay S (2015) Exposure to As-, Cd-, and Pb-mixture induces Aβ, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol Sci 143(1):64–80
Da Costa P, Gonçalves J, Baldissarelli J, Mann T, Abdalla F, Fiorenza A, da Rosa M, Carvalho F, Gutierres J, de Andrade C, Rubin M, Schetinger M, Morsch V (2017) Curcumin attenuates memory deficits and the impairment of cholinergic and purinergic signaling in rats chronically exposed to cadmium. Environ Toxicol 32(1):70–83
Yuan Y, Wang Y, Hu F, Jiang C, Zhang Y, Yang J, Zhao S, Gu J, Liu X, Bian J, Liu Z (2016) Cadmium activates reactive oxygen species-dependent AKT/mTOR and mitochondrial apoptotic pathways in neuronal cells. Biomed Environ Sci 29(2):117–126
Almeer R, Kassab R, AlBasher G, Alarifi S, Alkahtani S, Ali D, Abdel Moneim A (2019) Royal jelly mitigates cadmium-induced neuronal damage in mouse cortex. Mol Biol Rep 46(1):119–131
Tsentsevitsky A, Zakyrjanova G, Petrov A (2020) Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: key role of ROS. Free Radic Biol Med 155:19–28
Al Olayan E, Aloufi A, AlAmri O, El-Habit O, Abdel Moneim A (2020) Protocatechuic acid mitigates cadmium-induced neurotoxicity in rats: role of oxidative stress, inflammation and apoptosis. Sci Total Environ 723:137969
Elkhadragy M, Kassab R, Metwally D, Almeer R, Abdel-Gaber R, Al-Olayan E, Essawy E, Amin H, Abdel Moneim A (2018) Fragaria ananassa protective effects of methanolic extract in a rat model of cadmium chloride-induced neurotoxicity. Biosci Rep 38(6):BSR20180861
Chi Q, Hu X, Zhao B, Zhang Q, Zhang K, Li S (2020) Regulation of HS-induced necroptosis and inflammation in broiler bursa of Fabricius by the miR-15b-5p/TGFBR3 axis and the involvement of oxidative stress in this process. J Hazard Mater 406:124682
Kitur K, Parker D, Nieto P, Ahn D, Cohen T, Chung S, Wachtel S, Bueno S, Prince A (2015) Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog 11(4):e1004820
Wegner K, Saleh D, Degterev A (2017) Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci 38(3):202–225
Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 34(47):5796–5806
Zhang Y, Su S, Zhao S, Yang Z, Zhong C, Chen X, Cai Q, Yang Z, Huang D, Wu R, Han J (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8:14329
Han C, Guan Z, Zhang P, Fang H, Li L, Zhang H, Zhou F, Mao Y, Liu W (2018) Oxidative stress induced necroptosis activation is involved in the pathogenesis of hyperoxic acute lung injury. Biochem Biophys Res Commun 495(3):2178–2183
Li X, Xing M, Chen M, Zhao J, Fan R, Zhao X, Cao C, Yang J, Zhang Z, Xu S (2017) Effects of selenium-lead interaction on the gene expression of inflammatory factors and selenoproteins in chicken neutrophils. Ecotoxicol Environ Saf 139:447–453
Zhang Y, Liu Q, Yin H, Min Y, Li S (2020) Selenium deficiency causes immune damage by activating the DUSP1/NF-κB pathway and endoplasmic reticulum stress in chicken spleen. Food Funct 11(7):6467–6475
Kim Y, Ahn C, Je J (2016) Anti-inflammatory action of high molecular weight Mytilus edulis hydrolysates fraction in LPS-induced RAW264.7 macrophage via NF-κB and MAPK pathways. Food Chem 202:9–14
Shindo R, Kakehashi H, Okumura K, Kumagai Y, Nakano H (2013) Critical contribution of oxidative stress to TNFα-induced necroptosis downstream of RIPK1 activation. Biochem Biophys Res Commun 436(2):212–216
Zhang H, Liu Q, Kong L, Xu S (2019) Mucin 1 downregulation impairs the anti-necroptotic effects of glucocorticoids in human bronchial epithelial cells. Life Sci 221:168–177
Zhang J, Wang S, Hao X, Sun G, Xu S (2020) The antagonistic effect of selenium on lead-induced necroptosis via MAPK/NF-κB pathway and HSPs activation in the chicken spleen. Ecotoxicol Environ Saf 204:111049
Protopapas A, Vradelis S, Karampitsakos T, Steiropoulos P, Chatzimichael A, Paraskakis E (2019) Elevated levels of alveolar nitric oxide may indicate presence of small airway inflammation in patients with inflammatory bowel disease. Lung 197(5):663–670
Dejban P, Rahimi N, Takzare N, Dehpour A (2020) Biochemical and histopathological evidence for the beneficial effects of modafinil on the rat model of inflammatory bowel disease: involvement of nitric oxide pathway. Pharmacol Rep 72(1):135–146
Tang K, Li H, Qu K, Fan R (2019) Selenium alleviates cadmium-induced inflammation and meat quality degradation via antioxidant and anti-inflammation in chicken breast muscles. Environ Sci Pollut Res 26(23):23453–23459
Guo K, Ge J, Zhang C, Lv M, Zhang Q, Talukder M, Li J (2020) Cadmium induced cardiac inflammation in chicken (Gallus gallus) via modulating cytochrome P450 systems and Nrf2 mediated antioxidant defense. Chemosphere 249:125858
Chi X, Shi G, Zhang Q, Liu Q, Yin H, Zhang Y, Li S (2020) Astilbin protects chicken peripheral blood lymphocytes from cadmium-induced necroptosis via oxidative stress and the PI3K/Akt pathway. Ecotoxicol Environ Saf 190:110064
Xing M, Jin X, Wang J, Shi Q, Cai J, Xu S (2018) The antagonistic effect of selenium on lead-induced immune dysfunction via recovery of cytokine and heat shock protein expression in chicken neutrophils. Biol Trace Elem Res 185(1):162–169
Wang L, Shi X, Zheng S, Xu S (2020) Selenium deficiency exacerbates LPS-induced necroptosis by regulating miR-16-5p targeting PI3K in chicken tracheal tissue. Metallomics 12(4):562–571
Zhang J, Hao X, Xu S (2020) Selenium prevents lead-induced necroptosis by restoring antioxidant functions and blocking MAPK/NF-κB pathway in chicken lymphocytes. Biol Trace Elem Res 198(2):644–653
D'Andrea G (2015) Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia 106:256–271
Yang H, Song Y, Liang Y, Li R (2018) Quercetin treatment improves renal function and protects the kidney in a rat model of adenine-induced chronic kidney disease. Med Sci Monit 24:4760–4766
Gupta R, Shukla R, Chandravanshi L, Srivastava P, Dhuriya Y, Shanker J, Singh M, Pant A, Khanna V (2017) Protective role of quercetin in cadmium-induced cholinergic dysfunctions in rat brain by modulating mitochondrial integrity and MAP kinase signaling. Mol Neurobiol 54(6):4560–4583
Nna V, Usman U, Ofutet E, Owu D (2017) Quercetin exerts preventive, ameliorative and prophylactic effects on cadmium chloride-induced oxidative stress in the uterus and ovaries of female Wistar rats. Food Chem Toxicol 102:143–155
Fan H, Tang H, Shan L, Liu S, Huang D, Chen X, Chen Z, Yang M, Yin X, Yang H, Hao D (2019) Quercetin prevents necroptosis of oligodendrocytes by inhibiting macrophages/microglia polarization to M1 phenotype after spinal cord injury in rats. J Neuroinflammation 16(1):206
Tang K, Liu X, Wang Z, Qu K, Fan R (2019) Trehalose alleviates cadmium-induced brain damage by ameliorating oxidative stress, autophagy inhibition, and apoptosis. Metallomics 11(12):2043–2051
Ma J, Li Z, Xie W, Liu C, Liu S (2015) Quercetin protects mouse liver against CCl4-induced inflammation by the TLR2/4 and MAPK/NF-κB pathway. Int Immunopharmacol 28(1):531–539
Alidadi H, Khorsandi L, Shirani M (2018) Effects of quercetin on tubular cell apoptosis and kidney damage in rats induced by titanium dioxide nanoparticles. Malaysian J Med Sci 25(2):72–81
Alnahdi H, Sharaf I (2019) Possible prophylactic effect of omega-3 fatty acids on cadmium-induced neurotoxicity in rats’ brains. Environ Sci Pollut Res Int 26(30):31254–31262
Galluzzi L, Kepp O, Chan F, Kroemer G (2017) Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol 12:103–130
Chaudhary G, Yadav P, Yadav A, Tiwari M, Gupta A, Sharma A, Pandey A, Pandey A, Chaube S (2019) Necroptosis in stressed ovary. J Biomed Sci 26(1):11
Wang Y, Chen H, Chang W, Chen R, Xu S, Tao D (2020) Protective effects of selenium yeast against cadmium-induced necroptosis via inhibition of oxidative stress and MAPK pathway in chicken liver. Ecotoxicol Environ Saf 206:111329
Krumschnabel G, Ebner H, Hess M, Villunger A (2010) Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat Toxicol 99(1):73–85
Wang J, Zhu H, Liu X, Liu Z (2014) N-acetylcysteine protects against cadmium-induced oxidative stress in rat hepatocytes. J Vet Sci 15(4):485–493
Zhang L, Feng Q, Wang T (2018) Necrostatin-1 protects against paraquat-induced cardiac contractile dysfunction via RIP1-RIP3-MLKL-dependent necroptosis pathway. Cardiovasc Toxicol 18(4):346–355
Wang H, Matsushita M, Zhang L, Abel G, Mommer B, Huddy T, Storm D, Xia Z (2020) Inducible and conditional stimulation of adult hippocampal neurogenesis rescues cadmium-induced impairments of adult hippocampal neurogenesis and hippocampus-dependent memory in mice. Toxicol Sci 177(1):263–280
Wang X, Wang T, Pan T, Huang M, Ren W, Xu G, Amin H, Kassab R, Abdel Moneim A (2020) Senna alexandrina extract supplementation reverses hepatic oxidative, inflammatory, and apoptotic effects of cadmium chloride administration in rats. Environ Sci Pollut Res 27(6):5981–5992
Sinha M, Manna P, Sil P (2009) Induction of necrosis in cadmium-induced hepatic oxidative stress and its prevention by the prophylactic properties of taurine. J Trace Elem Med Biol 23(4):300–313
Qin L, Zhang Y, Wan C, Wang Z, Cong Y, Li S (2020) MiR-196-5p involvement in selenium deficiency-induced immune damage via targeting of NFκBIA in the chicken trachea. Metallomics 12(11):1679–1692
Zhang Y, Cui J, Lu Y, Huang C, Liu H, Xu S (2020) Selenium deficiency induces inflammation via the iNOS/NF-κB pathway in the brain of pigs. Biol Trace Elem Res 196(1):103–109
Nakazawa H, Chang K, Shinozaki S, Yasukawa T, Ishimaru K, Yasuhara S, Yu Y, Martyn J, Tompkins R, Shimokado K, Kaneki M (2017) iNOS as a driver of inflammation and apoptosis in mouse skeletal muscle after burn injury:possible involvement of Sirt1 S-nitrosylation-mediated acetylation of p65 NF-κB and p53. PLoS One 12(1):e0170391
Jiang X, Feng X, Huang H, Liu L, Qiao L, Zhang B, Yu W (2017) The effects of rotenone-induced toxicity via the NF-κB-iNOS pathway in rat liver. Toxicol Mech Methods 27(4):318–325
Liu S, Xu F, Fu J, Li S (2015) Protective roles of selenium on nitric oxide and the gene expression of inflammatory cytokines induced by cadmium in chicken splenic lymphocytes. Biol Trace Elem Res 168(1):252–260
Wang Y, Liu J, Chen R, Qi M, Tao D, Xu S (2020) The antagonistic effects of selenium yeast (SeY) on cadmium-induced inflammatory factors and the heat shock protein expression levels in chicken livers. Biol Trace Elem Res 198(1):260–268
Takano M, Ohkusa M, Otani M, Min K, Kadoyama K, Minami K, Sano K, Matsuyama S (2015) Lipid A-activated inducible nitric oxide synthase expression via nuclear factor-κB in mouse choroid plexus cells. Immunol Lett 167(2):57–62
Reuter S, Gupta S, Chaturvedi M, Aggarwal B (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49(11):1603–1616
Zhang R, Liu Y, Xing L, Zhao N, Zheng Q, Li J, Bao J (2018) The protective role of selenium against cadmium-induced hepatotoxicity in laying hens: expression of Hsps and inflammation-related genes and modulation of elements homeostasis. Ecotoxicol Environ Saf 159:205–212
Aja P, Izekwe F, Famurewa A, Ekpono E, Nwite F, Igwenyi I, Awoke J, Ani O, Aloke C, Obasi N, Udeh K, Ale B (2020) Hesperidin protects against cadmium-induced pancreatitis by modulating insulin secretion, redox imbalance and iNOS/NF-ĸB signaling in rats. Life Sci 259:118268
Yang T, Cao C, Yang J, Liu T, Lei X, Zhang Z, Xu S (2018) miR-200a-5p regulates myocardial necroptosis induced by Se deficiency via targeting RNF11. Redox Biol 15:159–169
Cui Y, Yin K, Gong Y, Qu Y, Liu H, Lin H (2019) Atrazine induces necroptosis by miR-181-5p targeting inflammation and glycometabolism in carp lymphocytes. Fish Shellf Immunol 94:730–738
Chang Y, Hsu S, Liu Y, Lin Y, Lin M, Huang S, Ho J, Wu L (2015) Gallic acid induces necroptosis via TNF-α signaling pathway in activated hepatic stellate cells. PLoS One 10(3):e0120713
Mao T, Han C, Wei B, Zhao L, Zhang Q, Deng R, Liu J, Luo Y, Zhang Y (2018) Protective effects of quercetin against cadmium chloride-induced oxidative injury in goat sperm and zygotes. Biol Trace Elem Res 185(2):344–355
Liu Y, Zhang X, Guan T, Jia S, Liu Y, Zhao X (2020) Effects of quercetin on cadmium-induced toxicity in rat urine using metabonomics techniques. Hum Exp Toxicol 39(4):524–536
Endale M, Park S, Kim S, Kim S, Yang Y, Cho J, Rhee M (2013) Quercetin disrupts tyrosine-phosphorylated phosphatidylinositol 3-kinase and myeloid differentiation factor-88 association, and inhibits MAPK/AP-1 and IKK/NF-κB-induced inflammatory mediators production in RAW 264.7 cells. Immunobiology 218(12):1452–1467
Bahar E, Kim J, Yoon H (2017) Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. Int J Mol Sci 18(9):1989
Zhang A, Guo Y, Zhang S, Fan X, Wang X, Zhou X, Yang K, Zhou H (2015) Cytokine effects and cellular signaling pathways of grass carp HSP70 in head kidney leukocytes. Fish Shellf Immunol 46(2):550–556
Zhao X, Chen Z, Zhao J, Zhang P, Pu Y, Jiang S, Hou J, Cui Y, Jia X, Zhang S (2016) Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis 7:e2089
Kıyga E, Şengelen A, Adıgüzel Z, Önay Uçar E (2020) Investigation of the role of quercetin as a heat shock protein inhibitor on apoptosis in human breast cancer cells. Mol Biol Rep 47(7):4957–4967
Zhou J, Fang L, Yao W, Zhao X, Wei Y, Zhou H, Xie H, Wang L, Chen L (2011) Effect of quercetin on heat shock protein expression in HepG2 cells determined by SILAC. Zhonghua Zhong Liu Za Zhi [Chin J Oncol] 33(10):737–741
Acknowledgments
The authors thank the members of the Cd-diet group from Northeast Agricultural University for the help they supplied in the research. This study was supported by the Natural Science Foundation of Heilongjiang Province (Project No. LH2019H111) and Foundation Item of Heilongjiang University of Chinese Medicine (Project No.15041200003).
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
All other authors have read the manuscript and have agreed to submit it in its current form for consideration for publication in the Journal.
Rights and permissions
About this article

Cite this article
Liu, L., Liu, Y., Cheng, X. et al. The Alleviative Effects of Quercetin on Cadmium-Induced Necroptosis via Inhibition ROS/iNOS/NF-κB Pathway in the Chicken Brain. Biol Trace Elem Res 199, 1584–1594 (2021). https://doi.org/10.1007/s12011-020-02563-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-020-02563-4