
Abstract
With the increasing evidences of cadmium-induced cognitive deficits associated with brain cholinergic dysfunctions, the present study aimed to decipher molecular mechanisms involved in the neuroprotective efficacy of quercetin in rats. A decrease in the binding of cholinergic–muscarinic receptors and mRNA expression of cholinergic receptor genes (M1, M2, and M4) was observed in the frontal cortex and hippocampus on exposure of rats to cadmium (5.0 mg/kg body weight, p.o.) for 28 days compared to controls. Cadmium exposure resulted to decrease mRNA and protein expressions of choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) and enhance reactive oxygen species (ROS) generation associated with mitochondrial dysfunctions, ultrastructural changes, and learning deficits. Enhanced apoptosis, as evidenced by alterations in key proteins involved in the pro- and anti-apoptotic pathway and mitogen-activated protein (MAP) kinase signaling, was evident on cadmium exposure. Simultaneous treatment with quercetin (25 mg/kg body weight, p.o.) resulted to protect cadmium-induced alterations in cholinergic–muscarinic receptors, mRNA expression of genes (M1, M2, and M4), and expression of ChAT and AChE. The protective effect on brain cholinergic targets was attributed to the antioxidant potential of quercetin, which reduced ROS generation and protected mitochondrial integrity by modulating proteins involved in apoptosis and MAP kinase signaling. The results exhibit that quercetin may modulate molecular targets involved in brain cholinergic signaling and attenuate cadmium neurotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cadmium, one of the trace elements present in the earth’s crust, has extensive uses in mining and smelting [1]. Use of cadmium is common in electroplating, and cadmium salts are frequently used as color pigments in metals and plastics and in the manufacture of batteries and phosphate fertilizers [2–4]. Besides anthropogenic uses, volcanic eruptions, burning of fossil fuel, incineration of municipal waste, recycling of cadmium-plated scrap, and electronic waste significantly contribute to the release of cadmium in the environment [1, 5]. High cadmium levels have been found in drinking water and crops grown on soil contaminated with the metal [6–11]. Human exposure to cadmium may therefore occur both in occupational and non-occupational settings and poses a serious risk to health and associated problems. Renal dysfunctions and decreased bone mineral density are frequently reported on exposure to cadmium in non-occupationally exposed individuals in cross-sectional studies [3, 12]. While monitoring health outcomes, increased urinary levels of cadmium have been associated with hypertension, diabetes, and diabetic nephropathy in different sets of population [3, 12]. A number of autopsy reports exhibit high accumulation of cadmium in body organs including the brain [13–15]. Satrug and Moore [12] suggested that high accumulation of cadmium in the liver and kidney is due to the poor accumulation of cadmium associated with its reabsorption in the kidney with age.
Although exposure to cadmium may affect physiological functioning [12] and enhance the risk of developing cancers of the body organs in humans, it has been found to affect the functioning of the central and peripheral nervous systems adversely [16, 17]. Headache, vertigo, and other neurological abnormalities including olfactory dysfunctions have been reported in cadmium-exposed individuals [18–21]. Cadmium exposure has been found to cause cognitive dysfunctions and affect the development of visual perception in children [20, 21]. Motor dysfunctions have also been reported in humans on cadmium exposure [22]. Chronic exposure to cadmium has been found to cause peripheral neuropathy and affect psychomotor functions in occupational workers [21, 23, 24]. Cadmium easily crosses the blood–brain barrier, and thus, the brain is one of the soft targets [17, 25, 26]. The toxic effects of cadmium are largely attributed to its poor elimination from the body and thus high accumulation in tissues including the brain [26–28]. Enhanced oxidative stress in the brain on cadmium exposure is frequently reported and considered to be one of the potential mechanisms in its neurotoxicity [26, 29–33]. Increased expression of metallothionein, a cysteine-rich protein, has been observed on cadmium exposure due to its high affinity in different organs including the brain [5, 34, 35]. Furthermore, cadmium may disrupt the metabolism of trace elements, especially copper and zinc, which play important roles in its metabolism and kinetics [35, 36]. A number of studies found that cadmium may decrease the calcium influx and inhibit the activity of Na+ and K+-ATPase in the brain and, thus, affect the process of neurotransmission and energy metabolism, respectively [37, 38]. Alterations in brain neurotransmitters following exposure to cadmium have been reported and associated with neurobehavioral abnormalities in experimental studies [39–43]. Brain cholinergic alterations have been frequently reported on cadmium exposure and associated with cognitive deficits [23]. There are a number of reports exhibiting that cadmium may influence the activity of brain acetylcholinesterase both in vivo and in vitro and disrupt the integrity of cholinergic functions [44, 45]. It has been found that cadmium easily targets mitochondria in the brain and other tissues and affects its functional integrity [46, 47]. The primary role of the mitochondria in the cell is to regulate bioenergetics via oxidative phosphorylation, and thus, alteration at any of the step in the electron transport chain (ETC) may result into energy failure. Furthermore, the mitochondria are the main sites of reactive oxygen species (ROS) generation and contribute in apoptosis via caspase cascade and the cell survival pathway including mitogen-activated protein (MAP) kinases [48]. MAP kinases are highly regulated serine threonine kinases and include c-Jun N-terminal kinases (JNKs), p38, and extracellular signal-regulated kinases (ERKs) as important members in the family which contribute extensively to neuronal cell death [49, 50]. The role of MAP kinase and mechanistic target of rapamycin (mTOR) in cadmium-mediated cell death in neurons has been reported [51, 52]. Mitochondrial dysfunctions in neurodegenerative diseases are well established [53]. In an interesting study, the activation of muscarinic–cholinergic receptors was found to protect mitochondrial damage and inhibit mitochondrial-mediated cell death, thus suggesting that there is a close link of muscarinic–cholinergic receptor alteration with the integrity of the mitochondria and apoptotic cell death [54]. In view of the increasing risks of human exposure to neurodegenerative disorders, including Parkinson’s, Alzheimer’s, and Huntington’s diseases, due to increased cadmium levels in the environment [5, 22, 43, 55–57], there is a great thrust to understand its impact on the functioning of the brain and investigate whether cadmium-induced neurotoxicity could be protected.
Quercetin, a class of bioflavonoid, is widely present in a variety of vegetables and fruits, while high levels are present in onions, apples, and berries. The presence of quercetin in medicinal plants like Ginkgo biloba and Hypericum perforatum (St. John’s wort) has also been reported [58, 59]. The broad pharmacological spectrum of quercetin due to its anti-inflammatory, vasodilating, and metal chelating effects has attracted much attention to assess its therapeutic potential in a number of diseases [60]. Interestingly, the bioactivity of quercetin due to its antioxidant and radical scavenging activity has been associated with its neuroprotective potential in experimental models of neurodegenerative disorders [61–63]. Experimental studies carried out recently found that cadmium exposure could affect memory and cause anxiogenic changes associated with alterations in brain acetylcholinesterase activity and impaired antioxidant defense in rats, and these changes were protected by quercetin [44, 64]. While cadmium may affect the molecular targets in brain cholinergic signaling, the exact mechanism of the protective potential of quercetin in modulating cadmium-induced neurotoxicity is not understood. The present study has therefore been carried out to decipher the molecular mechanisms associated with cadmium-induced cognitive deficits and the role of the mitochondria and MAP kinase in such alteration and the protective efficacy of quercetin.
Materials and Methods
Experimental Animals and Housing Conditions
The experiments in the present study were performed on adult male rats of Wistar strain (180 ± 20 g) obtained from the central animal house of CSIR-Indian Institute of Toxicology Research (CSIR-IITR), Lucknow. Rats were acclimatized for 7 days before the start of the experiment. The experimental animal rooms had controlled temperature (25 ± 2 °C) with a 12-h light/dark cycle. Standard hygiene conditions were ensured in the experimental animal rooms, and pellet diet and purified drinking water were available to the animals ad libitum. The study was approved by the institutional animal ethics committee of CSIR-IITR, Lucknow, and official guidelines formed by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests, Government of India, were followed for carrying out the experiments all throughout.
Experimental Design and Treatment Procedure
There were four treatment groups as per the following details:
-
Group I
—Rats treated with cadmium as cadmium chloride (dissolved in distilled water, 5 mg/kg body weight, p.o., once daily for 28 days)
-
Group II
—Rats treated with quercetin (suspended in distilled water containing 0.1 % Tween 20, 25 mg/kg body weight, p.o., once daily for 28 days)
-
Group III
—Rats treated with cadmium and quercetin simultaneously as in groups I and II, respectively
-
Group IV
—Rats treated with vehicle (distilled water containing 0.1 % Tween-20) for the duration of the treatment and served as the controls
There were 42 rats in each treatment group. A separate set of five rats from each treatment group was used to assess spatial memory and learning by passive avoidance test and Y-maze. For the assay of cholinergic–muscarinic receptors, mitochondrial complexes, and estimation of ROS and mitochondrial membrane potential (MMP) in the frontal cortex and hippocampus, a set of five rats each from different treatment groups was used following standard protocols. Furthermore, a separate set of three rats from each treatment group was used for Western blotting, quantitative real-time polymerase chain reaction (qRT-PCR), and histological and ultrastructural studies. For neurochemical studies, a set of rats from each treatment group was killed 24 h after the last dose. The brain was quickly removed, washed in ice-cold saline, and dissected into the frontal cortex and hippocampus following standard procedure [65]. For ultrastructural and histological studies, rats were perfused and the brains were taken out.
Behavioral Studies
Assessment of Learning and Memory
Effects on the learning and memory in rats treated with cadmium or quercetin alone or in combination were assessed by passive avoidance test using a shuttle box. However, spatial memory and learning and continuous alternation test were assessed by the Y-maze in a separate set of rats.
Assessment of Spatial Memory and Continuous Alternation
Spatial memory was assessed through novelty seeking behavior using the Y-maze and following the procedure described by Wang et al. [66]. Briefly, the test consists of two trials separated by an inter-trial interval of 4 h to assess spatial recognition memory test. Out of three arms of the Y-maze, one arm was blocked and termed as the novel arm. During the first trial, rats were placed in the start arm and allowed to explore the start arm and the other arm for 15 min as the third arm (novel arm) was blocked. After an inter-trial interval of 4 h, rats were placed in the same arm (start arm) and allowed to explore all arms for duration of 5 min. During this time, rats were free to visit all three arms. The number of entries and time spent in the novel arm versus other arm were recorded on a computer. The results are expressed as percent of time spent and percent of entries in the novel arm versus other arm.
Furthermore, continuous alternation in rats was assessed using the Y-maze as described by Yamada et al. [67]. Briefly, rats were placed at the end of one arm of the Y-maze and allowed to move freely for 5 min. The series and sequence of entry into each arm of the Y-maze were recorded automatically on the computer. The alternation percentage was calculated as the ratio of actual to possible alternation multiplied by 100.
Passive Avoidance Response
The passive avoidance response was monitored using the shuttle box (Techno, India) following the procedure described by Yadav et al. [68]. Briefly, the shuttle box consists of two chambers—a lighted and a dark chamber separated by a guillotine door. Rats were placed in the lighted chamber of the shuttle box, and after acclimatization for 30 s, the guillotine door was opened. As the rats crossed into the dark chamber, the guillotine door was closed and a low-intensity foot shock (0.5 mA, 10 s) was given. The first trial was for acquisition; retention was assessed in subsequent trials carried out 24 h after the first trial. The transfer of rat from the light to the dark compartment was recorded as transfer latency time (TLT) in seconds. The criterion for improved cognitive activity was considered as an increase in the TLT on the retention trial (second trial and more) as compared to the acquisition trial (first trial). The shock was not given to the rats in the retention trials to avoid reacquisition.
Neurochemical Studies
Assay of Cholinergic–Muscarinic Receptors
Assay of cholinergic–muscarinic receptors in the frontal cortex and hippocampus was performed by radioligand receptor binding following the procedure described earlier by Khanna et al. [69]. In brief, the frontal cortex or hippocampus isolated from the brain was processed for the preparation of synaptic membranes. The brain region (frontal cortex or hippocampus) was homogenized in 19 volumes of Tris–HCl buffer (5 mM, pH 7.4) and the homogenate centrifuged (40,000×g) for 15 min at 4 °C. The sedimented pellet was suspended in homogenization buffer (5 mM Tris–HCl, pH 7.4) and recentrifuged (40,000×g) for 15 min at 4 °C. The pellet thus obtained was finally suspended in Tris–HCl buffer (40 mM, pH 7.4) and stored at −20 °C.
For the assay of cholinergic–muscarinic receptors, the reaction mixture in a final volume of 1 ml contained Tris–HCl buffer (40 mM, pH 7.4), 3H-quinuclidinyl benzilate (3H-QNB, 1 × 10−9 M), and membrane protein either from the frontal cortex or the hippocampus (around 200–250 μg). Binding incubations were carried out in triplicate for 15 min at 37 °C. A set of tubes containing atropine sulfate (1 × 10−6 M), a competitor, was also run simultaneously to assess nonspecific binding. Soon after incubation, the contents of the binding tubes were rapidly filtered on glass fiber discs (25-mm diameter, 1.0-μm pore size; Whatman GF/B). The filter discs were washed twice rapidly with cold Tris–HCl buffer (40 mM) to remove unbound radioligand. They were then dried and counted in 5 ml of scintillation mixture (PPO, POPOP, naphthalene, toluene, and methanol) over a β-scintillation counter at an efficiency of 30–40 % for 3H. Specific binding was calculated by subtracting the nonspecific binding (in the presence of atropine sulfate) from the total binding and expressed as picomoles of ligand bound per gram protein. Furthermore, Scatchard analysis was carried out using different concentrations of 3H-QNB to determine whether change in the binding is due to alteration in the affinity (K d) or number of receptor binding sites (B max).
Expression of Cholinergic–Muscarinic Receptors, Choline Acetyltransferase, and Acetylcholinesterase Genes
The frontal cortex and hippocampus dissected out from the brain were processed for RNA isolation using Trizol (Life Technologies, USA) following the recommended protocol. qRT-PCR for the different receptor subtypes was carried out as described earlier [70]. The sequence of primers used for cholinergic–muscarinic receptors (CHRM1, CHRM2, CHRM3, CHRM4, and CHRM 5), choline acetyltransferase, acetylcholinesterase, and β-actin used in the present study has been described earlier [71–73].
Isolation of the Mitochondria
The activity of enzyme complexes involved in the electron transport chain was measured in the mitochondria isolated from the frontal cortex and hippocampus following the recipe as described in detail [74]. Briefly, the frontal cortex or hippocampus was homogenized in buffer (pH 7.4) that contained HEPES (5 mM), sucrose (75 mM), mannitol (225 mM), EGTA (1 mM), and bovine serum albumin (BSA, 1 mg/ml). The homogenate was centrifuged (2000×g) for 3 min at 4 °C. The supernatant thus obtained was separated and centrifuged (12,000×g) for 10 min at 4 °C. The pellet was suspended in the same buffer used for homogenization, and digitonin (0.02 %) was added for the lysis followed by centrifugation (12,000×g) for 10 min at 4 °C. The mitochondrial pellet was isolated, washed, and suspended in isotonic buffer and used for biochemical assay. Furthermore, the purity of the mitochondria isolated from the frontal cortex and hippocampus was assessed by running separate blots of voltage-dependent anion channel (VDAC), a mitochondrial protein, and β-actin, a cytosolic protein.
Assay of Mitochondrial Complexes in Brain Regions
To assess complex I (NADH-ferricyanide reductase) activity, ferricyanide was used as the electron acceptor following the method of Hatefi et al. [75]. Briefly, the reaction mixture (final volume, 1 ml) contained phosphate buffer (50 mM, pH 7.4), NADH (0.17 mM), ferricyanide (0.6 mM), and Triton X-100 (0.1 %, v/v) at 30 °C. Mitochondrial suspension (10–30 μg protein) was added to the reaction mixture in the cuvette to start the reaction. The rate of oxidation of NADH was assessed using a spectrophotometer at 340 nm. The activity of complex I enzyme has been expressed as nanomoles of NADH oxidized per minute per milligram protein.
For the assay of complex II–III (succinate-cytochrome c reductase) activity, reduction of ferricytochrome c to ferrocytochrome c was monitored in the presence of succinate following the method described by Clark et al. [76]. The assay mixture in a total volume of 1 ml contained phosphate buffer (100 mM), succinate (2 mM), KCN (1 mM), EDTA (0.3 mM), and cytochrome c (1.2 mg/ml). Mitochondrial suspension (around 10–30 μg) was added to initiate the reaction and reduction of ferricytochrome c was monitored at 550 nm. The activity of complexes II–III has been expressed as nanomoles of oxidized cytochrome c reduced per minute per milligram protein.
The activity of complex IV (cytochrome c oxidase) was assessed by monitoring the oxidation of reduced cytochrome c (ferrocytochrome c) following standard procedure [77]. Briefly, ferricyanide (1 mM) in phosphate buffer (10 mM, pH 7.4) was added to oxidized ferrocytochrome c in a final volume of 1 ml at room temperature. The reaction was initiated by adding mitochondrial suspension (around 10–30 μg protein) and the rate of oxidation was recorded at 550 nm. The activity of complex IV has been expressed as nanomoles of reduced cytochrome c oxidized per minute per milligram protein.
Estimation of Reactive Oxygen Species
The generation of ROS in the frontal cortex and hippocampus was assessed following the method of Rush et al. [78]. Briefly, mitochondrial suspension either from the frontal cortex or hippocampus was incubated with DCFH-DA dye (10 μl, 100 μM final concentration) at room temperature for 30 min. DCFH-DA is oxidized to fluorescent DCF by intracellular ROS during the course of reaction. The generation of ROS was measured using a fluorescence reader at excitation 485 nm/emission 520 nm, and the results are expressed as percent of the control.
Assessment of Mitochondrial Membrane Potential
The MMP in the frontal cortex and hippocampus was estimated using JC-1 dye as it is a sensitive indicator to assess change in fluorescence from red to green following standard procedure [79]. Briefly, mitochondrial cell suspension from the hippocampus and frontal cortex was incubated with JC-1 dye (10 μM) at 37 °C for 15 min and washed with phosphate-buffered saline (PBS). The cells were finally suspended in 0.5 ml PBS (106 cells/ml) and analyzed by flow cytometer (FACS CantoTM II, BD Bio-Sciences, San Jose, CA, USA).
Immunoexpression of Choline Acetyltransferase, Acetylcholinesterase, PKCβ1, and Key Proteins Associated with Apoptosis and MAP Kinase Signaling
Immunoexpression of selected proteins was assessed by Western blotting following the procedure described by Jamal et al. [80]. Briefly, the frontal cortex/hippocampus was lysed and equal amount of protein (30 μg protein/lane) was electrophoresed on SDS-PAGE (12 %) and transferred onto nitrocellulose membrane followed by blocking with buffer containing BSA (5 %). The membrane was incubated overnight at 4 °C with antibodies—acetylcholinesterase (AChE; 1:1000, Millipore), choline acetyltransferase (ChAT; 1:1000, Sigma), protein kinase C (PKC) β1 (1:1000, Sigma), Bcl-2 (1:1000, CST), Bax (1:1000, CST), caspase-3 (1:1000, CST), pJNK1/2 and pJNK3 (1:1000, CST), pp38 (1:1000, Sigma), AP1 (1:1000, Sigma), cytochrome C (Cyt C; 1:1000, Millipore), and β-actin (1:1000, CST) followed by incubation with horseradish peroxidase-linked secondary antibody (anti-mouse IgG, 1:4000; anti-rabbit IgG, 1:4000) at room temperature for 60 min and detected by the chemiluminescent method. Densitometric measurements of bands in the immunoblots were carried out using digital gel image analysis system (ImageQuant LAS 500) and normalized by β-actin to correct variations, if any, in protein loading.
Histological Studies
Histological studies were carried out in the hippocampus and frontal cortex using Nissl staining as described by Veena et al. [81]. Briefly, thin sections of the hippocampus and frontal cortex were cut from the brain using Cryotome (Microm, HM520 USA). The sections were stained with cresyl violet (0.1 %) and dehydrated through graded series of alcohol. Finally, the sections were coverslipped with DPX mounting media and the intensity of Nissl-stained neurons was determined using a computerized image analysis system (ImageJ).
Ultrastructural Studies
For ultrastructural studies, a separate set of rats was used. Rats were anesthetized with ketamine (30 mg/kg) and perfused with paraformaldehyde (4 %) and glutaraldehyde (0.1 %). The perfused brain was dissected into the frontal cortex and hippocampus and cut into fine pieces (approx. 2 mm). Primary fixation of sections was carried out for 2 h in glutaraldehyde (2.5 %) prepared in sodium cacodylate buffer (0.1 M, pH 7.2). Subsequently, post-fixation was carried out in osmium tetraoxide (1 %) for 1–2 h followed by dehydration and embedding in araldite and DDSA medium. The tissues were baked at 65 °C for 48 h and cut into thin sections (60–90 nm) using ultramicrotome (Leica EM UC 67). The thin sections on copper mesh grids were stained with uranyl acetate and lead citrate (2 %) for contrast. Examination of the brain sections was carried out through transmission electron microscope (Tecnai G2 spirit transmission electron microscope equipped with Gatan CCD/Orius camera at 60 KV).
Protein Estimation
Protein content was determined following the method of Lowry et al. [82] using BSA as a reference standard.
Statistical Analysis
One-way analysis of variance using the GraphPad Prism software was used to analyze the data. Average of five or three observations were taken for behavioral and neurochemical parameters for analyzing the data statistically. To assess the levels of significance comparing all the pair of columns, Newman–Keuls test was employed and values up to p < 0.05 considered significant. Values have been expressed as the mean ± SEM. While studying the expression of certain proteins, especially caspase and PKCβ1 by Western blotting, experiments were repeated twice.
Results
Effect on Learning and Memory
As cholinergic dysfunctions are associated with impairment in learning and memory, both spatial memory and learning were assessed using shuttle box and Y-maze, respectively.
Effect on Spatial Memory and Continuous Alternation
To assess the effect of cadmium on spatial memory, percentage of alternation was monitored using Y-maze. A significant impairment (F (3,16) = 4.505, 43 %, p < 0.05) in alternation was observed in rats on cadmium exposure as compared to those in the control group. Simultaneous exposure with quercetin in cadmium-exposed rats resulted to causing an improvement in alternation as compared to rats treated with cadmium (F (3,16) = 4.505, 50 %, p < 0.05) alone. No significant changes in continuous alternation were observed in rats treated with quercetin alone (Fig. 1a).

Effect on spatial learning and memory assessed by the Y-maze following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. a Continuous alternation test. b Percent entries in other/novel arms. c Percent time spent in other/novel arms. Values are the mean ± SEM of five animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. In Fig. 1b, c compared to novel vs other arm. CONT control, Cd cadmium, QUR quercetin
Furthermore, effect on the novelty seeking behavior on cadmium exposure and protective effect of quercetin was also assessed by Y-maze. Percent entries and time spent in the novel arm and other arms were found to be similar in rats on cadmium exposure (p > 0.05). Rats simultaneously exposed to cadmium and quercetin were found to spend more time in the novel arm, with an increase in percentage of entries (p < 0.05) as compared to those treated with cadmium alone (Fig. 1b, c). Furthermore, frequency of visits and time spent in the novel arm were found to be higher (p < 0.01) as compared to other arms in rats in the control and quercetin-exposed groups.
Effect on Passive Avoidance Response
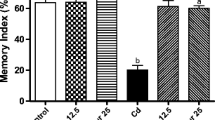
Exposure of rats to cadmium resulted to decrease transfer latency time (TLT) in the retention trials (p < 0.05) as compared to the acquisition trials. These rats crossed into the dark chambers early and thus indicate impairment in learning and memory. Simultaneous treatment with quercetin in cadmium-treated rats was found to protect these changes, as evidenced by the increase in the transfer latency time (p < 0.01) in the retention trials as compared to cadmium-treated rats. Interestingly, the transfer latency time was found to be increased in rats in the control (p < 0.001) and quercetin-exposed (p < 0.001) groups, suggesting that there was no impairment in learning and memory. Furthermore, no significant difference in the transfer latency time on the acquisition trial was observed in rats in any of the treatment groups (Fig. 2).

Effect on learning and memory assessed by shuttle box following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of five animals in each group. Significantly different **p < 0.05, ***p < 0.01, as compared to acquisition trial. CONT control, Cd cadmium, QUR quercetin
Neurochemical Studies
Effect on the Binding of Cholinergic–Muscarinic Receptors and Expression of Cholinergic–Muscarinic Receptor Genes in the Frontal Cortex and Hippocampus
A significant decrease in the binding of 3H-QNB was observed both in the frontocortical (F (3,16) = 4.963, 40 %, p < 0.05) and hippocampal (F (3,16) = 17.25, 56 %, p < 0.001) membranes of rats on cadmium exposure as compared to the controls. Alteration in the binding was found to be due to the decreased number of binding sites (B max), with no significant effect on the affinity (K d) in both the brain regions as revealed by Scatchard analysis (Table 1). Interestingly, simultaneous exposure to quercetin was found to protect cadmium-induced decrease in the binding of cholinergic–muscarinic receptors both in the frontal cortex (F (3,16) = 4.963, 42 %, p < 0.05) and hippocampus (F (3,16) = 17.25, 56 %, p < 0.05). No significant effect on the binding of cholinergic–muscarinic receptors was observed in either of the brain regions of rats exposed to quercetin alone as compared to rats in the control group (Fig. 3).

Effect on 3H-QNB binding in the frontocortical and hippocampal membranes of rats following exposure to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of five animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
The impact of cadmium exposure on the mRNA expression of the cholinergic receptor gene in the frontal cortex and hippocampus was assessed by qRT-PCR. Cadmium exposure for 28 days in rats caused a decrease in the expressions of CHRM1 (F (3,8) = 6.471, 55 %, p < 0.01; F (3,8) = 11.45, 54 %, p < 0.01); CHRM2 (F (3,8) = 9.107, 45 %, p < 0.01; F (3,8) = 10.98, 58 %, p < 0.01); CHRM3 (F (3,8) = 0.7009, 15 %, p > 0.05; F (3,8) = 0.8996, 18 %, p > 0.05); CHRM4 (F (3,8) = 6.839, 45 %, p < 0.05; F (3,8) = 5.606, 46 %, p < 0.05); and CHRM5 (F (3,8) = 1.296, 16 %, p > 0.05; F (3,8) = 1.125, 20 %, p > 0.05) receptor genes both in the frontal cortex and hippocampus as compared to the controls. Interestingly, changes in the mRNA expressions of CHRM1 (F (3,8) = 6.471; F (3,8) = 11.45, 95 %, p < 0.05); CHRM2 (F (3,8) = 9.107, 93 %, p < 0.01; F (3,8) = 10.98, 92 % p < 0.01); CHRM3 (F (3,8) = 0.7009, 78 %, p < 0.05; 21 %); CHRM4 (F (3,8) = 6.839, 70 %, p < 0.05); and CHRM5 (F (3,8) = 1.296, 82 %, p < 0.01; 31 %, p > 0.05) genes on cadmium exposure were protected in rats on simultaneous treatment with quercetin both in the frontal cortex and hippocampus (Fig. 4a, b).

Effect on the expression of muscarinic–cholinergic receptor genes in the frontal cortex (a) and hippocampus (b) following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
Effect on the Expression of Choline Acetyltransferase and Acetylcholinesterase in the Frontal Cortex and Hippocampus
The effect of cadmium on the expression of ChAT, a marker for the integrity of cholinergic neurons, and AChE, an enzyme involved in the metabolism of acetylcholine, and the protective efficacy of quercetin were also assessed both in the frontal cortex and hippocampus. Exposure of rats to cadmium resulted to decreased mRNA expressions of ChAT and AChE in the frontal cortex (F (3,8) = 5.479, 42 %, p < 0.05; F (3,8) = 8.528, 50 %, p < 0.01) and hippocampus (F (3,8) = 11.25, 55 %, p < 0.05; F (3,8) = 10.91, 60 %, p < 0.01) as compared to the controls, indicating alterations in the cholinergic system. Contrary to this, simultaneous exposure with quercetin in cadmium-exposed rats caused significant protection in the mRNA expressions of ChAT and AChE both in the frontal cortex (F (3,8) = 5.479, 82 % p < 0.01; F (3,8) = 8.528, 68 %, p < 0.05) and hippocampus (F (3,8) = 11.25, 56 %, p < 0.05; F (3,8) = 10.91, 74 %, p < 0.01) as compared to those exposed to cadmium alone (Fig. 5a).

Effect on the expressions of ChAT and AChE genes (a) and their proteins (b) following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01,***p < 0.001. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin, ChAT choline acetyltransferase, AChE acetylcholinesterase
A decrease in the protein expressions of ChAT and AChE in the frontal cortex (F (3,8) = 8.594, 1.74-fold, p < 0.01; F (3,8) = 19.65, 1.65-fFold, p < 0.01) and hippocampus (F (3,8) = 8.740, 1.61-fold, p < 0.01; F (3,8) = 28.69, 1.58-fold, p < 0.001) was observed in rats on cadmium exposure as compared to the controls. Simultaneous exposure to cadmium and quercetin was found to cause an increase in the expressions of ChAT and AChE proteins both in the frontal cortex (F (3,8) = 8.594, 1.38-fold, p < 0.05; F (3,8) = 19.65, 1.42-fold, p < 0.05) and hippocampus (F (3,8) = 8.740, 1.20-fold, p < 0.05; F (3,8) = 28.69, 1.54-fold, p < 0.01) as compared to rats exposed to cadmium alone. No significant change in the mRNA and protein expressions of ChAT and AChE was observed either in the frontal cortex or hippocampus of rats exposed to quercetin alone as compared to the controls (Fig. 5a).
Effect on the Expression of PKCβ1
Exposure to cadmium in rats significantly decreased the expression of PKCβ1, an important protein involved in synaptic signaling both in the frontal cortex (F (3,8) = 18.83, 1.55-fold, p < 0.001) and hippocampus (F (3,8) = 28.72, 1.40-fold, p < 0.001) as compared to rats in the control group. Simultaneous exposure with quercetin in cadmium-exposed rats was found to protect the changes on the expression of PKCβ1 both in the frontal cortex (F (3,8) = 18.83, 1.28-fold, p < 0.05) and hippocampus (F (3,8) = 28.72, 1.70-fold, p < 0.01) as compared to cadmium-exposed rats. No significant change in the expression of PKCβ1 was observed either in the frontal cortex or hippocampus of rats exposed to quercetin alone compared to the controls (Fig. 6).

Effect on the expression of PKCβ1 following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
Effect of Cadmium or Quercetin or their Co-treatment in Rats on Mitochondrial Dysfunctions
To understand the molecular mechanisms underlying cadmium-induced cholinergic deficits and the protective potential of quercetin in cholinergic neurons, the focus was on mitochondrial integrity and associated activation of caspase cascade which leads to neuronal cell death. The purity of the mitochondria was found to be high both in the frontal cortex and hippocampus. Expression of VDAC, a mitochondrial protein, was high in the mitochondrial pellet, while the expression of β-actin was found to be negligible (Fig. 8a).
Effect on the Activity of Mitochondrial Complexes
The effect of cadmium on the activity of enzyme complexes involved in the electron transport chain and cellular bioenergetics and the protective efficacy of quercetin were assessed in the frontal cortex and hippocampus, the cholinergic-rich areas of the brain. Exposure to cadmium in rats for 28 days resulted to decreased activity of complex I (F (3,16) = 4.697, 30 %, p < 0.05; F (3,16) = 4.611, 28 %, p < 0.05), complexes II–III (F (3,16) = 27.87, 58 %, p < 0.001; F (3,16) = 12.25, 53 %, p < 0.01), and complex IV (F (3,16) = 1.935, 22 %, p < 0.05; F (3,16) = 4.892, 26 %, p < 0.05) both in the frontal cortex and hippocampus as compared to the controls. Interestingly, cadmium-induced decrease in the activity of mitochondrial complexes was found to be protected as evident from the increase in the activity of complex I (F (3,16) = 4.697, 28 %, p < 0.05; F (3,16) = 4.611, 18 %, p < 0.05), complexes II–III (F (3,16) = 27.87, 71 %, p < 0.01; F (3,16) = 12.25, 55 %, p < 0.05), and complex IV (F (3,16) = 1.935, 17 %, p < 0.05; F (3,16) = 4.892, 28 %, p < 0.05) in rats simultaneously treated with quercetin. No significant change in the activity of any of the complexes was observed either in the frontal cortex or hippocampus treated with quercetin as compared to the controls (Fig. 7).

Effect on the activity of mitochondrial complexes in the frontal cortex and hippocampus following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of five animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. a Complex I activity. b Complex II–III activity. c Complex IV activity. CONT control, Cd cadmium, QUR quercetin
Effect on the Generation of Reactive Oxygen Species
Exposure of rats to cadmium resulted to increase the generation of ROS in the frontal cortex (F (3,16) = 9.577, 69 %, p < 0.01) and hippocampus (F (3,16) = 10.36, 75 %, p < 0.01), as evidenced by the enhanced fluorescence of DCHF dye as compared to rats in the control group (Fig. 8). Simultaneous exposure to quercetin in cadmium-exposed rats was found to protect ROS generation both in the frontal cortex (F (3,16) = 9.577, 37 %, p < 0.01) and hippocampus (F (3,16) = 10.36, 28 %, p < 0.01) as compared to rats treated with cadmium alone. No significant change in ROS levels was observed both in the frontal cortex and hippocampus of rats treated with quercetin alone as compared to control rats (Fig. 8).

Purity of the mitochondria isolated from the brain frontal cortex and hippocampus (a) and effect on reactive oxygen species generation (b) following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days Values are the mean ± SEM of five animals in each group. Significantly different: *p < 0.05, **p < 0.01, ***p < 0.001. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
Effect on Mitochondrial Membrane Potential
Cadmium exposure for 28 days resulted to cause a significant impairment in the mitochondria containing green JC-1 dye detected at the FL1 channel. Functional mitochondria with healthy cells displayed red JC-1-aggregates and were detected at the FL2 channel. Exposure of rats to cadmium increased the percentage of depolarized cells, suggesting a decrease in the mitochondrial membrane potential in the frontal cortex (F (3,8) = 14.52, 14.10 %, p < 0.01) and hippocampus (F (3,8) = 30.28, 12.47 %, p < 0.001) as compared to the controls. Simultaneous exposure to cadmium and quercetin was found to attenuate the percentage of depolarized cells in the frontal cortex (F (3,8) = 14.52, 8.4 %, p < 0.01) and hippocampus (F (3,8) = 30.28, 7.7 %, p < 0.01), suggesting an increase in the mitochondrial membrane potential as compared to rats exposed to cadmium alone (Fig. 9). No significant change in the mitochondrial membrane potential was observed in the frontal cortex and hippocampus of rats exposed to quercetin as compared to the controls.

Effect on the mitochondrial membrane potential following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of five animals in each group. Significantly different: **p < 0.05, ***p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. Representative dot plot of individual experiment in the frontal cortex (a) and in the hippocampus (b). CONT control, Cd cadmium, QUR quercetin, FC frontal cortex, HP hippocampus
Effect on the Expression of Pro- and Anti-apoptotic Proteins
Exposure of rats to cadmium resulted to an increased expression of Bax, a pro-apoptotic protein, in the frontal cortex (F (3,8) = 4.967, 1.34-fold, p < 0.01) and hippocampus (F (3,8) = 19.58, 2.08-fold, p < 0.001) associated with a decrease in the expression of Bcl2, an anti-apoptotic protein, both in the frontal cortex (F (3,8) = 13.97, 1.54-fold, p < 0.01) and hippocampus (F (3,8) = 32.42, 1.49-fold, p < 0.001) as compared to control rats. Increase in the ratio of Bax/Bcl2 was distinct both in the frontal cortex and hippocampus, suggesting enhanced apoptosis following cadmium exposure. An increase in the expression of caspase-3, an executer protein, was also evident in the frontal cortex (F (3,8) = 5.671, 1.93-fold, p < 0.05) and hippocampus (F (3,8) = 7.811, 2.40-fold, p < 0.01) on cadmium exposure in rats. Simultaneous exposure of rats to cadmium and quercetin resulted to a decreased expression of Bax in the frontal cortex (F (3,8) = 4.967, 1.84-fold, p < 0.05) and hippocampus (F (3,8) = 19.58, 1.71-fold, p < 0.01) and increased expression of Bcl-2 in the frontal cortex (F (3,8) = 13.97, 1.44-fold, p < 0.05) and hippocampus (F (3,8) = 32.42, 1.59-fold, p < 0.01) as compared to cadmium-exposed rats. A decrease in the expression of caspase-3 in the frontal cortex (F (3,8) = 5.671, 1.82-fold, p < 0.05) and hippocampus (F (3,8) = 7.811, 1.76-fold, p < 0.05) was also evident in cadmium-treated rats simultaneously exposed to quercetin. No significant change in the expression of any of these proteins was observed either in the frontal cortex or hippocampus of rats exposed to quercetin as compared to rats in the control group (Fig. 10).

Effect on the expression of pro-, anti-apoptotic, and executer proteins in the frontal cortex and hippocampus of rats following exposure to cadmium, quercetin, and their co-exposure for 28 days Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin. Ratio of Bax/Bcl2 was determined to quantitate apoptosis
Effect on the Expression of Cytochrome C
Release of Cyt C from the inner mitochondrial spaces is the main key event in caspase-mediated apoptotic cell death. The effect of cadmium on cytochrome C release from the mitochondria was assessed through protein expression using Western blotting techniques. Cadmium exposure significantly increased the expression of Cyt C protein in the frontal cortex (F (3,8) = 41.13, 1.70-fold, p < 0.001) and hippocampus (F (3,8) = 54.62, 2.25-fold, p < 0.001) as compared to the controls. Simultaneous exposure to cadmium and quercetin was found to decrease the expression of Cyt C protein both in the frontal cortex (F (3,8) = 41.13, 1.75-fold, p < 0.01) and hippocampus (F (3,8) = 54.62, 1.73-fold, p < 0.001) as compared to rats treated with cadmium alone. No significant change in Cyt C expression was observed both in the frontal cortex and hippocampus of rats exposed to quercetin alone as compared to the controls (Fig. 11).

Effect on the expression of Cyt C in the frontal cortex and hippocampus following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
Effect on the Expression of MAPK Proteins
To further understand the mechanism of cadmium-induced apoptotic cell death, we further assessed the expression of MAP kinase proteins following cadmium exposure and the protective efficacy of quercetin in the frontal cortex and hippocampus. Exposure to cadmium resulted to the increased expressions of AP1, pp38, pJNK1/2, and pJNK3 both in the frontal cortex (F (3,8) = 9.465, 1.59-fold, p < 0.01; F (3,8) = 4.430, 1.25-fold, p < 0.05, F (3,8) = 3.962, 1.82-fold, p < 0.05; F (3,8) = 25.73, 1.80-fold, p < 0.001; F (3,8) = 10.43, 1.38-fold, p < 0.01) and in the hippocampus (F (3,8) = 18.71, 1.85-fold, p < 0.001; F (3,8) = 15.88, 1.49-fold, p < 0.01; F (3,8) = 17.82,1.94-fold, p < 0.01; F (3,8) = 25.83, 1.6-fold, p < 0.001; F (3,8) = 8.412, 1.52-fold, p < 0.01) as compared to the controls. Simultaneous exposure to cadmium and quercetin was found to decrease the expressions of AP1, p38, JNK1/2, and JNK3 proteins in the frontal cortex (F (3,8) = 9.465, 1.79-fold, p < 0.05; F (3,8) = 4.430, 1.90-fold, p < 0.05; F (3,8) = 3.962, 1.70-fold, p < 0.05; F (3,8) = 25.73, 1.84-fold, p < 0.01; F (3,8) = 10.43, 1.81-fold, p < 0.05) and hippocampus (F (3,8) = 18.71, 1.75-fold, p < 0.01; F (3,8) = 15.88, 1.87-fold, p < 0.05; F (3,8) = 17.82¸ 1.64-fold, p < 0.05; F (3,8) = 25.83, 1.71-fold, p < 0.01; F (3,8) = 8.412, 1.80-fold, p < 0.01) in comparison to rats exposed to cadmium alone (Fig. 12). No significant change in the expression of any of these proteins was observed either in the frontal cortex or hippocampus of rats exposed to quercetin as compared to rats in the control group (Figs. 12 and 13).

Effect on the expression of pp38 and AP1 in the frontal cortex and hippocampus following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin

Effect on the expression of stress proteins following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Values are the mean ± SEM of three animals in each group. Significantly different: *p < 0.05, **p < 0.01, ***p < .001. a Compared to the control group. b Compared to the cadmium-exposed group. CONT control, Cd cadmium, QUR quercetin
Histological Studies
Severe degeneration of neurons in the frontal cortex (F (3,8) = 63.01, 1.72-fold, p < 0.01) and the dentate gyrus area of the hippocampus (F (3,8) = 87.55, 1.65-fold, p < 0.001) was clearly visible in rats on exposure to cadmium for 28 days in comparison to the controls. Loss of synapse or the loss of neuron in the cholinergic-rich area is the major event of any cholinergic dysfunction and associated functional changes. A trend of recovery in the frontal cortex (F (3,8) = 63.01, 1.49-fold, p < 0.01) and dentate gyrus (F (3,8) = 87.55, 1.32-fold, p < 0.001) was evident in cadmium-exposed rats on simultaneous exposure with quercetin as compared to rats treated with cadmium alone. There was no significant change in the neuron density in any of the brain regions of rats exposed to quercetin alone as compared to rats in the control group (Fig. 14).

Photomicrographs of the frontocortical (a) and hippocampal sections (b) illustrating Nissl staining following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Fold change exhibits percent area of degenerated neurons. Values are the mean ± SEM of three sections from each group. Significantly different: **p < 0.05, ***p < 0.001. a Compared to the control group. b Compared to the cadmium-exposed group. Scale bar, 100 μm. CONT control, Cd cadmium, QUR quercetin
Effect of Cadmium, Quercetin, and their Co-treatment on Ultrastructural Changes
Cadmium exposure in rats resulted to disrupt the ultrastructures both in the frontal cortex and hippocampus as compared to rats in the control group. A marked deterioration in the hippocampus and frontal cortex, as visualized by loss in cell organelles, vacuole formation in the cytoplasm, mitochondrial damage, swollen mitochondria with loss in cristae (cracked or missing cristae), along with disruption in the mitochondrial membrane in unmyelinated axons, was observed on cadmium exposure. Furthermore, loss of myelin sheath was also clearly visible both in the frontal cortex and in the hippocampus on cadmium exposure as compared to the control rats. Interestingly, simultaneous exposure with quercetin was found to protect cadmium-induced ultrastructural changes and preserve mitochondrial integrity. Restoration in mitochondria with improvement in electron density and cristae structure was clearly evident. Furthermore, mitochondria with complete mitochondrial membrane and cristae were observed in rats simultaneously treated with cadmium and quercetin (Fig. 15).

Effect on ultrastructural changes in the frontal cortex and hippocampus following exposure of rats to cadmium, quercetin, and their co-exposure for 28 days. Focus is on the mitochondria, and electron micrographs (×30,000) and inset view (×110,000) have been presented. a, e Control group of the frontal cortex and hippocampus showing normal anatomy of unmyelinated and myelinated neurons with well-developed mitochondria. b, f Cadmium-exposed group exhibits loss in cell organelles and mitochondrial damage. c, g Quercetin-exposed group shows normal anatomy like the controls. d, h Simultaneous exposure to cadmium and quercetin exhibits protection
Discussion
A number of studies have been carried out to decipher the molecular mechanisms of cadmium neurotoxicity. The dose of cadmium used in these studies ranges from 1 to 10 mg/kg body weight, involving different routes of exposure including oral/intragastric, i.p., or through drinking water. The duration of exposure also varies from 4 to 45 days to assess the short- or long-term effect of cadmium on behavioral and neurochemical endpoints [37, 44, 83–86]. As most of the studies have been carried out at a dose of 5 mg/kg body weight of cadmium [30, 84, 87, 88], we selected the same dose in the present study. Furthermore, to assess the neuroprotective efficacy of quercetin, studies have been carried out at different doses (5–100 mg/kg body weight, p.o.) in experimental models of neurotoxicity and neurodegenerative diseases [44, 61, 89–93]. The dose, duration, and route of exposure vary in these studies and are largely dependent on the experimental design. In most of these experimental studies, quercetin has been found to exert neuroprotective potential at the dose of 25 mg/kg body weight [61, 93–95], and thus, we also opted the same dose in the present study. Furthermore, the duration of exposure in the present study was for 28 days to assess sub-chronic effects.
In view of the vulnerability of cholinergic neurons to cadmium, studies have been carried out to understand its impact on cholinergic receptors, AChE activity, and other parameters associated with the integrity of cholinergic functions. Decrease in AChE activity in the cerebral cortex and hippocampus, increase in hypothalamus, and no change in the striatum and cerebellum were observed on cadmium exposure (2.5 mg/kg body weight for 45 days) in rats [44]. In another study, Abdalla et al. [46] found an increase in synaptic AChE activity in rats exposed to cadmium (2.5 mg/kg body weight) for 45 days. Furthermore, an increase in brain AChE activity was also found in rats exposed to cadmium (1 mg/kg body weight) either intraperitoneally or intramuscularly for 14 days or 4 months, respectively [45, 96]. Cadmium-induced decrease in AChE activity has been reported in vitro [97]. While there are evidences exhibiting that cadmium exposure may cause alterations in brain AChE activity, consistent changes have not been observed. Variations in AChE activity on cadmium exposure have been largely attributed to differences in the dose, duration, and route of exposure. Use of different animal species, cellular preparations, approaches either in vivo or in vitro, and assay protocols could also be associated with the variability in the results [98]. Of the various factors suggested to be associated with the alteration of AChE activity, deactivation of the enzymatic site directly or occupation of the active site of the enzyme by cadmium are quite convincing [99]. It has been found that cadmium may affect the cholinergic transmission by inhibiting the activity of ChAT, a marker for the integrity of cholinergic neurons [100]. Del Pino et al. [101] found a dose-dependent decrease in ChAT activity in SN56 cells on cadmium exposure in vitro. Inhibition in the activity of ChAT was associated with a decrease in the synthesis and release of acetylcholine, an important neurotransmitter in brain cholinergic transmission [102, 103].
There are five distinct subtypes of muscarinic–cholinergic receptors which are distributed differentially in brain regions and play important roles in modulating pharmacological functions. The presence of M1, M2, and M4 cholinergic receptors has been found to be more in the hippocampus and cortex, and their role in modulating cognitive functions is well demonstrated in experimental and clinical studies [104]. Exposure to cadmium resulted to decreased muscarinic–cholinergic receptors in rat brain both in vivo and in vitro [105]. It was found that cadmium may inhibit the sensitivity of M1 receptors as the pharmacological effect in response to acetylcholine and pirenzapine, a selective antagonist for M1 receptors, was decreased. In an interesting study, del Pino et al. [76] found that cadmium-induced cell death in septal SN56 basal forebrain cholinergic neurons is mediated through the inhibition of M1 receptors. Furthermore, cadmium-induced inhibition on M1 receptors was associated with the overexpression of GSK-3b and AChE-S and decreased expression of AChE-R [101]. Effect on other muscarinic–cholinergic receptor subtypes, which could have given a complete picture of the effect of cadmium, was not studied by them. In the present study, a significant decrease in the mRNA expressions of M1, M2, and M4 and no change in M3 and M5 cholinergic receptors were observed both in the hippocampus and frontal cortex. Decrease in the binding of 3H-QNB, known to label muscarinic–cholinergic receptors in the hippocampus and frontal cortex, as observed in the present study, is consistent with transcriptional changes associated with the decreased mRNA expressions of M1, M2, and M4 cholinergic receptors. It is further interesting that the transcriptional changes in ChAT and AChE in the present study are consistent with the translational changes on cadmium exposure and, thus, exhibit the vulnerability of cholinergic neurons. The decreased expression of AChE in the frontal cortex and hippocampus on cadmium exposure in the present study indicate reduced activity and may have contributed to enhancing the acetylcholine levels, resulting in the downregulation of cholinergic–muscarinic receptors. Furthermore, reduction in the expression of ChAT both in the frontal cortex and hippocampus on cadmium exposure may be due to auto feedback regulation as a result of increased acetylcholine levels.
The role of brain cholinergic receptors in modulating learning and memory is well established [106]. Loss of cholinergic neurons associated with selective loss of AChE and ChAT activities and alterations in muscarinic–cholinergic receptors in the hippocampus and frontal cortex has been found to impair learning and memory [107–109]. Both the frontal cortex and hippocampus are cholinergic-rich areas, and their role in modulating learning and memory is well documented [110, 111]. Furthermore, muscarinic–cholinergic receptors which are important in regulating memory circuits are highly distributed in both these brain areas [106, 112]. In view of this, the present study is focused to assess the protective potential of quercetin in cadmium-induced cholinergic alterations in the hippocampus and frontal cortex.
Several isoforms of PKCs are distributed in the brain, and their role in modulating learning and memory is well accepted [113, 114]. Based on molecular cloning, 12 isoforms of PKC identified are broadly classified into three major classes—conventional (cPKCα, cPKCβ, cPKCβII, and cPKCγ), novel (nPKCδ, nPKCμ, nPKCε, nPKCθ, and nPKCη), and atypical (aPKCk and aPKCλ). It has been found that activation of PKC modulates learning involving conventional PKC isoforms. PKCα and PKCβ/βII are largely postsynaptic, while PKCγ is also presynaptic [115]. The role of PKCβ1 is well established in modulating learning and memory [116–118]. Involvement of PKCβ1 in early synaptic events and in modulating avoidance learning has also been demonstrated [118]. In an interesting study, a decrease in PKCβ resulted to impaired learning in mice [116]. Moreover, decreases in brain muscarinic–cholinergic receptors and expression of PKCβ1 on exposure of rats to arsenic or those subjected to stress and exposed to lambda-cyhalothrin have been correlated with decreased learning and memory [117, 119, 120]. Impairment in spatial memory and learning as assessed by the Y-maze and passive avoidance task in cadmium-exposed rats in the present study may be linked to decreased brain cholinergic–muscarinic receptors and expression of PKCβ1 and increased oxidative stress.
Like other heavy metals, cadmium may affect the integrity of the mitochondria, as observed by the decrease in the mitochondrial membrane potential and ATP levels. While studying the effect on mitochondrial functions, Belyaeva et al. [83] found that cadmium inhibited the FCCP-uncoupled respiration. Furthermore, cadmium resulted to cause mitochondrial membrane permeabilization associated with alterations in the activity of mitochondrial complexes, which are involved in modulating ETC [46]. Alterations in mitochondrial bioenergetics, important for energy production through oxidative phosphorylation, may enhance ROS generation, suggested to be one of the potential mechanisms in cadmium neurotoxicity. It has been found that cadmium-induced ROS generation may enhance lipid peroxidation in cortical neurons [32]. Alterations in the activity of antioxidant enzymes, which could counteract the generation of free radical species on cadmium exposure, have also been reported and associated with enhanced oxidative stress [121]. Thus, the increased ROS generation associated with impairment in the activity of complexes I–IV and decreased mitochondrial membrane potential on cadmium exposure in the frontal cortex and hippocampus in the present study are consistent with these reports and suggest that cadmium may affect mitochondrial integrity and associated functions.
Cadmium, being pro-oxidant in nature, has been found to enhance the expression of Bax, a pro-apoptotic protein, and Bcl2, an anti-apoptotic protein. Activation of caspase-3, an executor protein and a crucial mediator in apoptosis, has also been observed either independently by activating the death receptor pathway or releasing cytochrome C. It has been found that ERKs, JNKs, and p38 are important constituents of MAP kinases and play important roles in modulating cellular processes, including differentiation, survival, and apoptosis [52]. Of the three isoforms of JNKs identified, JNK3 is specific to neuronal cells while JNK1/2 is distributed in other tissues [122]. JNKs are stress-activated proteins and are known to trigger apoptotic signals by modulating the expression of mitochondrial pro- and anti-apoptotic proteins either by phosphorylation or indirectly modulating pro-apoptotic proteins by transactivation of the transcription factor c-Jun on N-terminal Ser-63 and Ser-73. Furthermore, JNK-dependent mitochondrial apoptosis requires the activation of JNK at phosphorylation sites at Thr-183 and Tyr-185 and is involved in neuronal death. While investigating the mechanism of cadmium-induced oxidative stress in pancreatic beta cells, cadmium was found to target the mitochondrial-dependent apoptotic pathway [47]. Enhanced release of Cyt C associated with the decreased expression of Bcl-2 resulted to activating caspases and affecting mitochondrial functions. Furthermore, enhanced oxidative stress was found to affect downstream JNK signaling and contributed to apoptosis. Cadmium-induced neuronal death has also been observed via targeting the JNK and PTEN-Akt/mTOR network [123]. In the present study, activation of pJNK and pp38 in the hippocampus and frontal cortex on cadmium exposure appears to contribute to the activation of transcription factor AP1. Interestingly, activation of JNK may cause Bax translocation associated with the release of Cyt C and contribute to neuronal apoptosis on cadmium exposure, as evident in the present study.
Ultrastructural and morphological changes in the brain have been frequently reported on exposure to environmental chemicals [124]. Blebbing of the mitochondria and damaged mitochondrial cristae, as observed on cadmium exposure, may be responsible for mitochondrial dysfunctions. Loss of synapses both in the hippocampus and frontal cortex further indicates that ultrastructural integrity could be affected on cadmium exposure. Furthermore, abnormalities in mitochondria in the frontal cortex and hippocampus, the cholinergic-rich areas in the brain, exhibit their vulnerability on cadmium exposure. Consistent with this, decreased Nissl staining in the hippocampus and frontal cortex provides histological evidence of damage of neurons in these cholinergic-rich areas of the brain on cadmium exposure.
Quercetin is preferred over other flavonoids in view of its broad pharmacological spectrum due to its strong radical scavenging potential and high antioxidant activity associated with anti-inflammatory and metal-chelating effects [125]. The protective role of quercetin in cardiovascular and metabolic disorders has been reported by a number of investigators and is associated with decreased oxidative stress [126]. In view of the fact that quercetin crosses the blood–brain barrier, a number of studies have been carried out to assess its protective efficacy in experimental models of neurological disorders and chemical-induced neurotoxicity. Interestingly, quercetin was found to attenuate the neurotoxic effects of rotenone and 6-OHDA in hemiparkinsonian rats by inhibiting oxidative stress [63]. Quercetin was found to reduce tauopathy and beta-amyloidosis and improve learning and memory in a triple-transgenic mouse model of Alzheimer’s disease due to a decrease in oxidative stress [127]. Brain dopaminergic dysfunctions due to polychlorinated biphenyl-induced oxidative stress were also found to be attenuated by quercetin in rats [90]. The protective role of quercetin in aluminum-induced cholinergic deficits was found to be associated with decreased oxidative damage [91]. The metal chelating property of quercetin was associated with decreased levels of aluminum in brain and resulted to inhibiting oxidative damage in these studies [94]. Unsal et al. [128] also found that quercetin may protect cadmium-induced neuronal damage by inhibiting oxidative stress and apoptosis. The protective effect of quercetin was associated with scavenging of hydroxyl radicals associated with an increase in the capacity of antioxidant enzymes. Alteration in the activity of brain AChE on cadmium exposure was found to be protected by quercetin earlier [64]. Although effect on other important targets of the cholinergic system was not studied, the pharmacological restoration in AChE activity by quercetin was attributed to its antioxidant potential.
As the role of mitochondrial dysfunctions in the etiology of neurodegenerative diseases is well demonstrated, a number of studies have been carried out to assess the effect of quercetin on mitochondrial bioenergetics. Quercetin has been found to improve mitochondrial dysfunctions in a rat model of Huntington’s disease induced by nitropropionic acid, as evidenced by reversal in the activity of mitochondrial complexes associated with decreased oxidative stress [61]. Chakraborty et al. [95] further found that quercetin could protect from inflammatory changes in a rat model of Huntington’s disease, as evidenced by the decreased microglia proliferation and increased astrocytic numbers, although striatal lesion induced by nitropropionic acid was persistent. Quercetin at a low dose was found to enhance mitochondrial biogenesis both in the brain and muscles associated with increased maximal endurance and physical running activity in mice. Based on this, it was suggested that the use of quercetin may be explored in neurodegenerative and metabolic disorders involving mitochondrial dysfunctions [94]. Interestingly, quercetin was found to upregulate complex I activity in the mitochondria in rotenone-induced hemiparkinsonian rats [129]. In the present study, cadmium-induced alterations in brain cholinergic receptors and expression of ChAT and AChE were protected on simultaneous treatment with quercetin. The protective effect on brain cholinergic targets appears to be associated with the antioxidant potential of quercetin which could reduce ROS generation and protect the integrity of the mitochondria, as evidenced by the increase in the activity of mitochondrial complexes I–IV and mitochondrial membrane potential. Furthermore, preserving the integrity of the mitochondria may have reduced oxidative stress. The protective changes of quercetin may further be attributed to its chelating effect, resulting to decreased cadmium levels both in the hippocampus and frontal cortex, as observed by us in an ongoing study.
Quercetin has been found to modulate the mitochondrial-dependent apoptotic pathway. While investigating the protective effect of quercetin in focal cerebral ischemia in rats, Yao et al. [130] found increased expression of Bcl-2 and decreased expression of Bax and cleaved caspase-3 proteins in cortex, suggesting decreased apoptosis. It was found that quercetin enhanced the levels of BDNF and Trk-β which could stimulate the PI3K/AKT neuronal survival pathway. Quercetin was also found to protect oligodendrocyte progenitor cells from oxygen glucose deprivation-induced apoptosis in vitro. Enhanced expressions of Bax and caspase-3 and decreased expression of Bcl-2 due to oxygen glucose deprivation were found to activate the PI3K/AKT signaling pathway [131]. Programmed cell death in hemiparkinsonian rats was also found to be protected by quercetin. Simultaneous treatment with quercetin resulted to decreasing the expression of Bax and enhancing the expressions of Bcl-2 and caspase-3, which were found to be altered on cadmium exposure both in the hippocampus and frontal cortex. Furthermore, increases in the expressions of proteins associated with MAP kinase signaling, especially JNKs, pp38, and AP1, in the hippocampus and frontal cortex of rats on cadmium exposure were found to be reduced in rats simultaneously treated with quercetin, suggesting that quercetin may decrease apoptosis. Interestingly, quercetin has been found to exhibit anti-apoptotic properties and modulate MAPK signaling and the PI3/Akt pathway in a number of studies on chemical-induced neurotoxicity and experimental models of neurodegenerative diseases [61, 130, 131]. It is possible that in normal situations, quercetin maintains cellular homeostasis because of its antioxidant properties and thus may not interfere with the cell signaling pathways. However, in the case of neurotoxicity or in neurodegenerative state, it modulates pathways and molecular targets including those associated with MAP kinases, cell survival, and apoptosis. This could possibly be the reason that treatment with quercetin alone in rats had no effect on the levels of cytochrome c and expression of MAP kinases both in the hippocampus and frontal cortex in the present study.
It is further interesting to note that blebbing in the mitochondria and disrupted cristae in the hippocampus and frontal cortex of rats on cadmium exposure were found to be protected by quercetin. Also, loss of synapses as evident on cadmium exposure both in the hippocampus and frontal cortex, was found to be reduced, suggesting that quercetin could protect the ultrastructural integrity of the mitochondria.
While the role of the brain cholinergic system in regulating learning and memory is well accepted, quercetin has been found to protect spatial memory by increasing the antioxidant capacity [132]. Memory impairment by scopolamine was also found to be prevented by quercetin [133]. Decreased expression of PKCβ1 on cadmium exposure was also found to be protected by quercetin.
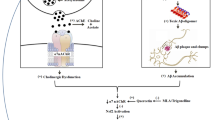
The results of the present study demonstrate that mitochondrial dysfunctions associated with enhanced oxidative stress and apoptosis significantly contribute to cadmium-induced brain cholinergic dysfunctions. More interestingly, the data provide evidence that quercetin has the ability to modulate the molecular targets involved in brain cholinergic signaling and attenuate cadmium-induced neurotoxicity (Fig. 16). Although the protective efficacy of quercetin appears to be attributed to its antioxidant capacity, the data have relevance to implore its protective potential in neurological and neurotoxicological disorders involving cholinergic dysfunctions.

Mechanism and targets associated with cadmium-induced cholinergic alterations and the protective potential of quercetin. Exposure to cadmium resulted to the decreased expression of ChAT and AChE and affected the integrity of cholinergic–muscarinic receptors associated with a decrease in the expression of PKCβ1, a postsynaptic signaling protein. Brain cholinergic alterations on cadmium exposure may be linked to learning and memory deficits in rats. Cadmium exposure also caused mitochondrial dysfunctions associated with enhanced ROS generation and apoptosis involving activation of caspase cascade and MAP kinases. Disruption in the ultrastructures and loss of neurons both in the frontal cortex and hippocampus were evident on cadmium exposure. Simultaneous treatment with quercetin in cadmium-exposed rats reduced ROS generation and protected mitochondrial integrity by modulating proteins involved in apoptosis and MAP kinase signaling and protected cholinergic integrity both in the frontal cortex and hippocampus
References
Bernhoft RA (2013) Cadmium toxicity and treatment. Sci World J 2013:7
Schoeters G, HOND ED, Zuurbier M, Naginiene R, HAZEL P, Stilianakis N, Ronchetti R, Koppe JG (2006) Cadmium and children: exposure and health effects. Acta Paediatr 95(s453):50–54
Satarug S, Garrett SH, Sens MA, Sens DA (2011) Cadmium, environmental exposure, and health outcomes. Ciencia Saude Coletiva 16(5):2587–2602
Méndez-Armenta M, Ríos C (2007) Cadmium neurotoxicity. Environ Toxicol Pharmacol 23(3):350–358
Wang B, Du Y (2013) Cadmium and its neurotoxic effects. Oxidative Med Cell Longev 2013:12
Khade SW, Adholeya A (2009) Arbuscular mycorrhizal association in plants growing on metal-contaminated and noncontaminated soils adjoining Kanpur tanneries, Uttar Pradesh, India. Water Air Soil Pollut 202(1–4):45–56
Radha RV, Kumutha K, Marimuthu P (2014) Assessment of cadmium contamination of soils in sewage disposal areas of Coimbatore district, Tamil Nadu, India. Curr World Environ 9(2):379–386
Borah K, Bhuyan B, Sarma H (2009) Heavy metal contamination of groundwater in the Tea Garden Belt of Darrang District, Assam, India. J Chem 6(S1):S501–S507
Simmons R, Pongsakul P, Saiyasitpanich D, Klinphoklap S (2005) Elevated levels of cadmium and zinc in paddy soils and elevated levels of cadmium in rice grain downstream of a zinc mineralized area in Thailand: implications for public health. Environ Geochem Health 27(5–6):501–511
Wang Y, Björn LO (2014) Heavy metal pollution in Guangdong Province, China, and the strategies to manage the situation. Front Environ Sci 2(9):1–12
Cai S, Yue L, Shang Q, Nordberg G (1995) Cadmium exposure among residents in an area contaminated by irrigation water in China. Bull World Health Organ 73(3):359–367
Satarug S, Moore MR (2004) Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect 112(10):1099–1103
Hayashi C, Koizumi N, Nishio H, Koizumi N, Ikeda M (2012) Cadmium and other metal levels in autopsy samples from a cadmium-polluted area and non-polluted control areas in Japan. Biol Trace Elem Res 145(1):10–22
Y-f C, J-f W, J-f C, Xiao-Ying W, Yang L, Guo Y-d (2012) An investigation and pathological analysis of two fatal cases of cadmium poisoning. Forensic Sci Int 220(1):e5–e8
Mari M, Nadal M, Schuhmacher M, Barbería E, García F, Domingo JL (2014) Human exposure to metals: levels in autopsy tissues of individuals living near a hazardous waste incinerator. Biol Trace Elem Res 159(1–3):15–21
Waalkes MP, Coogan TP, Barter RA (1992) Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit Rev Toxicol 22(3–4):175–201
Wong K-L, Klaassen CD (1982) Neurotoxic effects of cadmium in young rats. Toxicol Appl Pharmacol 63(3):330–337
Järup L, Berglund M, Elinder CG, Nordberg G, Vanter M (1998) Health effects of cadmium exposure—a review of the literature and a risk estimate. Scand J Work Environ Health 24:1–51
Pihl R, Parkes M (1977) Hair element content in learning disabled children. Science 198(4313):204–206
Thatcher R, Lester M, McAlaster R, Horst R (1982) Effects of low levels of cadmium and lead on cognitive functioning in children. Arch Environ Health: Int J 37(3):159–166
Marlowe M, Errera J, Jacobs J (1983) Increased lead and cadmium burdens among mentally retarded children and children with borderline intelligence. Am J Ment Defic 87(5):477–483
Okuda B, Iwamoto Y, Tachibana H, Sugita M (1998) Parkinsonism after acute cadmium poisoning. Occup Health Ind Med 5(38):232–265
Viaene M, Masschelein R, Leenders J, De Groof M, Swerts L, Roels H (2000) Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occup Environ Med 57(1):19–27
Viaene M, Roels H, Leenders J, De Groof M, Swerts L, Lison D, Masschelein R (1999) Cadmium: a possible etiological factor in peripheral polyneuropathy. Neurotoxicology 20(1):7–16
Choudhuri S, Liu WL, Berman NE, Klaassen CD (1996) Cadmium accumulation and metallothionein expression in brain of mice at different stages of development. Toxicol Lett 84(3):127–133
Shukla A, Shukla GS, Srimal R (1996) Cadmium-induced alterations in blood–brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum Exp Toxicol 15(5):400–405
Gonçalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG, Stefanello N, Rubin MA, Dressler VL, Morsch VM (2010) N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact 186(1):53–60
Zalups RK, Ahmad S (2003) Molecular handling of cadmium in transporting epithelia. Toxicol Appl Pharmacol 186(3):163–188
Attia A, Ibrahim F, EL-Latif NAA, Aziz SW (2014) Antioxidant effects of curcumin against cadmium chloride-induced oxidative stress in the blood of rats. J Pharmacogn Phytother 6(3):33–40
Shagirtha K, Muthumani M, Prabu SM (2011) Melatonin abrogates cadmium induced oxidative stress related neurotoxicity in rats. Eur Rev Med Pharmacol Sci 15(9):1039–1050
Nemmiche S, Chabane-Sari D, Guiraud P (2007) Role of α-tocopherol in cadmium-induced oxidative stress in Wistar rat’s blood, liver and brain. Chem Biol Interact 170(3):221–230
Lopez E, Arce C, Oset-Gasque M, Canadas S, Gonzalez M (2006) Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med 40(6):940–951
Monroe RK, Halvorsen SW (2006) Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med 41(3):493–502
Méndez-Armenta M, Villeda-Hernández J, Barroso-Moguel R, Nava-Ruı́ C, Jiménez-Capdeville ME, Rı́ C (2003) Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicol Lett 144(2):151–157
Hidalgo J, Aschner M, Zatta P, Vašák M (2001) Roles of the metallothionein family of proteins in the central nervous system. Brain Res Bull 55(2):133–145
Nishimura N, Nishimura H, Ghaffar A, Tohyama C (1992) Localization of metallothionein in the brain of rat and mouse. J Histochem Cytochem 40(2):309–315
Antonio M, Corpas I, Leret M (1999) Neurochemical changes in newborn rat’s brain after gestational cadmium and lead exposure. Toxicol Lett 104(1):1–9
Antonio MT, Corredor L, Leret ML (2003) Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicol Lett 143(3):331–340
Minami A, Takeda A, Nishibaba D, Takefuta S, Oku N (2001) Cadmium toxicity in synaptic neurotransmission in the brain. Brain Res 894(2):336–339
Andersson H, Petersson-Grawe K, Lindqvist E, Luthman J, Oskarsson A, Olson L (1997) Low-level cadmium exposure of lactating rats causes alterations in brain serotonin levels in the offspring. Neurotoxicol Teratol 19(2):105–115
Lafuente A, Fenàndez-Rey E, Seara R, Pérez-Lorenzo M, Esquifino A (2001) Alternate cadmium exposure differentially affects amino acid metabolism within the hypothalamus, median eminence, striatum and prefrontal cortex of male rats. Neurochem Int 39(3):187–192
Lafuente A, Gonzalez-Carracedo A, Romero A, Esquifino A (2003) Effect of cadmium on 24-h variations in hypothalamic dopamine and serotonin metabolism in adult male rats. Exp Brain Res 149(2):200–206
Ashok A, Rai NK, Tripathi S, Bandyopadhyay S (2015) Exposure to As-, Cd-, and Pb-mixture induces Aβ, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol Sci 143:64–80
Abdalla FH, Schmatz R, Cardoso AM, Carvalho FB, Baldissarelli J, de Oliveira JS, Rosa MM, Nunes MAG, Rubin MA, da Cruz IB (2014) Quercetin protects the impairment of memory and anxiogenic-like behavior in rats exposed to cadmium: possible involvement of the acetylcholinesterase and Na+, K+-ATPase activities. Physiol Behav 135:152–167
Carageorgiou H, Tzotzes V, Pantos C, Mourouzis C, Zarros A, Tsakiris S (2004) In vivo and in vitro effects of cadmium on adult rat brain total antioxidant status, acetylcholinesterase, (Na+, K+)‐ATPase and Mg2+-ATPase activities: protection by L‐cysteine. Basic Clin Pharmacol Toxicol 94(3):112–118
Belyaeva EA, Sokolova TV, Emelyanova LV, Zakharova IO (2012) Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: effects of cadmium, mercury, and copper. Sci World J 2012:14
Chang K-C, Hsu C-C, Liu S-H, Su C-C, Yen C-C, Lee M-J, Chen K-L, Ho T-J, Hung D-Z, Wu C-C (2013) Cadmium induces apoptosis in pancreatic β-cells through a mitochondria-dependent pathway: the role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS One 8(2), e54374
Beal MF (1996) Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol 6(5):661–666
Pearson G, Robinson F, Beers Gibson T, Xu B-e, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions 1. Endocr Rev 22(2):153–183
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81(2):807–869
Chen L, Liu L, Luo Y, Huang S (2008) MAPK and mTOR pathways are involved in cadmium‐induced neuronal apoptosis. J Neurochem 105(1):251–261
Chen L, Liu L, Huang S (2008) Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45(7):1035–1044
Leuner K, Hauptmann S, Abdel-Kader R, Scherping I, Keil U, Strosznajder JB, Eckert A, Müller WE (2007) Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxid Redox Signal 9(10):1659–1676
De Sarno P, Shestopal SA, King TD, Zmijewska A, Song L, Jope RS (2003) Muscarinic receptor activation protects cells from apoptotic effects of DNA damage, oxidative stress, and mitochondrial inhibition. J Biol Chem 278(13):11086–11093
Li X, Lv Y, Yu S, Zhao H, Yao L (2012) The effect of cadmium on Aβ levels in APP/PS1 transgenic mice. Exp Ther Med 4(1):125–130
Panayi A, Spyrou N, Iversen B, White M, Part P (2002) Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J Neurol Sci 195(1):1–10
Johnson S (2001) Gradual micronutrient accumulation and depletion in Alzheimer’s disease. Med Hypotheses 56(6):595–597
Dajas F, Rivera-Megret F, Blasina F, Arredondo F, Abin-Carriquiry J, Costa G, Echeverry C, Lafon L, Heizen H, Ferreira M (2003) Neuroprotection by flavonoids. Braz J Med Biol Res 36(12):1613–1620
Havsteen BH (2002) The biochemistry and medical significance of the flavonoids. Pharmacol Ther 96(2):67–202
Larson AJ, Symons JD, Jalili T (2010) Quercetin: a treatment for hypertension?—A review of efficacy and mechanisms. Pharmaceuticals 3(1):237–250
Sandhir R, Mehrotra A (2013) Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid: implications in Huntington’s disease. Biochim Biophys Acta (BBA)-Mol Basis Dis 1832(3):421–430
Yang T, Kong B, Gu J-W, Kuang Y-Q, Cheng L, Yang W-T, Xia X, Shu H-F (2014) Anti-apoptotic and anti-oxidative roles of quercetin after traumatic brain injury. Cell Mol Neurobiol 34(6):797–804
Haleagrahara N, Siew CJ, Mitra NK, Kumari M (2011) Neuroprotective effect of bioflavonoid quercetin in 6-hydroxydopamine-induced oxidative stress biomarkers in the rat striatum. Neurosci Lett 500(2):139–143
Abdalla FH, Cardoso AM, Pereira LB, Schmatz R, Gonçalves JF, Stefanello N, Fiorenza AM, Gutierres JM, da Silva Serres JD, Zanini D (2013) Neuroprotective effect of quercetin in ectoenzymes and acetylcholinesterase activities in cerebral cortex synaptosomes of cadmium-exposed rats. Mol Cell Biochem 381(1–2):1–8
Glowinski J, Iversen LL (1966) Regional studies of catecholamines in the rat brain—I. J Neurochem 13(8):655–669
Wang W, Li S, Dong H-p, Lv S, Y-y T (2009) Differential impairment of spatial and nonspatial cognition in a mouse model of brain aging. Life Sci 85(3):127–135
Yamada K, Noda Y, Hasegawa T, Komori Y, Nikai T, Sugihara H, Nabeshima T (1996) The role of nitric oxide in dizocilpine-induced impairment of spontaneous alternation behavior in mice. J Pharmacol Exp Ther 276(2):460–466
Yadav RS, Chandravanshi LP, Shukla RK, Sankhwar ML, Ansari RW, Shukla PK, Pant AB, Khanna VK (2011) Neuroprotective efficacy of curcumin in arsenic induced cholinergic dysfunctions in rats. Neurotoxicology 32(6):760–768
Khanna VK, Husain R, Seth PK (1994) Effect of protein malnutrition on the neurobehavioural toxicity of styrene in young rats. J Appl Toxicol 14(5):351–356
Singh A, Mudawal A, Shukla RK, Yadav S, Khanna VK, Sethumadhavan R, Parmar D (2014) Effect of gestational exposure of cypermethrin on postnatal development of brain cytochrome P450 2D1 and 3A1 and neurotransmitter receptors. Mol Neurobiol 52(1):741–756
Morley BJ, Warr WB, Rodriguez–Sierra J (2004) Transient expression of acetylcholinesterase in the posterior ventral cochlear nucleus of rat brain. J Assoc Res Otolaryngol 5(4):391–403
Madziar B, Shah S, Brock M, Burke R, Lopez‐Coviella I, Nickel AC, Cakal EB, Blusztajn JK, Berse B (2008) Nerve growth factor regulates the expression of the cholinergic locus and the high‐affinity choline transporter via the Akt/PKB signaling pathway. J Neurochem 107(5):1284–1293
Borges MO, Abreu ML, Porto CS, Avellar MCW (2001) Characterization of muscarinic acetylcholine receptor in rat Sertoli cells. Endocrinology 142(11):4701–4710
Bagh MB, Maiti AK, Jana S, Banerjee K, Roy A, Chakrabarti S (2008) Quinone and oxyradical scavenging properties of N-acetylcysteine prevent dopamine mediated inhibition of Na+, K+-ATPase and mitochondrial electron transport chain activity in rat brain: implications in the neuroprotective therapy of Parkinson’s disease. Free Radic Res 42(6):574–581
Hatefi Y (1978) Preparation and properties of NADH: ubiquinone oxidoreductase (complex I), EC 1.6. 5.3. Methods Enzymol 53:11–16
Clark J, Bates T, Boakye P, Kuimov A, Land J (1997) Investigation of mitochondrial defects in brain and skeletal muscle. In: Turner AJ, Bachelard HS (eds) Neurochemistry: a practical approach. Oxford University Press, Oxford, pp 151–174
Wharton DC (1967) Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 10:245–250
Rush JW, Quadrilatero J, Levy AS, Ford RJ (2007) Chronic resveratrol enhances endothelium-dependent relaxation but does not alter eNOS levels in aorta of spontaneously hypertensive rats. Exp Biol Med 232(6):814–822
Kane CJ, Chang JY, Roberson PK, Garg TK, Han L (2008) Ethanol exposure of neonatal rats does not increase biomarkers of oxidative stress in isolated cerebellar granule neurons. Alcohol 42(1):29–36
Jamal M, Ameno K, Ameno S, Morishita J, Wang W, Kumihashi M, Ikuo U, Miki T, Ijiri I (2007) Changes in cholinergic function in the frontal cortex and hippocampus of rat exposed to ethanol and acetaldehyde. Neuroscience 144(1):232–238
Veena J, Srikumar B, Mahati K, Raju T, Rao BS (2011) Oxotremorine treatment restores hippocampal neurogenesis and ameliorates depression-like behaviour in chronically stressed rats. Psychopharmacology 217(2):239–253
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Kim W, Kim DW, Yoo DY, Jung HY, Nam SM, Kim JW, Hong S-M, Kim D-W, Choi JH, Moon SM (2014) Dendropanax morbifera Léveille extract facilitates cadmium excretion and prevents oxidative damage in the hippocampus by increasing antioxidant levels in cadmium-exposed rats. BMC Complement Altern Med 14(1):428
El-Demerdash FM, Yousef MI, Kedwany FS, Baghdadi HH (2004) Cadmium-induced changes in lipid peroxidation, blood hematology, biochemical parameters and semen quality of male rats: protective role of vitamin E and β-carotene. Food Chem Toxicol 42(10):1563–1571
Haider S, Anis L, Batool Z, Sajid I, Naqvi F, Khaliq S, Ahmed S (2015) Short term cadmium administration dose dependently elicits immediate biochemical, neurochemical and neurobehavioral dysfunction in male rats. Metab Brain Dis 30(1):83–92
Winiarska-Mieczan A (2015) The potential protective effect of green, black, red and white tea infusions against adverse effect of cadmium and lead during chronic exposure—a rat model study. Regul Toxicol Pharmacol 73(2):521–529
Gong D, Liu B, Tan X (2015) Genistein prevents cadmium-induced neurotoxic effects through its antioxidant mechanisms. Drug Res 65(2):65–69
Karaca S, Eraslan G (2013) The effects of flaxseed oil on cadmium-induced oxidative stress in rats. Biol Trace Elem Res 155(3):423–430
Halder S, Kar R, Mehta AK, Bhattacharya SK, Mediratta PK, Banerjee BD (2016) Quercetin modulates the effects of chromium exposure on learning, memory and antioxidant enzyme activity in F1 generation mice. Biol Trace Elem Res 171:391–398
Bavithra S, Selvakumar K, Kumari RP, Krishnamoorthy G, Venkataraman P, Arunakaran J (2012) Polychlorinated biphenyl (PCBs)-induced oxidative stress plays a critical role on cerebellar dopaminergic receptor expression: ameliorative role of quercetin. Neurotox Res 21(2):149–159
Sharma DR, Wani WY, Sunkaria A, Kandimalla RJ, Verma D, Cameotra SS, Gill KD (2013) Quercetin protects against chronic aluminum-induced oxidative stress and ensuing biochemical, cholinergic, and neurobehavioral impairments in rats. Neurotox Res 23(4):336–357
Tota S, Awasthi H, Kamat PK, Nath C, Hanif K (2010) Protective effect of quercetin against intracerebral streptozotocin induced reduction in cerebral blood flow and impairment of memory in mice. Behav Brain Res 209(1):73–79
Joseph KD (2015) Combined oral supplementation of fish oil and quercetin enhances neuroprotection in a chronic rotenone rat model: relevance to Parkinson’s disease. Neurochem Res 40(5):894–905
Davis JM, Murphy EA, Carmichael MD, Davis B (2009) Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Phys Regul Integr Comp Phys 296(4):R1071–R1077
Chakraborty J, Singh R, Dutta D, Naskar A, Rajamma U, Mohanakumar KP (2014) Quercetin improves behavioral deficiencies, restores astrocytes and microglia, and reduces serotonin metabolism in 3‐nitropropionic acid‐induced rat model of Huntington’s disease. CNS Neurosci Ther 20(1):10–19
Carageorgiou H, Tzotzes V, Sideris A, Zarros A, Tsakiris S (2005) Cadmium effects on brain acetylcholinesterase activity and antioxidant status of adult rats: modulation by zinc, calcium and L‐cysteine co‐administration. Basic Clin Pharmacol Toxicol 97(5):320–324
Gkanti V, Stolakis V, Kalafatakis K, Liapi C, Zissis KM, Zarros A, Tsakiris S (2014) Postnuclear supernatants of rat brain regions as substrates for the in vitro assessment of cadmium-induced neurotoxicity on acetylcholinesterase activity. Biol Trace Elem Res 158(1):87–89
Zarros A, Kalopita K, Tsakiris S, Baillie GS (2013) Can acetylcholinesterase activity be considered as a reliable biomarker for the assessment of cadmium-induced neurotoxicity? Food Chem Toxicol 56:406–410
Casalino E, Sblano C, Landriscina C (1997) Enzyme activity alteration by cadmium administration to rats: the possibility of iron involvement in lipid peroxidation. Arch Biochem Biophys 346(2):171–179
Dwivedi C (1983) Cadmium-induced sterility: possible involvement of the cholinergic system. Arch Environ Contam Toxicol 12(2):151–156
Del Pino J, Zeballos G, Anadon MJ, Capo MA, Díaz MJ, García J, Frejo MT (2014) Higher sensitivity to cadmium induced cell death of basal forebrain cholinergic neurons: a cholinesterase dependent mechanism. Toxicology 325:151–159
Hayashi H, Takayama K (1978) Inhibitory effects of cadmium on the release of acetylcholine from cardiac nerve terminals. Jpn J Physiol 28(3):333–345
Alberts P, Ögren V, Sellström Å (1985) Cadmium inhibition of [3H]acetylcholine secretion in guinea‐pig ileum myenteric plexus. Acta Physiol Scand 124(2):313–316
Abrams P, Andersson KE, Buccafusco JJ, Chapple C, Groat WC, Fryer AD, Kay G, Laties A, Nathanson NM, Pasricha PJ (2006) Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol 148(5):565–578
Hedlund B, Gamarra M, Bartfai T (1979) Inhibition of striatal muscarinic receptors in vivo by cadmium. Brain Res 168(1):216–218
Levey AI (1996) Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci 93(24):13541–13546
Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PG (2011) The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-β42 with memantine. Behav Brain Res 221(2):594–603
O’Reilly RC, Rudy JW (2001) Conjunctive representations in learning and memory: principles of cortical and hippocampal function. Psychol Rev 108(2):311
Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221(2):555–563
Whishaw IQ (1989) Dissociating performance and learning deficits on spatial navigation tasks in rats subjected to cholinergic muscarinic blockade. Brain Res Bull 23(4–5):347–358
Jarrard LE (1993) On the role of the hippocampus in learning and memory in the rat. Behav Neural Biol 60(1):9–26
Van der Zee E, Luiten P (1999) Muscarinic acetylcholine receptors in the hippocampus, neocortex and amygdala: a review of immunocytochemical localization in relation to learning and memory. Prog Neurobiol 58(5):409–471
Bank B, DeWeer A, Kuzirian AM, Rasmussen H, Alkon DL (1988) Classical conditioning induces long-term translocation of protein kinase C in rabbit hippocampal CA1 cells. Proc Natl Acad Sci 85(6):1988–1992
Alkon DL, Sun M-K, Nelson TJ (2007) PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol Sci 28(2):51–60
Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S (1993) Modified hippocampal long-term potentiation in PKCγ-mutant mice. Cell 75(7):1253–1262
Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD (2000) A role for the β isoform of protein kinase C in fear conditioning. J Neurosci 20(16):5906–5914
Wu J, Song T-B, Li Y-J, He K-S, Ge L, Wang L-R (2007) Prenatal restraint stress impairs learning and memory and hippocampal PKCbeta1 expression and translocation in offspring rats. Brain Res 1141:205–213
Paratcha G, Furman M, Bevilaqua L, Cammarota M, Vianna M, de Stein ML, Izquierdo I, Medina JH (2000) Involvement of hippocampal PKCβI isoform in the early phase of memory formation of an inhibitory avoidance learning. Brain Res 855(2):199–205
Shukla RK, Gupta R, Srivastava P, Dhuriya YK, Singh A, Chandravanshi LP, Kumar A, Siddiqui MH, Parmar D, Pant AB (2015) Brain cholinergic alterations in rats subjected to repeated immobilization or forced swim stress on lambda-cyhalothrin exposure. Neurochem Int 0186(15):30081–30084
Chandravanshi LP, Yadav RS, Shukla RK, Singh A, Sultana S, Pant AB, Parmar D, Khanna VK (2014) Reversibility of changes in brain cholinergic receptors and acetylcholinesterase activity in rats following early life arsenic exposure. Int J Dev Neurosci 34:60–75
Kumar R, Agarwal AK, Seth PK (1996) Oxidative stress-mediated neurotoxicity of cadmium. Toxicol Lett 89(1):65–69
Jing L, Anning L (2005) Role of JNK activation in apoptosis: a double-edged sword. Cell Res 15(1):36–42
Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q, Guo M, Huang S, Chen L (2014) Celastrol prevents cadmium‐induced neuronal cell death via targeting JNK and PTEN‐Akt/mTOR network. J Neurochem 128(2):256–266
L-l L, J-l Z, Z-w Z, H-d Y, Sun G, Xu S-w (2014) Protective roles of selenium on nitric oxide-mediated apoptosis of immune organs induced by cadmium in chickens. Biol Trace Elem Res 159(1–3):199–209
Morikawa K, Nonaka M, Narahara M, Torii I, Kawaguchi K, Yoshikawa T, Kumazawa Y, Morikawa S (2003) Inhibitory effect of quercetin on carrageenan-induced inflammation in rats. Life Sci 74(6):709–721
Rivera L, Morón R, Sánchez M, Zarzuelo A, Galisteo M (2008) Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity 16(9):2081–2087
Ansari MA, Abdul HM, Joshi G, Opii WO, Butterfield DA (2009) Protective effect of quercetin in primary neurons against Aβ(1–42): relevance to Alzheimer’s disease. J Nutr Biochem 20(4):269–275
Unsal C, Kanter M, Aktas C, Erboga M (2015) Role of quercetin in cadmium-induced oxidative stress, neuronal damage, and apoptosis in rats. Toxicol Ind Health 31:1106–1115. doi:10.1177/0748233713486960
Karuppagounder S, Madathil S, Pandey M, Haobam R, Rajamma U, Mohanakumar K (2013) Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience 236:136–148
Yao R-Q, Qi D-S, Yu H-L, Liu J, Yang L-H, Wu X-X (2012) Quercetin attenuates cell apoptosis in focal cerebral ischemia rat brain via activation of BDNF–TrkB–PI3K/Akt signaling pathway. Neurochem Res 37(12):2777–2786
Wang X-Q, Yao R-Q, Liu X, Huang J-J, Qi D-S, Yang L-H (2011) Quercetin protects oligodendrocyte precursor cells from oxygen/glucose deprivation injury in vitro via the activation of the PI3K/Akt signaling pathway. Brain Res Bull 86(3):277–284
Sun SW, Yu HQ, Zhang H, Zheng YL, Wang JJ, Luo L (2007) Quercetin attenuates spontaneous behavior and spatial memory impairment in d-galactose-treated mice by increasing brain antioxidant capacity. Nutr Res 27(3):169–175
Richetti S, Blank M, Capiotti K, Piato A, Bogo M, Vianna M, Bonan C (2011) Quercetin and rutin prevent scopolamine-induced memory impairment in zebrafish. Behav Brain Res 217(1):10–15
Acknowledgments
The study has been carried out as a part of the INDEPTH programme of the Council of Scientific and Industrial Research (CSIR), New Delhi. Richa Gupta and Pranay Srivastava acknowledges the support of CSIR, New Delhi, for providing Research Fellowship. The authors thank the Director, CSIR–Indian Institute of Toxicology Research (CSIR-IITR), Lucknow, for his interest in the present study and providing necessary support. Technical assistance provided by Mr. B.S. Pandey is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gupta, R., Shukla, R.K., Chandravanshi, L.P. et al. Protective Role of Quercetin in Cadmium-Induced Cholinergic Dysfunctions in Rat Brain by Modulating Mitochondrial Integrity and MAP Kinase Signaling. Mol Neurobiol 54, 4560–4583 (2017). https://doi.org/10.1007/s12035-016-9950-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9950-y