
Abstract
The performance of crop plants is critically affected by biotic and abiotic stress. These stressors threaten food availability by reducing overall crop yield and productivity. Changes in chromatin state by epigenetic modification are part of plant adaptive and survival responses and are considered pivotal for improving agronomic traits. Epigenetics is an exciting field that involves heritable gene expression changes that do not require changes in DNA sequence. Epigenetic modification is well known as a crucial player in plant phenotypic diversity and defense against pathogens. Hence, there is a growing interest in unlocking the epigenome for crop improvement. Herein, we highlight the epigenetic modifications implicated in plant biotic stress response and their contributions to important agronomic traits. We also discussed adopting epigenetics to expand phenotypic diversity and produce desired characteristics in crop plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the United Nations’ 2030 envisioned sustainable development goals is the alleviation of hunger, termed “End Hunger,” a process achievable through sustainable food production (Nations and Affairs 2015). Food availability is highly challenged by the constant adjustments in environmental and climatic conditions. An additional significant challenge for crop plants is biotic stress elicited by pests and pathogens. These factors directly or indirectly affect crop yield, development, growth, and nutritional values, thereby threatening food security. For instance, pathogens and pests alone account for approximately 30% of the global yield loss of major staple crops (Savary et al. 2019). In the mid-twentieth century, banana commercialization was drastically reduced due to the epidemic of banana Fusarium wilt disease (Dita et al. 2018). In addition, to yield loss, pests and diseases reduce the quality of crops and make plants serve as vectors of food pathogens (Rizzo et al. 2021). Hence, the management of pests and diseases remains indispensable for crop improvement and sustainable food production.
In the course of rising food security, the use of fertilizers and pesticides was adopted; however, their contribution to environmental hazards and health risks limited their application; thus, a better alternative become very necessary (Aktar et al. 2009; Nicolopoulou-Stamati et al. 2016). Although genomic selection and genetic engineering are currently making waves in improving agronomic phenotypes and disease resistance, those approaches rely on alterations of DNA sequence (Datta and Security 2013). Therefore, to conserve genetic sequence and prevent the potential loss of essential genes, epigenetics emerged as a powerful source of phenotypic diversity that only influences gene function (Deans and Maggert 2015). The term epigenetics was coined by Conrad Waddington, denoting “in addition to genetics” (Tronick and Hunter 2016). It is a heritable alteration in gene expression that does not result from changes in DNA sequences. Natural epigenetic variation can occur spontaneously due to errors in the epigenetic maintenance system. It can also be triggered by environmental factors such as stress and genomic changes, including transposon insertion and paramutation (Springer and Schmitz 2017; Angers et al. 2020).
Epigenetics can be artificially induced using chemical agents and a targeted or non-targeted mutation in the epigenetic machinery (Springer and Schmitz 2017). Plants show robust epigenetic variation, which accounts for their phenotypic plasticity, i.e., their ability to produce varying phenotypes from one genotype (Agarwal et al. 2020, Sun et al. 2021). DNA methylation, histone modification, and RNA silencing are critical epigenetic mechanisms that underpin several phenotypic changes in the plant. These epigenetic modifications have been found to pass across generations, creating a platform for exploiting the epigenetic changes for improved crop production (Akimoto et al. 2007; He and Li 2018; Zhi and Chang 2021). Studies have shown that epigenetic modifications contribute to important agronomic phenotypes and act as key players in plant defense responses against pathogens (Gupta and Salgotra 2022; Hannan Parker et al. 2022). To this end, a detailed understanding of epigenetics’ involvement in disease resistance and other important agronomic traits remains indispensable for its adoption in growing resilient crops that meet agronomic demands.
Epigenetics shapes the plant immune/defense systems
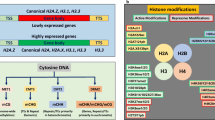
Plants have a long history of interaction with pathogenic soil microbes. This interaction takes the form of an action-reaction response, described by Jones and Dangl as a zigzag model, whereby the action of a pathogen triggers a plant immune reaction for survival (Jones and Dangl 2006). The primary line of defense in plant immunity is the pathogen/microbe-associated molecular pattern (PAMP or MAMP)-triggered immunity (PTI). PTI is elicited by the recognition of specific molecular patterns (such as bacteria flagellin, peptidoglycan, lipo-oligosaccharides, and elongation factor, TU; fungi chitin and xylanase; oomycetes glucan) by pattern-recognition receptors (PRR) localized on the plant plasma membrane (Boller and Felix 2009; Zipfel and Robatzek 2010; Choi and Klessig 2016). The perception of PAMP activates downstream signaling components, including mitogen-activated protein kinase (MAPK), reactive oxygen species (ROS), and defense hormones (salicylic acid, ethylene, and jasmonic acid) for the elimination of invading pathogens (Ramirez-Prado et al. 2018; Saijo et al. 2018). Pathogens strategize and secrete effector molecules that subvert PTI (Göhre and Robatzek 2008). Plants, in turn, counter this effector activity through the secondary defense line, effector-triggered immunity (ETI). Plants use their resistance (R) gene product, particularly the intracellular nucleotide binding and leucine-rich repeat (NLR) receptor, to detect pathogen effectors and elicit ETI that induce activation of disease-responsive genes for pathogen resistance (McDowell and Simon 2008; Cui et al. 2015). Plant immunity and defense response are fine-tuned by epigenetic mechanisms. Evidence has shown that defense-associated hormone signaling, transcription factors, and defense-responsive gene expression are under tight epigenetic control (Zhu et al. 2016; Ramirez-Prado et al. 2018). Plants utilize this epigenetic control to balance active disease resistance and fitness costs associated with constitutive activation of immune the response (Richard et al. 2018a, b). Table 1 summarizes several epigenetic changes that shape the defense response.
DNA methylation in plant defense framework
DNA methylation is an epigenetic mechanism involving the methylation of the DNA base, cytosine in the Carbon-5 position. It occurs in three DNA sequence contexts: CG, CHG, and CHH (where H could be A, C, or T) (Cokus et al. 2008; Zhang et al. 2018). In plants, de novo DNA methylation is triggered by small interfering RNA (siRNA), mediated via the RNA-directed DNA polymerase (RdDP) pathway, and catalyzed by domain-rearranged DNA methyltransferase 2 (DRM2) (Erdmann and Picard 2020; Gallego-Bartolomé 2020). The canonical RdDP process involves the synthesis of 24-nucleotide small interfering RNA (siRNA) by the combined activity of Pol IV, RNA-dependent RNA polymerase 2 (RDRP2), and dicer-like 3 (DCL3) (Zhang et al. 2018). The siRNAs are loaded into Argonaute (AGO) proteins and bind complementary Pol V RNA transcript for directing DNA methylation Liu et al. 2018a, b). While DRM2 catalyzes de novo DNA methylation, other enzymes such as methyltransferase 1 (MET1), Chromomethylase 3 (CMT3), and CMT2 maintain DNA methylation in the CG, CHG, and CHH contexts, respectively (Erdmann and Picard 2020). Notably, DRM2 also plays a crucial role in CHH DNA methylation maintenance (He et al. 2011). The removal of the C-5 methyl group on cytosine of the DNA sequence (demethylation) in plants is mediated by DNA glycosylase members such as Demeter (DME), Repressor of silencing 1 (ROS1), Demeter-like 2 (DML2) and DML3 through base excision repair mechanisms (Li et al. 2018).
Myriads of studies have shown that DNA (de)methylation regulates the expression of defense responsiveness. As DNA methylation is attributed to gene silencing and DNA demethylation to gene activation, it is found that treatment of rice plants with DNA methylase inhibitors such as 5-azacytidine or 5-aza-deoxycytidine causes activation of disease-resistance genes and the development of disease resistance in rice plants (Akimoto et al. 2007; Atighi et al. 2020). In the same line, studies in the model plant, Arabidopsis thaliana reveal that mutant strains bearing defective DNA demethylation exhibit compromised MAMP-triggered immunity, resulting in susceptibility to bacteria pathogens (Yu et al. 2013; Huang et al. 2022). In contrast, mutants with impaired RdDM and DNA methylation maintenance exhibit high resistance to the Pseudomonas syringae pv tomato DC300 (pst) (biotrophic pathogen) compared to the wild type (Dowen et al. 2012; Yu et al. 2013). Correspondingly, salicylic acid (SA) signaling-related genes, including pathogenesis-related (PR) genes are downregulated in former mutants and upregulated in the later mutants, demonstrating the crucial role of DNA demethylation in PTI and activation of defense-responsive genes. More so, partners in the RdDM pathway, including AGO4, PiolV, and DCL, have also been shown to be involved in plant immunity (Agorio and Vera 2007; Zhu et al. 2016).
Genome-wide DNA methylation profiling in plants shows that DNA methylation is conserved mainly in transposable elements (TE) and repeat sequences and correlates with the silencing of nearby genes in cis (Bender 2004; Tirnaz and Batley 2019a, b). Hence, (de)methylation of promoter TE of defense-related genes upon biotic stress promotes their expression/suppression to modulate immune response, as shown in Fig. 1. For instance, TE insertion upstream of ZmCCT, a resistance gene for maize Gibberella stalk rot disease, causes enrichment of methylated GC, resulting in ZmCCT suppression and disease susceptibility (Wang et al. 2017a, b, c). DNA demethylases mainly target the promoter transposons and repeat elements of the stress-responsive genes, triggering their activation for enhanced disease resistance (Le et al. 2014). Studies show that the promoter transposable and repeat elements of Xa21G (Oryzae resistance gene), RMG1 (Arabidopsis bacteria resistance gene), and RLP43 gene (Orphan immune receptor gene) are hypomethylated and expressed upon pathogen infection for enhanced disease resistance (Akimoto et al. 2007; Halter et al. 2021). DNA methylation of the promoter in the Miniature inverted-repeat TEs (MITEs) of PigmS (susceptible to rice blast, Magnaporthe oryzae) represses PigmS expression. This methylation hinders PigmS-mediated inhibition of PigmR, a gene known to confer resistance to rice blast diseases (Zhai et al. 2017). Moreover, the constitutive expression of PigmR has an associated yield loss; hence, DNA methylation-induced regulation of PigmS expression is essential to balance the PigmR-incurred yield cost and associated disease resistance (Zhai et al. 2017). Although promoter cytosine methylation is often attributed to the repression of defense-responsive genes, it is crucial to note that this does not apply to all cases. The enhanced expression of the rice blast resistance gene, pib, under hypermethylation of the promoter region (Li et al. 2011) is a typical example revealing that DNA methylation of the promoter sequence can also function as a positive regulator of defense gene expression.

Epigenetic involvement in plant immune response and disease resistance. Biotic stress induces epigenetic modifications at defense-related genes, resulting in gene expression changes and increased disease resistance
In contrast to promoter TE DNA methylation, the expression of genes bearing cytosine methylation in the gene body is context-dependent, where CG methylation positively regulates gene expression and CHG/CHH methylation negatively regulates gene expression (Zhang et al. 2006; You et al. 2012; Wang et al. 2017a; b, c). CG methylation is predominant in the gene body, however, whole-genome methylation analysis of the NLR genes of common bean (Phaseolus vulgaris) shows surprisingly high gene body methylation in the three sequences, CG, CHG, and CHH, correlating with low NLR gene expression (Richard et al. 2018a, b). In general, it can be deduced that DNA (de)methylation regulates several aspects of plant immune response to biotic stress. With the growing evidence of target manipulation of DNA methylome and their transgenerational inheritance, DNA methylation represents a promising approach for improving disease-resistance phenotype of crop plants.
Histone modification regulates plant immunity
DNA is packaged in the chromatin as a bead-like repeat unit called nucleosome, comprising eight core histone proteins (two H2A/H2B dimers and one H3/H4 tetramer) in which DNA bases wrap (Jansen and Verstrepen 2011). An external linker histone H1 binds nucleosome 10 bp at both entry and exit site of the core nucleosome to form the chromatosome complex (van Holde and Zlatanova 2007; Cutter and Hayes 2015). The function and architecture of the chromatin system is regulated by post-translational modification in histone tails. Histone modification entails adding one or more chemical groups, including acetyl, methyl, ubiquitin, phosphoryl, SUMO, carbonyl, and glycosyl, to the histone tail by histone writers. These modifications regulate the function of chromatin by determining the transcriptional conditions of genes (Kouzarides 2007; Gelato and Fischle 2008). Acetylation and methylation of core histone proteins are the key chromatin modifications that regulate plant defense against biotic and abiotic stress (Kumar 2018; Varotto et al. 2020). Histone ubiquitination and Linker histone H1 have also been implicated in stress response at the epigenetic level (Rutowicz et al. 2015; Zarreen et al. 2022).
Histone acetylation and defense response
The histone writer, histone acetyltransferase (HAT), is responsible for adding an acetyl mark to the lysine residue of histone. In contrast, histone deacetylase (HDAC) is the eraser that removes the acetyl mark from histone. Histone acetylation is associated with euchromatin formation (transcription activation), which has been shown to regulate plant immunity and defense response. For, instance the NLR genes (SNC1) of Arabidopsis thaliana demonstrates high expression with enhanced pathogen resistance in histone deacetylase HDA9 loss of function mutation upon pathogen pst infection (Yang et al. 2019). The Chip-Seq analyses reveal that HDA9 deacetylates H3K9 in the gene loci, suggesting that acetylation in this locus (due to the HDAC mutation) is linked to the NLR gene expression and the pathogen resistance observed. More so, H4K12ac has been shown to modulate the expression of defense response genes such as the R protein families during common bean interaction with fungal rust (Uromyces appendiculatus) pathogen (Ayyappan et al. 2015). Interestingly, histone acetylation mediates resistance against pathogens by inducing SA defensive signaling and the PTI response. For example, histone acetylation defective mutants, HAC1/5, exhibit impaired induction of PR1/2 genes (SA-responsive genes), leading to reduced basal resistance to bacterial infection (Jin et al. 2018). A loss of function mutations of the histone acetylation enzyme Histone Acetyl Transferase 1 (HAC1) reduce PTI priming, thereby enhancing susceptibility to bacterial infection in environmentally challenged plants (Singh et al. 2014). It was found that environmental stress induces the enrichment of the histone acetylation mark on PTI-responsive genes, which keep the chromatin in an open state such that, upon bacterial infection, these genes are transcribed to inhibit bacteria growth. HACI mutants did not resist bacterial infection even though they underwent repeated stress induction, suggesting that environmental stress promotes plant immunity by inducing histone acetylation (Singh et al. 2014).
Similarly, silencing of histone deacetylase701 (HDT701), a histone H4 deacetylase in transgenic rice, results in elevated transcription of PTI-related genes, ROS production, and resistance to the rice fungal pathogen Magnaporthe oryzae and the bacterial pathogen, Xanthomonas oryzae pv oryzae (Ding et al. 2012). In addition to the proven implication of histone acetylation in plant defense, the finding that the soya bean pathogen, Phytophthora sojae, produces an effector, PsAvh23, that disrupts the acetylation function of HAT as a counter-defense mechanism to promote disease susceptibility (Kong et al. 2017) supports the positive correlation between histone acetylation and PTI induction. In addition, loss of function mutations of the histone deacetylases, HDA19 and HDA6, promote increased resistance to bacteria and activation of SA-responsive genes (PR genes) (Choi et al. 2012; Wang et al. 2017). A contrasting report by Kim et al. related HDA19 loss of function mutation to bacteria susceptibility and reduced induction of SA-responsive genes (Kim et al. 2008). However, a separate study by Choi and partners demonstrated that HDA19 mutants display enhanced expression of SA defense-responsive genes (PR1 and PR2) with a boost in bacterial resistance (Choi et al. 2012). The Chip-assay in this later study revealed a higher level of H3Ac in the promoter regions of PR1 and PR2 of HDA19 mutants than in the wild type. Given this evidence, the difference in these studies could be attributed to varying experimental conditions. Correspondingly, exogenous treatment of plants with SA or its analogue (2,6-dichloroisonicotinic acid) evokes the acetylation of histones 3 and 4 at PR1 and PR2 promoters to facilitate gene expression (Choi et al. 2012; Jin et al. 2018; Chen et al. 2020). Since similar SA defense signaling is elicited upon plant challenge by a biotrophic pathogen, it implies that histone acetylation is necessary for defense against biotrophs (Alvarez-Venegas et al. 2007; Yang et al. 2015; Jin et al. 2018). Therefore, artificial modulation of HAT could enhance plant resistance to biotrophs. On the other hand, jasmonic acid (JA) and ethylene are integral to plant defense against the necrotrophic pathogen (Berr et al. 2010). HDA19 and HDA6 transcripts are induced by JA and ethylene treatment and mediate resistance against the necrotrophic pathogen; thus, they could be required for defense priming against necrotrophic pathogens (Ding and Wang 2015; Zhu et al. 2016; Ramirez-Prado et al. 2018).
Histone methylation and defense response
Histone methylation occurs both in lysine and arginine. The histone methylation writer includes the lysine writer, histone lysine methyltransferase (HKMT), and the arginine writer, protein arginine methyltransferase (PRMT). Methyl marks on histone lysine residues are removed in plants by two histone demethylation enzymes, lysine-specific demethylase-1 and Jumonji C (JmjC) (Bannister and Kouzarides 2011; Ding and Wang 2015). The HKMT of plants is a SET domain-containing protein involved in various phenotypic changes in response to both biotic and abiotic stress (Huang et al. 2016; Lee et al. 2016). Histone methylation could be a repressive or active mark depending on the lysine residue involved and the number of methyl groups involved; for instance, methylation of lysine 4 residues of histone 3 (H3K4me) is associated with transcription activation and induction of different readouts that induce gene expression. H3K36me3 (Histone 3 lysine 36 trimethylation) is also a transcriptional activating mark, while H3K9, H3K27 and H4K20 di- and tri-methylations are linked to gene silencing (Li et al. 2007; Liu et al. 2010; Jørgensen et al. 2013).
Histone lysine methylation is a conserved mechanism that regulates defense responses against pathogens by modulating the expression of defense marker genes and SA, JA, and ethylene signaling. Arabidopsis SET Domain Group 8 (SDG8), an HKMT that catalyzes H3K36me3, has been shown to promote plant defense against necrotrophic fungi via the induction subset genes of the JA and ethylene signaling (Berr et al. 2010). SDG8 loss of function mutants maintain the same level of JA/ethylene as wild type but exhibit impaired induction of JA/Ethylene pathway genes and reduced fungal resistance. The Chip analysis reveals that SDG8 targets and methylates H3K4 of defense marker genes downstream of JA/Ethylene pathway. As such, the induction of these genes is impaired in the SDG8 mutants, suggesting that SDG8-induced H3KA methylation of the JA/ethylene pathway genes is critical for defense against necrotrophic fungi (Berr et al. 2010).
Another plant HKMT important in regulating the expression of disease-resistance genes is Arabidopsis trithorax 1 (ATX1). A Loss of function ATX1 mutants demonstrates increased P. syringae susceptibility and downregulation of WRKY70 genes and PR genes (Alvarez-Venegas et al. 2007). WRKY70 is a transcription factor at the node of convergence between two antagonist pathways (the SA and JA pathways), mediating activation of the SA and repression of the JA defense response. ATX1 establishes H3K4 methylation mark on WRKY70, thereby driving activation of the downstream PR genes of SA defense signaling. Interestingly, the ATX1-induced H3K4me3 mark also exists in both the active PR genes and the repressed JA-responsive genes, suggesting that this mark could function to keep the genes prepared for rapid transcriptional change (Alvarez-Venegas et al. 2007). In addition, the histone methyltransferase, KRYPTONITE, coordinates with CMT3 and mediates transcriptional gene silencing of viral genomes (Sun et al. 2015).
Besides methylation, demethylation of repressive histone methylation marks induces activation of the defense response, as has been demonstrated using the histone lysine demethylase, Jumonji C domain protein JMJ705, which triggers removal of the repressive methyl mark H3K27me2/3 under pathogen (Xanthomonas oryzae) infection, causing increased rice resistance to Xanthomonas bacterial blight disease (Li et al. 2013). Similarly, JmjC domain-containing protein 27 (JMJ27), an H3K9 demethylase, is expressed under pst infection, and its loss-of-function mutants show weakened PR gene expression and poor resistance to the bacterial pathogen, demonstrating that JMJ27 is involved in mediating defense against the bacterial pathogen (Dutta et al. 2017).
Histone ubiquitination and defense response
Ubiquitin is a small regulatory protein that is marked on the lysine residue of substrate protein by the coordinated action of three enzymes; ubiquitin-activating enzyme E1, ubiquitin conjugase E2, and ubiquitin ligase E3 (Neutzner and Neutzner 2012). This modification is reversed by the ubiquitin-specific proteases or deubiquitinases. Ubiquitination process including monoubiquitination (Single ubiquitin per lysine residue) and polyubiquitination (chain of ubiquitin per lysine residues) regulate diverse cellular processes in both plant and animals (Sadanandom et al. 2012; Sampson et al. 2023). While polyubiquitination is known to target proteins for proteasome degradation, monoubiquitination is involved in non-proteolytic function including chromatin modification, protein translocation, and protein interactions hence histone monoubiquitination is more predominant than polyubiquitination (Nakagawa and Nakayama 2015; Mattiroli and Penengo 2021; Magits and Sablina 2022).
H2A and H2B monoubiquitination identified as H2Aub and H2Bub, respectively, is the most studied histones ubiquitination. H2Aub is catalyzed by polycomb group (PcG) repressive complex 1 (PRC1) and is associated with a heterochromatin state (Bratzel et al. 2010; Kalb et al. 2014; Barbour et al. 2020). H2Aub colocalizes with the transcriptional repressive histone mark HK3me to regulate several developmental and abiotic stress responses by mediating gene silencing (Lee et al. 2015). In plants, H2Bub is catalyzed by E2 ubiquitin-conjugating enzymes (UBC1, UBC2 and UBC3) and E3 histone monoubiquitination 1 (HUB1) and HUB2 (Fleury et al. 2007; Cao et al. 2008; Xu et al. 2009). Contrary to H2Aub, H2Bub is associated with an active chromatin state, and its diverse role including photomorphogenesis, circadian clock regulation, development and stress responses is linked to its ability to promote transcription activation (Zarreen et al. 2022). Although the role of histone ubiquitination in plant immunity and biotic stress response is still emerging, studies using HUB and UBC mutants demonstrate that H2Bub facilitate the expression of defense-responsive gene during pathogen invasion (Dhawan et al. 2009; Hu et al. 2014; Zhang et al. 2015a, 2015b).
Linker histone H1 in epigenetic and defense response
Unlike core nucleosome histones, the role of H1 in chromatin structural modification and overall epigenetic state is poorly characterized. H1 proteins are known to facilitate chromatin compaction and stability (Fyodorov et al. 2018; Willcockson et al. 2021) They are found to be enriched in the heterochromatin region thus, they are associated with transcription repression (Zlatanova 1990). However, the growing interest in dissecting the role of H1 at the epigenetic level shows that H1 is not a global transcriptional repressor as H1 depletion does not lead to significant expression of some genes (Fan et al. 2005). Hence, H1 modulates epigenetic state by altering the transcription of specific genes. Studies show that H1 regulates gene expression by interfering with histone modification and DNA methylation (Yang et al. 2013; Willcockson et al. 2021).
In plants, H1 regulates development and gene imprinting in a manner that is linked to DNA methylation changes at specific gene loci (Wierzbicki and Jerzmanowski 2005; Rea et al. 2012; Rutowicz et al. 2019). Some H1 variants have been associated with adaptive stress response in plants. For instance, H1-3 variant of Arabidopsis thaliana, H1-S variant of tomato, H1-C and H1-D variant of tobacco and H1-D variant of wild tomato L. pennellii are identified as variants that show high expression under abiotic stress induction, thus they are called stress-induced variants (Wei and O'Connell 1996, Scippa et al. 2004, Wang, Wang et al. 2014, Rutowicz et al. 2015). Compared to abiotic stress response, the role of H1 variants in plant immunity and biotic stress response is under-examined. A recent study by Sheik et al. investigated the implication of H1 variant mutants in plant immunity and defense priming (Sheikh et al. 2023). It is found that H1 variants h1-1, h1-2, and h1-3 triple mutants are totally resistance to bacteria pst DC3000 and fungal Botrytis cinerea infections but not the single mutants. The expression of PR1 genes and the levels of defense-related hormones and enzymes are elevated in the h1 triple mutant plants showing an increase in basal immunity (Sheikh et al. 2023). Interestingly, the h1 triple mutant plants are insensitive to flg22 defense priming and this is attributed to changes in DNA methylation and histone acetylation implying that H1 influences the epigenetic landscape of defense genes to modulate plant immunity response.
Antiviral defense and RNA silencing
Plants have developed multiple defense mechanisms against viruses. Their fundamental line of antiviral defense is through RNA silencing (Wang et al. 2012; Moon and Park 2016; Akhter et al. 2021). Induction of resistant (R) genes and PTI-mediated defense response against the invading virus has also been identified (Soosaar et al. 2005; Calil and Fontes 2017; Sett et al. 2022). However, the mechanism underpinning extracellular virus recognition for PTI-mediated antiviral defense is not fully understood, as viruses do not encode PAMP (Leonetti et al. 2021). RNA silencing, which is the dominant virus defense mechanism in plants, is a conserved mechanism for regulating gene expression under the direction of small RNAs (sRNA). Two major sRNAs employed in plant antiviral defense mechanisms via RNA silencing are micro-RNA (miRNA) and small interfering RNA (siRNA) (Wang et al. 2012). RNA silencing regulates gene expression at the transcriptional level via siRNA and post-transcriptional level via either siRNA or miRNA (Sijen et al. 2001). The mechanism of RNA silencing involves the dicing of precursor sRNAs by DCL and loading of the synthesized sRNA into an AGO containing RNA-induced silencing complex (RISC), from where they are directed to the target genome for degradation or translational repression (Baulcombe 2004; Ding and Voinnet 2007). Perfectly paired long dsRNA serves as a precursor for siRNA synthesis while imperfectly paired short hairpin RNA serves as a precursor for miRNA synthesis (Guleria et al. 2011). miRNAs originate endogenously and target different loci from their source of generation. siRNAs in contrast, originate either endogenously or exogenously (virus, transposons or transgene precursors) and target the same loci from where they are generated (Tang et al. 2003; Ding et al. 2004). This feature makes siRNA well suited for antiviral defense hence siRNA has been historically associated with plant natural antiviral immunity. Recent studies have postulated the involvement of miRNA in plant antiviral immunity (Llave 2004; Pérez-Quintero et al. 2010). The success of artificial miRNA in plant viral defense also confirms the antiviral properties of miRNA (Satish et al. 2021).
The synthesis of siRNA is accompanied by secondary amplification via RNA-dependent-RNA polymerase. Through secondary amplification, more siRNAs are produced and spread to unaffected parts, providing immunity against the virus in those parts. This phenomenon explains one of Wingard’s (1928) observations while studying the symptoms of tobacco ringspot disease. He found that newly formed leaves in a tobacco ringspot virus-infected plant display a symptomless phenotype, implying the spread of antiviral immunity to these leaves (Wingard 1928). Similarly, host recovery from virus infection has been reported in several other plants (Chellappan et al. 2004; Palukaitis 2011; Nie and Molen 2015). SiRNA secondary amplification is also important in maintaining defense against pathogens under artificially induced gene silencing (Song et al. 2018). Defense against DNA viruses such as geminivirus is mediated via the RdDM pathway-associated DNA methylation and silencing of the viral genome by siRNA (Raja et al. 2008; Butterbach et al. 2014). Studies have shown that invading viruses counter RNA silencing defense by producing viral RNA silencing suppressors (vRSS) (Roth et al. 2004; Burgyán and Havelda 2011). Geminiviruses encode proteins that hijack RdDM machinery to impede transcriptional gene silencing (TGS). This process enables viral accumulation, and a typical example is a geminivirus-encoded transactivator, AC2, which inhibits KYP-induced DNA methylation and transcriptional gene silencing (Sun et al. 2015). Following the ETI-mediated immune response, plants can in turn counter virus secondary pathogenicity by exploiting suppression of TGS, to upregulate expression of R genes under virus infection (Pumplin and Voinnet 2013; Moon and Park 2016; Diezma-Navas et al. 2019). Through this mechanism, the fitness cost associated with a long-term activation of defense genes is reduced, as R genes are activated only during infection.
The growing advances in RNA technology have facilitated the large-scale engineering of dsRNA, siRNA, hpRNA for improving plant immunity (Taliansky et al. 2021). In practice, exogenous treatment of plants with virus-derived dsRNA has been shown to confer resistance against viruses in several plants (Konakalla et al. 2021; Patil et al. 2021). RNA silencing presents a promising approach in agricultural biotechnology as the knowledge of RNA silencing has been extrapolated for improving other agronomic characteristics. However, RNA silencing technology still faces some limitations like off-target effect and decreasing effectivity across generations, which needs improvement for optimum application.
Epigenetics contribute to plant morphology, stress response, and nutritional value
The phenotypic diversity of important agronomic traits, such as flowering time, growth, nutritional value, yield, and others, has often been attributed to DNA sequence polymorphism. However, evidence has revealed that genetic and epigenetic modifications contribute to these traits. This contribution is evident in epialleles, which are loci with altered chromatin states due to DNA methylation variation (Srikant and Wibowo 2021). Epialleles display phenotypes that vary from those of their wild type which can be passed across generations (Weigel and Colot 2012). Cases of phenotypic reversion to the wild-type phenotype are observed among epiallelic populations and correlate with reversion in the chromatin modification at the concerned locus (Jacobsen and Meyerowitz 1997; Cubas et al. 1999), clearly revealing that the phenotypic variations arise from epigenetic could only modification. Such reversion infers that epialleles are not stable across generations like genomic alleles. Naturally occurring epialleles were first reported in the Linaria vulgaris Lcyc gene. The Lcyc epimutants, unlike the wild types, are heavily methylated and transcriptionally silent, resulting in floral morphology (radial symmetry) different from the wild type (bilateral symmetry) (Cubas et al. 1999). Similar spontaneous epimutation resulting from hypermethylation of the SBP-box promoter at the Colorless non-ripening (Cnr) locus has been reported in tomatoes and found to cause ripening defects (Manning et al. 2006).
In rice plants, a related situation has been reported, where the promoter of the gene encoding adenylate kinase is hypermethylated, resulting in epialleles (Epi-ak1) that exhibit the albino phenotype in leaves and panicles (Wei et al. 2017). Besides hypermethylated epialleles, hypomethylation of the transcription termination region of the epigenetic short panicle (Esp) gene and Fertilization-Independent Endosperm1 (FIE1) gene, respectively, results in a gain of a function epialleles, causing short panicle architecture and dwarf stature in rice (Zhang et al. 2012; Luan et al. 2019). Several other naturally occurring epialleles of agronomic value are associated with genetic influence, mainly from the spread of DNA methylation from the transposable element and repeat sequences. These include epialleles causing a transition from male to female sex in melon (Martin et al. 2009), epialleles causing dwarf phenotype and small grain size in rice (Miura et al. 2009; Zhang et al. 2015) and epialleles causing vitamin E accumulation in tomato (Quadrana et al. 2014). Paramutation is another source of epialleles that has been shown to affect pigmentation and phosphate accumulation in maize (Chandler 2007; Pilu et al. 2009).
In addition to naturally occurring epialleles, experimentally induced changes in DNA methylation produce heritable phenotypic changes in complex traits, as demonstrated among epigenetic recombinant inbred lines (EpiRILs) (Springer and Schmitz 2017). EpiRILs are genetically identical homozygous lines that segregate at the DNA methylation level. EpiRIL population has been successfully constructed in A. thaliana by crossing homozygous DNA methylation defective mutants with the isogenic wild type (Johannes et al. 2009; Cortijo et al. 2014). The EpiRIL population shows heritable variations in complex traits, which are accounted for by differential DNA methylation induced by the parent mutants (Johannes et al. 2008). Using the differentially methylated regions as biomarkers, epigenetic quantitative trait loci (QTLepi) associated with phenotypic variation are mapped (Kooke et al. 2015). This process has been used to demonstrate the influence of epigenetics on complex traits such as flowering time, plant height, root length, and abiotic stress response (Johannes et al. 2009; Cortijo et al. 2014; Kooke et al. 2015).
Another line of evidence can be obtained by phenotyping plants with defective DNA methylation emanating from 5-azacytidine treatment or mutation in the DNA (de)methylation enzymes. Studies have shown that 5-azacytidine treatment produces early flowering phenotypes, premature ripening of tomatoes, red pigmentation in apples, and reduced somatic embryogenesis in Arabidopsis (Kondo et al. 2006; Zhong et al. 2013; Grzybkowska et al. 2018; Ma et al. 2018). Related morphological effects, including aberrant developmental phenotypes, are observed among DNA (de)methylation mutants (Kakutani et al. 1996; Li et al. 2018; Zhang et al. 2018). Recently, Liu et al. identified the thickened aleurone mutants (ta2-1) in rice that emanate from a mutation in the DNA demethylase OsROS1 (Liu et al. 2018). Aleurone is a cell layer in seed endosperm, rich in protein, vitamins, and minerals; hence, the thickened aleurone of ta2-1 mutants depicts high nutritional content. It is observed that the ta2-1 mutants exhibit elevated DNA methylation in the CG and CHG of the endosperm when compared with the wild-type. In addition, two putative transcription factors for aleurone differentiation are hypermethylated and underexpressed in the ta2-1 mutants, implying that they are the target of OsROS1-induced DNA demethylation that hinders the increase in several aleurone layers in rice (Liu et al. 2018). In effect, altering OsROS1 activity could be a valuable approach for improving the nutritional value of rice.
Similarly, studies have shown that DNA demethylation is critical in accumulating prolamine, a seed storage protein in wheat and barley (Wen et al. 2012). Although prolamines are a rich source of plant dietary proteins, they are harmful to people with celiac disease because they contain autoimmune epitopes that trigger autoimmune reactions in these people (Gil-Humanes et al. 2010; Osorio et al. 2012). Analysis of the methylation status of the barley B-hordein gene, a prolamine gene of barley, shows that the promoter CpG of the endosperm is demethylated (Sørensen 1992), thus proving that hypomethylation is necessary for prolamine accumulation. Since DME is primarily expressed in the central cell of the female gametophyte that forms the endosperm, it is likely that DME-induced hypomethylation is responsible for prolamine gene activation in the endosperm. Hence, to develop celiac-tolerable wheat, Wen and associates show that RNAi-induced silencing of DME causes a reduction in prolamine level of about 67% (Wen et al. 2012). With the growing evidence of the contribution of DNA (de)methylation to agronomic traits, it follows that manipulating the activity of DNA (de)methylation enzymes could be a powerful means of developing desired phenotypes in crops.
More interesting is the active contribution of histone modification to phenotypic variations in the plant, especially in the regulation of flowering time. This is well noted in vernalization, a situation in which flowering is enhanced under prolonged cold conditions. Vernalization-induced flowering is mediated by silencing of the Flower Locus C (FLC) gene through modification of FLC locus with the H3K9 and H3K27 di- and trimethylation marks (Bastow et al. 2004). This silencing epigenetic mark is maintained even after vernalization has ended but becomes erased during embryogenesis to ensure activation of FLC and continuous requirement of vernalization in every season (Sheldon et al. 2008). The epigenetic memory created during vernalization is maintained by the polycomb repressor complex and is mitotically transferred to the next generation of cells (De Lucia et al. 2008). Similarly, several adverse environmental conditions that cause abiotic stress, such as high temperature, drought, flood, and high salinity, among others, induce an epigenetic response. This response is somatically memorable, enabling improved tolerability and performance of plants under subsequent exposure to such stress (He and Li 2018).
Epigenetics for expanding phenotypic variation and epi-modifications in plants
The completion of the genome mapping of many important plants has provided plant breeders with new or improved tools such as genome-wide association studies, which allow for a clearer understanding of the relationship between many genes and their phenotypes, encouraging more intense gene-based breeding. This knowledge has significantly reduced the choice of high-performing cultivars and has significantly narrowed genetic diversity (Esquinas-Alcázar 2005; Morrell et al. 2011; Palmgren et al. 2015). Dense monocropping of such high-performing cultivars is often a means to improve the value of the cultivated land in large commercial farmyards (Bruce 2012). Pitiably, though, such dense monocropping of genetically identical plants is highly vulnerable to host-adapted pests and diseases (Zhi and Chang 2021). It then becomes imperative to introduce advantageous disease-relevant phenotypic variations while maintaining genetic integrity or to create enabling conditions for variations to appear when necessary (Forsman 2014). Disease-relevant phenotypes of interest may include the differential accumulation of specific compounds, changes in photosynthetic activity, transpiration rate, leaf surface temperature, gas exchange, chlorophyll, and carotenoid concentration (Oerke et al. 2006; Bürling et al. 2011; Mahlein 2016; Cahon et al. 2018; Reynolds et al. 2020). It may be possible to modify these phenotypes for crop pathogen resistance through epigenetic manipulations. Studies assert that changes in the “chromatin landscape” may be an important determinant of plant phenotypic variation and form a basis for rapid response or evolution under stress (Zhang et al. 2013; Diez et al. 2014).
The role of epigenetic mechanisms—DNA methylation, histone modifications—in controlling disease-relevant and other agronomic phenotypic variations through transcriptome reprogramming when plants are subjected to biotic or abiotic stress has been demonstrated in several studies (Walley et al. 2008; Dowen et al. 2012; Yu et al. 2013; Johnson et al. 2015; Rambani et al. 2015; Kellenberger et al. 2016; López Sánchez et al. 2016; Muñoz-Viana et al. 2017; Annacondia et al. 2018; Wang et al. 2018; Geng et al. 2019; Yang et al. 2019; Atighi et al. 2020). Although the effects of modification of the epigenetic profile are not fully understood, it is possible to artificially control the induction of transcriptome reprogramming towards the formation of desirable disease-resistance phenotypes capable of transgenerational inheritance. In this case, genetically identical epi-modified plants imbued with disease-relevant phenotypic variations would be produced (Cubas et al. 1999; Manning et al. 2006; Baubec et al. 2009; Martin et al. 2009; Long et al. 2011; Mirouze and Paszkowski 2011; Telias et al. 2011; Chen and Zhou 2013; Liu et al. 2015; Griffin et al. 2016). Such induced transgenerational epialleles, together with naturally occurring epialleles associated with disease-resistant phenotypes can be used for epi-breeding in which genetically identical plants with different epigenetic profiles are crossed to obtain a desired phenotype variation while preserving the genotype (Gallusci et al. 2017; Springer and Schmitz 2017; Latutrie et al. 2019; Tirnaz and Batley 2019a, b; Varotto et al. 2020).
Exploiting epigenetics for crop improvement
Environmental pressure and chemicals to which plants are constantly exposed generally affect the performance of plants by altering gene expression. Environmentally induced phenotypic changes are likened to epigenetics rather than genetic variation. For instance, a study showed that two populations of mangrove plant species grown under different environmental pressure (salt Marsh and Riverside) show distinct phenotypes and extensive DNA methylation variation but little genetic variation; implying that their phenotypic differences are more a consequence of epigenetics than genetics (Lira-Medeiros et al. 2010). It is well established that plants can memorize past stress and use such memory to increase adaptive advantages against future stress (Odoh 2017; He and Li 2018; Kenneth et al. 2018; Sun et al. 2021). In fact, plants previously exposed to stress perform better on subsequent exposure to the same stress than the native plants (Lämke and Bäurle 2017). In addition, exposure to one stress can induce adaptive advantages for another different stress, as exemplified in the enhanced herbicide resistance of grass weed Alopecurus myosuroides exposed to drought stress (Mohammad et al. 2022) and enhanced biotic stress resistance of A. thaliana plant exposed to cold, heat and salt stress (Singh et al. 2014). This phenomenon referred to as “priming against future stress” is attributed to epigenetic modifications in the stress-responsive genes (Conrath 2011; Jaskiewicz et al. 2011; Sun et al. 2021).
More interestingly, the stress-induced epigenetic state is somatically heritable, necessitating the application of this approach in training crops for smart performance under stress (Sani et al. 2013). Although there is limited evidence of transgenerational inheritance of these environmentally induced epigenetic traits as they are mostly reverted during meiosis, few studies have, however, shown that offspring of primed plants acquire differential DNA methylation of their parents and demonstrate enhanced stress tolerance (Paszkowski and Grossniklaus 2011; Cong et al. 2019; Feiner et al. 2022). Therefore, effectors of DNA methylation should be considered in employing this approach for breeding stress-resilient crops. It is important to note that maintenance of stress-induced epigenetic memory is costly, especially when the stress is no more; hence, resetting the epigenetic state is an essential phenomenon adopted by plants to reduce associated fitness costs.
As discussed in previous paragraphs, DNA methylation loss of function mutation and the chemical inhibitors of DNA methylation such as 5-azacytidine and zebularine are valuable sources of epigenetic variation. They cause genome-wide perturbation of DNA methylation and produce several phenotypic variations of agronomic interest. However, their non-specific and broad effect (i.e., targeting varieties of genes) could produce undesired phenotypes such as developmental abnormality, limiting their application in breeding specific traits (see Fig. 2). Nevertheless, they are employed in epigenome-wide studies to investigate the effect of DNA modification in plants and identify good candidate genes for targeted epigenome modification (Fieldes and Amyot 2000; Griffin et al. 2016; Agarwal et al. 2020). For instance, In EpiRIL studies, the differentially methylated regions serve as biomarkers for mapping quantitative trait loci which are potential sites for target-specific epigenome editing (Cortijo et al. 2014).

Approaches to epigenome editing. The genome contains methylated and non-methylated regions. Targeted epigenome modification such as DNA demethylation of gene A using epigenetic editing tools (CRISPR-dCas9, ZF, TALE) triggers specific DNA demethylation and results in the expression of gene A. DNA methylation mutants and chemical methylation inhibitor cause genome-wide DNA demethylation
Target manipulation of the epigenome to alter the expression of a gene of interest, as shown in Fig. 2, is critical. This precise epigenome engineering is made possible by the use of genome editing tools; zinc finger protein (ZF), transcription activator-like effector (TALE), and clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR associated (Cas) (CRISPR/Cas) system (Waryah et al. 2018; Gallego-Bartolomé 2020; Shin et al. 2022). In simple modifications of these systems, the gene editing tools which serve as tailor-made DNA binding domains are linked with effector epigenome modifiers, likely an epigenetic modifying enzyme or the recruiter for site-directed epigenetic modification. These tools have been successfully utilized to alter the expression of disease-related genes in animals, particularly inducing the re-expression of tumor suppressor genes (Huisman et al. 2016). Several studies have demonstrated the application of ZF and TALE in producing specific desired phenotypes in plants. A typical example is targeted DNA methylation of FLOWER WAGENINGEN (FWA) epiallele using zinc finger protein fused with SUVH2, a SET and ring-associated (SRA) domain-containing protein (Johnson et al. 2014). The FWA gene is normally methylated and silenced, displaying an early flowering phenotype, whereas FWA epimutants are gain-of-function mutants exhibiting DNA methylation loss with ectopic expression of FWA and late flowering phenotype (Soppe et al. 2000). In those studies, it is found that ZF-SUVH2 construct, directed to FWA epialleles restores the early flowering phenotype of the wild-type FWA gene due to targeted DNA methylation at the FWA locus that causes silencing of this gene (Johnson et al. 2014).
In another study, targeted demethylation of FWA using ZF fused with human demethylase, TET1 (Ten-Eleven Translocation 1) causes FWA upregulation and heritable late flowering phenotype like that of FWA epialleles (Gallego-Bartolomé et al. 2018). Recently, Veley et al. showed the relevance of epigenome editing in improving cassava resistance to cassava bacterial blight disease. In the study, ZF combined with RdDM protein DMS3 (defective in meristem silencing 3) is used for targeted methylation of effector binding element (EBE) of cassava susceptibility gene meSWEET10a. The targeted methylation prevents the expression of meSWEET10a and inhibits the binding of the pathogenic bacteria effector, TAL20, to this site, yielding cassava plants with increased resistance to bacterial blight (Veley et al. 2022).
Although ZF and TALE tools have proven valuable for epigenome editing, their labor-intensive, cost and time-consuming nature necessitate the adoption CRISPR-Cas9 system for targeted epigenome modification. CRISPR-Cas9 is a robust and widely used genetic editing tool, composed of single-guide RNA that binds complementary DNA and a Cas9 endonuclease that produces a double-stranded break on the homologous DNA (Han and Kim 2019). This tool has been repurposed for epigenome modification using deactivate/dead Cas9 (dCas9) which has been fused with an effector enzyme (Waryah et al. 2018). The CRISPR-dCas9 system is currently used to produce targeted transcriptional modulation, histone modification, and DNA de/Methylation in both plants and animals (Selma and Orzáez 2021). For instance, the direct fusion of Arabidopsis histone acetyltransferase 1 (AtHAT1) to dCas9 improves drought-resistance stress by promoting targeted gene expression activation of AREB1 a drought-responsive gene (Roca Paixão et al. 2019). dCas9 fused with ROS1 demethylase has also been shown to induce targeted reactivation of methylated-silenced genes in Arabidopsis (Devesa-Guerra et al. 2020). Because the direct fusion of effector to dCas9 is associated with low modulation level, a modified version whereby effectors are recruited to CRISPR-dCas9 through aptamers such as Sun Tag, SAM (Synergistic Association Mediator), ScRNA (Scaffolding RNA) is developed (Konermann et al. 2015; Papikian et al. 2019). This new model not only produced amplified expression but also accommodate the use of different effectors within a single CRISPR-dCas9 system. The aptamer-dCas9 model has been demonstrated in plants with Sun Tag-TET1-dCas9 targeted on FWA genes and MS2-dCas9 system targeted on FT gene of Arabidopsis thaliana (Gallego-Bartolomé et al. 2018; Lee et al. 2019). In the latter construct, the MS2 is linked to effector VP64 transcriptional activator, p300 HAT1 domain or KYPTONITE, and produces altered flowering time phenotype based on the activation/repression of FT genes by each construct (Lee et al. 2019). These studies clearly validate the competence of this approach in generating desired characteristics in plants. Although epigenome editing is still emerging, several positive outcomes from these studies prove that epigenome editing serves as an effective and reliable means of creating desired agronomic phenotypes for crop improvement. Nevertheless, there are technological shortcomings that need to be overcome for the effective translation of this approach in industries. One challenge is the inherent substrate promiscuity of some effector enzymes which likely affect the study of specific substrate modification. Another critical challenge is the off-target DNA binding site observed when the concentration of the effector remains high following target site saturation. Optimizing the epigenome editing technology to overcome these limitations would improve the overall biotechnological application of this fascinating technology.
Future perspective
DNA methylation profiles respond quickly to environmental stimuli and can direct the evolutionary path of an entire genome (Tirnaz and Batley 2019a, b). Different DNA methylation profiles of introns, exons, and intron–exon boundaries are strongly associated with the regulation of DNA splicing events, which can generate novel functional or non-functional genes or even inactivate genes (Zilberman et al. 2007; Regulski et al. 2013; Tirnaz and Batley 2019a, b). In addition, the movement of TE, which mediates the evolution of resistance genes through transposon-mediated gene modifications such as copy number variations (CNV), segmental and tandem gene recombination, or whole-genome multiplications, is determined by DNA methylation and other epigenetic marks, and the observation has been demonstrated in several model plants, including rice, wheat, and cassava (Walker et al. 1995; Franzke et al. 2011; Lisch 2013; Saijo and Reimer-Michalski 2013; Sun et al. 2014; Wang et al. 2015; Wang et al. 2017; Neupane et al. 2018). It may be possible to epigenetically preprogram splicing events or control the movements of TE to create or destroy functional or dysfunctional genes or to inactivate genes in response to specific environmental stimuli, such as pathogen infection, in the whole plant or tissues exposed to pathogens, thus epigenetically creating genetically dynamic plants. This idea could be termed epigenetically directed genetic recombination if it is achieved someday.
While TE movements may be beneficial for increasing evolutionary plasticity, TE insertions are not always desirable as such movements may disrupt essential genes, especially artificially inserted genes that may not have indigent genes' epigenetic marks. Studies show that, in nature, DNA methylation has been used to control TE movements for genome stability through selective CG methylations. Such sequence-specific methylation guides can be used to protect artificially inserted genes (Biémont and Vieira 2006; Ito et al. 2013; Cavrak et al. 2014). However, more studies involving more plants are required to sufficiently understand the concept.
Some studies suggest that the epigenetic profile induced by certain biotic and abiotic stress is characteristic, consistent, replicable, and stable down several generations of a plant (Manning et al. 2006; Tricker et al. 2013; Cortijo et al. 2014). Rodriguez and Wilkinson termed the epigenetic profile characteristic of certain biotic or abiotic stressors, the epigenetic fingerprint of the stressor and proposed the use of DNA cytosine methylation pattern—especially 5mC methylations as a possible biomarker for epigenetic fingerprinting (Rodríguez López and Wilkinson 2015). If stable epigenetic biomarkers are identified that accurately predict a phenotypic state, such epigenetic biomarkers could be considered the epi-fingerprints of the phenotype, then epi-fingerprinting could find application in breeding and varietal selection—where epi-fingerprinting can be used to quickly identify individuals that will manifest a desired epi-dependent phenotype. It will be possible to diagnose diseases or pest activities at the early stages of infection or infestation and to also diagnose asymptomatic pathologies using disease epi-fingerprints—epigenetic profiles resulting from pathogen activity (Rodríguez López and Wilkinson 2015).
The relationship between epigenetic variations and plant phenotypes can be explored using statistical or process-based epigenetic models built by codifying existing knowledge on epigenetic mechanisms and related biological processes. Such models can reveal the relationship between DNA methylomes and transcriptomes, epigenetic variations, and plant phenotypes (Angel et al. 2011; Buck-Sorlin 2013; Colicchio et al. 2015; Gallusci et al. 2017). Such tools can be applied in epi-breeding to predict the link between epi-variations and plant phenotypes or to guide the decision to activate or deactivate certain epi-variations to improve disease resistance (Zhi and Chang 2021).
Conclusion
Global food security is besieged by the constantly increasing threat of pests and diseases. These pests and diseases project their impact on the socioeconomic progress of nations and are not without environmental implications. It is estimated that 20 to 40% of crop production efficiency is lost due to pests and diseases, with economic implications of up to $220 billion annually (Zeng et al. 2022). The need to develop improved crops with enhanced resistance to pests and diseases, among other desirable characteristics, cannot be over-emphasized. Epigenetics is a promising complement, if not an alternative, to current efforts in the fight against crop pathogens. The rapid growth of interest in epigenetics is evidenced by the enormous growth of literature in the area. This interest is undoubtedly due to the growing knowledge of epigenetic mechanisms and their applications. The potentials of epigenetics lie in the transgenerational stability and heritability of some epigenetic markers such as DNA methylation patterns, certain histone modifications, and chromatin assemblies. The pattern of occurrence of these markers has been linked to the pattern of expression of disease-resistance genes in plants and, consequently, a change of disease-resistance phenotype. In terms of crop improvement, epigenetic mechanisms provide a more dynamic, less invasive approach to breeding disease-resistant, environmentally responsive crops compared to genetic mechanisms. Epigenetics present a vast, intriguing source of phenotypic variations and can be exploited in the production of epi-modified plants, epi-breeding, genome stabilization, epi-modeling, and epi-fingerprinting, all of which can be used for crop improvement.
However, epigenetics is still faced with many challenges and limitations, and many questions remain unanswered. Epigenetic marks are not as stable as genetic transformation as they may be altered by biotic and abiotic stress and are usually erased during meiosis or can spontaneously be lost after several generations. It then becomes necessary to determine which regions of the genome are stably epi-modified and which are not. A mechanism for ensuring the stability of desirable epialleles or for predicting unstable epialleles needs to be developed to ensure perpetuation where such is desired. In addition, most studies have been on model plants and as such may not be freely extrapolated to all plants. It then becomes important to determine the relationship between the epigenetic profiles of genes of the same family across different plants.
Data availability
The data sets for this study are available from the corresponding author upon reasonable request.
References
Agarwal G et al (2020) Epigenetics and epigenomics: underlying mechanisms, relevance, and implications in crop improvement. Funct Integr Genomics 20(6):739–761
Agorio A, Vera P (2007) ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 19(11):3778–3790
Akhter MS et al (2021) Resistance induction based on the understanding of molecular interactions between plant viruses and host plants. Virol J 18(1):176
Akimoto K et al (2007) Epigenetic inheritance in rice plants. Ann Bot 100(2):205–217
Aktar MW et al (2009) Impact of pesticides use in agriculture: their benefits and hazards. Interdiscip Toxicol 2(1):1–12
Alvarez-Venegas R et al (2007) Epigenetic control of a transcription factor at the cross section of two antagonistic pathways. Epigenetics 2(2):106–113
Angel A et al (2011) A Polycomb-based switch underlying quantitative epigenetic memory. Nature 476(7358):105–108
Angers B et al (2020) Sources of epigenetic variation and their applications in natural populations. Evol Appl 13(6):1262–1278
Annacondia ML et al (2018) Stress response regulation by epigenetic mechanisms: changing of the guards. Physiol Plant 162(2):239–250
Atighi MR et al (2020) Genome-wide DNA hypomethylation shapes nematode pattern-triggered immunity in plants. New Phytol 227(2):545–558
Ayyappan V et al (2015) Genome-wide profiling of histone modifications (H3K9me2 and H4K12ac) and gene expression in Rust (Uromyces appendiculatus) inoculated common bean (Phaseolus vulgaris L.). PLoS ONE 10(7):e0132176
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Barbour H et al (2020) Polycomb group-mediated histone H2A monoubiquitination in epigenome regulation and nuclear processes. Nat Commun 11(1):5947
Bastow R et al (2004) Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427(6970):164–167
Baubec T et al (2009) Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J 57(3):542–554
Baulcombe D (2004) RNA silencing in plants. Nature 431(7006):356–363
Bender J (2004) DNA methylation and epigenetics. Annu Rev Plant Biol 55:41–68
Berr A et al (2010) Arabidopsis histone methyltransferase SET DOMAIN GROUP8 mediates induction of the jasmonate/ethylene pathway genes in plant defense response to necrotrophic fungi. Plant Physiol 154(3):1403–1414
Biémont C, Vieira CJN (2006) Junk DNA as an evolutionary force. Nature 443(7111):521–524
Boller T, Felix G (2009) A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol 60:379–406
Bratzel F et al (2010) Keeping cell identity in Arabidopsis requires PRC1 RING-finger homologs that catalyze H2A monoubiquitination. Curr Biol 20(20):1853–1859
Bruce TJ (2012) GM as a route for delivery of sustainable crop protection. J Exp Bot 63(2):537–541
Buck-Sorlin G (2013) Process-based model. In: Dubitzky W, Wolkenhauer O, Cho K-H, Yokota H (eds) Encyclopedia of systems biology. Springer, New York, pp 1755–1755
Burgyán J, Havelda Z (2011) Viral suppressors of RNA silencing. Trends Plant Sci 16(5):265–272
Bürling K et al (2011) Use of blue-green and chlorophyll fluorescence measurements for differentiation between nitrogen deficiency and pathogen infection in winter wheat. J Plant Physiol 168(14):1641–1648
Butterbach P et al (2014) Tomato yellow leaf curl virus resistance by Ty-1 involves increased cytosine methylation of viral genomes and is compromised by cucumber mosaic virus infection. Proc Natl Acad Sci U S A 111(35):12942–12947
Cahon T et al (2018) Do aphids alter leaf surface temperature patterns during early infestation? InSects 9(1):34
Calil IP, Fontes EPB (2017) Plant immunity against viruses: antiviral immune receptors in focus. Ann Bot 119(5):711–723
Cao Y et al (2008) Histone H2B monoubiquitination in the chromatin of flowering locus C regulates flowering time in Arabidopsis. Plant Cell 20(10):2586–2602
Cavrak VV et al (2014) How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genet 10(1):e1004115
Chandler VL (2007) Paramutation: from maize to mice. Cell 128(4):641–645
Chellappan P et al (2004) Short interfering RNA accumulation correlates with host recovery in DNA virus-infected hosts, and gene silencing targets specific viral sequences. J Virol 78(14):7465–7477
Chen X, Zhou DX (2013) Rice epigenomics and epigenetics: challenges and opportunities. Curr Opin Plant Biol 16(2):164–169
Chen J et al (2020) Reprogramming and remodeling: transcriptional and epigenetic regulation of salicylic acid-mediated plant defense. J Exp Bot 71(17):5256–5268
Choi HW, Klessig DF (2016) DAMPs, MAMPs, and NAMPs in plant innate immunity. BMC Plant Biol 16(1):232
Choi SM et al (2012) HDA19 is required for the repression of salicylic acid biosynthesis and salicylic acid-mediated defense responses in Arabidopsis. Plant J 71(1):135–146
Cokus SJ et al (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452(7184):215–219
Colicchio JM et al (2015) DNA methylation and gene expression in Mimulus guttatus. BMC Genomics 16(1):507
Cong W et al (2019) Transgenerational memory of gene expression changes induced by heavy metal stress in rice (Oryza sativa L.). BMC Plant Biol 19(1):282
Conrath U (2011) Molecular aspects of defence priming. Trends Plant Sci 16(10):524–531
Cortijo S et al (2014) Mapping the epigenetic basis of complex traits. Science 343(6175):1145–1148
Cubas P et al (1999) An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401(6749):157–161
Cui H et al (2015) Effector-triggered immunity: from pathogen perception to robust defense. Annu Rev Plant Biol 66:487–511
Cutter AR, Hayes JJ (2015) A brief review of nucleosome structure. FEBS Lett 589(20 PT A):2914–2922
Datta AJA, Security F (2013) Genetic engineering for improving quality and productivity of crops. Agric Food Security 2(1):1–3
De Lucia F et al (2008) A PHD-polycomb repressive complex 2 triggers the epigenetic silencing of FLC during vernalization. Proc Natl Acad Sci U S A 105(44):16831–16836
Deans C, Maggert KA (2015) What do you mean, “epigenetic”? Genetics 199(4):887–896
De-La-Peña C et al (2012) Regulation of disease-responsive genes mediated by epigenetic factors: interaction of Arabidopsis–Pseudomonas. Mol Plant Pathol 13(4):388–398
Deng Y et al (2017) Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355(6328):962–965
Devesa-Guerra I et al (2020) DNA methylation editing by CRISPR-guided excision of 5-methylcytosine. J Mol Biol 432(7):2204–2216
Dhawan R et al (2009) HISTONE MONOUBIQUITINATION1 interacts with a subunit of the mediator complex and regulates defense against necrotrophic fungal pathogens in Arabidopsis. Plant Cell 21(3):1000–1019
Diez CM et al (2014) Epigenetics and plant genome evolution. Curr Opin Plant Biol 18:1–8
Diezma-Navas L et al (2019) Crosstalk between epigenetic silencing and infection by tobacco rattle virus in Arabidopsis. Mol Plant Pathol 20(10):1439–1452
Ding SW, Voinnet O (2007) Antiviral immunity directed by small RNAs. Cell 130(3):413–426
Ding B, Wang GL (2015) Chromatin versus pathogens: the function of epigenetics in plant immunity. Front Plant Sci 6:675
Ding SW et al (2004) RNA silencing: a conserved antiviral immunity of plants and animals. Virus Res 102(1):109–115
Ding B et al (2012) HDT701, a histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell 24(9):3783–3794
Dita M et al (2018) Fusarium wilt of banana: current knowledge on epidemiology and research needs toward sustainable disease management. Front Plant Sci 9:1468
Dowen RH et al (2012) Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci U S A 109(32):E2183-2191
Dutta A et al (2017) JMJ27, an Arabidopsis H3K9 histone demethylase, modulates defense against Pseudomonas syringae and flowering time. Plant J 91(6):1015–1028
Erdmann RM, Picard CL (2020) RNA-directed DNA methylation. PLoS Genet 16(10):e1009034
Esquinas-Alcázar J (2005) Protecting crop genetic diversity for food security: political, ethical and technical challenges. Nat Rev Genet 6(12):946–953
Fan Y et al (2005) Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123(7):1199–1212
Feiner N et al (2022) Environmentally induced DNA methylation is inherited across generations in an aquatic keystone species. iScience 25(5):104303
Fieldes MA, Amyot LM (2000) Evaluating the potential of using 5-azacytidine as an epimutagen. Can J Bot 77(11):1617–1622
Fleury D et al (2007) The Arabidopsis thaliana homolog of yeast BRE1 has a function in cell cycle regulation during early leaf and root growth. Plant Cell 19(2):417–432
Forsman A (2014) Effects of genotypic and phenotypic variation on establishment are important for conservation, invasion, and infection biology. Proc Natl Acad Sci U S A 111(1):302–307
Franzke A et al (2011) Cabbage family affairs: the evolutionary history of Brassicaceae. Trends Plant Sci 16(2):108–116
Fyodorov DV et al (2018) Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 19(3):192–206
Gallego-Bartolomé J (2020) DNA methylation in plants: mechanisms and tools for targeted manipulation. New Phytol 227(1):38–44
Gallego-Bartolomé J et al (2018) Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc Natl Acad Sci USA 115(9):E2125–E2134
Gallusci P et al (2017) Epigenetics for plant improvement: current knowledge and modeling avenues. Trends Plant Sci 22(7):610–623
Gelato KA, Fischle W (2008) Role of histone modifications in defining chromatin structure and function. Biol Chem 389(4):353–363
Geng S et al (2019) DNA methylation dynamics during the interaction of wheat progenitor Aegilops tauschii with the obligate biotrophic fungus Blumeria graminis f. sp. tritici. New Phytol 221(2):1023–1035
Gil-Humanes J et al (2010) Effective shutdown in the expression of celiac disease-related wheat gliadin T-cell epitopes by RNA interference. Proc Natl Acad Sci U S A 107(39):17023–17028
Göhre V, Robatzek S (2008) Breaking the barriers: microbial effector molecules subvert plant immunity. Annu Rev Phytopathol 46:189–215
Griffin PT et al (2016) A comparative analysis of 5-azacytidine- and zebularine-induced DNA demethylation. G3 Bethesda 6(9):2773–2780
Grzybkowska D et al (2018) Azacitidine (5-AzaC)-treatment and mutations in DNA methylase genes affect embryogenic response and expression of the genes that are involved in somatic embryogenesis in Arabidopsis. Int J Mol Sci 85(2):243–256
Guleria P et al (2011) Plant small RNAs: biogenesis, mode of action and their roles in abiotic stresses. Genomics Proteomics Bioinformatics 9(6):183–199
Gupta C, Salgotra RK (2022) Epigenetics and its role in effecting agronomical traits. Front Plant Sci 13:925688
Halter T et al (2021) The Arabidopsis active demethylase ROS1 cis-regulates defence genes by erasing DNA methylation at promoter-regulatory regions. Elife 10:e62994
Han Y-J, Kim J-I (2019) Application of CRISPR/Cas9-mediated gene editing for the development of herbicide-resistant plants. Plant Biotechnology Reports 13(5):447–457
Hannan Parker A et al (2022) Epigenetics: a catalyst of plant immunity against pathogens. New Phytol 233(1):66–83
He Y, Li Z (2018) Epigenetic environmental memories in plants: establishment, maintenance, and reprogramming. Trends Genet 34(11):856–866
He XJ et al (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21(3):442–465
Hu M et al (2014) Histone H2B monoubiquitination is involved in regulating the dynamics of microtubules during the defense response to Verticillium dahliae toxins in Arabidopsis. Plant Physiol 164(4):1857–1865
Huang Y et al (2016) Identification of SET domain-containing proteins in Gossypium raimondii and their response to high temperature stress. Sci Rep 6:32729
Huang M et al (2022) Active DNA demethylation regulates MAMP-triggered immune priming in Arabidopsis. J Genet Genomics 49:796–809
Huisman C et al (2016) Re-expression of selected epigenetically silenced candidate tumor suppressor genes in cervical cancer by TET2-directed demethylation. Mol Ther 24(3):536–547
Ito H et al (2013) Evolution of the ONSEN retrotransposon family activated upon heat stress in Brassicaceae. Gene 518(2):256–261
Jacobsen SE, Meyerowitz EM (1997) Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277(5329):1100–1103
Jansen A, Verstrepen KJ (2011) Nucleosome positioning in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 75(2):301–320
Jaskiewicz M et al (2011) Chromatin modification acts as a memory for systemic acquired resistance in the plant stress response. EMBO Rep 12(1):50–55
Jin H et al (2018) Salicylic acid-induced transcriptional reprogramming by the HAC-NPR1-TGA histone acetyltransferase complex in Arabidopsis. Nucleic Acids Res 46(22):11712–11725
Johannes F et al (2008) Epigenome dynamics: a quantitative genetics perspective. Nat Rev Genet 9(11):883–890
Johannes F et al (2009) Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 5(6):e1000530
Johnson LM et al (2014) SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 507(7490):124–128
Johnson KC et al (2015) The chromatin remodeler SPLAYED negatively regulates SNC1-mediated immunity. Plant Cell Physiol 56(8):1616–1623
Jones JD, Dangl JL (2006) The plant immune system. Nature 444(7117):323–329
Jørgensen S et al (2013) Histone H4 Lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 41(5):2797–2806
Kakutani T et al (1996) Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci U S A 93(22):12406–12411
Kalb R et al (2014) Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21(6):569–571
Kellenberger RT et al (2016) Herbivore-Induced DNA demethylation changes floral signalling and attractiveness to pollinators in Brassica rapa. PLoS ONE 11(11):e0166646
Kenneth OC et al (2018) Plant growth promoting Rhizobacteria (PGPR): A novel agent for sustainable food production. Am J Agric Biol Sci 14(1):35–54
Kim KC et al (2008) Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 20(9):2357–2371
Konakalla NC et al (2021) Induction of plant resistance in tobacco (Nicotiana tabacum) against tomato spotted wilt orthotospovirus through foliar application of dsRNA. Viruses 13(4):662
Kondo H et al (2006) Flowering induced by 5-azacytidine, a DNA demethylating reagent in a short-day plant Perilla frutescens var. crispa. Physiol Plant 127(1):130–137
Konermann S et al (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583–588
Kong L et al (2017) A phytophthora effector manipulates host histone acetylation and reprograms defense gene expression to promote infection. Curr Biol 27(7):981–991
Kooke R et al (2015) Epigenetic basis of morphological variation and phenotypic plasticity in Arabidopsis thaliana. Plant Cell 27(2):337–348
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Kumar SJE (2018) Epigenomics of plant responses to environmental stress. Epigenomes 2(1):6
Lämke J, Bäurle I (2017) Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol 18(1):124
Latutrie M et al (2019) Epigenetic variation for agronomic improvement: an opportunity for vegetatively propagated crops. Am J Bot 106(10):1281–1284
Le TN et al (2014) DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol 15(9):458
Lee HG et al (2015) Genome-wide activities of Polycomb complexes control pervasive transcription. Genome Res 25(8):1170–1181
Lee S et al (2016) Global regulation of plant immunity by histone lysine methyl transferases. Plant Cell 28(7):1640–1661
Lee JE et al (2019) CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 14(9):e0222778
Leonetti P et al (2021) Regulation of plant antiviral defense genes via host RNA-silencing mechanisms. Virol J 18(1):194
Li B et al (2007) The role of chromatin during transcription. Cell 128(4):707–719
Li Y et al (2011) Induced Pib expression and resistance to Magnaporthe grisea are compromised by cytosine demethylation at critical promoter regions in rice. J Integr Plant Biol 53(10):814–823
Li T et al (2013) Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 25(11):4725–4736
Li Y et al (2018) Active DNA demethylation: mechanism and role in plant development. Plant Cell Rep 37(1):77–85
Lira-Medeiros CF et al (2010) Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 5(4):e10326
Lisch D (2013) How important are transposons for plant evolution? Nat Rev Genet 14(1):49–61
Liu C et al (2010) Histone methylation in higher plants. Annu Rev Plant Biol 61:395–420
Liu R et al (2015) A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc Natl Acad Sci U S A 112(34):10804–10809
Liu J et al (2018a) Mutations in the DNA demethylase OsROS1 result in a thickened aleurone and improved nutritional value in rice grains. Proc Natl Acad Sci U S A 115(44):11327–11332
Liu W et al (2018b) RNA-directed DNA methylation involves co-transcriptional small-RNA-guided slicing of polymerase V transcripts in Arabidopsis. Nat Plants 4(3):181–188
Llave C (2004) MicroRNAs: more than a role in plant development? Mol Palnt Pathol 5(4):361–366
Long Y et al (2011) Epigenetic QTL mapping in Brassica napus. Genetics 189(3):1093–1102
López Sánchez A et al (2016) The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J 88(3):361–374
Luan X et al (2019) Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice. Rice (NY) 12(1):20
Ma C et al (2018) The effect of promoter methylation on MdMYB1 expression determines the level of anthocyanin accumulation in skins of two non-red apple cultivars. BMC Plant Biol 18(1):108
Magits W, Sablina AA (2022) The regulation of the protein interaction network by monoubiquitination. Curr Opin Struct Biol 73:102333
Mahlein AK (2016) Plant disease detection by imaging sensors—parallels and specific demands for precision agriculture and plant phenotyping. Plant Dis 100(2):241–251
Manning K et al (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38(8):948–952
Martin A et al (2009) A transposon-induced epigenetic change leads to sex determination in melon. Nature 461(7267):1135–1138
Mattiroli F, Penengo L (2021) Histone ubiquitination: an integrative signaling platform in genome stability. Trends Genet 37(6):566–581
McDowell JM, Simon SA (2008) Molecular diversity at the plant–pathogen interface. Dev Comp Immunol 32(7):736–744
Mirouze M, Paszkowski J (2011) Epigenetic contribution to stress adaptation in plants. Curr Opin Plant Biol 14(3):267–274
Miura K et al (2009) A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc Natl Acad Sci U S A 106(27):11218–11223
Mohammad VH et al (2022) Drought exposure leads to rapid acquisition and inheritance of herbicide resistance in the weed Alopecurus myosuroides. Ecol Evol 12(2):e8563
Moon JY, Park JM (2016) Cross-talk in viral defense signaling in plants. Front Microbiol 7:2068
Morrell PL et al (2011) Crop genomics: advances and applications. Nat Rev Genet 13(2):85–96
Muñoz-Viana R et al (2017) Arabidopsis chromatin assembly factor 1 is required for occupancy and position of a subset of nucleosomes. Plant J 92(3):363–374
Nakagawa T, Nakayama K (2015) Protein monoubiquitylation: targets and diverse functions. Genes Cells 20(7):543–562
Nations, Department of Economic and S. Affairs (2015) Transforming our world: The 2030 agenda for sustainable development
Neupane S et al (2018) Genome-wide identification of NBS-encoding resistance genes in Sunflower (Helianthus annuus L.). Genes (basel) 9(8):384
Neutzner M, Neutzner A (2012) Enzymes of ubiquitination and deubiquitination. Essays Biochem 52:37–50
Nicolopoulou-Stamati P et al (2016) Chemical pesticides and human health: the urgent need for a new concept in agriculture. Front Public Health 4:148
Nie X, Molen TA (2015) Host recovery and reduced virus level in the upper leaves after Potato virus Y infection occur in tobacco and tomato but not in potato plants. Viruses 7(2):680–698
Odoh CK (2017) Plant growth promoting rhizobacteria (PGPR): a bioprotectant bioinoculant for sustainable agrobiology. A Review. Int J Adv Res Biol Sci (IJARBS) 4(5):123–142
Oerke EC et al (2006) Thermal imaging of cucumber leaves affected by downy mildew and environmental conditions. J Exp Bot 57(9):2121–2132
Osorio C et al (2012) Targeted modification of wheat grain protein to reduce the content of celiac causing epitopes. Funct Integr Genomics 12(3):417–438
Palmgren MG et al (2015) Are we ready for back-to-nature crop breeding? Trends Plant Sci 20(3):155–164
Palukaitis P (2011) The road to RNA silencing is paved with plant-virus interactions. The Plant Pathol J 27(3):197–206
Papikian A et al (2019) Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat Commun 10(1):729
Paszkowski J, Grossniklaus U (2011) Selected aspects of transgenerational epigenetic inheritance and resetting in plants. Curr Opin Plant Biol 14(2):195–203
Patil BL et al (2021) Exogenous dsRNA-mediated field protection against Pigeonpea sterility mosaic emaravirus. J Plant Biochem Biotechnol 30(2):400–405
Pérez-Quintero AL et al (2010) Plant microRNAs and their role in defense against viruses: a bioinformatics approach. BMC Plant Biol 10:138
Pilu R et al (2009) A paramutation phenomenon is involved in the genetics of maize low phytic acid1-241 (lpa1-241) trait. Heredity (edinb) 102(3):236–245
Pumplin N, Voinnet O (2013) RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nat Rev Microbiol 11(11):745–760
Quadrana L et al (2014) Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat Commun 5:3027
Raja P et al (2008) Viral genome methylation as an epigenetic defense against geminiviruses. J Virol 82(18):8997–9007
Rambani A et al (2015) The methylome of soybean roots during the compatible interaction with the soybean cyst Nematode. Plant Physiol 168(4):1364–1377
Ramirez-Prado JS et al (2018) Plant immunity: from signaling to epigenetic control of defense. Trends Plant Sci 23(9):833–844
Rea M et al (2012) Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant J 71(5):776–786
Regulski M et al (2013) The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res 23(10):1651–1662
Reynolds M et al (2020) Breeder friendly phenotyping. Plant Sci 295:110396
Richard MMS et al (2018a) Molecular mechanisms that limit the costs of NLR-mediated resistance in plants. Mol Plant Pathol 19(11):2516–2523
Richard MMS et al (2018b) Genomic and epigenomic immunity in common bean: the unusual features of NB-LRR gene family. DNA Res 25(2):161–172
Rizzo DM et al (2021) Plant health and its effects on food safety and security in a One Health framework: four case studies. One Health Outlook 3:6
Roca Paixão JF et al (2019) Improved drought stress tolerance in Arabidopsis by CRISPR/dCas9 fusion with a Histone Acetyl Transferase. Sci Rep 9(1):8080
Rodríguez López CM, Wilkinson MJ (2015) Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front Plant Sci 6:397
Roth BM et al (2004) Plant viral suppressors of RNA silencing. Virus Res 102(1):97–108
Rutowicz K et al (2015) A specialized histone H1 Variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol 169(3):2080–2101
Rutowicz K et al (2019) Linker histones are fine-scale chromatin architects modulating developmental decisions in Arabidopsis. Genome Biol 20(1):157
Sadanandom A et al (2012) The ubiquitin-proteasome system: central modifier of plant signalling. New Phytol 196(1):13–28
Saijo Y, Reimer-Michalski E-M (2013) Epigenetic control of plant immunity. Epigenetic Memory and Control in Plants, Springer, Berlin, pp 57–76
Saijo Y et al (2018) Pattern recognition receptors and signaling in plant–microbe interactions. Plant J 93(4):592–613
Sampson C et al (2023) The roles of E3 ubiquitin ligases in cancer progression and targeted therapy. Clin Transl Med 13(3):e1204
Sani E et al (2013) Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol 14(6):R59
Satish D et al (2021) The landscape of microRNAs in plant viral infections. Plant Gene 26:100293
Savary S et al (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3(3):430–439
Scippa GS et al (2004) The histone-like protein H1-S and the response of tomato leaves to water deficit. J Exp Bot 55(394):99–109
Selma S, Orzáez D (2021) Perspectives for epigenetic editing in crops. Transgenic Res 30(4):381–400
Sett S et al (2022) Resistance genes on the verge of plant–virus interaction. Trends Plant Sci 27(12):1242–1252
Sheikh AH et al (2023) Linker histone H1 modulates defense priming and immunity in plants. Nucleic Acids Res 51(9):4252–4265
Sheldon CC et al (2008) Resetting of FLOWERING LOCUS C expression after epigenetic repression by vernalization. Proc Natl Acad Sci U S A 105(6):2214–2219
Shin H et al (2022) Epigenome editing: targeted manipulation of epigenetic modifications in plants. Genes Genomics 44(3):307–315
Sijen T et al (2001) Transcriptional and posttranscriptional gene silencing are mechanistically related. Curr Biol 11(6):436–440
Singh P et al (2014) Environmental history modulates arabidopsis pattern-triggered immunity in a histone acetyltransferase1-dependent manner. Plant Cell 26(6):2676–2688
Song XS et al (2018) Secondary amplification of siRNA machinery limits the application of spray-induced gene silencing. Mol Plant Pathol 19(12):2543–2560
Soosaar JLM et al (2005) Mechanisms of plant resistance to viruses. Nat Rev Microbiol 3(10):789–798
Soppe WJ et al (2000) The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6(4):791–802
Sørensen MB (1992) Methylation of B-hordein genes in barley endosperm is inversely correlated with gene activity and affected by the regulatory gene Lys3. Proc Natl Acad Sci U S A 89(9):4119–4123
Springer NM, Schmitz RJ (2017) Exploiting induced and natural epigenetic variation for crop improvement. Nat Rev Genet 18(9):563–575
Sun H et al (2014) DNA methylation pattern of Photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum). New Phytol 204(3):682–692
Sun YW et al (2015) Attenuation of histone methyltransferase KRYPTONITE-mediated transcriptional gene silencing by Geminivirus. Sci Rep 5:16476
Sun C et al (2021) Exploration of epigenetics for improvement of drought and other stress resistance in crops: a review. Plants (basel) 10(6):1226
Taliansky M et al (2021) RNA-based technologies for engineering plant virus resistance. Plants (basel) 10(1):82
Tang G et al (2003) A biochemical framework for RNA silencing in plants. Genes Dev 17(1):49–63
Telias A et al (2011) Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol 11:93
Tirnaz S, Batley J (2019a) DNA methylation: toward crop disease resistance improvement. Trends Plant Sci 24(12):1137–1150
Tirnaz S, Batley J (2019b) Epigenetics: potentials and challenges in crop breeding. Mol Plant 12(10):1309–1311
Tricker PJ et al (2013) Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int J Mol Sci 14(4):6674–6689
Tronick E, Hunter RG (2016) Waddington, dynamic systems, and epigenetics. Front Behav Neurosci 10:107
van Holde K, Zlatanova J (2007) Chromatin fiber structure: Where is the problem now? Semin Cell Dev Biol 18(5):651–658
Varotto S et al (2020) Epigenetics: possible applications in climate-smart crop breeding. J Exp Bot 71(17):5223–5236
Veley KM et al (2022) Improving disease resistance in plants by editing the epigenome. Nat Commun 14:85
Walker EL et al (1995) Transposon-mediated chromosomal rearrangements and gene duplications in the formation of the maize R-r complex. EMBO J 14(10):2350–2363
Walley JW et al (2008) The chromatin remodeler SPLAYED regulates specific stress signaling pathways. PLoS Pathog 4(12):e1000237
Wang C et al (2010) Arabidopsis putative deacetylase AtSRT2 regulates basal defense by suppressing PAD4, EDS5 and SID2 expression. Plant Cell Physiol 51(8):1291–1299
Wang MB et al (2012) RNA silencing and plant viral diseases. Mol Plant Microbe Interact 25(10):1275–1285
Wang W et al (2014) Overexpression of Camellia sinensis H1 histone gene confers abiotic stress tolerance in transgenic tobacco. Plant Cell Rep 33(11):1829–1841
Wang H et al (2015) CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc Natl Acad Sci U S A 112(44):13729–13734
Wang C et al (2017a) A transposon-directed epigenetic change in ZmCCT underlies quantitative resistance to Gibberella stalk rot in maize. New Phytol 215(4):1503–1515
Wang X et al (2017b) Gene-body CG methylation and divergent expression of duplicate genes in rice. Sci Rep 7(1):2675
Wang Y et al (2017c) Histone deacetylase 6 6 represses pathogen defence responses in Arabidopsis thaliana. Plant Cell Environ 40(12):2972–2986
Wang C et al (2018) Epigenetic changes in the regulation of Nicotiana tabacum response to cucumber mosaic virus infection and symptom recovery through single-base resolution methylomes. Viruses 10(8):402
Waryah CB et al (2018) Zinc fingers, TALEs, and CRISPR systems: a comparison of tools for epigenome editing. Methods Mol Biol 1767:19–63
Wei T, O’Connell MA (1996) Structure and characterization of a putative drought-inducible H1 histone gene. Plant Mol Biol 30(2):255–268
Wei X et al (2017) An epiallele of rice AK1 affects photosynthetic capacity. J Integr Plant Biol 59(3):158–163
Weigel D, Colot V (2012) Epialleles in plant evolution. Genome Biol 13(10):249
Wen S et al (2012) Structural genes of wheat and barley 5-methylcytosine DNA glycosylases and their potential applications for human health. Proc Natl Acad Sci U S A 109(50):20543–20548
Wierzbicki AT, Jerzmanowski A (2005) Suppression of histone H1 genes in Arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics 169(2):997–1008
Willcockson MA et al (2021) H1 histones control the epigenetic landscape by local chromatin compaction. Nature 589(7841):293–298
Xu L et al (2009) The E2 ubiquitin-conjugating enzymes, AtUBC1 and AtUBC2, play redundant roles and are involved in activation of FLC expression and repression of flowering in Arabidopsis thaliana. Plant J 57(2):279–288
Yang SM et al (2013) H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc Natl Acad Sci USA 110(5):1708–1713
Yang L et al (2015) Salicylic acid biosynthesis is enhanced and contributes to increased biotrophic pathogen resistance in Arabidopsis hybrids. Nat Commun 6:7309
Yang X et al (2019) Downregulation of nuclear protein h2b induces salicylic acid mediated defense against PVX infection in Nicotiana benthamiana. Front Microbiol 10:1000
Yang L et al (2020) HOS15 and HDA9 negatively regulate immunity through histone deacetylation of intracellular immune receptor NLR genes in Arabidopsis. New Phytol 226(2):507–522
You W et al (2012) Atypical DNA methylation of genes encoding cysteine-rich peptides in Arabidopsis thaliana. BMC Plant Biol 12:51
Yu A et al (2013) Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc Natl Acad Sci U S A 110(6):2389–2394
Zarreen F et al (2022) The diverse roles of histone 2B monoubiquitination in the life of plants. J Exp Bot 73(12):3854–3865
Zeng C et al (2022) Global dynamics of a new huanglongbing transmission model with quarantine measures. J Appl Math Phys 10(2):360–371
Zhang X et al (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126(6):1189–1201
Zhang L et al (2012) Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24(11):4407–4421
Zhang YY et al (2013) Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol 197(1):314–322
Zhang Y et al (2015) Tomato histone H2B monoubiquitination enzymes SlHUB1 and SlHUB2 contribute to disease resistance against Botrytis cinerea through modulating the balance between SA- and JA/ET-mediated signaling pathways. BMC Plant Biol 15:1–20
Zhang X et al (2015) Epigenetic mutation of RAV6 affects leaf angle and seed size in rice. Plant Physiol 169(3):2118–2128
Zhang H et al (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8):489–506
Zhi P, Chang C (2021) Exploiting epigenetic variations for crop disease resistance improvement. Front Plant Sci 12:692328
Zhong S et al (2013) Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 31(2):154–159
Zhu QH et al (2016) Epigenetic mechanisms: an emerging player in plant–microbe interactions. Mol Plant Microbe Interact 29(3):187–196
Zilberman D et al (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39(1):61–69
Zipfel C, Robatzek S (2010) Pathogen-associated molecular pattern-triggered immunity: veni, vidi...? Plant Physiol 154(2):551–554
Zlatanova J (1990) Histone H1 and the regulation of transcription of eukaryotic genes. Trends Biochem Sci 15(7):273–276
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sampson, C., Ikenwugwu, T.H., Okagu, I.U. et al. Epigenetics: Toward improving crop disease resistance and agronomic characteristics. Plant Biotechnol Rep 18, 1–20 (2024). https://doi.org/10.1007/s11816-023-00876-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-023-00876-z