
Abstract
In this research, stearic acid and propylene oxide are used to synthesize biodegradable heterocyles (pyrazole, pyran, chromene, and pyrimidine) to improve their solubility and lower their toxicity. The chemical structure of these surfactants was confirmed using IR, 1H, and 13C NMR spectra. Some of these compounds have higher activity than commercial antibiotics and may be useful in the pharmaceutical industry.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Enaminone derivatives are polydentate reagents and useful raw materials for the synthesis of many classes of organic compounds and heterocyclic systems [1–8]. In addition, many enaminone derivatives exhibit biological activities and are used as antitumor, antibacterial and anticonvulsant agents [9–11]. For this reason, photochemical, reduction, oxidation, nucleophilic and electrophilic interaction in these materials leads to various biologically active compounds [12, 13]. The family of nitrogen heterocyclic compounds such as pyrazole and pyrimidine derivaties are an important class of compounds in medicinal chemistry and have contributed to the understanding of biological processes [14–16]. Moreover, enaminone derivatives have attracted considerable attention in the design of biologically active pyrone and pyridone derivatives [17, 18].
Oils and fats have been used to produce customized products and new chemical reactions for the synthesis of new heterocyclic compounds. Such environmentally friendly operations are particularly favored in green chemistry, clean energy, renewable resources, and enzyme reaction research [19–21]. Oleochemicals with heterocyclic systems are organic compounds that contain both hydrophobic and hydrophilic groups. Due to their high solubility, foaming, wetting, and aggregation properties, they have a wide range of applications from consumer products to industrial uses, such as cleaning, detergents, cosmetics, emulsifiers, toiletries, and pharmaceuticals and petrochemicals [22–27]. In view of the above-mentioned properties, a variety of new heterocycles derivatives were synthesized to identify new selective and less toxic antimicrobial agents [28–31].
Experimental Section
Uncorrected melting points were measured using a Gallen Kamp melting point apparatus. The infrared spectra were recorded using KBr discs on FTIR 8300 Shimadzu spectrophotometer and the results were expressed in wave number (cm−1). 1H and 13C NMR spectra were recorded with Bruker AC300 spectrometer (Fällanden, Switzerland) operating at 600 MHz for 1H and 150 MHz for 13C, using deuterochloroform (CDCl3) as a solvent. Chemical shifts are expressed in δ (ppm) using TMS as the internal standard. The mass spectra were recorded on an Agilent GC/MS MSD (7890A/5975C) at 70 eV at the Core Labs and Major Facilities, King Abdullah University of Science and Technology. Elemental microanalysis was carried out using a CHNS elemental analyzer (model EA3000; EURO VECTOR Instruments). Biological activity was screened at the Microbiology Department, Faculty of Applied Science, Umm Al-Qura University, Saudi Arabia.
Synthesis of N-(4-Acetylphenyl)stearamide (3)
To a cold solution of p-aminoacetophenone (0.2 g, 1.5 mmol) in dry acetone (15 mL) containing triethylamine (0.5 mL), stearoyl chloride 2 (0.45 g, 1.5 mmol) in dry acetone (15 mL) was added dropwise with stirring over a period of 1 h. The mixture was stirred overnight, and then poured into a beaker containing crushed ice with a few drops of hydrochloric acid. The solids were collected by filtration and recrystallized from benzene. White-yellow crystals were formed. Yield (85 %), m.p. 100–102 °C, IR (ν/cm−1); 3328 (NH), 2916–2849 (CH-aliph) and 1678, 1656 (C = O). 1H NMR (δ, ppm): 0.85 (t, 3H, terminal CH3), 1.19–1.57 (s, 32H, CH2 aliphatic), 2.51 (s, 3H, COCH3), 6.97–7.87 (m, 4H, ArH), 9.77 (s, 1H, NH). Anal. Calc. (%) for C26H43NO2 (401.63): C, 77.75; H, 10.79; N, 3.49. Found C, 77.31; H, 10.47; N, 3.22.
Synthesis of N-(4-(3-(Dimethylamino)acryloyl)phenyl)stearamide (4)
A mixture of compound 3 (0.6 g, 1.5 mmol) and dimethylformamide dimethylacetal (DMF-DMA) (0.19 g, 1.5 mmol) in dry xylene (15 mL) was heated to reflux for 3 h, allowed to cool, and the solid product was collected by filtration and recrystallized from xylene. Yellow crystals were formed. Yield (0.46 g, 77 %), m.p. 81–83 °C, IR (ν/cm−1); 3326 (NH), 3073 (CH-arom), 2915–2848 (CH-aliph), 1685,1656 (C=O) and 1613 (C=C).1H NMR (δ, ppm): 0.88 (t, 3H, terminal CH3), 1.25–1.72 (s, 32H, CH2 aliph), 2.92, 3.12 (2 s, 6H, N(CH3)2), 5.70, 5.71 (d, 1H, J = 7.2 Hz, CH), 7.26–7.93 (m, 5H, ArH and CH), 9.65 (s, 1H, NH). 13C-NMR (δ, ppm): 14.03, 22.16, 29.07, 39.08, 39.22, 39.35, 39.49, 39.63, 39.77, 39.91, 40.03, 96.23, 126.41, 126.46, 128.46, 128.55, 128.65, 129.32, 132.96, 143.03, 154.31, 167.38, 187.83, 197.17. Anal. Calc. (%) for C29H48N2O2 (456.70): C, 76.27; H, 10.59; N, 6.13. Found C, 76.05; H, 10.31; N, 6.36 %.
Synthesis of N-(4-(Isoxazol-5-yl)phenyl)stearamide (5)
A solution of enaminone 4 (0.68 g, 1.5 mmol) and hydroxylamine hydrochloride (0.1 g, 1.5 mmol) in a mixture of triethylamine (0.5 mL) and ethanol (15 mL) was heated to reflux for 8 h, and then allowed to cool. The product was collected by filtration, dried and recrystallized from ethanol. A pale yellow solid was formed. Yield (0.5 g, 74 %); m.p. 96–98 °C; IR (υ/cm−1): 3314 (NH), 2916–2848 (CH-aliph), 1678 (C = O), 1601 (C = C), 1552 (C = N), 1169 (C–O–C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal, CH3), 1.39–1.72 (s, 32H, CH2 aliph), 5.66 (s, 1H, C3-H isoxazole), 6.67 (s, 1H, C4-H isoxazole), 7.26–7.92 (m, 4H, ArH), and 8.45 (s, 1H, NH). 13C-NMR (δ, ppm): 14.02, 22.15, 28.76, 29.07, 31.34, 33.73, 39.08, 39.22, 39.36, 39.49, 39.63, 39.77, 39.91, 40.03, 102.12, 125.15, 128.26, 128.54, 137.03, 158.54, 166.62. Anal.Calc. (%) for C27H42N2O2 (426.63): C, 76.01; H, 9.92; N, 6.57. Found C, 76.31; H, 10.20; N, 6.84.
Synthesis of N-(4-(1H-pyrazol-5-yl)phenyl)stearamide (6)
To a boiling solution of enaminone 4 (0.68 g, 1.5 mmol) in a mixture of ethanol (10 mL) and acetic acid (10 mL), hydrazine hydrate (0.07 g, 1.7 mmol) was added. The reaction mixture was heated to reflux for 6 h, and then cooled. The solid product was collected by filtration and recrystallized from dioxane. An orange powder was formed. Yield (0.53 g, 78 %); m.p. 101–103 °C, IR (ν/cm−1): 3320, 3287 (NH), 2916–2849 (CH-aliph) 1654 (C=O), 1616 (C=C), and 1544 (C=N). 1H NMR (δ, ppm): 0.87 (t, 3H, terminal CH3), 1.24–1.70 (s, 32H, CH2 aliph), 5.71, 6.91 (2d, 2H, J = 8.2 Hz, pyrazol-H4, H5), 7.26–7.90 (m, 5H, Ar–H and NH), 10.56 (s, 1H, 1NH). 13C-NMR (δ, ppm): 14.78, 25.47, 26.44, 29.26, 29.37, 29.48, 29.62, 29.66, 29.68, 29.70, 31.93, 37.92, 108.34, 118.82, 129.77, 132.77, 138.87, 140.30, 142.39, 159.01. Anal. Calc. (%) for C27H43N3O (425.65): C, 76.19; H, 10.18; N, 9.87. Found C, 76.43; H, 10.38; N, 9.52.
Synthesis of N-(4-(3,5-Dihydro-2H-1,4-oxathiepin-7-yl)phenyl)stearamide (7)
To a solution of enaminone 4 (0.68 g, 1.5 mmol) in ethanol (20 mL) with few drops of triethylamine, 2-mercaptoethanol (0.12 g, 1.5 mmol) was added. The reaction mixture was refluxed for 8 h. After cooling to room temperature, the solid was collected by filtration and recrystallized from ethanol. A brown powder was formed. Yield (0.49 g, 72 %); m.p. 111–113 °C; IR (υ/cm−1): 3319 (NH), 2916–2849 (CH-aliph), 1656 (C = O), 1606 (C = C), 1160 (C–O–C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.24–1.72 (s, 32H, CH2 aliph), 2.88 (t, 2H, J = 8.0 Hz, C3-H oxathiepin ring), 3.10 (d, 2H, J = 7.2 Hz, C5-H oxathiepin ring), 3.90 (t, 2H, C2-H oxathiepin ring), 5.70 (t, 1H, C6-H oxathiepin ring), and 7.26–8.92 (m, 5H, ArH and NH). 13C-NMR (δ, ppm): 14.09, 22.15, 28.76, 29.07, 33.73, 39.08, 39.22, 39.36, 39.49, 39.63, 39.77, 39.91, 40.03, 73.13, 91.99, 125.15, 128.26, 128.54, 136.36, 138.35, 160.95, 182.18. Anal. Calc. (%) for C29H47NO2S (473.75): C, 73.52; H, 10.00; N, 2.96; S, 6.77. Found C, 73.80; H, 10.27; N, 3.26; S, 6.98.
Synthesis of N-(4-(5-Acetyl-6-methyl-4H-pyran-2-yl)phenyl)stearamide (8)
An equimolar amount of acetylacetone (0.15 g, 1.5 mmol) was added to a solution of enaminone 4 (0.68 g, 1.5 mmol) in glacial acetic acid (20 mL). The reaction mixture was refluxed for 12 h, left to cool and poured onto ice-cold water containing a few drops of 1 N HCl. The precipitate that formed was collected by filtration and recrystallized from ethanol. A light brown solid was formed. Yield (0.46 g, 68 %), mp 114–116 °C. IR (ν/cm−1): 3321 (NH), 2916–2849 (CH-aliph), 1692, 1656 (C = O), 1079 (C–O–C); 1H NMR (δ, ppm): 0.89 (t, 3H, terminal CH3), 1.24–1.62 (s, 32H, CH2 aliph), 2.30 (s, 3H, CH3), 2.56 (s, 1H, COCH3), 4.11 (d, 2H, J = 7.0 Hz, CH2 pyran), 6.64 (t, 1H, J = 8.2 Hz, C-H pyran), 7.26–7.93 (m, 5H, ArH and NH). 13C-NMR (δ, ppm): 13.57, 14.02, 19.16, 22.15, 26.43, 29.25, 29.37, 29.47, 29.61, 29.66, 29.68, 29.70, 30.94, 31.93, 37.94, 95.29, 115.39, 118.79, 129.77, 132.80, 142.35, 159.30, 179.60, 196.95. Anal. Calc. (%) for C32H49NO3 (495.74): C, 77.53; H, 9.96; N, 2.83. Found C, 77.21; H, 9.68; N, 2.60.
Synthesis of N-(4-(2-Thioxo-1,2-dihydropyrimidin-4-yl)phenyl)stearamide (9)
A mixture of enaminone 4 (0.68 g, 10 mmol) and thiourea (0.11 g, 1.5 mmol) in ethanol (20 mL) with few drops of triethylamine was heated to reflux for 8 h. After cooling to room temperature, the obtained solid was collected by filtration and recrystallized from EtOH/DMF. Yellow crystals were formed. Yield (0.54 g, 80 %); m.p. 125–127 °C; IR (ν/cm−1): 3338, 3278 (NH), 2914–2849 (CH-aliph.), 1657 (C = O), 1600 (C = C), 1563 (C = N), 1274 (C = S); 1H NMR (δ, ppm): 0.87 (t, 3H, terminal CH3), 1.22–1.69 (s, 32H, CH2 aliph), 5.71 (d, 1H, C5-pyrimidine), 7.26–7.88 (m, 5H, ArH and C4-pyrimidine), 8.28 (s, 1H, NH), and 8.57 (s, 1H, NH); 13C-NMR (δ, ppm): 14.13, 22.69, 25.53, 26.43, 29.31, 29.37, 29.43, 29.53, 29.67, 29.68, 29.71, 31.93, 37.77, 45.09, 92.04, 118.92, 128.60, 129.67, 132.42, 135.62, 143. 03, 141.09, 154.31, 172.11, 172.37, 187.83, 197.17. Anal. Calc. (%) for C28H43N3OS (469.73): C, 71.59; H, 9.23; N, 8.95; S, 6.83. Found C, 71.83; H, 9.47; N, 8.71; S, 6.60.
General Procedure for Synthesis of Pyrimidines Derivatives (10a, b)
A mixture of enaminone 4 (0.68 g, 1.5 mmol) and urea (0.1 g, 15 mmol) or guanidine hydrochloride (0.14 g, 1.5 mmol) in ethanol (15 mL) catalyzed by triethylamine or glacial acetic acid (15 mL) in the presence of freshly fused sodium acetate was heated to reflux for 4 h, and allowed to cool and then poured onto ice-cold water containing a few drops of 1 N HCl. The precipitate was collected by filtration, dried, and recrystallized from the mixture of EtOH/DMF.
N-(4-(2-Oxo-1,2,3,6-tetrahydropyrimidin-4-yl)phenyl)stearamide (10a)
Brown powder; yield (0.45 g, 67 %); m.p. 116–118 °C; IR (ν/cm−1): 3310–3246 (NH), 3072 (CH-arom), 2915–2848 (CH-aliph), 1685, 1654 (C = O), 1610 (C = C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.24–1.73 (s, 32H, CH2 aliphatic), 3.66 (d, 2H, pyrimidine ring), 5.26 (t, 1H, CH pyrimidine), 6.44 (s, 2H, 2NH), 7.26–7.92 (m, 5H, ArH and NH). Anal. Calc. (%) for C28H45N3O2 (455.68): C, 73.80; H, 9.95; N, 9.22. Found C, 73.61; H, 9.74; N, 9.02.
N-(4-(2-Imino-1,2,3,6-tetrahydropyrimidin-4-yl) phenyl)stearamide (10b)
Dark yellow solid; yield (0.54 g, 79 %); m.p. 120–122; IR (ν/cm−1): 3318–3176 (NH), 3056 (CH-arom), 2915–2848 (CH-aliph),1688 (C = O), 1623 (C = C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.24–1.70 (s, 32H, CH2 aliph), 4.12 (d, 2H, CH2 pyrimidine), 5.69 (t, 1H, CH pyrimidine), 7.26–7.90 (m, 6H, ArH and 2NH), 10.92 (s, 1H, NH). Anal. Calc. (%) for C28H46N4O (454.69): C, 73.96; H, 10.20; N, 12.32. Found C, 73.71; H, 10.54; N, 12.09.
Synthesis of ethyl 3-(4-Stearamidophenyl)-2, 5-dihydro-1H-pyrrole-2-carboxylate (11)
A mixture of enaminone 4 (0.68 g, 1.5 mmol), in ethanol (15 mL) containing a catalytic amount of triethylamine and ethyl glycinate hydrochloride (0.21 g, 1.5 mmol) was heated to reflux for 8 h. The formed solid was filtered and recrystallized from benzene. A pink powder was formed. Yield (0.53 g, 78 %); m.p. 127–129 °C; IR (ν/cm−1): 3316, 3225 (NH), 2915–2849 (CH-aliph), 1710, 1655 (C = O), 1613 (C = C); 1H NMR (δ, ppm): 0.85 (t, 3H, terminal CH3), 1.24–1.70 (s, 32H, CH2 aliph), 1.72 (t, 3H, J = 8.6 Hz, CH3), 3.72, 3.99 (2d, 2H, J = 8.2 Hz, CH2 pyrrole), 4.20 (s, 2H, CH2), 4.24 (s, 1H, C2-H pyrrole), 5.77 (t, 1H, J = 8.4 Hz C4-H pyrrole), 7.26–7.89 (m, 5H, ArH and NH) and 8.81 (s, H, NH); 13C NMR (δ, ppm): 14.13, 22.70, 25.57, 26.44, 29.27, 29.29, 29.37, 29.39, 29.49, 29.63, 29.67, 29.71, 31.93, 37.86, 49.77, 58.46, 61.75, 91.78, 118.83, 118.93, 128.38, 128.66, 129.56, 129.74, 132.65, 142.58, 169.35, 174.95. Anal. Calc. (%) for C31H50N2O3 (498.74): C, 74.65; H, 10.10; N, 5.62. Found C, 74.33; H, 9.78; N, 5.41.
Synthesis of N-(4-(7H-thiazolo[3,2-a]pyrimidin-5-yl)phenyl)streamside (12)
An equimolar amount of enaminone 4 (0.68 g, 1.5 mmol) in a mixture of acetic acid/ethanol (15 mL:15 mL) and 2-aminothiazole (0.15 g, 1.5 mmol) was heated to reflux for 3 h. The formed solid was collected and recrystallized from methanol. A brown powder was formed. Yield (0.46 g, 68 %); m.p. 112–114 °C, IR (ν/cm−1): 3315 (NH), 3073 (CH-aromatic), 2915–2848 (CH-aliph), 1663 (C = O), 1632 (C = C) and 1565 (C = N). 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.28–1.75 (s, 32H, CH2 aliphatic), 2.39, 2.57 (2d, 2H, J = 8.2 Hz, CH2 pyrimidine), 4.86 (t, 1H, J = 8.7 Hz CH pyrimidine), 6.26, 6.86 (2 s, 2H, thiazol-H4, H5), 7.26–7.93 (m, 5H, ArH and NH). MS m/z (%): 494 (M+−1, 4.17). Anal. Calc. (%) for C30H45N3OS (495.76): C, 72.68; H, 9.15; N, 8.48; S, 6.47. Found C, 72.89; H, 9.36; N, 8.70; S, 6.69.
Synthesis of N-(4-(9aH-pyrido[1,2-a]pyrimidin-4-yl)phenyl)stearamide (13)
A mixture of compound 4 (0.68 g, 1.5 mmol) and 2-aminopyridine (0.14 g, 1.5 mmol) in a mixture of acetic acid/ethanol (15 mL:15 mL) was heated to reflux for 4 h. The obtained solid was collected by filtration and recrystallized from ethanol. White yellow crystals were formed. Yield (0.48 g, 72 %); m.p. 119–121 °C, IR (ν/cm−1); 3305 (NH), 3073 (CH-arom), 2915–2848 (CH-aliph), 1673 (C=O), 1631 (C=C) and 1564 (C=N). 1H NMR (δ, ppm): 0.79 (t, 3H, CH3), 1.28–1.79 (s, 32H, CH2 aliph), 5.70 (t, 1H, CH pyrimidine), 2.78 (d, 2H, CH2 pyrimidine), 7.29–7.85 (m, 8H, ArH), 8.19 (s, 1H, NH) 13C NMR (δ, ppm): 14.13, 22.70,25.47, 26.44, 29.26, 29.37, 29.48, 29.62, 29.66, 29.68, 29.70, 31.93, 83.24, 83.82, 109.48, 118.82, 127.64, 128.68, 129.77, 131.80, 132.48, 137.92, 139.92, 142.36, 153.73, 171.76, 187.01. MS m/z (%): 450 (M++1, 0.81). Anal. Calc. (%) for C32H47N3O (489.74): C, 78.48; H, 9.67; N, 8.58. Found C, 78.68; H, 9.89; N, 8.80.
Synthesis of N-(4-(2H-benzo [4, 5] thiazolo[3,2-a]pyrimidin-4-yl)phenyl)stearamide (14)
To a solution of enaminone 4 (0.68 g, 1.5 mmol) in ethanol (20 mL), 2-aminobenzothiazole (0.22 g, 1.5 mmol) was added. The reaction mixture was heated to reflux for 8 h. The solid was filtered and recrystallized from dioxane. A brown powde was formed. Yield (0.5 g, 74 %); m.p. 135–137 °C; IR (υ/cm−1): 3294 (NH), 3073 (CH-arom), 2915–2848 (CH-aliph),1676 (C = O), 1631 (C = C), 1564 (C = N); 1H NMR (300 MHz) δ: 0.87 (t, 3H, terminal CH3), 1.28–1.75 (s, 32H, CH2 aliph), 2.33 (d, 2H, J = 7.2 Hz, CH2 pyrimidine), 5.57 (t, H, J = 8.4 Hz, CH pyrimidine), 7.26–7.93 (m, 8H, ArH), 8.28 (s, 1H, NH); 13C NMR (δ, ppm): 14.13, 22.70, 25.45, 26.43, 29.25, 29.37, 29.47, 29.61, 29.66, 29.68, 29.69, 29.70, 30.94, 31.93, 37.94, 96.95, 118.10, 123.26, 126.37, 128.66, 129.67, 129.68, 132.70, 134.71, 142.96, 153.94, 178.68. MS m/z (%): 545 (M+, 0.95). Anal. Calc. (%) for C34H47N3OS (545.82): C, 74.82; H, 8.68; N, 7.70; S, 5.87. Found C, 74.53; H, 8.44; N, 7.48; S, 5.56.
Synthesis of N-(4-(benzo [4, 5] imidazole[1,2-a]pyrimidin-4-yl)phenyl)stearamide (15)
A mixture of enaminone 4 (0.68 g, 1.5 mmol) in ethanol (20 mL) and 2-aminobenzoimidazole (0.21 g, 1.5 mmol) was heated to reflux for 8 h. The separated solid was filtered and recrystallized from ethanol. A brown powde was formed. Yield (0.46 g, 68 %); m.p. 123–125 °C; IR (υ/cm−1): 3366 (NH), 3072 (CH-arom), 2915–2848 (CH-aliph),1666 (C = O), 1622 (C = C), 1565 (C = N); 1H NMR (δ, ppm): 0.88 (t, 3H, terminal CH3), 1.31–1.75 (s, 32H, CH2 aliph), 2.40 (s, 2H, CH2 pyrimidine),5.36 (s, 1H, CH pyrimidine), 7.26–7.94 (m, 8H, ArH), and 9.17 (s, 1H, NH). MS m/z (%): 524 (M+−2, 0.98). Anal. Calc. (%) for C34H48N4O (526.76): C, 77.52; H, 8.80; N, 10.64. Found C, 77.21; H, 8.57; N, 10.38.
General Procedure for the Synthesis of pyrano[2,3-d]pyrimidine derivatives (16a, b)
A mixture of compound 4 (0.68 g, 1.5 mmol) in acetic acid (20 mL) and barbituric acid (0.2 g, 1.5 mmol) or thiobarbituric acid (0.22 g, 1.5 mmol) was heated to reflux for 12 h, allowed to cool and poured onto ice-cold water containing a few drops of 1 N HCl. The precipitate that formed was collected by filtration, and recrystallized from ethanol.
N-(4-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-pyrano[2,3-d]pyrimidin-7-yl)phenyl)stearamide (16a)
Reddish yellow powder; yield (0.54 g, 80 %); m.p. 111–113 °C; IR (υ/cm−1): 3341–3277 (NH), 2916–2848 (CH-aliph), 1678, 1660 (C = O), 1601 (C = C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.16–1.75 (s, 32H, CH2 aliph), 2.45 (d, 2H, J = 7.2 Hz, CH2), 5.85 (t, 1H, C3-H pyran), 6.85 (s, 1H, NH), 7.26–7.47 (m, 4H, ArH), 8.08, 8.82 (2 s, 2H, 2NH). Anal. Calc. (%) for C31H45N3O4 (523.71): C, 71.10; H, 8.66; N, 8.02. Found: C, 71.37; H, 8.85; N, 8.22.
N-(4-(4-Oxo-2-thioxo-2,3,4,5-tetrahydro-1H-pyrano[2,3-d]pyrimidin-7-yl)phenyl)stearamide (16b)
Brown powder; yield (0.52 g, 77 %); m.p. 129–131 °C; IR (υ/cm−1): 3332–3270 (NH), 2916–2849 (CH-aliph), 1683, 1656 (C = O), 1613 (C = C), 1249 (C = S), 1074 (C–O–C); 1H NMR (δ, ppm): 0.88 (t, 3H, CH3), 1.29–1.75 (s, 32H, CH2 aliph), 2.39 (d, 2H, J = 7.2 Hz, CH2), 5.68 (t, 1H, J = 8.4 Hz, C3-H pyran), 7.26–7.94 (m, 5H, ArH and NH), 8.90, 12.15 (2 s, 2H, 2NH). Anal. Calc. (%) for C31H45N3O3S (539.77): C, 68.98; H, 8.40; N, 7.78; S, 5.94. Found: C, 68.68; H, 8.19; N, 7.52; S, 6.22.
General Procedure for the Synthesis of Chromene Derivatives (17–21)
A mixture of enaminone 4 (0.68 g, 1.5 mmol) in acetic acid (20 mL) and resorcinol (0.17 g, 1.5 mmol), α-naphthol (0.22 g, 1.5 mmol), β-naphthol (0.22 g, 1.5 mmol), salicaldehyde (0.18 g, 1.5 mmol), or α-tetralone (0.22 g, 1.5 mmol), in each case, was heated to reflux 10–12 h, and then poured onto crushed ice with a few drops of hydrochloric acid. The solid was collected by filtration and recrystallized from acetic acid.
N-(4-(7-Hydroxy-4H-chromen-2-yl)phenyl)stearamide (17)
Brown powder; yield (0.56 g, 83 %); m.p. 117–119 °C; IR (υ/cm−1): 3446, 3286 (OH and NH), 3073 (CH-arom), 2916–2849 (CH-aliph), 1656 (C = O), 1077 (C–O–C); 1H NMR (δ, ppm): 0.85 (t, 3H, terminal CH3), 1.31–1.74 (s, 32H, CH2 aliph), 2.39 (d, 2H, J = 7.2 Hz, CH2), 5.96 (t, 1H, J = 8.7 Hz, CH), 6.41 (s, 1H, NH), 7.26–7.93 (m, 7H, ArH), and 9.98 (s, 1H, OH); MS m/z (%): 505 (M+, 0.99). Anal. Calc. (%) for C33H47NO3 (505.73): C, 78.37; H, 9.37; N, 2.77. Found: C, 78.58; H, 9.61; N, 2.98.
N-(4-(4H-benzo[h]chromen-2-yl)phenyl)stearamide (18)
Dark brown powder; yield (0.51 g, 75 %); m.p. 126–128 °C; IR (v/cm−1): 3342 (NH), 3078 (CH-arom), 2916–2848 (CH-aliph), 1678 (C = O), 1621 (C = C); 1H NMR (δ, ppm): 0.86 (t, 3H, terminal CH3), 1.25–1.74 (s, 32H, CH2 aliph), 2.40 (d, 2H, J = 7.2 Hz, CH2), 5.54 (t, 1H, J = 8.8 Hz, C3-H chromen), 6.85–8.09 (m, 10H, ArH), 8.82 (s, 1H, NH). Anal. Calc. (%) for C37H49NO2 (539.79): C, 82.33; H, 9.15; N, 2.59. Found: C, 82.61; H, 9.42; N, 2.85.
N-(4-(1H-benzo[f]chromen-3-yl)phenyl)stearamide (19)
Brown powder; yield (0.56 g, 83 %); 123–125 °C; IR (v/cm−1): 3345 (NH), 3075 (CH-arom), 2915–2848 (CH-aliph), 1667 (C = O), 1617 (C = C); 1H NMR (δ, ppm) δ: 0.86 (t, 3H, terminal CH3), 1.25–1.74 (s, 32H, CH2 aliph), 3.73 (s, 2H, CH2), 5.65 (t, 1H, J = 8.8 Hz, C3-H chromen), 7.11–7.94 (m, 10H, ArH and NH). 13C NMR (δ, ppm): 14.13, 22.70, 25.48, 26.44, 29.10, 29.26, 29.37, 29.48, 29.62, 29.66, 29.67, 29.68, 29.70, 29.71, 30.69, 31.93, 37.86, 106.48, 117.92, 118.87, 123.47, 126.35, 127.75, 128.85, 129.75, 129.80, 132.65, 134.66, 142.36, 153.73, 178.88. Anal. Calc. (%) for C37H49NO2: (539.79): C, 82.33; H, 9.15; N, 2.59. Found: C, 82.58; H, 9.34; N, 2.81.
N-(4-(8-Formyl-4H-chromen-2-yl)phenyl)stearamide (20)
Brown powder; yield (0.58 g, 85 %); m.p. 125–127 °C; IR (v/cm−1): 3324 (NH), 3054 (CH-arom.), 2915–2848 (CH-aliph), 1714, 1671 (C = O); 1H NMR (δ, ppm): 0.88 (t, 3H, terminal CH3), 1.27–1.73 (s, 32H, CH2 of aliph), 2.39 (d, 2H, CH2), 5.35 (t, 1H, J = 8.6 Hz, CH pyran), 7.26–8.40 (m, 7H, ArH), 8.92 (s, 1H, NH), 10.15 (s, 1H, CHO). Anal. Calcd. (%) for C34H47NO3 (517.74): C, 78.87; H, 9.18; N, 2.71. Found: C, 78.54; H, 8.87; N, 2.96.
N-(4-(5,6-Dihydro-1H-benzo[f]chromen-3-yl)phenyl)stearimidamide (21)
Gray powder; yield (0.57 g, 84 %); m.p. 132–134 °C; IR (υ/cm−1): 3344 (NH), 3069 (CH-arom), 2915–2848 (CH-aliph), 1687 (C = O), 1625 (C = C), 1120 cm−1 (C–O–C); 1H NMR (δ, ppm): 0.87 (t, 3H, terminal CH3), 1.24–1.63 (s, 32H, CH2 aliph), 2.29 (t, 2H, J = 8.6 Hz, CH2), 2.57 (d, 2H, J = 7.2 Hz, C4-H chromen), 2.98 (t, 2H, J = 8.8 Hz, CH2), 4.13 (t, 1H, J = 8.6 Hz, C3-H chromen), 7.25–8.03 (m, 8H, ArH), 8.04 (s, 1H, NH). Anal. Calc. (%) for C37H50NO2 (540.82): C, 82.17; H, 9.69; N, 5.18. Found: C, 82.32; H, 9.84; N, 5.34.
Synthesis of N-(4-(5-oxo-4,5-dihydroindeno[1,2-b]pyran-2-yl)phenyl)stearamide (22)
A solution of compound 4 (0.68 g, 1.5 mmol) in acetic acid (30 mL) and 1,3-indandione (0.22 g, 1.5 mmol) was heated to reflux for 12 h, and then poured onto ice/water. The solid was collected by filtration and recrystallized from acetic acid. A back powder was formed. Yield (0.57, 84 %); m.p. 122–124 °C; IR (υ/cm−1): 3334 (NH), 3074 (CH-arom.), 2916–2848 (CH-aliph), 1705,1672 (C = O); 1H-NMR (δ, ppm): 0.82 (t, 3H, terminal CH3), 1.16–1.55 (s, 32H, CH2 aliphatic), 2.48, 2.51 (2d, 2H, J = 7.1 Hz, C4-H pyran), 4.03 (t, 1H, J = 8.7 Hz, C3-H pyran), 7.19–7.87 (m, 8H, ArH), 8.56 (s, 1H, NH). 13C NMR (δ, ppm): 25.53, 26.43, 29.31, 29.33, 29.37, 29.43, 29.53, 29.67, 29.68, 29.71, 31.93, 37.77, 45.09, 92.04, 107.07, 118.92, 122.88, 128.60, 129.67, 132.42, 135.62, 141.09, 143.03, 154.31, 172.11, 187.83, 197.17. Anal. Calc. (%) for C36H47NO3 (541.76): C, 79.81; H, 8.74; N, 2.59. Found: C, 79.54; H, 8.48; N, 2.22.
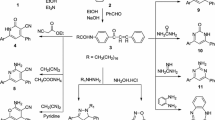
Preparation of Surface-Active Agents
Addition of 5 mol of propylene oxide to the active hydrogen atoms in the newly synthesized compounds 5–22 was carried out according to the Morgos procedure [32]. An amount of 0.5 wt% KOH solution containing 0.01 mol of the synthesized compound was heated to 70 °C with stirring while passing a slow stream of nitrogen through the system to flush out the oxygen and remove the water from the catalyst. The nitrogen purge was discontinued and propylene oxide was added dropwise with continuous stirring and heating under reflux to retain the propylene oxide. The alkoxylation reaction was conducted for different time intervals ranging from 1 to 10 h after which the apparatus was filled with nitrogen and cooled. The reaction vessel was weighed and the amount of reacted propylene oxide and the average degree of propoxylation were determined from the increment in the mass of the reaction mixture. Addition of propylene oxide gave a mixture of propoxylated products, and their structures were confirmed based on IR and 1H NMR spectra. The IR spectra reveled a broad band in the region of 3500–2500 cm−1 (OH) and two other bands at 1170–1048 and 950–840 cm−1 (C–O–C ether linkage of the polypropoxy chain) in addition to the original bands of these compounds. 1H NMR-spectra showed the protons of the propoxy groups, which appeared as broad multiple signals in the region of 3.2–3.8, in addition to the other signals of these compounds.
Antibacterial Activity
The new surface-active agents was investigated in vitro for their antibacterial activities against Staphylococcus aureus (ATCC 25923) and Bacillus cereus (ATCC 10987) as Gram-positive bacteria and Serratia marcesens (ATCC 274) and Proteus mirabilis (SM514) as Gram-negative bacteria using an agar diffusion method [33] and filter paper disc-diffusion technique [34].
Surface and Interfacial Tension
Surface and interfacial tensions were measured according to Findlay [35] with a Krüss K6 tensiometer [36] (Krüss, Hamburg, Germany) for different concentrations of the synthesized surfactants using a platinum iridium ring at constant temperature (25 °C). Paraffin oil was used for the interfacial tension measurements and the tensiometer was calibrated using the method described in ASTM D1331-01 [37].
Cloud Point
In a temperature-controlled bath, a 1.0 wt% solution of the tested compound was gradually heated until the clear or nearly clear solution became definitely turbid [38]. The temperature was measured and the solution allowed to cool until it became clear again. This process was then repeated to check the reproducibility of the recorded temperature.
Wetting Time
The time of wetting was measured by immersing a cotton skein (1 g) in a 1 wt% solution of the prepared surfactants in distilled water at 25 °C according to the Draves technique [39]. The sinking time was measured in seconds.
Foaming Properties
The foaming height was measured by the Ross Miles method [40]. The surfactant solution was allowed to fall from a set height into the same surfactant solution in a volumetric cylinder, hence creating foam. The height of the foam was visually assessed.
Emulsification Stability
The emulsifying property of the prepared surfactants was determined as follows: in a 100-mL graduated stoppered tube, an aqueous solution of the surfactant (10 mL, 20 mol) was mixed with light paraffin oil (6 mL). The mixture was mixed vigorously by Cimarec™ magnetic stirring (Thermo Scientific) using an estimated stirring speed of 1100 rpm for 2 min at 25 °C. The tube was placed upright and the separation of the emulsion was observed. The time taken for the separation of 9 mL of the aqueous layer indicates the emulsion stability of the surfactant [41].
Biodegradability
The biodegradation of the synthesized surfactants was determined using the River Water Die-Away method [42]. The river water for testing was sampled from the River Nile. In this test, a stirred solution contained the tested surfactant (1000 ppm) incubated at 25 °C. Samples were withdrawn daily, filtered using Whatman filter paper and the surface tension measured using a Du-Nouy tensiometer (Kruss type K6). This process was repeated for 7 days.
Results and Discussion
Enaminone derivatives are useful raw materials for the synthesis of different heterocyclic compounds as shown in Scheme 1 [43]. Stirring stearoyl chloride [44] with 4-aminoacetophenone in dry acetone containing a catalytic amount of triethyl amine gave the acetyl derivative 3, in good yield. The structure of compound 3 was confirmed from the spectroscopic data. The IR spectrum revealed two absorption bands at 3322 and 1668 cm−1 due to NH and CO groups. In addition, the 1H NMR spectrum showed a multiplet in the region of 1.19–1.57 corresponding to CH2 aliphatic protons and 9.77 ppm for NH group. Treatment of compound 3 with dimethylformamide-dimethylacetal (DMF-DMA) in boiling xylene gave the enaminone derivative 4. The structure of enaminone 4 was established based on the spectral data. The IR spectrum showed absorption bands at 3326 (NH), and 1685 and 1656 cm−1 (CO), while the 1H NMR spectrum revealed two singlet signals at 2.92 and 3.12 due to two methyl groups on the amine and two doublet signals at 5.70 and 5.71 for the olefinic protons.

Synthesis of N-(4-(3-(dimethylamino)acryloyl) phenyl)stearamide (4)
The reactivity of enaminone 4 towards different carbon nucleophiles was investigated to produce new heterocyclic systems with surface and biological activities as shown in Scheme 2. Thus, the reaction of compound 4 with hydroxylamine hydrochloride in boiling ethanol containing a few drops of triethyl amine furnished N-(4-(isoxazol-5-yl)phenyl)stearamide (5) in good yields. The spectral data of the isolated product was in complete agreement with structure 5, in which the IR spectrum appeared to lack of an absorption band corresponding to the function of CO conjugated at 1678 cm−1 and showed absorption band at 1552 cm−1 due to C = N. The 1HNMR spectrum showed signals at 5.66 and 6.67 for 2CH protons of isoxazole ring, and 8.45 ppm for NH proton. Similarly, compound 4 was heated with hydrazine hydrate in boiling ethanol and gave the pyrazole derivative 6. The structure of pyrazole 6 was established by analytical and spectroscopic data. The 1H NMR spectrum displayed signals at 5.71 and 6.91 ppm due to H4 and H5 of pyrazole and 10.56 ppm for the NH group. Continung the reaction of enaminone 4 with bifunctional reagents, it was found that the reflux of enaminone 4 with mercapto ethanol in boiling ethanol with a few drops of triethyl amine produced N-(4-(3,5-dihydro-2H-1,4-oxathiepin-7-yl)phenyl)stearamide (7).

Synthesis of isoxazole 5, pyrazole 6, oxathiepin 7, pyran 8, pyrimidines 9, 10a, b and pyrrole 11 derivatives
Extension of the reactivity of enaminone 4 towards different nucleophiles was carried out to obtain the heterocyclic derivatives. Thus, the reaction of compound 4 with acetylacetone in glacial acetic acid produced the pyran derivative 8. The structure of pyran 8 was inferred from the spectral data. The 1HNMR spectrum showed signals at 2.30 and 2.56 ppm due to CH3 group protons, acetyl protons, 4.11 ppm for CH2 protons of the pyran ring and 6.64 ppm for methine proton of the pyran ring. Moreover, the reaction of enaminone 4 with thiourea in boiling ethanol containing a few drops of triethylamine gave N-(4-(2-thioxo-1,2-dihydropyrimidin-4-yl)phenyl)stearamide 9. Its 1HNMR spectrum exhibited signals at 5.71 for pyrimidine protons, and 8.28 and 8.57 ppm due to the two amino groups. Similarly, treatment of enaminone 4 with urea and/or guanidine hydrochloride in boiling ethanol containing a few drops of pyridine or glacial acetic acid with sodium acetate furnished the pyrimidine derivatives 10a, b. The structure of compounds 10a, b was assigned based on the elemental analysis and spectral data (see “Experimental”).
It is interesting in this connection that the reaction of enaminone 4 with ethyl glycinate hydrochloride in a boiling ethanol containing a catalytic amount of triethylamine afford ethyl 3-(4-stearamidophenyl)-2, 5-dihydro-1H-pyrrole-2-carboxylate 11. The structure of pyrrole 11 was identified on the basis of the analytical and spectral data. The 1H NMR spectrum showed bands at 1.72 ppm as a triplet signal corresponding to the methyl group of ester, doublets at 3.72 and 3.99 ppm due to CH2 of the pyrrole ring, a singlet at 4.24 ppm for C2-H of the pyrrole ring, and a multiplet at 5.77 ppm for C4-H of the pyrrole ring.
In view of the growing biological important of fused thiazoles [45], it was of interest to investigate the reactivity of enaminone 4 towards fused heterocyclic compounds for a facile synthesis of a bicyclic system, as shown in Scheme 3. Thus, condensation of enaminone 4 with 2-aminothiazole and/or 2-aminopyridine in boiling glacial acetic acid/ethanol furnished the thiazolo[3,2-a]pyrimidine and/or the pyrido[1,2-a] pyrimidine derivatives 12 and/or 13, respectively. Also, the reaction of enaminone 4 with 2-aminobenzothiazole and/or 2-aminobenzoimidazole in boiling glacial acetic acid produced the pyrimido[2,1-b]benzo-thiazole and/or pyrimido[1,2-a]benzoimidazole derivatives 14 and/or 15, respectively. The behavior of enaminone 4 towards an active methylene group incorporated into the heterocyclic ring has also been studied. Thus, cycloaddition of enaminone 4 with barbituric acid and/or thiobarbituric acid in glacial acetic acid afforded pyrano[2,3-d]pyrimidine derivatives 16a, b (Scheme 3). In addition, the reaction of enaminone 4 with resorcinol, α-naphthol, β-naphthol and/or α-tetralone, in boiling glacial acetic acid gave the chromene derivatives 17–21. All collected data for compounds 14–21 were consistent with the proposed structures (see “Experimental”).

Synthesis of fused pyrimidine 12–16 and chromene 17 derivatives
Finally, treatment of 4 with 1,3-indandione in boiling glacial acetic acid yielded pyran derivative 22 (Scheme 4). The assignment of structure 22 was based on the analytical and spectral data. The 1H NMR spectrum displayed doublet and triplet signals at 2.48 and 4.03 ppm for C4-H and C3-H of the pyran ring, respectively.

Synthesis of chromene 18–21 and pyran 22 derivatives
Surface-Active Agents
The aim of this work was to synthesis nonionic surface active agents bearing heterocyclic moieties with an intermediate fatty chain in order to obtain high surface and biological activity. Thus, treatment of the synthesized compounds 5–22 with 5 mol of propylene oxide in the presence of KOH produced nonionic surface active agents 23–40 having a higher degree of antimicrobial and surface activity, which can serve in the manufacture of drugs, cosmetic, antibacterial and antifungal compounds. This additional process is one of the important processes used to introduce hydrophilic groups into a hydrophobic moiety. The reaction conditions are illustrated in Table 1. The addition of propylene oxide for compounds 9 and 16a is shown in Scheme 5 as an example. The structure of the prepared nonionic surfactants was confirmed using spectroscopic tools.

Propoxylation of compounds 9 and 16a as a general example
Antibacterial Activity
The newly synthesized targeted compounds 5–22 were evaluated for their in vitro antibacterial activity against Staphylococcus aureus (ATCC 25923) and Bacillus cereus (ATCC 10987) as examples of Gram-positive bacteria and Serratia marcesens (ATCC 274) and Proteus mirabilis (SM514) as examples of Gram-negative bacteria, using an agar diffusion method and filter paper disc-diffusion technique. Chloramphenicol® and Ampicilin® were used as reference drugs.
The agar diffusion method was used for the determination of the preliminary antibacterial. The results depicted in Table 2 revealed that most of the tested compounds displayed variable inhibitory effects on the growth of the tested Gram-positive and Gram-negative bacterial strains. The results revealed that compounds 30 and 34a exhibited broad spectrum antibacterial profile against the tested organisms.
On the other hand, for the filter paper disc-diffusion technique, the results summarized in Table 3 revealed, that the compounds 30 and 34b exhibited strongly inhibitory to some of the tested bacteria. While, compounds 23, 24, 25, 26, 27, 28a, 28b, 35, 37, 38, 39 and 40 showed moderate activities against the tested bacteria.
Evaluation of the Surface-Active Agents 23–40
The construction of surfactant molecules contain the heterocyclic moiety considered as an important class of surface-active agents due to double characters, one due to disagreement between the similarity of the hydrophobic and hydrophilic structures, which gave surface properties, and the other one to the heterocyclic moiety with the aid of the hydrophilic moiety (propylene oxide), which gave biological activity. Moreover, the widespread use of these compounds is due to easy rinsing, good detergency and low foam in cleaning beer and milk bottles. The surface properties such as surface and interfacial tension, wetting time, cloud point, emulsification properties and foaming were measured in neutral medium in order to evaluate the possible application of these products in different industrial fields and are depicted in Table 4.
Surface and Interfacial Tension
The results reflected in Table 4 showed that the tested compounds produced a reduction of surface tension ranged from 29 to 37 mN/m. Compounds 32 and 41 have the maximum ability to reduce surface tension of aqueous system while compound 27 has the minimum ability. In general, the results showed that all the prepared compounds have exhibited pronounced surface and interfacial tension and surface activity.
Cloud Point
The cloud point helps us to determine the storage stability because storing at temperatures significantly greater than the cloud point may result in phase separation and instability. The results in Table 4 indicated that compounds 32, 38 and 41 have high cloud points, while compounds 26 and 27 have low cloud points. In general, the compounds that showed high cloud points will give a good performance in hot water, which reflects the fact that they can be used over on a wide range of temperatures.
Wetting Time
Examination of wetting properties for the synthesized compounds showed that they possess a potent wetting-inducing efficiency. The dilute solution of all compounds could wet the cotton skeins in periods ranging from 28 to 41 s. The results summarized in Table 4 showed that compound 41 exhibited the shortest sinking time and consequently is the most efficient wetting agent among the studied group. Generally, the results showed that the products were very effective wetting agents in distilled water solutions. In this respect, these compounds may be potentially useful in a variety of applications where wetting is desired, e.g., dyeing processing, paints, cosmetics and many other operations.
Foaming height
Foams may be applied or encountered at all stages in the processing industries and have important properties that may be desirable in some process contexts and undesirable in others [46]. The results outlined in Table 4 showed that the investigated compounds poduce low to moderate foaming. The low-foaming power compounds have applications in the dyeing and auxiliary industries [47]. Compound 33 exhibited the highest foaming height. Commonly, these low-foaming effects may be attributed to the presence of many hydrophilic groups, which cause a considerable increase in the area per molecule and produce less cohesive forces at the surface.
Emulsion Stability
In many industrial processes, it is necessary to use surfactants to remove oily impurities via emulsification. [48] The emulsifying ability of the prepared nonionic compounds was determined and are listed in Table 4. The results indicated that all the compounds exhibit adequate emulsification stability especially compounds 31 and 45. Members of this series could produce oil/water emulsions of considerable stability. This shows that the compounds under investigation could be useful in cosmetics, formulations, pesticides, textile processing and dye baths.
Biodegradability
To ensure that the synthesized compounds are ecofriendly, the river water die-away test was used to give a measure of biodegradability. The results shown in Table 5 revealed that on the first day 40–50 % of the surfactants was biodegradable. After the first day, they decreased by 15–20 %. At 6 days, over 90 % die-away was observed. This means that these compounds are safe for human beings as well as for the environment.
Conclusion
The objective of the present study was to synthesize and investigate the antimicrobial and surface activities of isoxazole, pyrazole, oxathiin, pyran, pyrrole, chromene, and pyrimidine derivatives. This work is a valuable addition to the synthetic methodology available for the synthesis of heterocyclic derivatives from fatty acids. The newly synthesized compounds 23–40 exhibit varying degrees of microbial inhibition and surface activity, and show potential as insecticides or pesticides as well as in the manufacturing of drugs and pharmaceuticals.
References
Zaghloul EK, Khadija MA, Ebtisam AH, Mohamed HE (2002) Enaminones in heterocyclic synthesis: a novel route to polyfunctionalized substituted thiophene, 2,3-dihydro-1,3,4-thiadiazole and naphtho[1,2-b]furan derivatives. Phosphorus Sulfur 177:105–114
Zeba NS, Nayeem A, Farheen F, Kulsum K (2013) Highly efficient solvent-free synthesis of novel pyranyl pyridine derivatives via β-enaminones using ZnO nanoparticles. Tetrahedron Lett 54(28):3599–3604
Ahmed AH (2014) Heterocyclic synthesis via enaminones: synthesis and molecular docking studies of some novel heterocyclic compounds containing sulfonamide moiety. Int J Org Chem 4:68–81
Nouria AA, Maher RI, Mohamed HE, Elizabeth J, Yehia AI (2012) Enaminones in a multicomponent synthesis of 4-aryldihydropyridines for potential applications in photo induced intramolecular electron-transfer systems. Beilstein J Org Chem 8:441–447
Silvio C, Lourenço LB (2014) Synthesis of quinolinediones by catalyst-free formal aza-[3 + 2+1] cycloaddition of enaminones, aldehydes and meldrum’s acid. J Braz Chem Soc 25(7):1311–1316
Mahmoud MA (2014) Synthesis of 1,3,4-thiadiazoles, α-pyranone, pyridine, polysubstituted benzene from 1, 3, 4-thiadiazolyl ethanone and testing against tuberculosis based on molecular docking studies. Orient J Chem 30(3):1099–1109
Jehane IE, Aliaa RM, Nahed AH, Amal ME, Magda MN, Mohamed A (2014) In vivo antioxidant and antigenotoxic evaluation of an enaminone derivative BDHQ combined with praziquantel in uninfected and Schistosoma mansoni infected mice. J Appl Pharm Sci. 4(05):025–033
Viktor OI, Satenik M, Ashot G, Mariia M, Alexander V, Dmytro V, Vyacheslav YS, Peter L (2012) 2, 3-Unsubstituted chromones and their enaminone precursors as versatile reagents for the synthesis of fused pyridines. Org Biomol Chem 10:890–894
Riyadh SM (2011) Enaminones as building blocks for the synthesis of substituted pyrazoles with antitumor and antimicrobial activities. Molecules 16:1834–1853
Riyadh SM, Abdelhamid IA, Al-Matar HM, Hilmy NM, Elnagdi MH (2008) Enamines as precursors to polyfunctional heteroaromatic compounds; a decade of development. Heterocycles 75:1849–1905
Heikhshoaie I, Fabian WMF (2009) Theoretical insights into material properties of Schiff bases and related azo compounds. Curr Org Chem 13:149–171
El-Apasery MA, Al-Mousawi SM, Mahmoud H, Elnagdi MH (2011) Novel routes to biologically active enaminones, dienoic acid amides, arylazonicotinates and dihydro-pyridazines under microwave irradiation. Int Res J Pure Appl Chem 1(3):69–83
Oddeti G, Bhagavathula SD, Yellajyosula LNM (2012) A brief review on synthesis and applications of β-enamino carbonyl compounds. Org Commun 5(3):105–119
Kateb AA, Abd El-Rahman NM, Saleh TS, Ali MH, Elhaddad AS, El-Dosoky AY (2012) Microwave mediated facile synthesis of some novel pyrazole, pyrimidine, pyrazolo[1,5-a]pyrimidine, triazolo[1,5-a]pyrimidine and pyrimido[1,2-a]benzimidazole derivatives under solvent less condition. Nat Sci 10(11):77–86
Oshinori T, Hiroto O, Shinya K, Hisako M (2009) Reaction of enaminones with carbon disulfide: synthesis of heterocycles using enamino dithiocarboxylates. J Heterocycl Chem 28(5):1245–1255
Hanadi YM, Abdellatif MS (2014) Enamines in heterocyclic synthesis: route to amino-pyrazolo pyrimidines and arylpyrazolopyrimidiens derivatives. Mod Chem 2(1):1–5
Fadda AA, Amine MS, Areif MMH, Kh Farahat E (2014) Novel synthesis, antimicrobial evaluation and reactivity of dihydroacetic acid with N, C-nucleophiles. Pharmacologia 5:1–11
Kibou Z, Cheikh N, Choukchou-Braham N, Mostefakara B, Benabdellah M, Villemin D (2011) New methodology for the synthesis of 2-pyridones using basic Al2O3 as catalyst. J Mater Environ Sci 2(3):293–298
Himani V, Aiman A, Abdul R, Fohad MH, Iqbal A (2014) Synthesis and antimicrobial evaluation of fatty chain substituted 2,5-dimethyl pyrrole and 1,3-benzoxazin-4-one derivatives. J Saudi Chem Soc 26:290–299
Jubie S, Ramesh PN, Dhanabal P, Kalirajan R, Muruganantham N, Antony AS (2012) Synthesis, antidepressant and antimicrobial activities of some novel stearic acid analogues. Eur J Med Chem 54:931–935
Khan AA, Alam M, Tufail S, Mustafa J, Owais M (2011) Synthesis and characterization of novel PUFA esters exhibiting potential anticancer activities: an in vitro study. Eur J Med Chem 46:4878–4886
Xu LL, Zheng CJ, Sun LP, Miao J, Piao HR (2012) Synthesis of novel 1,3-diaryl pyrazole derivatives bearing rhodanine-3-fatty acid moieties as potential antibacterial agents. Eur J Med Chem 48:174–178
Dongqing Xu, Qi Banghua, Di F, Xuemei Z, Ting Z (2016) Preparation, characterization and properties of novel cationic gemini surfactants with rigid amido spacer groups. J Surfact Deterg 19:91–99
Vinayika S, Rashmi T (2016) Surface and aggregation properties of synthesized cetyl alcohol based bis-sulfosuccinate gemini surfactants in aqueous solution. J Surfact Deterg 19:111–118
Wiesław H, Karolina D, Arkadiusz C, Agata S, Katarzyna M (2016) 2-Ethylhexanol derivatives as nonionic surfactants: synthesis and properties. J Surfact Deterg 19:155–164
Farshori NN, Banday MR, Ahmad A, Khan AU, Rauf A (2011) 7-Hydroxycoumarin derivatives: synthesis, characterization and preliminary antimicrobial activities. Med Chem Res 20:535–541
Varshney H, Ahmad A, Farshori NN, Ahmad A, Khan AU, Rauf A (2013) Synthesis and evaluation of in vitro antimicrobial activity of novel 2,3-disubstituted-4-thiazolidinones from fatty acid hydrazides. Med Chem Res 22:3204–3212
El-Sayed R, Khairou SK (2015) Propoxylated fatty thiazole, pyrazole, triazole, and pyrrole derivatives with antimicrobial and surface activity. J Surfact Deterg 18:661–673
El-Sayed R (2013) Substituted thiadiazole, oxadiazole, triazole and triazinone as antimicrobial and surface activity compounds. J Surfact Deterg 16(1):39–47
El-Sayed R, Amani S, Al Mazrouee L (2015) Synthesis and evaluation of polyfunctionally substituted heterocyclic compounds derived from 2-cyano-n-(5-pentadecyl-1,3,4-thiadiazol-2-yl)acetamide. Synth Commun 1(45):886–897
El-Sayed R, Asghar BH (2014) Synthesis of various nitrogen heterocycles as antimicrobial surface agents. J Heterocycl Chem 51:1245–1251
Morgo´s J, Sallay P, Farkas L, Ruszna´k I (1986) A new approach of ethoxylation catalyzed by bridge head nitrogen containing compounds. J Am Oil Chem Soc 63:1209–1210
Carrod LP, Grady FD (1972) Antibiotic and chemotherapy, 3rd edn. Churchill Livingstone, Edinburgh
Coffen DL, Korzan DG (1971) Synthetic quinine analogs. III. Frangomeric and anchimeric processes in the preparation and reactions of α, β-epoxy ketones. J Org Chem 36:390–395
Findlay A (1963) Practical physical chemistry, 6th edn. Longmans, London, pp 1039–1040
Weil JK, Stirton AJ, Nunez-Ponzoa MV (1966) Ether alcohol sulfates. The effect of oxypropylation and oxybutylation on surface active properties. J Am Oil Soc 43:603–609
ASTM D1331-01 (2001) Standard Test method for surface and interfacial tension of solutions of surface active agents
Durham K (1961) Properties of detergent solutions-amphipathy and adsorption. Surf activity deterg, vol 1. MacMillan, London, pp 1–28
Draves CZ, Clarkson R (1931) A new method for the evaluation of wetting agents. J Am Dye Stuff Rep 20:201
Ross J, Milles GD (1941) Apparatus for comparison of foaming properties of soaps and detergents. Oil Soap 18:99–102
Eter ET, Richard RE, Darid A (1974) Biodegradable surfactants derived from corn starch. J Am Oil Chem Soc 51:486–494
Falbe J (1986) Surfactants for consumer, chap 4. Springer, Heidelberg
Ghodile NG, Rajput PR (2014) Synthesis and characterization of some new n, o and s containing chlorosubstituted heterocycles and their antimicrobial screening against some common causative organisms. Int J Innov Res Sci Eng Technol 3(4):373–377
El-Sayed R (2008) Surface active properties and biological activity of novel nonionic surfactants containing pyrimidines and related nitrogen heterocyclic ring systems. Grasas Aceites 59(2):110–120
Samir B, Abd El-Gaber T, Fadda AA (2011) Behavior of 2-cyano-3-(dimethylamino)-N-(4-phenylthiazol-2-yl)-acrylamide towards some nitrogen nucleophiles. Arkivoc 2:227–239
Laurier LS, Elaine NS, Gerrard DM (2003) Surfactants and their applications. Annu Rep Prog Chem Sect C 99:3–48
Somaya EA, Eissa AEMF, Ahmed MN (2015) Synthesis, physicochemical properties and biological evaluation of some peptide candidates by use of liquid phase method as potential antimicrobial and surface active agents. Int J Pharm 11(7):726–731
Ismail T (2013) Evaluation of some non-ionic surfactants prepared from olive oil having antiseptic activity. 3rd International Conference on Medical, Biological and Pharmaceutical Sciences (ICMBPS’2013) January 4–5, Bali (Indonesia)
Acknowledgments
The authors are greatly appreciative to the staff of the Microbiology Department, Faculty of Science, Umm Al-Qura University for facilitating in carrying out the antibacterial investigation for the newly synthesized compounds.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
El-Sayed, R. Synthesis of Biodegradable Pyrazole, Pyran, Pyrrole, Pyrimidine and Chromene Derivatives having Medical and Surface Activities. J Surfact Deterg 19, 1153–1167 (2016). https://doi.org/10.1007/s11743-016-1881-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11743-016-1881-0