
Abstract
The plasma membrane redox system (PMRS) containing NADH-dependent reductases is known to be involved in the maintenance of redox state and bioenergetics. Neuronal cells are very vulnerable to oxidative stress and altered energy metabolism linked to mitochondrial dysfunction. However, the role of the PMRS in these pathways is far from clear. In this study, in order to investigate how cytochrome b5 reductase (b5R), one of the PM redox enzymes, regulates cellular response under stressed conditions, human neuroblastoma cells transfected with b5R were used for viability and mitochondrial functional assays. Cells transfected with b5R exhibited significantly higher levels of the NAD+/NADH ratio, consistent with increased levels of b5R activity. Overexpression of b5R made cells more resistant to H2O2 (oxidative stress), 2-deoxyglucose (metabolic stress), rotenone and antimycin A (energetic stress), and lactacystin (proteotoxic stress), but did not protect cells against H2O2 and serum withdrawal. Overexpression of b5R induced higher mitochondrial functions such as ATP production rate, oxygen consumption rate, and activities of complexes I and II, without formation of further reactive oxygen species, consistent with lower levels of oxidative/nitrative damage and resistance to apoptotic cell death. In conclusion, higher NAD+/NADH ratio and consequent more efficient mitochondrial functions are induced by the PMRS, enabling them to maintain redox state and energy metabolism under conditions of some energetic stresses. This suggests that b5R can be a target for therapeutic intervention for aging and neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aging is a complex and irreversible degeneration process and associated with a wide range of age-related diseases including neurodegenerative disorders, cardiovascular diseases, and cancer (Blasco 2005). Aging causes changes in antioxidant capacity followed by impairment of the mitochondrial function and oxidative damage. In fact, oxidative stress and mitochondrial dysfunction are known to be two of the causes of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease (Beal 2004, 2005; Jenner 2003).
The mitochondria are a main microorganelle, which produces ATP. However, a major portion of free radicals is produced inevitably during oxidative phosphorylation (Mattson et al. 2008; Murphy 2009). Mitochondrial dysfunction and shortage of ATP supply are commonly identified as early events in normal aging process and in many age-related diseases (Kim et al. 2008; Luft and Landau 1995). The brain requires a large amount of glucose and oxygen to produce ATP. Neuronal cells are very vulnerable to oxidative and energetic stresses (e.g., glucose limitation, drastic energy demand, etc.) because the brain has relatively low antioxidant capacity compared with other tissues such as the liver (Mattson et al. 2008; Sims 1992). A variety of neurodegenerative diseases such as Alzheimer’s disease (AD) (Mattson 2004; Mosconi et al. 2008), Parkinson’s disease (PD) (Ferrer 2009), and Huntington’s diseases (Browne 2008) are known to occur by metabolic alteration and oxidative stress during the aging process. The mechanisms by which neurons survive under conditions of metabolic and oxidative stress can be a therapeutic target for aging intervention and prevention of neurodegenerative diseases.
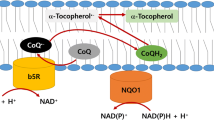
When ATP supply by mitochondrial respiration is not sufficient, cells can survive through stimulation of alternative pathways such as glycolysis, fermentation, and plasma membrane redox system (PMRS) if they are supplemented with pyruvate and uridine (Miller et al. 1996). In fact, the plasma membrane has many cellular functions such as maintenance of cell homeostasis, cell signaling, and protection of cells against oxidative stress. The PM exhibits a compensatory mechanism in response to mitochondrial dysfunction via the PMRS, which is also involved in electron transport and energy metabolism (Hyun et al. 2006b; Villalba and Navas 2000). Previously, we reported that ρo cells without functional mitochondria can survive and are more resistant to oxidative stress via enhanced activities of PMRS redox enzymes (Hyun et al. 2007). The PMRS contains NAD(P)H-dependent redox enzymes including cytochrome b5 reductase (b5R) and NAD(P)H-quinone oxidoreductase 1 (NQO1), electron shuttles such as coenzyme Q (CoQ) and cytochrome c, and electron donors (e.g., NAD(P)H) (Hyun et al. 2006b). PM redox enzymes convert an oxidized form of CoQ to a reduced state by transferring electrons from NAD(P)H.
b5R is a monomeric flavoprotein of 32 kDa NADH-dependent reductase, which is noncovalently bound to FAD and located on the inner surface of the PM (Bewley et al. 2001; Rao et al. 1992; Sparla et al. 1997). A reduced form of CoQ is a lipid-soluble antioxidant molecule, which protects membrane lipids from oxidative damage by scavenging superoxide or other radicals alone or in association with α-tocopherol (Crane 2001; Turunen et al. 2004). The PMRS can maintain metabolic balance through modulating the NAD+/NADH ratio in response to metabolic fluctuation. In fact, the PMRS plays a key role in the regulation of pyrimidine nucleotide balance, making cells survive, grow, and differentiate under various physiological conditions (Buron et al. 1993; De Luca et al. 2005; Medina et al. 1997). For example, NADH-coenzyme Q reductase 1 (NQR1) in yeast, a homologue of NQO1, is coupled to the transition from fermentation to respiration, resulting in the extension of life span (Jimenez-Hidalgo et al. 2009). b5R can play a dual role in the regulation of oxidative and metabolic stresses through interaction between forkhead box class O 3a (FOXO3a) and nuclear factor (erythroid-derived 2)-like2 (Nrf2) (Siendones et al. 2014).
b5R is suggested to use plant polyphenols (e.g., quercetin, catechin, resveratrol) as well as NADH as an electron donor (Kesharwani et al. 2012). Although b5R can be a more potent therapeutic target for validating bioactive phytochemicals, the mechanisms of b5R responsible for aging intervention and neuroprotection are yet to be investigated. Other studies showed that PM-bound b5R is a component of protein complexes tightly associated with lipid raft microdomains of neuronal PM (Marques-da-Silva et al. 2010; Samhan-Arias et al. 2009). b5R is stimulated and clustered in the lipid microdomain enriched with caveolin in cerebellar granule neurons at an early stage of apoptosis induced by oxidative stress (Samhan-Arias et al. 2012). Interestingly, in aged mice, b5R activated by calorie restriction (CR) and aerobic exercise can attenuate oxidative damage (Rodriguez-Bies et al. 2015). These findings suggest a potentially essential role of b5R in cellular adaptation in response to metabolic alteration and oxidative stress known to occur in neurodegenerative diseases.
In this study, we used human neuroblastoma cells transfected with b5R to investigate how b5R protects neuronal cells. We found that elevated levels of b5R make cells more resistant to metabolic and oxidative stress. Cells overexpressing b5R maintained a higher NAD+/NADH ratio and enhanced mitochondrial functions without further production of reactive oxygen species (ROS), suggesting that this PMRS enzyme improves cellular bioenergetics.
Methods
Cell culture, transfection, and differentiation
SH-SY5Y human neuroblastoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (Invitrogen, Carlsbad, CA), 100 IU/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) in a humidified 5 % CO2/95 % air atmosphere. The cells were transfected with pCB6 vector containing the full-length b5R, as described previously (Hyun et al. 2010). Six different clones were selected using G-418. In order to induce cell differentiation, cells were treated with 10 μM retinoic acid (RA) and cultured in the DMEM containing 3 % FBS for 3 days.
Characterization of the transfected cells
Cells were lysed and levels of cytochrome b5 reductase proteins were determined by immunoblot analysis using a b5R monoclonal antibody, as described previously (Hyun et al. 2010; Seow et al. 2005). Levels of b5R proteins in the PM of the transfected cells were measured following isolation of the PMs by a two-phase partition, as described previously (De Cabo et al. 2004; Hyun et al. 2010).
Isolation of the plasma membranes
The PM isolation procedure was carried out using the two-phase partition, as described previously (De Cabo et al. 2004; Navas et al. 1989). Cells were homogenized in the ice-cold homogenization buffer with 37 mM Tris-maleate, 10 mg/ml dextran T-500, 50 mM magnesium chloride, 0.5 M sucrose, 28 mM sodium hydroxide, 1 mM ethylene diamine trichloroacetic acid (EDTA), 1 mM phenylmethyl sulfonyl fluoride (PMSF), and 5 mM dithiothreitol (DTT) (pH 6.4) using a Dounce homogenizer (Golgi grind). This homogenate was centrifuged at 5000g for 15 min. Then, one half of the pellet produced was resuspended in 1 mM sodium bicarbonate, homogenized in a Teflon glass tissue homogenizer, and centrifuged at 5000g for 15 min. The light brown, top portion of the pellet was combined with a mixture of 6.6 % (w/w) dextran and 6.6 % (w/w) polyethylene glycol in 0.1 M sucrose and 5 mM potassium phosphate (pH 7.2). The mixture was inverted vigorously 40 times at 4 °C and the two phases were separated by centrifugation at 550g for 3 min. The upper phase was then diluted with 1 mM sodium bicarbonate (pH 7.2) and centrifuged at 20,000g for 30 min. The PM fractions (pellets) were resuspended in the suspension buffer with 25 mM Tris, 10 % glycerol, 0.1 mM DTT, 1 mM EDTA, and 1 mM PMSF (pH 7.6).
Protein concentration in the PM fraction was determined by the Bradford assay (Bradford 1976). Immunoblotting assay using enzyme markers for PM and mitochondria was performed to measure the purity of the isolated fractions as described previously (De Cabo et al. 2004). For immunoblotting, anti-Na+/K+-ATPase α-subunit monoclonal antibody (1:1000 dilution, Affinity BioReagents, Golden, CO) and anticytochrome c oxidase subunit I monoclonal antibody (1:1000 dilution, Molecular Probe, Eugene, OR) were used.
Activity of b5R
The activity of NADH-ascorbate free radical (NADH-AFR) reductase (EC 1.6.5.4), which was mainly due to b5R, was determined using a spectrophotomer, as described previously (De Cabo et al. 2004). A decrease in absorbance at 340 nm was read following the addition of 66 × 10−3 units of ascorbate oxidase to the assay buffer (50 mM Tris-HCl buffer, 0.2 mM NADH, and 0.4 mM fresh ascorbate, pH 7.6) and 50 μg of PM. The extinction coefficient used to calculate specific activity was 6.22 mM−1 cm−1.
Levels of metabolites
NAD+ and NADH levels were determined separately using a NAD/NADH Quantitation Kit (BioVision, Mountain View, CA). Briefly, in order to measure the total levels of NAD and NADH, lysates were transferred into a 96-well plate, and 100 μl NAD cycling buffer and 2 μl NAD cycling enzyme mix were added. The mixtures were incubated at room temperature for 5 min to convert NAD to NADH, and NADH developer was then added to each well. The plate was incubated for 2 h and absorbance was read at 450 nm. In order to measure NADH, NAD was decomposed by heating the lysates at 60 °C for 30 min, followed by the same procedures described above. A standard curve was generated by serial dilution of standard NADH solution, and the NAD+/NADH ratio was calculated as [(NADtotal − NADH) / NADH].
Cell viability assay
Cells were exposed to a normal culture medium containing 2-deoxyglucose (5, 10, 15, 20 mM) (Sigma, St. Louis, MO), rotenone (50, 100, 150, 200 μM) (Sigma), antimycin A (50, 100, 150, 200 μM) (Sigma), and lactacystin (10, 20, 30, 40 mM) (Sigma), or to a serum-free medium, for 24 h. In the case of H2O2 treatment, the cells were exposed to hydrogen peroxide (50, 100, 150, 200 μM) in the absence of serum for 1 h and then incubated with normal culture medium for 24 h. Cell viability was measured using 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma), and intensity was read at 450 nm and by the trypan blue exclusion.
Isolation of the mitochondria and confirmation of the purity of the mitochondrial fractions
Mitochondrial fractions were isolated from the cells by centrifugation and kept at 4 °C, as described previously with minor modifications (Shim et al. 2011). Briefly, cells were washed with ice-cold PBS and homogenized in 10 mM Tris buffer (pH 7.6) with a protease inhibitor cocktail (1.5 mM) (Sigma). The homogenates were centrifuged at 600g for 10 min at 4 °C, and then the supernatants were centrifuged again at 14,000g for 10 min at 4 °C. The resulting pellets were carefully removed and resuspended in the assay buffer.
Immunoblot assays using enzyme markers for the cytosol and mitochondria were performed to establish the relative purity of the isolated mitochondrial fractions using antibodies against peroxiredoxin II (cytosol marker, 1:2000, AbFrontier, South Korea), peroxiredoxin III (mitochondrial marker, 1:2000, AbFrontier), and complex I (1:2000, Invitrogen). Briefly, the isolated mitochondrial fractions were lysed in PBS (pH 7.4) containing 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml pepstastin A, and 0.1 % Triton X-100 and placed on ice for 5 min. The lysates were centrifuged at 12,000g for 10 min, and the supernatants were transferred into new Eppendorf tubes. Protein levels were measured using the Bradford reagent (Bradford 1976), and 10 μg of protein was separated by SDS-PAGE, as described previously (Hyun et al. 2006a, 2007). The separated proteins were transferred electrophoretically to a nitrocellulose membrane (Whatman GmbH, Dassel, Germany), which was then incubated with the primary antibodies. Immune complexes were detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents (1:5000, AbFrontier).
ATP production rate
Mitochondria were isolated from the cells and the ATP production rate (APR) was determined by the luciferase assay, as previously described, with minor modifications (Vives-Bauza et al. 2007). Isolated mitochondria (10 μg) were suspended in 200 μl of reaction buffer A (0.1 % BSA, 150 mM KCl, 0.1 mM MgCl2, 25 mM Tris-HCl, 10 mM potassium phosphate, pH 7.4) containing 160 μM diadenosine pentaphosphate, 1 mM pyruvate, 100 μM ADP, and either 5 mM NADH or 5 mM glutamate/malate or 5 mM succinate. Then, 10 μl of buffer B (500 mM Tris-acetate, pH 7.75) containing 20 μg/ml luciferase (Invitrogen) and 0.8 mM D-luciferin (Invitrogen) was added to the reaction mixture. The light emission was monitored using a luminometer (20/20n, Turner Biosystems, Sunnyvale, CA) for 5 min at 10 s intervals. A standard curve for luminescence was made by using solutions containing increasing concentrations of ATP (Sigma).
Oxygen consumption rate
Oxygen consumption rate (OCR) of living cells was assessed using an Instech Two Channel Fiber Optic Oxygen Monitor System (Instech Laboratories, Inc., Plymouth Meeting, PA, USA), as described previously (Hyun et al. 2007). Briefly, cell chambers were maintained at 37 °C in a circulating water bath. Cells were trypsinized and centrifuged at 1500g for 2 min at 4 °C. Cells were then resuspended in DMEM. Respiration rates were determined in the presence of 5 mM NADH, 5 mM glutamate/malate, or 5 mM succinate, and the results were calculated as nanomoles of oxygen consumed per minute per milligram of protein. Activated OCR was measured following the addition of ADP. All measurements were assessed with Fiber Optic Oxygen Monitor Operating Software and Slope Calculator Software (Instech Laboratories). Resting respiration rate was determined as the OCR under conditions where no inhibitors or uncouplers were administered to the mitochondria. Nonmitochondrial oxygen consumption was determined as the respiration rate following incubation of cells with 100 mM potassium cyanide (KCN) (Fluka BioChemika, Bruchs, Germany).
Activities of complexes I and II
Activities of mitochondrial complexes I and II were assessed using decylubiquinone and dichloroindolphenol (DCIP), as described earlier (Janssen et al. 2007). Briefly, 10 μg of the isolated mitochondrial fractions was preincubated in the reaction buffer for complex I (70 μM decylubiquinone, 60 μM DCIP, 1 μM antimycin A, 0.35 % BSA, 25 mM potassium phosphate, pH 7.4) or complex II (70 μM decylubiquinone, 60 μM DCIP, 1 μM antimycin A, 50 μM rotenone, 500 μM EDTA, 200 μM ATP, 0.1 % BSA, 80 mM potassium phosphate, pH 7.4) at 37 °C. The reaction was initiated by the addition of substrates (complex I: 5 mM NADH or 5 mM glutamate/malate and complex II: 5 mM succinate and 300 μM potassium cyanide) to the mixture, and absorbance was read at 600 nm for 5 min with 20 s intervals.
Production of ROS
Mitochondrial fractions isolated freshly from isolated freshly were analyzed. Hydrogen peroxide (H2O2) released from the mitochondria was measured using the fluorescent dye Amplex Red (Molecular Probes, Eugene, OR) which reacts with H2O2 with a 1:1 stoichiometry in the presence of horseradish peroxidase and thereby produces the highly fluorescent chemical resorufin (Li and May 2002; Zhou et al. 1997). Briefly, a reaction buffer (50 mM Tris, pH 7.4) was supplemented with 5 μM Amplex Red, 0.5 U/ml HRP, and 20 U/ml Cu,Zn-superoxide dismutase to prevent the auto-oxidation of Amplex Red and interference with quantitative assessment of low rates of H2O2 production (Votyakova and Reynolds 2004). The supplemented buffers were preincubated at 37 °C and mitochondrial fractions and electron donors (5 mM succinate or 5 mM glycerol 3-phosphate) were added to the reaction mixture. The detection of H2O2 in mitochondrial suspensions (0.1 mg protein/ml) was recorded as an increase in Amplex Red fluorescence using excitation and emission wavelengths of 560 and 590 nm, respectively. The response of Amplex Red to H2O2 was calibrated by incubation with known amounts of H2O2 solution from 100 to 1000 pmol. The concentration of commercial 30 % H2O2 solution (Sigma) was calculated from light absorbance at 240 nm using E 240 mM = 43.6 cm−1, and the stock solution was diluted to 100 μM with water and used for calibration immediately.
Markers of oxidative/nitrative damage
Lipid peroxidation levels were assessed using the 8-Isoprostane Assay Kit (OxisResearch, Portland, OR). Briefly, following exposure to each of the mitochondrial toxins, cells were lysed and cell extracts (100 μl) were added to a 96-well plate and incubated with 100 μl horseradish peroxidase-conjugated antibody at room temperature for 1 h; a 200-μl substrate was added to the plate and it was incubated for 30 min. Absorbance was read at 450 nm after stopping the reaction by adding 50 μl 3 M sulfuric acid. Protein carbonyl content was determined as described previously (Lyras et al. 1997), except that the final cell extracts were dissolved in 1 ml of 6 M guanidinium hydrochloride (Sigma). Carbonyl content was calculated as nanomoles per milligram of protein (Reznick and Packer 1994). Measurement of protein-bound nitrotyrosine content of isolated plasma membranes was performed using the Nitrotyrosine Assay Kit (OxisResearch).
Apoptotic features
Following treatment with rotenone or antimycin A for 24 h, cell membrane damage and chromatin condensation were determined using propidium iodide or Hoechst 33258, as described previously (Kelner et al. 2000; Lee et al. 2001).
Statistical analysis
Statistical differences were analyzed by one- or two-way ANOVA test. Multiple comparisons were followed with post hoc Bonferroni t test where necessary.
Results
b5R is stably overexpressed in human neuroblastoma cells
Six different clones of SH-SY5Y cell lines stably overexpressing b5R were established, and the levels of b5R expression and enzymatic activity in these clones were examined. Among these clones, two expressed b5R at levels three- to fourfold greater than in vector-transfected control cell clones (Fig. 1). A clone with the highest levels of b5R expression and biochemical activity was used for further evaluation of the subcellular localization and functional activity of b5R in this study. Overexpressed b5R was present in both PM and cytosols, as confirmed by immunoblot analysis using a b5R antibody. Measurements of b5R enzyme activity also showed that b5R transfectants exhibited an approximately threefold greater NQO1 activity compared with control cells, consistent with its expression level (Fig. 2a).

Characterization of human neuroblastoma cells transfected with b5R. Control SH-SY5Y cells and the indicated clones of cells overexpressing b5R were cultured under normal culture conditions and lysed. Immunoblot analysis was performed using a b5R monoclonal antibody.

Levels of b5R activity in the PMs and cytosols and cytoplasmic NAD+/NADH ratio are enhanced in cells overexpressing b5R. Plasma membranes (PMs) were isolated by a two-phase partition. a b5R activity was determined using menadione and cytochrome c as substrates. b Cells were lysed and the NAD+/NADH ratio was measured using a NAD/NADH Quantitation Kit. Values are the mean and SEM (n = 6). # p <0.01 compared with the value between untransfected control cells and b5R transfectants in the same fractions
Overexpressed b5R increased the NAD+/NADH ratio
In order to assess the impact of b5R on cellular metabolic balance, the NAD+/NADH ratio was measured in neuroblastoma cells expressing basal or increased levels of b5R. The NAD+/NADH ratio was significantly elevated, by more than 800 %, in cells transfected with b5R compared with control cells (p < 0.01) (Fig. 2b). However, the NAD+/NADH ratio in the mitochondria of b5R transfectants was decreased by about 50 %, compared with control cells.
Cells transfected with b5R protected cells against toxic insults
In order to investigate whether overexpressed b5R could make cell more resistant under stressed conditions, viability of the control and b5R transfectants was determined following exposure to six different toxic insults for 24 h: H2O2 (an oxidative stress inducer), 2-deoxyglucose (a glycolysis inhibitor), rotenone (a mitochondrial complex I inhibitor), antimycin A (a mitochondrial complex III inhibitor), lactacystin (a proteasome inhibitor), and serum-free medium (an apoptosis trigger).
In neuroblastoma cells with basal levels of b5R, cell viability was decreased following exposure to increasing concentrations of H2O2 (50–200 μM) (Fig. 3a).Overexpressed b5R tended to protect the cells against H2O2, but it was not significant. In control cells, cell viability was greatly reduced when treated with 2-deoxyglucose (10–20 mM), compared with normal culture conditions (Fig. 3b). However, overexpressed b5R made cells more resistant to being killed by 2-deoxyglucose. Cell viability was declined by the addition of rotenone in a dose-dependent manner with significant differences between control and cells overexpressing b5R (Fig. 3c). Antimycin A caused a decrease in cell viability in a dose-dependent manner, and cells transfected with b5R were less vulnerable to rotenone, compared with control cells (Fig. 3d). Treatment of control cells with lactacystin also induced cell viability loss, but it was attenuated by overexpression of b5R (Fig. 3e). Finally, the vulnerability of SH-SY5Y cells to serum withdrawal, a way of inducing apoptosis (Pedersen et al. 2002), was not significantly different between control cells and cells transfected with b5R (Fig. 3f). Following the addition of H2O2, rotenone, and antimycin A, cell viability assessed by the trypan blue exclusion (Supplementary Fig. 2A–C) also showed similarities to that assessed by the MTT assay.

Neuroblastoma cells overexpressing b5R exhibit reduced vulnerability to metabolic, oxidative, and proteotoxic insults, but are not protected against trophic factor withdrawal. Cells were exposed to H2O2 (a), 2-deoxyglucose (b), rotenone (c), antimycin A (d), lactacystin (e), or serum-free medium (f) for 24 h, and cell viability was measured using the WST-8 assay (see “Methods” for details). Values are the mean and SEM (n = 6). *p <0.01 compared with the value for untransfected control cells under normal culture conditions. # p <0.01 compared with the value between untransfected control cells and b5R transfectants under the same culture conditions
Cells overexpressing b5R exhibited enhanced mitochondrial electron transport without further production of ROS
Previously, we published that mitochondrial activity is enhanced by NQO1, a PM redox enzyme (Kim et al. 2013), suggesting that mitochondrial function could be modulated by another PM enzyme, b5R. In order to verify the protective effects of b5R on cell viability and to examine the effects of b5R on bioenergetics, mitochondrial functions and ROS production were assessed in both control and b5R transfectants. Mitochondrial APR was determined by a luciferase assay. Mitochondrial APR was significantly increased by overexpression of b5R in the presence of NADH or glutamate/malate (p < 0.01) (Fig. 4a, b). APR tended to be elevated in cells transfected with b5R treated with succinate, but it was not significant (Fig. 4c). OCR was dramatically elevated in cells transfected with b5R when supplemented with NADH or glutamate/malate (Fig. 4d, e). The addition of succinate also showed increased OCR, but it was not significant (Fig. 4f).

ATP production and O2 consumption rate are increased by overexpression of b5R. Intact mitochondria were prepared, and ATP production and O2 consumption rate were assessed in the presence of appropriate electron donors. ATP production rates in the presence of NADH (a and d), glutamate/malate (b and e), and succinate (c and f). Values are the mean ± SEM (n = 6). *p <0.01 compared with the value for untransfected control cells under normal culture conditions. # p <0.01 compared with the value between untransfected control cells and b5R transfectants under the same culture conditions
In order to further elucidate the mechanism by which b5R influences mitochondrial electron transport in SH-SY5Y cells, activities of mitochondrial complexes I and II and ROS production were assessed. Mitochondrial complex I activity in cells overexpressing b5R was significantly higher than control cells (p < 0.01) (Fig. 5a, b). Cell differentiation induced a higher activity of the complex I activity (Supplementary Fig. 3B and C). Complex I activity was much higher in b5R transfectants. Complex II activity was also enhanced in b5R transfectants, compared with control cells (p < 0.01) (Fig. 5c). Although there was an increase in electron transport, ROS production (in the presence of NADH, glutamate/malate, or succinate) was slightly lower in b5R transfectants, compared with control cells (Fig. 5d–f).

Activities of mitochondrial complexes I and II are elevated, and production of ROS is reduced in human neuroblastoma cells overexpressing b5R. Cells were cultured and then mitochondrial fractions were isolated by centrifugal fractionation. Mitochondrial complex activities were measured using appropriate electron donors. Complex I activity in the presence of NADH (a) and glutamate/malate (b). Complex II activity in the presence of succinate (c). ROS production in the presence of NADH (d), glutamate/malate (e), and succinate (f). Values are the mean ± SEM (n = 6). *p <0.01 compared with the value for untransfected control cells under normal culture conditions. # p <0.01 compared with the value between untransfected control cells and b5R transfectants under the same culture conditions
Overexpressed b5R attenuated oxidative/nitrative damage
In order to investigate whether overexpressed b5R could protect cells against mitochondrial toxins, the levels of oxidative/nitrative damage were measured following exposure to rotenone or antimycin A. The levels of protein carbonyls, a biomarker of protein oxidation, were slightly lower in cells transfected with b5R, compared with control cells, under normal culture conditions. The levels of protein carbonyls in control cells were greatly elevated following treatment with rotenone (Fig. 6a). These levels were significantly attenuated in cells overexpressing b5R (p < 0.01). Similarly, the levels of 3-nitrotyrosine, a marker of protein nitration, were dramatically increased in control cells after exposure to rotenone (Fig. 6b). This effect of rotenone was attenuated in cell transfected with b5R (p < 0.01). Antimycin A also caused elevated levels of both protein carbonyls (Fig. 6c) and 3-nitrotyrosine (Fig. 6d). These levels of both damages induced by antimycin A in b5R transfectants were greatly lower than those of control cells.

Levels of oxidative damage to lipids and proteins are attenuated in cells with elevated b5R levels compared with control cells. Cell extracts were used to measure levels of protein carbonyls (a, b) and nitrotyrosine (c, d) following exposure to 50 μM rotenone (a, c) or 100 μM antimycin A (b, d) for 24 h. Values are the mean ± SEM (n = 6). *p <0.01 compared with the value for untransfected control cells under normal culture conditions. # p <0.01 compared with the value between untransfected control cells and b5R transfectants under the same culture conditions
Overexpressed b5R protected cells against apoptotic cell death
In order to investigate whether overexpressed b5R renders cells more resistant to apoptotic cell death induced by mitochondrial toxins, apoptotic features were examined. Following exposure to rotenone, the levels of propidium iodide-positive cells (Fig. 7a) and chromatin condensation (Fig. 7c) were decreased in cells transfected with b5R, compared with control cells. Similar patterns of resistance to the apoptotic features were also shown in cells overexpressing b5R after treatment with antimycin A (Fig. 7b, d).

Apoptotic cell death induced by mitochondria toxins is attenuated by overexpressed b5R. Levels of cell membrane permeabilization (propidium iodide-positive cells) (a, b) and chromatin condensation (c, d) were also measured after the addition of 50 μM rotenone (a, c) or 100 μM antimycin A (b, d) for 24 h. Values are the mean ± SEM (n = 6). *p <0.01 compared with the value for untransfected control cells under normal culture conditions. # p <0.01 compared with the value between untransfected control cells and b5R transfectants under the same culture conditions
Discussion
Previously, we showed that overexpressed NQO1, another PM redox enzyme, can protect cells against metabolic and proteotoxic stress through sustaining bioenergetics by increased mitochondrial function (Hyun et al. 2012; Kim et al. 2013). b5R is also one of the PM redox enzymes in the PMRS and may be involved in cell survival under stressed conditions. However, its protective mechanism is yet to be investigated.
This study shows that b5R can play an important role in the survival of human neuroblastoma cells under metabolic and oxidative stresses (Fig. 3). As expected, the NAD+/NADH ratio was dramatically increased by overexpressed b5R (Fig. 2). Neural tumor cells (Nahimana et al. 2009) and normal neurons (Alano et al. 2010; Kruman et al. 2000; Liu et al. 2009) are very sensitive to a decline in the NAD+/NADH ratio because of their high energy demand. Impairment of energy metabolism and formation of protein aggregates are known to be responsible for a variety of neurodegenerative diseases. This study suggests a potential protective role for b5R in neurodegenerative diseases, consistent with a previous result (Incerpi et al. 2007). Failure of energy metabolism and altered abnormal protein handling are closely associated with an age-related decrease in PM redox enzymes (Hyun et al. 2006a).
Previous studies showed that NQO1 overexpression via a keap1-Nrf2 pathway using NQO1 inducers (e.g., sulforaphane, curcumine) renders neuroblastoma cells and primary neurons more resistant to metabolic and oxidative stress (Jia et al. 2008; Soane et al. 2010; Son et al. 2010). We also published that cells overexpressing NQO1 are more resistant to metabolic and proteotoxic insults (Hyun et al. 2012). In this study, overexpression of b5R made neuroblastoma cells more resistant to several toxic insults such as 2-deoxyglucose, rotenone, antimycin A, and lactacystin (Fig. 3). We identified that b5R transfectants were more resistant to oxidative/nitrative stress and apoptosis following exposure to mitochondrial toxins.
Higher resistance of b5R transfectants to mitochondrial inhibitors (Fig. 3) suggests that b5R may also be involved in bioenergetics, because similar results have been published recently using another PM redox enzyme, NQO1 (Kim et al. 2013). NADH (a direct source of electron donor for complex I), glutamate/malate (an electron donor mediated by malate-aspartate shuttle for complex I), and succinate (an electron donor supplied in the TCA cycle for complex II) were used as the best electron donors, as reported earlier (Panov et al. 2004). These phenomena were consistent with the data obtained from SH-SY5Y cells differentiated by RA (Supplementary Fig. 2). The complex I activity in b5R transfectants was further increased by RA. These data are consistent with a previous report that SH-SY5Y cells differentiated by RA exhibit increased OCR and higher mitochondrial respiratory capacity (Xun et al. 2012). In fact, RA can stimulate oxidative phosphorylation and mitochondrial biogenesis (Tourniaire et al. 2015).
Complexes I and III are a main site of mitochondrial ROS generation (Barja 1999; James et al. 2004). Levels of mitochondrial ROS are enhanced by treatment with mitochondrial inhibitors (e.g., rotenone, antimycin A) (Fig. 5) and in pathological conditions (e.g., hypoxia) (Kushnareva et al. 2002; Muller et al. 2004), consistent with the elevated levels of oxidative/nitrative damage (Fig. 6) and resistance to apoptotic cell death (Fig. 7). Usually, the mitochondria can produce ROS as much as they consume oxygen and produce ATP (Murphy 2009). We found that neuroblastoma cells transfected with b5R did not produce further ROS, whereas they generated more ATP through enhanced mitochondrial electron transport under normal culture conditions, compared with control cells (Figs. 4 and 5). These phenomena can be explained by some possibilities. Firstly, as stated earlier, ROS (e.g., O2 •−) generated during oxidative phosphorylation can be scavenged directly by b5R. In fact, b5R can convert an oxidized form of CoQ to a reduced state by transferring one electron one by one. This might account for no significant decrease in ROS production in b5R transfectants, compared with cells overexpressing NQO1, which induces attenuated levels of ROS while higher ATP production and activity of mitochondrial complexes I and II (Kim et al. 2013). Secondly, there could be causal relationship between higher QH2/Q− and NAD+/NADH ratio. Thirdly, b5R would induce electron transport from complex I or II to complex III without leakage of electrons, resulting in a more efficient production of ATP. This explanation is supported by increased NAD+/NADH ratio (Fig. 2) and attenuated ROS production (Fig. 4) in cells overexpressing b5R.
CoQ is identified at significant levels in the PM and existed as three different states (Lopez-Lluch et al. 2010). Semi-ubiquinone is a state between oxidized and reduced forms of CoQ and transitionally formed by one electron transfer (Andreeva and Crompton 1994; James et al. 2004). Therefore, some studies showed that semi-ubiquinone can be a source of superoxide (O2 •−) production using purified b5R fractions (Samhan-Arias and Gutierrez-Merino 2014) and O2 •− in the PM can be locally increased by b5R along with the formation of lipid rafts with caveolins (Samhan-Arias et al. 2012). However, another study reported that b5R is responsible for maintaining basal levels of the reduced form of CoQ (Lopez-Lluch et al. 2010). Although semi-ubiquinone is formed transiently by one electron transfer from NADH, it can immediately be converted to a fully reduced form of CoQ by consequent one electron transfer (Crane and Bottger 2001). This is supported by our finding that b5R can attenuate the levels of ROS following exposure to toxins (Fig. 5). These suggest that the PM b5R can neutralize overproduced O2 •− by transferring one electron to oxidized CoQ. b5R can regulate CoQ levels and prevent oxidative damage in aged mice muscle through being stimulated by CR and aerobic exercise (Rodriguez-Bies et al. 2015). Also, phase I (e.g., b5R) and II (NQO1) enzymes can be activated by Aegle marmelos and protect against damage mediated by free radicals in mice (Singh et al. 2000). Consistent with our findings, these studies suggest a possible protective effect of b5R against oxidative stress.
Many studies demonstrated that impairment of the ubiquitin-proteasome system (UPS) is closely associated with many types of cell death identified in neurodegenerative diseases and cancer (Cecarini et al. 2007; Hoeller and Dikic 2009; Huang and Figueiredo-Pereira 2010; Sohns et al. 2010). Therefore, the UPS has been a hot spot in research to find therapeutic targets. This study found that b5R protects SH-SY5Y cells against cell death by lactacystin, a potent proteasome inhibitor (Fig. 3f), suggesting a new model of proteotoxicity, even though its protective mechanisms are still not clear. Some reports showed that heat shock proteins (e.g., HSP70) can be upregulated in models of proteotoxicity-mediated ATP depletion (Barja 1999) and cancer (Kushnareva et al. 2002). Previously, we also presented that overexpressed NQO1 is also involved in the protection of neuronal cells against proteotoxicity (Hyun et al. 2012). Because many neurodegenerative diseases are related to the accumulation of abnormal proteins such as amyloid β, α-synuclein, prions, and other damaged proteins, it suggests that upregulated PM redox enzymes (e.g., b5R, NQO1) can play a key role in removing abnormal proteins by increasing chaperon proteins during pathogenic processes in AD, PD, and prion disease.
Taken together, this study found that b5R can be a therapeutic target for neurodegenerative diseases focused on the regulation of mitochondrial function. Further work is required to improve mitochondrial energy metabolism using other cell culture and animal models.
Abbreviations
- AD:
-
Alzheimer’s disease
- ANOVA:
-
Analysis of variance
- b5R:
-
Cytochrome b5 reductase
- CoQ:
-
Coenzyme Q
- DCIP:
-
Dichloroindolphenol
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- NAD:
-
Nicotinamide adenine dinucleotide
- NQO1:
-
NADH-quinone oxidoreductase 1
- PD:
-
Parkinson’s disease
- PMRS:
-
Plasma membrane redox system
- ROS:
-
Reactive oxygen species
- UPS:
-
Ubiquitin-proteasome system
References
Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30:2967–2978
Andreeva L, Crompton M (1994) An ADP-sensitive cyclosporin-A-binding protein in rat liver mitochondria. Eur J Biochem 221:261–268
Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31:347–366
Beal MF (2004) Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J Bioenerg Biomembr 36:381–386
Beal MF (2005) Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 26:585–586
Bewley MC, Marohnic CC, Barber MJ (2001) The structure and biochemistry of NADH-dependent cytochrome b5 reductase are now consistent. Biochemistry 40:13574–13582
Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6:611–622
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Browne SE (2008) Mitochondria and Huntington’s disease pathogenesis: insight from genetic and chemical models. Ann N Y Acad Sci 1147:358–382
Buron MI, Rodriguez-Aguilera JC, Alcain FJ, Navas P (1993) Transplasma membrane redox system in HL-60 cells is modulated during TPA-induced differentiation. Biochem Biophys Res Commun 192:439–445
Cecarini V, Ding Q, Keller JN (2007) Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res 41:673–680
Crane FL (2001) Biochemical functions of coenzyme Q10. J Am Coll Nutr 20:591–598
Crane FL, Bottger M (2001) Plasma membrane redox systems. Protoplasma 217:1–2
De Cabo R, Cabello R, Rios M, Lopez-Lluch G, Ingram DK, Lane MA, Navas P (2004) Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp Gerontol 39:297–304
De Luca T, Morre DM, Zhao H, Morre DJ (2005) NAD+/NADH and/or CoQ/CoQH2 ratios from plasma membrane electron transport may determine ceramide and sphingosine-1-phosphate levels accompanying G1 arrest and apoptosis. BioFactors 25:43–60
Ferrer I (2009) Early involvement of the cerebral cortex in Parkinson’s disease: convergence of multiple metabolic defects. Prog Neurobiol 88:89–103
Hoeller D, Dikic I (2009) Targeting the ubiquitin system in cancer therapy. Nature 458:438–444
Huang Q, Figueiredo-Pereira ME (2010) Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis 15:1292–1311
Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R (2006a) Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A 103:19908–19912
Hyun DH, Hernandez JO, Mattson MP, de Cabo R (2006b) The plasma membrane redox system in aging. Ageing Res Rev 5:209–220
Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R (2007) Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem 100:1364–1374
Hyun DH, Kim J, Moon C, Lim CJ, de Cabo R, Mattson MP (2012) The plasma membrane redox enzyme NQO1 sustains cellular energetics and protects human neuroblastoma cells against metabolic and proteotoxic stress. Age (Dordr) 34:359–370
Hyun DH, Mughal MR, Yang H, Lee JH, Ko EJ, Hunt ND, de Cabo R, Mattson MP (2010) The plasma membrane redox system is impaired by amyloid beta-peptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp Neurol 225:423–429
Incerpi S, Fiore AM, De Vito P, Pedersen JZ (2007) Involvement of plasma membrane redox systems in hormone action. J Pharm Pharmacol 59:1711–1720
James AM, Smith RA, Murphy MP (2004) Antioxidant and prooxidant properties of mitochondrial coenzyme Q. Arch Biochem Biophys 423:47–56
Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–36, discussion S36-28
Jia Z, Zhu H, Misra HP, Li Y (2008) Potent induction of total cellular GSH and NQO1 as well as mitochondrial GSH by 3H-1,2-dithiole-3-thione in SH-SY5Y neuroblastoma cells and primary human neurons: protection against neurocytotoxicity elicited by dopamine, 6-hydroxydopamine, 4-hydroxy-2-nonenal, or hydrogen peroxide. Brain Res 1197:159–169
Jimenez-Hidalgo M, Santos-Ocana C, Padilla S, Villalba JM, Lopez-Lluch G, Martin-Montalvo A, Minor RK, Sinclair DA, de Cabo R, Navas P (2009) NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast. Aging Cell 8:140–151
Kelner GS, Lee M, Clark ME, Maciejewski D, McGrath D, Rabizadeh S, Lyons T, Bredesen D, Jenner P, Maki RA (2000) The copper transport protein Atox1 promotes neuronal survival. J Biol Chem 275:580–584
Kesharwani RK, Singh DV, Misra K, Rizvi SI (2012) Plant polyphenols as electron donors for erythrocyte plasma membrane redox system: validation through in silico approach. Org Med Chem Lett 2:12
Kim J, Kim SK, Kim HK, Mattson MP, Hyun DH (2013) Mitochondrial function in human neuroblastoma cells is up-regulated and protected by NQO1, a plasma membrane redox enzyme. PLoS One 8:e69030
Kim JA, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414
Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP (2000) Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci 20:6920–6926
Kushnareva Y, Murphy AN, Andreyev A (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J 368:545–553
Lee M, Hyun D, Jenner P, Halliwell B (2001) Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J Neurochem 76:957–965
Li X, May JM (2002) Catalase-dependent measurement of H2O2 in intact mitochondria. Mitochondrion 1:447–453
Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP (2009) Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromol Med 11:28–42
Lopez-Lluch G, Rodriguez-Aguilera JC, Santos-Ocana C, Navas P (2010) Is coenzyme Q a key factor in aging? Mech Ageing Dev 131:225–235
Luft R, Landau BR (1995) Mitochondrial medicine. J Intern Med 238:405–421
Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B (1997) An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J Neurochem 68:2061–2069
Marques-da-Silva D, Samhan-Arias AK, Tiago T, Gutierrez-Merino C (2010) L-type calcium channels and cytochrome b5 reductase are components of protein complexes tightly associated with lipid rafts microdomains of the neuronal plasma membrane. J Proteome 73:1502–1510
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Medina MA, del Castillo-Olivares A, Nunez de Castro I (1997) Multifunctional plasma membrane redox systems. BioEssays 19:977–984
Miller SW, Trimmer PA, Parker WD Jr, Davis RE (1996) Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. J Neurochem 67:1897–1907
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195
Muller FL, Liu Y, Van Remmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279:49064–49073
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, Tjornelund J, Dawson KM, Dupuis M, Duchosal MA (2009) The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 113:3276–3286
Navas P, Nowack DD, Morre DJ (1989) Isolation of purified plasma membranes from cultured cells and hepatomas by two-phase partition and preparative free-flow electrophoresis. Cancer Res 49:2147–2156
Panov AV, Andreeva L, Greenamyre JT (2004) Quantitative evaluation of the effects of mitochondrial permeability transition pore modifiers on accumulation of calcium phosphate: comparison of rat liver and brain mitochondria. Arch Biochem Biophys 424:44–52
Pedersen WA, Chan SL, Zhu H, Abdur-Rahman LA, Verdi JM, Mattson MP (2002) Numb isoforms containing a short PTB domain promote neurotrophic factor-induced differentiation and neurotrophic factor withdrawal-induced death of PC12 cells. J Neurochem 82:976–986
Rao PV, Krishna CM, Zigler JS Jr (1992) Identification and characterization of the enzymatic activity of zeta-crystallin from guinea pig lens. A novel NADPH:quinone oxidoreductase. J Biol Chem 267:96–102
Reznick AZ, Packer L (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 233:357–363
Rodriguez-Bies E, Navas P, Lopez-Lluch G (2015) Age-dependent effect of every-other-day feeding and aerobic exercise in ubiquinone levels and related antioxidant activities in mice muscle. J Gerontol A Biol Sci Med Sci 70:33–43
Samhan-Arias AK, Garcia-Bereguiain MA, Martin-Romero FJ, Gutierrez-Merino C (2009) Clustering of plasma membrane-bound cytochrome b5 reductase within ‘lipid raft’ microdomains of the neuronal plasma membrane. Mol Cell Neurosci 40:14–26
Samhan-Arias AK, Gutierrez-Merino C (2014) Purified NADH-cytochrome b5 reductase is a novel superoxide anion source inhibited by apocynin: sensitivity to nitric oxide and peroxynitrite. Free Radic Biol Med 73:174–189
Samhan-Arias AK, Marques-da-Silva D, Yanamala N, Gutierrez-Merino C (2012) Stimulation and clustering of cytochrome b5 reductase in caveolin-rich lipid microdomains is an early event in oxidative stress-mediated apoptosis of cerebellar granule neurons. J Proteome 75:2934–2949
Seow HA, Belcourt MF, Penketh PG, Hodnick WF, Tomasz M, Rockwell S, Sartorelli AC (2005) Nuclear localization of NADPH:cytochrome c (P450) reductase enhances the cytotoxicity of mitomycin C to Chinese hamster ovary cells. Mol Pharmacol 67:417–423
Shim JH, Yoon SH, Kim KH, Han JY, Ha JY, Hyun DH, Paek SH, Kang UJ, Zhuang X, Son JH (2011) The antioxidant Trolox helps recovery from the familial Parkinson’s disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 11:707–715
Siendones E, SantaCruz-Calvo S, Martin-Montalvo A, Cascajo MV, Ariza J, Lopez-Lluch G, Villalba JM, Acquaviva-Bourdain C, Roze E, Bernier M, de Cabo R, Navas P (2014) Membrane-bound CYB5R3 is a common effector of nutritional and oxidative stress response through FOXO3a and Nrf2. Antioxid Redox Signal 21:1708–1725
Sims NR (1992) Energy metabolism and selective neuronal vulnerability following global cerebral ischemia. Neurochem Res 17:923–931
Singh RP, Banerjee S, Rao AR (2000) Effect of Aegle marmelos on biotransformation enzyme systems and protection against free-radical-mediated damage in mice. J Pharm Pharmacol 52:991–1000
Soane L, Li Dai W, Fiskum G, Bambrick LL (2010) Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J Neurosci Res 88:1355–1363
Sohns W, van Veen TA, van der Heyden MA (2010) Regulatory roles of the ubiquitin-proteasome system in cardiomyocyte apoptosis. Curr Mol Med 10:1–13
Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP (2010) Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem 112:1316–1326
Sparla F, Bagnaresi P, Scagliarini S, Trost P (1997) NADH:Fe(III)-chelate reductase of maize roots is an active cytochrome b5 reductase. FEBS Lett 414:571–575
Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet ML, Ribot J, Landrier JF (2015) All-trans retinoic acid induces oxidative phosphorylation and mitochondria biogenesis in adipocytes. J Lipid Res 56:1100–1109
Turunen M, Olsson J, Dallner G (2004) Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660:171–199
Villalba JM, Navas P (2000) Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal 2:213–230
Vives-Bauza C, Yang L, Manfredi G (2007) Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol 80:155–171
Votyakova TV, Reynolds IJ (2004) Detection of hydrogen peroxide with Amplex Red: interference by NADH and reduced glutathione auto-oxidation. Arch Biochem Biophys 431:138–144
Xun Z, Lee DY, Lim J, Canaria CA, Barnebey A, Yanonne SM, McMurray CT (2012) Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech Ageing Dev 133:176–185
Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 253:162–168
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A2039477 to D.H.), South Korea.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 514 kb)
About this article

Cite this article
Hyun, DH., Lee, GH. Cytochrome b5 reductase, a plasma membrane redox enzyme, protects neuronal cells against metabolic and oxidative stress through maintaining redox state and bioenergetics. AGE 37, 122 (2015). https://doi.org/10.1007/s11357-015-9859-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11357-015-9859-9