
Abstract
The plasma membrane redox system (PMRS) of nicotinamide adenine dinucleotide (NADH)-related enzymes plays a key role in the maintenance of cellular energetics. During the aging process, neural cells are particularly sensitive to impaired energy metabolism and oxidative damage, but the involvement of the PMRS in these processes is unknown. Here, we used human neuroblastoma cells with either elevated or reduced levels of the PMRS enzyme NADH-quinone oxidoreductase 1 (NQO1) to investigate how the PMRS regulates neuronal stress responses. Cells with elevated NQO1 levels were more resistant to death induced by 2-deoxyglucose, potassium cyanide (energetic stress), and lactacystin (proteotoxic stress), but were not protected from being killed by H2O2 and serum withdrawal. The NAD+(an oxidized form of NADH)/NADH ratio was maintained at a significantly higher level in cells overexpressing NQO1, consistent with enhanced levels of NQO1 activity. Levels of the neuroprotective transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells and nuclear factor (erythroid-derived 2)-like 2, and the protein chaperone HSP70 were elevated in cells overexpressing NQO1. Cells in which NQO1 levels were decreased by RNA interference exhibited increased vulnerability to death induced by 2-deoxyglucose and lactacystin. Thus, a higher NAD+/NADH ratio and activation of adaptive stress response pathways are enhanced by the PMRS in neuroblastoma cells, enabling them to maintain redox homeostasis under conditions of energetic and proteotoxic stress. These findings have implications for the development of therapeutic interventions for neural tumors and neurodegenerative conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A preponderance of research on cellular energy metabolism and oxidative stress has focused on mitochondria because this organelle generates large amounts of both adenosine triphosphate (ATP) and free radicals (Mattson et al. 2008; Murphy 2009). Impaired mitochondrial function and cellular energy metabolism are implicated in the normal aging process and in diseases of the cardiovascular, nervous, and musculoskeletal systems (Kim et al. 2008; Luft and Landau1995). Neurons rely on a constant supply of glucose and so are particularly vulnerable to conditions in which glucose availability is limited (e.g., ischemic stroke and hypoglycemia) or energy demand is increased greatly (e.g., epileptic seizures; Mattson et al. 2008; Sims 1992). The oxidative and metabolic alterations that occur in neurons during aging are believed to contribute to their dysfunction and degeneration in Alzheimer’s disease (AD; Mattson 2004; Mosconi et al. 2008), Parkinson’s disease (PD; Ferrer 2009), and Huntington’s diseases (Browne 2008). A better understanding of the mechanisms by which neurons cope with metabolic and oxidative stress may therefore provide opportunities for the development of novel interventions to prevent and treat neurodegenerative diseases.
The plasma membrane (PM) regulates numerous aspects of cell physiology and signaling, and also protects cells against oxidative stress. The PM contains enzymes involved in electron transport and energy metabolism, the so-called PM redox system (PMRS; Hyun et al. 2006b; Villalba and Navas 2000). Coenzyme Q (CoQ) can be reduced at the PM by either NAD(P)H-quinone oxidoreductase 1 (NQO1) or NADH-cytochrome b5 reductase. NQO1 is a nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)-dependent reductase that is translocated to the inner surface of the PM under stress conditions. (Li and Jaiswal 1992; Rushmore et al. 1991). CoQ is an important component of the PMRS where it functions as an antioxidant that protects lipids from oxidative damage either directly or by maintaining the active reduced forms of ascorbate and α-tocopherol (Crane 2001; Kagan et al. 1998; Turunen et al. 2004).
The PMRS is also important for modulating the cellular NAD+/NADH ratio in response to fluctuations in energy demand. Indeed, PMRS activity has been linked to processes such as cell survival, growth, and differentiation where changes in pyridine nucleotide balance play important roles (Buron et al. 1993; De Luca et al. 2005; Medina et al. 1997). In addition, we recently demonstrated that the PMRS can compensate for the dysfunction of mitochondria; mitochondria-deficient cells exhibit increased activities of multiple PMRS enzymes and, though more sensitive to glycolysis inhibitors, are more resistant to oxidative stress (Hyun et al. 2007). The contributions of individual PMRS to cytoprotection are unknown. Among the PMRS enzymes, NQO1 is of particular interest because its expression is induced by Nrf2, a transcription factor involved in adaptive cellular responses to oxidative and metabolic stress (Jaiswal 2004). Recent findings suggest that activation of Nrf2 can protect neurons against oxidative and metabolic insults (Johnson et al. 2008; Son et al. 2010), but whether NQO1 plays a pivotal role in such neuroprotection is unknown. Other studies have associated changes in NQO1 expression with the pathogenesis of AD (Raina et al. 1999), suggesting a potential role for NQO1 in cellular reactions to the metabolic impairment and increased oxidative stress known to occur in this disease (Mattson 2004).
In the present study, we used molecular genetic technologies to selectively increase or decrease levels of NQO1 in human neuroblastoma cells. We found that higher NQO1 levels resulted in increased resistance of the cells to energy deprivation and proteotoxicity but did not protect the cells against direct oxidative insults. Cells with higher NQO1 levels maintained a higher NAD+/NADH ratio, suggesting that this PMRS enzyme improves cellular energetics.
Methods
Cell culture and transfection
SH-SY5Y human neuroblastoma cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 IU/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) in a humidified 5% CO2/95% air atmosphere. The cells were transfected with pBE8 vector containing the full-length NQO1 cDNA (a generous gift from Alan Sartorelli at the Yale University School of Medicine) as described previously (Seow et al. 2004). Six different clones were selected using G-418. To reduce NQO1 levels, the cells were transfected with NQO1-specific short hairpin RNA (shRNA) in the pLKO.1 vector according the manufacturer’s protocol (Open Biosystems, Rockford, IL), and puromycin-resistant clones were selected.
Characterization of the transfected cells
Cells were lysed, and NQO1 protein levels were determined by immunoblotting using NQO1 monoclonal antibody provided by David Ross (University of Colorado, Denver, CO). Levels of NQO1 proteins in the PM of the transfected cells were measured following isolation of the PMs by a two-phase partition, as described previously (de Cabo et al. 2004; Hyun et al. 2006a). NQO1 activity was determined in vitro using electron donors, as described previously (de Cabo et al. 2004).
Isolation of plasma membranes
The PM isolation procedure was carried out using the two-phase partition, as described previously (de Cabo et al. 2004; Navas et al. 1989). Protein concentration in PM fraction was determined by the Bradford Assay (Bradford 1976). Immunoblotting assay using enzyme markers for PM and mitochondria was performed to measure the purity of the isolated fractions as described previously (de Cabo et al. 2004). For immunoblotting, anti-Na+/K+-ATPase α-subunit monoclonal antibody (1:1,000 dilution, Affinity BioReagents, Golden, CO) and anti-cytochrome c oxidase subunit I monoclonal antibody (1:1,000 dilution, Molecular Probe, Eugene, OR, USA) were used.
Levels of metabolites
NAD+ and NADH levels were determined separately using a NAD/NADH Quantitation Kit (BioVision, Mountain View, CA). Briefly, in order to measure total levels of NAD+ and NADH, lysates were transferred into a 96-well plate, and 100 μl NAD+ cycling buffer and 2 μl NAD+ cycling enzyme mix were added. The mixtures were incubated at room temperature for 5 min to convert NAD+ to NADH, and NADH developer was then added to each well. The plate was incubated for 2 h, and absorbance was read at 450 nm. To measure NADH, NAD+ was decomposed by heating the lysates at 60°C for 30 min, followed by the same procedures described above. A standard curve was generated by serial dilution of standard NADH solution and the NAD+/NADH ratio was calculated as [(NADtotal–NADH)/NADH].
Cell viability assay
Cells were exposed to normal culture medium containing 2-deoxyglucose (5, 10, 15, 20 mM), potassium cyanide (KCN; 5, 10, 15, 20 mM), lactacystin (10, 20, 30, 40 mM), or to serum-free medium for 24 h. In the case of H2O2 treatment, the cells were exposed to hydrogen peroxide (50, 100, 150, 200 μM) in the absence of serum for 1 h and then incubated with normal culture medium for 24 h. Cell viability was measured using WST-8 (Dojindo, Japan), and intensity was read at 450 nm.
Stress-response proteins
Levels of stress-response molecules were examined by immunoblot analysis using antibodies against the following proteins: p65 (1:1,000 dilution, Cell Signaling Technology, Danvers, MA), p50 (1:1,000 dilution, Cell Signaling Technology), heat shock protein 70 (HSP70) (1:1,000 dilution, Cell Signaling Technology), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (1:1,000 dilution, Abcam, Cambridge, MA). Briefly, the samples were lysed using phosphate buffered saline (pH 7.4) containing 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml pepstastin A, and 0.1% Triton X-100 and placed on ice for 5 min. The lysates were centrifuged at 12,000×g for 10 min, and the supernatants were transferred into new Eppendorf tubes. Protein levels were measured using the Bradford reagent (Bradford 1976), and a total of 20 μg of protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The separated proteins were transferred electrophoretically to a nitrocellulose membrane (Whatman GmBH, Dassel, Germany), which was then incubated with the primary antibodies. Immune complexes were detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ, USA).
Statistical analysis
Statistical differences were analyzed by one-way ANOVA, and pairwise comparisons were performed with a post hoc Bonferroni t test.
Results
NQO1 protects human neuroblastoma cells against energetic but not oxidative stress
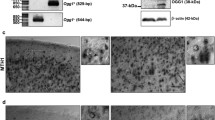
We first generated six different clones of SH-SY5Y human neuroblastoma cells stably overexpressing NQO1 and measured levels of NQO1 protein and enzymatic activity in these clones. Among these clones, four expressed NQO1 at levels three- to fivefold greater than in vector-transfected control cell clones (Fig. 1). These four clones were used for further evaluation of the subcellular localization and functional activity of NQO1. Immunoblot analysis of PM and cytosolic subcellular fractions demonstrated that NQO1 was present in both cellular compartments with levels being approximately threefold greater in samples from neuroblastoma cells overexpressing NQO1 (Fig. 2a). Measurements of NQO1 enzyme activity showed that clones overexpressing NQO1 possessed an approximately threefold greater NQO1 activity compared with control clones (Fig. 2b).

Characterization of human neuroblastoma cells overexpressing NQO1. Cells from the indicated clones of untransfected control SH-SY5Y cells and NQO1 transfected cells were lysed, and immunoblot analysis was performed using NQO1 monoclonal antibody

NQO1 enzymatic activity and the NAD+/NADH ratio are elevated in neuroblastoma cells overexpressing NQO1. Plasma membranes (PMs) were isolated by a two-phase partition. a NQO1 protein levels in cytosolic and PM fractions of control and NQO1-overexpressing cells were examined by immunoblot analysis. b NQO1 activity was determined using menadione and cytochrome c as substrates. c Cells were lysed, and the NAD+/NADH ratio was measured. Values are the mean and SEM (n = 6). *p < 0.01 compared with the value for untransfected control cells under normal culture conditions
To determine the impact of NQO1 on cellular bioenergetics, we determined levels of NAD+ and NADH in neuroblastoma cells expressing basal or elevated levels of NQO1. The NAD+/NADH ratio was significantly elevated by more than ninefold in cells overexpressing NQO1 compared with control cells (Fig. 2c).
We next performed experiments in which cells with basal or elevated levels of NQO1 were exposed to five different cytotoxic conditions: 2-deoxyglucose, an inhibitor of glycolysis; KCN, a mitochondrial toxin that inhibits complex IV in the electron transport chain; H2O2, a reactive oxygen species (ROS) that induces oxidative stress; the proteasome inhibitor lactacystin; and serum-free medium, which triggers apoptosis. Cell viability was quantified at 24 h after exposure to the insults. Because their primary energy substrate utilized by neurons is glucose, they are particularly vulnerable to being killed by 2-deoxyglucose (Cater et al. 2001). However, the severe restriction of glucose availability imposed by 2-deoxyglucose can induce a severe endoplasmic reticulum stress response that, in turn, triggers apoptosis in many types of normal and tumor cells (Dwarakanath 2009). In neuroblastoma cells with basal levels of NQO1, cell viability was significantly reduced in cultures exposed to 2-deoxyglucose at concentrations of 10–20 mM, compared with untreated cells or cells exposed to a lower concentration of 2-deoxyglucose (Fig. 3a). In contrast, cells overexpressing NQO1 were resistant to being killed by 10–20 mM 2-deoxyglucose. Cell viability was decreased by KCN in a concentration-dependent manner, with no significant differences in cells with basal or elevated levels of NQO1, although there was a trend towards greater cell viability in cells overexpressing NQO1 (Fig. 3b). We next exposed cells with basal or elevated levels of NQO1 to increasing concentrations of H2O2 (50–200 μM) and found that NQO1 did not protect the cells from being killed by this oxidative insult (Fig. 3c). The proteasome inhibitor lactacystin reduced neural cell viability in a concentration-dependent manner, and cells with elevated levels of NQO1 were significantly less vulnerable to lactacystin compared with cells with basal levels of NQO1 (Fig. 3d). Finally, we determined whether cells with elevated levels of NQO1 would be more or less vulnerable to serum withdrawal, a classic method of inducing neuronal apoptosis (Pedersen et al. 2002). The vulnerability of human neuroblastoma cells to serum withdrawal was not different in cells with basal or elevated NQO1 levels (Fig. 3e), suggesting that this PMRS enzyme does not modify this form of programmed cell death.

Neuroblastoma cells overexpressing NQO1 exhibit reduced vulnerability to metabolic and proteotoxic insults, but are not protected against oxidative stress and trophic factor withdrawal. Cells were exposed to 2-deoxyglucose (a), KCN (b), H2O2 (c), lactacystin (d), or serum-free medium (e) for 24 h, and cell viability was measured using the WST-8 assay (see Methods for details). Values are the mean and SEM (n = 6)
Cells overexpressing NQO1 exhibit elevated levels of proteins involved in cell survival signaling and protein stabilization
To further elucidate the mechanism by which NQO1 protects neural cells against metabolic and proteotoxic insults, we measured levels of several proteins known to protect neurons against various stressors including the transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (p65 and p50 subunits) and Nrf2, and the protein chaperone HSP70. Immunoblot analysis showed that levels of p65, p50, Nrf2, and HSP70 were all greater in cells overexpressing NQO1 (Fig. 4).

Levels of the stress-response transcription factors Nrf2 and NF–κB, and the protein chaperone HSP70 are elevated in neuroblastoma cells overexpressing NQO1. Cells were lysed, and levels of the indicated proteins (p65 and p50 subunits of NF–κB, Nrf2, and HSP70) were determined by immunoblot analysis
Depletion of NQO1 using RNA interference renders neuroblastoma cells vulnerable to energetic stress
To determine whether basal levels of endogenous NQO1 influenced the vulnerability of neural cells to metabolic stress we transfected neuroblastoma cells with a vector to express NQO1 shRNA. Immunoblot analysis showed that two different clones of neuroblastoma cells transfected with NQO1 shRNA exhibited less than 20% of the NQO1 protein levels compared with control untransfected cells (Fig. 5a). In contrast, levels of b5-reductase were similar in cells expressing NQO1 shRNA and untransfected control cells. Levels of NQO1 enzyme activity were significantly reduced in NQO1 shRNA-expressing cells to less than 15% of the NQO1 activity level in control cells (Fig. 5c), whereas levels of b5-reductase activity were unaffected by expression of NQO1 shRNA.

Characterization of human neuroblastoma cells in which NQO1 levels were depleted using RNA interference technology. a Protein levels of NQO1 and b5 reductase (b5R) were examined by immunoblot analysis. b NQO1 activity levels in cells with normal and reduced levels of NQO1. c Levels of b5R activity in cells with normal and reduced levels of NQO1
Cells in which NQO1 was depleted by RNA interference were significantly more vulnerable to being killed by 2-deoxyglucose compared with cells with basal levels of NQO1 (Fig. 6a). Cell viability was decreased by KCN in a concentration-dependent manner, with cells depleted of NQO1 exhibiting more cell death than cells with basal levels of NQO1 (Fig. 6b). We next exposed cells with basal or reduced levels of NQO1 to increasing concentrations of H2O2 (50–200 μM) and found that NQO1 depletion increased the vulnerability of the cells to the highest concentrations of H2O2 (150 and 200 μM; Fig. 6c). The proteasome inhibitor lactacystin reduced neural cell viability in a concentration-dependent manner, and cells with reduced levels of NQO1 were somewhat more vulnerable to lactacystin compared with cells with basal levels of NQO1 (Fig. 6d). Finally, the vulnerability of human neuroblastoma cells to serum withdrawal was not different in cells with basal or reduced NQO1 levels (Fig. 6e), suggesting that this PMRS enzyme does not modify this form of programmed cell death.

Neuroblastoma cells with reduced NQO1 levels exhibit increased vulnerability to metabolic and proteotoxic insults but are not more vulnerable to oxidative stress and trophic factor withdrawal. Control cells with normal levels of NQO1 and cells in which NQO1 was depleted using RNA interference were exposed to 2-deoxyglucose (a), KCN (b), H2O2 (c), lactacystin (d), or serum-free medium (e) for 24 h, and cell viability was measured using the WST-8 assay (see Methods for details). Values are the mean and SEM (n = 6)
Discussion
The major finding in the present study is that NQO1 plays a pivotal role in the survival of human neuroblastoma cells under conditions of metabolic and proteotoxic stress. The NAD+/NADH ratio was significantly greater in cells overexpressing NQO1 and significantly lower in cells deficient in NQO1. Both neural tumor cells (Nahimana et al. 2009) and normal neurons (Alano et al. 2010; Kruman et al. 2000; Liu et al. 2009) have high cellular energy requirements and so are sensitive to a drop in the NAD+/NADH ratio. By maintaining NAD+ levels, NQO1 may therefore promote the growth and survival of some tumor cells. Consistent with a role for NQO1 in tumor cell survival, NAD+ synthesis inhibitors were recently shown to induce the regression of neuroblastomas in vivo (Fuchs et al. 2010) and suppress hematologic cancers (Nahimana et al. 2009). Altered intracellular redox balance caused by knocking-down of NQO1 can impair synthesis of pyridine nucleotides and metabolism of glucose and fatty acids (Gaikwad et al. 2001). On the other hand, apoptosis of non-small-cell lung cancer cells can be promoted by B-lapachone, which is bioactivated by NQO1 (Bey et al. 2007), suggesting complex roles for this PMRS in cancers that may depend on factors such as the cell type, and the metabolic and redox states of the cells.
Because they exhibit some features of differentiated neurons, neuroblastoma cells are widely used as a model of neurons and, indeed, results obtained in studies of neuroblastoma cell lines have often been extended to primary neurons (Xie et al. 2010). For example, SH-SY5Y cells exhibit properties of dopaminergic neurons and are vulnerable to being killed by dopaminergic neurotoxins (Elkon et al. 2001; Li et al. 2010b; Sadan et al. 2009) and α-synuclein, a proteotoxic peptide of fundamental importance in PD (Bellucci et al. 2008; Hettiarachchi et al. 2009). Recently, a report showed that overexpressed N-ribosyldihydronicotineamide-quinone oxidoreductase 2 (NQO2), an isozyme of NQO1, causes elevated production of ROS, leading to neuronal cell death (Wang et al. 2008). NQO2 plays an important role in dopamine metabolism (Wang et al. 2008) and can generate ROS; inhibition of NQO2 can protect cells from neuronal damage (Mailliet et al. 2004; Nosjean et al. 2000). In contrast, NQO1 is involved in reduction of oxidized CoQ (Crane 2001; Kagan et al. 1998; Turunen et al. 2004). Human neuroblastoma cells have also been used to elucidate the molecular underpinnings of the neurotoxic actions of amyloid β-peptide (Aβ), findings relevant to the pathogenesis of AD (Datki et al. 2004; Lambert et al. 1994; Li et al. 2010a). Because impaired cellular energy metabolism and abnormal accumulation and self-aggregation of pathogenic peptides are believed to play pivotal roles in the dysfunction and degeneration of neurons in several major neurodegenerative disorders, our findings suggest a potential for NQO1 in protecting neurons against such disorders, consistent with a previous result (Incerpi et al. 2007). On the other hand, age-related declines in NQO1 and other PMRS enzymes (Hyun et al. 2006a) may predispose neurons to energy failure and proteotoxicity. Although the amount of NQO1 in the plasma membrane fraction was much greater than in the cytosolic fraction in neuroblastoma cells overexpressing NQO1, NQO1 in the cytosol may also contribute to the cytoprotective effect of NQO1.
Neuroblastoma cells overexpressing NQO1 exhibited elevated levels of two transcription factors (Nrf2 and NF–κB) and the protein chaperone HSP70, revealing a novel link between the PMRS and systems that exert cell survival-promoting effects elsewhere in the cell. Indeed, previous studies have shown that NQO1 is involved in activation of NF–κB (Ahn et al. 2006) and can interact with HSP70 (Anwar et al. 2002). Nrf2, a transcription factor responsive to oxidative and metabolic stress, induces the expression of multiple proteins that protect cells against stress including heme oxygenase 1 (HO-1), enzymes involved in glutathione metabolism, and NQO1 (Wakabayashi et al. 2010). We found that levels of Nrf2 are relatively low in non-transfected neuroblastoma cells and are elevated in cells that overexpress NQO1. These findings suggest that there is a positive feed-forward interaction between NQO1 and Nrf2 that greatly increases the resistance of the cells to oxidative stress. Stimuli that induce the expression of NQO1 via an Nrf2-mediated pathway (sulforaphane, plumbagin, and other chemicals, for example) also protect neuroblastoma cells and primary neurons against oxidative and metabolic insults (Jia et al. 2008; Soane et al. 2010; Son et al. 2010). However, Nrf2 induces the expression of multiple genes including HO-1 and phase 2 enzymes, and data in the present study suggest that NQO1 is particularly important in protecting cells against metabolic and proteotoxic stress.
NF–κB activation promotes the survival of cells by up-regulating the expression of antioxidant enzymes such as Mn-superoxide dismutase and the anti-apoptotic protein Bcl-2 (Mattson and Meffert 2006) but can also induce the expression of pro-inflammatory cytokines (Hoffmann and Baltimore 2006). The increased levels of p65 and p50, the prototypical NF–κB heterodimeric transcription factor, in response to NQO1 suggests a cooperative interaction of the Nrf2 and NF–κB adaptive stress-response pathways under conditions of energetic and proteotoxic stress.
The proteasome has received considerable attention in research on cell death (Sohns et al. 2010), cancer (Hoeller and Dikic 2009), and neurodegenerative disorders (Cecarini et al. 2007; Huang and Figueiredo-Pereira 2010). We found that NQO1 protects neuroblastoma cells from being killed by the proteasome inhibitor lactacystin, a model of proteotoxicity. The mechanism responsible for NQO1-mediated protection against proteotoxicity remains to be established. The up-regulation of HSP70 in neuroblastoma cells overexpressing NQO1 likely contributes to protection in the lactacystin proteotoxicity model. Indeed, a previous study showed that up-regulation of HSP70 expression by heat or metabolic preconditioning protects myoblasts against death induced by proteotoxicity-mediated ATP depletion (Kabakov et al. 2002). Moreover, many types of cancer cells exhibit elevated levels of protein chaperones including HSP70 (Mosser and Morimoto 2004). NQO1 is also responsible for the stabilization of proteins degraded by the 20S proteasome (Alard et al. 2010; Asher et al. 2005). By protecting against the proteotoxicity of neurodegenerative disease-associated proteins such as α-synuclein and Aβ and prions, up-regulation of NQO1 in neurons would be expected to protect neurons against pathogenic processes in AD, PD, and prion disorder, as explained in a previous report (SantaCruz et al. 2004). The latter possibility could be tested in future studies using cell culture and animal models.
References
Ahn KS, Sethi G, Jain AK, Jaiswal AK, Aggarwal BB (2006) Genetic deletion of NAD(P)H:quinone oxidoreductase 1 abrogates activation of nuclear factor-kappaB, IkappaBalpha kinase, c-Jun N-terminal kinase, Akt, p38, and p44/42 mitogen-activated protein kinases and potentiates apoptosis. J Biol Chem 281:19798–19808
Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30:2967–2978
Alard A, Fabre B, Anesia R, Marboeuf C, Pierre P, Susini C, Bousquet C, Pyronnet S (2010) NAD(P)H quinone-oxydoreductase 1 protects eukaryotic translation initiation factor 4GI from degradation by the proteasome. Mol Cell Biol 30:1097–1105
Anwar A, Siegel D, Kepa JK, Ross D (2002) Interaction of the molecular chaperone Hsp70 with human NAD(P)H:quinone oxidoreductase 1. J Biol Chem 277:14060–14067
Asher G, Tsvetkov P, Kahana C, Shaul Y (2005) A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev 19:316–321
Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P (2008) Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem 106:560–577
Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, Minna JD, Bornmann WG, Gao J, Boothman DA (2007) An NQO1-and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci USA 104:11832–11837
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Browne SE (2008) Mitochondria and Huntington’s disease pathogenesis: insight from genetic and chemical models. Ann NY Acad Sci 1147:358–382
Buron MI, Rodriguez-Aguilera JC, Alcain FJ, Navas P (1993) Transplasma membrane redox system in HL-60 cells is modulated during TPA-induced differentiation. Biochem Biophys Res Commun 192:439–445
Cater HL, Benham CD, Sundstrom LE (2001) Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J Physiol 531:459–466
Cecarini V, Ding Q, Keller JN (2007) Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res 41:673–680
Crane FL (2001) Biochemical functions of coenzyme Q10. J Am Coll Nutr 20:591–598
Datki Z, Papp R, Zadori D, Soos K, Fulop L, Juhasz A, Laskay G, Hetenyi C, Mihalik E, Zarandi M et al (2004) In vitro model of neurotoxicity of Abeta 1–42 and neuroprotection by a pentapeptide: irreversible events during the first hour. Neurobiol Dis 17:507–515
de Cabo R, Cabello R, Rios M, Lopez-Lluch G, Ingram DK, Lane MA, Navas P (2004) Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp Gerontol 39:297–304
De Luca T, Morre DM, Zhao H, Morre DJ (2005) NAD+/NADH and/or CoQ/CoQH2 ratios from plasma membrane electron transport may determine ceramide and sphingosine-1-phosphate levels accompanying G1 arrest and apoptosis. Biofactors 25:43–60
Dwarakanath BS (2009) Cytotoxicity, radiosensitization, and chemosensitization of tumor cells by 2-deoxy-D-glucose in vitro. J Cancer Res Ther 5(Suppl 1):S27–S31
Elkon H, Melamed E, Offen D (2001) 6-Hydroxydopamine increases ubiquitin-conjugates and protein degradation: implications for the pathogenesis of Parkinson’s disease. Cell Mol Neurobiol 21:771–781
Ferrer I (2009) Early involvement of the cerebral cortex in Parkinson’s disease: convergence of multiple metabolic defects. Prog Neurobiol 88:89–103
Fuchs D, Rodriguez A, Eriksson S, Christofferson R, Sundberg C, Azarbayjani F (2010) Metronomic administration of the drug GMX1777, a cellular NAD synthesis inhibitor, results in neuroblastoma regression and vessel maturation without inducing drug resistance. Int J Cancer 126:2773–2789
Gaikwad A, Long DJ 2nd, Stringer JL, Jaiswal AK (2001) In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem 276:22559–22564
Hettiarachchi NT, Parker A, Dallas ML, Pennington K, Hung CC, Pearson HA, Boyle JP, Robinson P, Peers C (2009) alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J Neurochem 111:1192–1201
Hoeller D, Dikic I (2009) Targeting the ubiquitin system in cancer therapy. Nature 458:438–444
Hoffmann A, Baltimore D (2006) Circuitry of nuclear factor kappaB signaling. Immunol Rev 210:171–186
Huang Q, Figueiredo-Pereira ME (2010) Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis 15:1292–1311
Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R (2006a) Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A 103:19908–19912
Hyun DH, Hernandez JO, Mattson MP, de Cabo R (2006b) The plasma membrane redox system in aging. Ageing Res Rev 5:209–220
Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R (2007) Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem 100:1364–1374
Incerpi S, Fiore AM, De Vito P, Pedersen JZ (2007) Involvement of plasma membrane redox systems in hormone action. J Pharm Pharmacol 59:1711–1720
Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36:1199–1207
Jia Z, Zhu H, Misra HP, Li Y (2008) Potent induction of total cellular GSH and NQO1 as well as mitochondrial GSH by 3H-1,2-dithiole-3-thione in SH-SY5Y neuroblastoma cells and primary human neurons: protection against neurocytotoxicity elicited by dopamine, 6-hydroxydopamine, 4-hydroxy-2-nonenal, or hydrogen peroxide. Brain Res 1197:159–169
Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, Chen PC (2008) The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann NY Acad Sci 1147:61–69
Kabakov AE, Budagova KR, Latchman DS, Kampinga HH (2002) Stressful preconditioning and HSP70 overexpression attenuate proteotoxicity of cellular ATP depletion. Am J Physiol Cell Physiol 283:C521–C534
Kagan VE, Arroyo A, Tyurin VA, Tyurina YY, Villalba JM, Navas P (1998) Plasma membrane NADH-coenzyme Q0 reductase generates semiquinone radicals and recycles vitamin E homologue in a superoxide-dependent reaction. FEBS Lett 428:43–46
Kim JA, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414
Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP (2000) Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci 20:6920–6926
Lambert MP, Stevens G, Sabo S, Barber K, Wang G, Wade W, Krafft G, Snyder S, Holzman TF, Klein WL (1994) Beta/A4-evoked degeneration of differentiated SH-SY5Y human neuroblastoma cells. J Neurosci Res 39:377–385
Li Y, Jaiswal AK (1992) Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem 267:15097–15104
Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D et al (2010a) GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis 19:1205–1219
Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH (2010b) Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem 113:1621–1631
Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP (2009) Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med 11:28–42
Luft R, Landau BR (1995) Mitochondrial medicine. J Intern Med 238:405–421
Mailliet F, Ferry G, Vella F, Thiam K, Delagrange P, Boutin JA (2004) Organs from mice deleted for NRH:quinone oxidoreductase 2 are deprived of the melatonin binding site MT3. FEBS Lett 578:116–120
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639
Mattson MP, Meffert MK (2006) Roles for NF–kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ 13:852–860
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Medina MA, del Castillo-Olivares A, Nunez de Castro I (1997) Multifunctional plasma membrane redox systems. Bioessays 19:977–984
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann NY Acad Sci 1147:180–195
Mosser DD, Morimoto RI (2004) Molecular chaperones and the stress of oncogenesis. Oncogene 23:2907–2918
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, Tjornelund J, Dawson KM, Dupuis M, Duchosal MA (2009) The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 113:3276–3286
Navas P, Nowack DD, Morre DJ (1989) Isolation of purified plasma membranes from cultured cells and hepatomas by two-phase partition and preparative free-flow electrophoresis. Cancer Res 49:2147–2156
Nosjean O, Ferro M, Coge F, Beauverger P, Henlin JM, Lefoulon F, Fauchere JL, Delagrange P, Canet E, Boutin JA (2000) Identification of the melatonin-binding site MT3 as the quinone reductase 2. J Biol Chem 275:31311–31317
Pedersen WA, Chan SL, Zhu H, Abdur-Rahman LA, Verdi JM, Mattson MP (2002) Numb isoforms containing a short PTB domain promote neurotrophic factor-induced differentiation and neurotrophic factor withdrawal-induced death of PC12 Cells. J Neurochem 82:976–986
Raina AK, Templeton DJ, Deak JC, Perry G, Smith MA (1999) Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer’s disease. Redox Rep 4:23–27
Rushmore TH, Morton MR, Pickett CB (1991) The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 266:11632–11639
Sadan O, Bahat-Stromza M, Barhum Y, Levy YS, Pisnevsky A, Peretz H, Ilan AB, Bulvik S, Shemesh N, Krepel D et al (2009) Protective effects of neurotrophic factor-secreting cells in a 6-OHDA rat model of Parkinson disease. Stem Cells Dev 18:1179–1190
SantaCruz KS, Yazlovitskaya E, Collins J, Johnson J, DeCarli C (2004) Regional NAD(P)H:quinone oxidoreductase activity in Alzheimer’s disease. Neurobiol Aging 25:63–69
Seow HA, Penketh PG, Belcourt MF, Tomasz M, Rockwell S, Sartorelli AC (2004) Nuclear overexpression of NAD(P)H:quinone oxidoreductase 1 in Chinese hamster ovary cells increases the cytotoxicity of mitomycin C under aerobic and hypoxic conditions. J Biol Chem 279:31606–31612
Sims NR (1992) Energy metabolism and selective neuronal vulnerability following global cerebral ischemia. Neurochem Res 17:923–931
Soane L, Li Dai W, Fiskum G, Bambrick LL (2010) Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J Neurosci Res 88:1355–1363
Sohns W, van Veen TA, van der Heyden MA (2010) Regulatory roles of the ubiquitin-proteasome system in cardiomyocyte apoptosis. Curr Mol Med 10:1–13
Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA et al (2010) Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem 112:1316–1326
Turunen M, Olsson J, Dallner G (2004) Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660:171–199
Villalba JM, Navas P (2000) Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal 2:213–230
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW (2010) When NRF2 talks, who’s listening? Antioxid Redox Signal 13:1649–1663
Wang W, Le WD, Pan T, Stringer JL, Jaiswal AK (2008) Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased susceptibility to Parkinson’s disease. J Gerontol A Biol Sci Med Sci 63:127–134
Xie HR, Hu LS, Li GY (2010) SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin Med J (Engl) 123:1086–1092
Acknowledgments
We thank Alan Sartorelli (Yale University School of Medicine, USA) and David Ross (University of Colorado at Denver, USA) for providing pBE8 and antibodies against NQO1, respectively. This study was supported, in whole and in part, by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (20100003064), South Korea.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Hyun, DH., Kim, J., Moon, C. et al. The plasma membrane redox enzyme NQO1 sustains cellular energetics and protects human neuroblastoma cells against metabolic and proteotoxic stress. AGE 34, 359–370 (2012). https://doi.org/10.1007/s11357-011-9245-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-011-9245-1