
Abstract
The ubiquitin/proteasome pathway is the major proteolytic quality control system in cells. In this review we discuss the impact of a deregulation of this pathway on neuronal function and its causal relationship to the intracellular deposition of ubiquitin protein conjugates in pathological inclusion bodies in all the major chronic neurodegenerative disorders, such as Alzheimer’s, Parkinson’s and Huntington’s diseases as well as amyotrophic lateral sclerosis. We describe the intricate nature of the ubiquitin/proteasome pathway and discuss the paradox of protein aggregation, i.e. its potential toxic/protective effect in neurodegeneration. The relations between some of the dysfunctional components of the pathway and neurodegeneration are presented. We highlight possible ubiquitin/proteasome pathway-targeting therapeutic approaches, such as activating the proteasome, enhancing ubiquitination and promoting SUMOylation that might be important to slow/treat the progression of neurodegeneration. Finally, a model time line is presented for neurodegeneration starting at the initial injurious events up to protein aggregation and cell death, with potential time points for therapeutic intervention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intracellular proteolysis is a very powerful mechanism that shapes the proteome following exposure to different stress conditions. The removal of misfolded or damaged proteins is critical to recovery from adverse conditions to ensure optimal cell survival. In the cytosol and nucleus, the major proteolytic pathway used by eukaryotic cells for disposing of misfolded or damaged proteins is the ubiquitin/proteasome pathway (UPP) [1]. Following re-translocation into the cytosol, misfolded endoplasmic reticulum (ER) proteins are also degraded by the UPP. In mammalian cells it is estimated that 80–90% of protein degradation is carried out by the UPP [2]. In addition, ubiquitin is likely to be used for targeting most substrates for degradation [3], since it is one of the most abundant proteins in cells present at 108 copies per cell (~5% of cell bulk protein) [4]. Mitochondria and endolysosomes have their own proteolytic systems, but they account for a small percentage (10–20%) of overall protein degradation [5].
Non-functional intracellular proteins must be disposed of quickly by the UPP to prevent cell damage caused by abnormal protein aggregation and/or inappropriate association with other proteins. In HeLa cells it was estimated that proteasomes are quite abundant constituting ~0.6% of the cell bulk protein and have a half life of approximately 5 days [6]. Proteasomes in HeLa cells degrade defective ribosomal products (DriPs) at a rate of 1 × 106/min, and proteins that make it to the cellular pool at a slower rate, i.e. 6 × 105/min [3]. Similar numbers were obtained for L929 cells [7], but proteasomal degradation rates for non-mitotic cells, such as neurons, remain to be established. It is clear that regardless of cell type, proteins defective in folding and/or intracellular location or that are not able to find a suitable partner must be turned over by the UPP quickly enough to prevent cellular demise. The UPP is thus an extremely dynamic pathway that plays a critical role in the intracellular quality control process to prevent cell death caused by proteotoxicity.
Besides removing abnormal and toxic proteins generated by a lifetime of environmental damage, the UPP controls many key cellular mechanisms critical for cell viability and function, such as cell cycle and division, signal transduction, development and transcription factor regulation [1]. In general, high levels of ubiquitinated proteins do not accumulate in healthy cells as they are rapidly degraded by the UPP. The function of the UPP can be impaired by many factors including the aging process, leading to the formation of ubiquitin protein aggregates detected in non-pathologic aging as well as in many neurodegenerative disorders, such as Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) diseases as well as amyotrophic lateral sclerosis (ALS) to name a few (reviewed in [8]). The inability to eliminate ubiquitinated proteins can result not only from a functional failure of the UPP but also from structural changes in the protein substrates which render them inaccessible to the degradation component. The UPP plays a critical role in cellular processes such as oxidative stress, inflammation and apoptosis, all of which are implicated in abnormal protein deposition and cell death in neurodegeneration.
Ubiquitin/proteasome pathway
There is a rising interest in the UPP as a pharmacological target to prevent/treat neurodegeneration. We summarize below the most important aspects of this pathway. Readers interested in more details are referred to several excellent recent reviews on the pathway in general [9] and in the nervous system in particular [10].
Ubiquitin and related enzymes
Ubiquitin is a small protein of 76 amino acids, which can form polyubiquitin chains at seven lysine residues: K6, K11, K27, K29, K33, K48 and K63. These chains are formed by the successive attachment of monomers by an isopeptide bond, most frequently formed between the side chain of Lys48 in one ubiquitin and the carboxyl group of the C-terminal Gly76 of a neighboring ubiquitin. Attachment of K48 polyubiquitin chains to lysine residues on a protein results in at least a 10-fold increase in its degradation rate [11]. Polyubiquitin chains with linkages involving lysine residues on ubiquitin other than K48 were found to play distinct roles, including DNA repair, activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), polysome stability and endocytosis (reviewed in [12]).
Ubiquitination of proteins is a complex process involving the following sequence of events: initially, a high energy thioester bond is formed between ubiquitin and the ubiquitin-activating enzyme E1, an ATP-dependent reaction. Secondly, a thioester bond is formed between the activated ubiquitin and ubiquitin-conjugating enzymes (E2). Thirdly, the carboxyl terminal of ubiquitin is covalently attached to the ε-amino group of a lysine residue on protein substrates via an isopeptide bond. This reaction is carried-out by ubiquitin ligases (E3) that confer substrate specificity to the UPP (review in [13]). Finally, another family of ubiquitination factors (E4) assembles the multiubiquitin chain generating longer ubiquitin-chains [14]. In some cases, E2, instead of E3 ubiquitin ligases, transfer directly ubiquitin to the substrate.
Removal of ubiquitin from ubiquitinated proteins and disassembly of polyubiquitin chains is mediated by de-ubiquitinating enzymes. There are more than 90 genes encoding de-ubiquitinating enzymes making them one of the largest family of enzymes of the ubiquitin pathway (reviewed in [15]). De-ubiquitinating enzymes are divided into two major classes: the first class includes the ubiquitin carboxyl-terminal hydrolases that remove small amides, esters, peptides and small proteins at the carboxyl terminus of ubiquitin. The second class comprises ubiquitin-specific processing proteases which disassemble the polyubiquitin chains and edit the ubiquitination state of proteins.
The state of substrate ubiquitination depends on the balance between ubiquitinating and de-ubiquitinating enzymes acting on it. Thus, cells developed a highly dynamic strategy based on a switch-on/switch-off type of mechanism that responds promptly to cellular requirements for proteolysis by the UPP.
26S proteasome and substrate recognition
Covalent binding of polyubiquitin chains to proteins targets them for degradation by the 26S proteasome, a large enzymatic complex with a native molecular mass of approximately 2,000 kDa (reviewed in [16]). The 26S proteasome contains ATPase and proteolytic components. The ATPase component, also known as the 19S particle, contains at least 19 subunits and can be further divided into two subcomplexes: the lid and the base (reviewed in [17]). The lid harbors receptors for polyubiquitin chains as well as de-ubiquitinating activity. The hexameric ATPases Associated with diverse cellular Activities (AAA+ ATPase) base unfolds the substrates and translocates the polypeptides into the proteolytic chamber. Unfolding and threading the substrate through the catalytic barrel are rate limiting steps in proteasomal degradation.
The proteolytic component of the 26S proteasome, known as the 20S proteasome, is a cylinder-like structure only long enough to accommodate ~50 residues of a fully extended polypeptide, although a 70 kDa globular protein can fit into the chamber [18]. The eukaryotic 20S proteasome consists of 28 subunits arranged in four heptameric stacked rings as α7β7β7α7 and forming a barrel-shaped structure (reviewed in [19]). The seven α-type subunits provide binding sites for regulatory particles and form the gated channel leading to the inner proteolytic chamber. Among the seven β-type constitutive subunits, only three of them bear active sites for peptide bond hydrolysis, namely β1, β2, and β5, which are responsible for the caspase-like, trypsin-like, and chymotrypsin-like activities, respectively [20].
During degradation of one substrate molecule by the 26S proteasome 300–400 ATP molecules are hydrolyzed. ATP hydrolysis is activated upon substrate binding and promotes three events: substrate unfolding, 20S proteasome gate opening, and protein translocation [21]. Substrate binding to the 19S particle stabilizes gate opening and facilitates substrate channeling and its access to active sites in the 20S proteasome [22]. ATP is also required for rapid association/dissociation of the 26S proteasome from/into the 19S particle and 20S proteasome [23]. While the 26S proteasome degrades polyubiquitinated proteins, the 20S proteasome may be sufficient to degrade oxidatively modified non-ubiquitinated proteins in an ATP-independent manner [24, 25].
Degradation of ubiquitinated proteins by the 26S proteasome is enhanced when more than one ubiquitin is attached to the target protein. The minimal signal for efficient degradation is a tetraubiquitin chain [26]. Removal of two ubiquitins from a tetraubiquitinated substrate by de-ubiquitinating enzymes can decrease substrate/26S proteasome affinity by approximately 100-fold, allowing the substrate to escape degradation. Longer chains do not increase substrate/26S proteasome affinity, but optimize their binding time. The interaction of the polyubiquitin chain with the 26S proteasome involves hydrophobic patches on the surface of the tetraubiquitin chain, generated by Leu8, Ile44, and Val70 in each ubiquitin moiety, and two hydrophobic sequences with the motif LeuAlaLeuAlaLeu in subunit Rpn10 of the 19S particle [27]. Rpn13 is another subunit of the 19S particle identified as a receptor for polyubiquitinated proteins [28]. Rpn13 is also a receptor for the deubiquitinating enzyme Uch37, suggesting that chain recognition and disassembly are coupled at the proteasome [28].
Three key factors target proteins for ubiquitination/degradation: (1) misfolding due to mutation or damaging events; (2) constitutively active ubiquitination signals; and (3) post-translational modifications such as phosphorylation/dephosphorylation events or co-factor binding. The unfolding of normal substrates precedes their degradation. This step is required to allow entry into the proteolytic chamber of the 20S proteasome through its narrow opening [26]. Unfolding activities are provided by ATPase subunits in the base of the 19S particle or by extraproteasomal chaperones. In fact, proteasomes can be recruited to ubiquitinated substrates in conjunction with molecular chaperones [29].
Delivery of substrates to the proteasome
One of the most extraordinary features of mammalian cells is how crowded they are, raising the question of how proteasomes encounter their substrates. In the cytoplasm, proteasomes can be found free, associated with cytoskeletal elements or bound to the endoplasmic reticulum (reviewed in [30]). Proteasomes are slowly and unidirectionally transported from the cytoplasm into the nucleus but can rapidly diffuse within each compartment (cytoplasm or nucleus) without encountering selective barriers [31]. Thus it was proposed that proteasomes perform quality control by continuously colliding with their substrates, degrading those that are specifically tagged or misfolded [31].
Polyubiquitinated proteins can be directly recognized by the Rpn10/Rpn13 subunits of the 19S particle. However, multiple shuttling factors other than proteasome subunits were shown to bind polyubiquitinated proteins and deliver them to proteasomes for degradation (reviewed in [32, 33]). Most of these shuttling receptors contain two important domains: (1) at the N-terminus a ubiquitin-like domain, such as UBL or UBX, that binds to proteasomes, and (2) at the C-terminus a ubiquitin-binding domain, such as ubiquitin-interacting motifs (UIM) or ubiquitin-associated domain (UBA), that binds polyubiquitinated proteins. The sequestosome 1/p62 (p62/sqstm1), Rad23 and DSK2 are examples of such shuttling factors. These UBL (or UBX)-UBA shuttling factors can alternatively deliver substrates to Cdc48 complexes for unfolding. Cdc48 complexes comprise six identical subunits with two AAA+ ATPase domains, where substrates can undergo unfolding before being shuttled to the proteasome for degradation. Other proteins, such as Parkin, VHL and CHIP, are ubiquitin ligases that have their own UBL domain for proteasome binding, or associate with partners that have one. This dual function (ubiquitination and/or proteasomal binding) within the same molecule or complex provides a means to respond promptly to specific cellular proteolytic requirements, by ubiquitinating and quickly delivering substrates to proteasomes. These multiple direct and indirect proteasome delivery mechanisms provide for another level of selectivity needed for regulated protein degradation by the UPP.
Paradox of protein aggregation: toxicity or protection
All major human chronic neurodegenerative diseases, such as PD, AD, HD and ALS, are characterized by ubiquitinated proteins that accumulate in abnormal intraneuronal inclusions in the respective affected areas of the CNS (reviewed in [34]). The mechanisms leading to the formation of such abnormal aggregates remain elusive. Surprisingly, the degradation rate of mature post-synthetically damaged proteins is not significantly affected by insults such as oxidative stress or increased temperature [35]. However, degradation of newly synthesized proteins is highly affected by stress. Newly synthesized proteins go through a “fragile period” during which they are highly sensitive to degradation by the UPP. Its impairment induces accumulation of ubiquitinated proteins that develop into protein aggregates reminiscent of those detected in many neurodegenerative diseases. The role of these abnormal protein deposits in the progression of neurodegeneration is controversial. Inclusions may arise from a cellular attempt to compartmentalize accumulated proteins preventing their interference with normal cell function. Their presence may also confer cytotoxic effects that can contribute to neuronal cell damage. We will discuss some aspects of this controversy below.
Protein overexpression concern
Many studies addressing the effects and mechanisms involved in protein aggregation in neurodegeneration do not focus on the endogenous proteome. Instead, they rely on high and unregulated overexpression of different kinds of proteins in cells as well as in transgenic animals, an approach that may lead to artifacts because it bypasses the internal mechanisms regulating the UPP. For example, expression of green fluorescent protein (GFP) or a GFP fused to ubiquitin caused by itself the formation of ubiquitin positive aggregates in transfected HeLa and RAW cells [36]. The transfection reagent (FuGene 6) alone was sufficient to induce aggregate formation, but to a lesser extent. Furthermore, GFP-like proteins transfected into mammalian cells show a tendency to oligomerize and aggregate into punctuate structures [37]. These structures are not cytosolic aggregates, instead they are lysosomes containing the aggregated proteins. Hence, formation of ubiquitin positive-aggregates caused by overexpressing non-resident proteins or by the transfection reagents themselves may be misleading.
In HEK 293 cells, protein aggregates generated by the transient overexpression of huntingtin or cystic fibrosis transmembrane conductance regulator (CFTR) aggregation-prone fragments were shown to directly inhibit the UPP [38]. It is possible that the UPP would be inhibited by the overexpressed proteins even if they did not aggregate. Furthermore, it is not clear how cells handle the expression of high levels of proteins that in many instances do not replicate any of the cell’s own proteome. For example, huntingtin is a very large protein. Its molecular weight is ~350 kDa. It is possible that expressing GFP (~30 kDa), which is less than one tenth of the size of huntingtin, with an expanded polyQ will not behave in the same manner as full length huntingtin. These experimental approaches may not replicate the actual mechanisms involved in highly specific recognition, ubiquitination, deubiquitination and delivery steps that work in concert and regulate the turnover of intracellular proteins by the UPP. It is possible that they do not entirely reflect the natural intracellular responses.
Accumulation predicts degree of cell death independently of aggregation
Studies addressing the role of aggregate formation on cell death rely, in many instances, on transfection and overexpression of wild type or mutant forms of proteins, such as tau [39], α-synuclein [40], parkin [41] or huntingtin [42], to list a few. In some cases the whole protein or its truncated fragments, as in the case of huntingtin, are fused to GFP. In addition the transfected cells, in many instances, are treated with proteasome inhibitors [40] or maintained in serum-free media which induces autophagy and/or apoptosis [42]. Some of these studies support the notion that aggregates are beneficial to the cell [43] and act as “neuron protection agencies” [44].
The above studies involve highly overexpressed proteins that in some cases, do not mimic any intracellular protein, thus data interpretation should take these circumstances into consideration. In addition, it is likely that neurons that exhibit protein aggregates are committed to the death pathway rather than to survival. Many of the dead neurons are cleared by microglia and those that remain with aggregates are probably waiting their turn. This view is supported by the progressive nature of these neurodegenerative disorders, suggesting that groups of neurons gradually die and are cleared. Furthermore, only a few neurons with protein aggregates are detected in brains of control individuals that do not have the disease. It is thus likely that neurons with aggregates reach a point of no return, die and are cleared by microglia.
Several studies demonstrate that aggregate formation does not correlate with neuronal survival. The amount of diffuse, non-aggregated, huntingtin-GFP fusion products accumulating in primary rat neuronal cultures maintained in serum free media was directly related to the degree of cell death [42]. In rat embryonic striatal cells, preventing the formation of intranuclear inclusions containing mutant huntingtin resulted in accelerated cell death [45]. Formation of intranuclear inclusions in the latter study was halted by inhibiting ubiquitination through overexpression of a dominant negative mutant of a ubiquitin conjugating enzyme (hCdc34). This approach most likely caused deregulation of intracellular protein degradation resulting in an abnormal accumulation of intracellular proteins that could not be degraded because they were not ubiquitinated. This phenomenon could lead to proteotoxicity and thus accelerate cell death even in the absence of protein aggregates. Preventing protein aggregation by using RNA interference (RNAi) to silence the expression of p62/sqstm1, a UBA-shuttling protein, failed to protect human neuroblastoma cells from cell death induced by a product of inflammation, prostaglandin J2 (PGJ2) [46]. P62/sqstm1 knockdown prevented protein aggregation without diminishing the accumulation of endogenous ubiquitinated proteins. Furthermore, the accumulation of ubiquitinated proteins was an early response, suggesting that if cells fail to degrade the high levels of ubiquitinated proteins induced by PGJ2-treatment, apoptosis follows triggering caspase activation and tau cleavage, known to precede tau aggregation and tau pathology [47].
Together these studies support the notion that intracellular accumulation of proteins, rather than their aggregation, is a pathological event that accelerates cell death. Therapeutic strategies that promote the degradation of the accumulated proteins rather than preventing their aggregation would be relevant to neurodegenerative disorders associated with protein misfolding.
Cytoskeletal collapse: an alternative to aggresomes
Pericentriolar structures within centrosomes are deposition sites for ubiquitinated proteins that escape UPP degradation and were named accordingly “aggresomes” [29]. The ubiquitinated proteins deposited in aggresomes resulted from overexpressing mutant CFTR or presenilin 1 or from impairing protein degradation by treating cells with proteasome inhibitors [29, 48, 49]. Centrosomes are associated with high levels of 26S proteasomes and with de-ubiquitinating activity [50]. While some studies suggest that the retrograde transport of ubiquitin protein aggregates to centrosomes is dependent on microtubule integrity [29] others indicate that this process does not require intact microtubules [50].
There is another possible interpretation for the deposition of aggregates at the centrosome, also known as the microtubule organizing center (MTOC). Formation of ubiquitin-protein aggregates throughout the cell could disrupt the microtubule network causing microtubules to collapse to their site of origin, i.e. the MTOC. This collapse would cause the re-distribution onto the MTOC of aggregates, proteins and complexes, such as proteasomes, that are associated with or neighbor the cytoskeletal elements. Such events were shown to be induced by the product of inflammation PGJ2 which disrupts the structural integrity of microtubules and actin filaments [51]. A similar model for the localization of protein aggregates at the MTOC could be applied to toxins such as 1-methyl-4-phenylpyridinium (MPP+) and rotenone that strongly hinder microtubule polymerization and induce PD symptoms in animal models [52–54]. These results suggest that preserving microtubule integrity is critical to neuronal homeostasis. Disruption of the microtubule network causes defects in axonal transport, a mechanism common to many neurodegenerative disorders (reviewed in [55]).
Reversible aggregates
Reversible intracellular protein aggregates form in maturing dendritic cells in response to protein damaging agents and are known as dendritic cell aggresome-like induced structures (DALIS) [56]. Upon stimulation with the pro-inflammatory agent lipopolysaccharide followed by treatment with the translation damaging agent puromycin, DRiPs are sorted into DALIS which are large reversible cytosolic aggregates. Unlike aggresomes, DALIS are not localized in the pericentriolar area and lack vimentin cages [56]. In addition, DALIS contain many components of the ubiquitination machinery, including E1, E2s and E3s [57]. When DRiPs are formed they are rapidly sequestered into DALIS where they are eventually ubiquitinated. This mechanism allows dendritic cells to regulate the degradation rate of DRiPs, an ability that is important for their immune functions [56].
DALIS-like structures are also present in non-dendritic cells and form in response to different types of stresses such as heat shock, the catalase inhibitor 3-amino-1,2,4-triazole (ATZ; causes oxidative stress) and sodium arsenate (uncouples oxidative phosphorylation) [36]. These reversible aggregates do not co-localize with proteasomes or γ-tubulin, are inhibited by blocking protein synthesis with cycloheximide, and form in a microtubule- and actin-independent manner confirming that they are not aggresomes.
It is possible that the formation of these two types of reversible aggregates is mediated by intracellular proteins that contain UBA domains (reviewed in [58, 59]). These proteins, such as Mud1, Rad23 and p62/sqstm1, non-covalently bind polyubiquitin chains 300-times more tightly than mono-ubiquitin [60, 61]. Due to their high affinity for polyubiquitin chains, these UBA-containing proteins could serve as receptors for binding and storing ubiquitinated proteins [62]. Reversibly “storing” excessive amounts of polyubiquitinated proteins generated after a stress event could provide an efficient mechanism to regulate substrate access to the proteasome. This “storage” mechanism could prevent proteasomal shutdown by excessive substrate levels. The shuttling of polyubiquitinated proteins from these “storing stations” to the proteasome could be under tight regulation by mechanisms such as phosphorylation and/or ubiquitination (mono and poly). These dynamic aggregates could serve as a microenvironment for the recruitment of multimeric signaling complexes. Notably, protein aggregates were shown to be dynamic structures [63]. Their composition, including transient association with chaperones, changes under a range of environmental and physiological stresses. These reversible intracellular storage aggregates could thus prevent cell damage and promote cell survival. However, aggregate size could determine their toxicity [64] as their expansion may interfere with cell function and thus confer fatal effects [65].
Cytoplasmic versus nuclear aggregates
Not all intracellular aggregates with ubiquitinated proteins are cytoplasmic like in PD. Some accumulate in the nucleus, such as in HD. The cause of this differential subcellular aggregate distribution is not clear.
An investigation of the nuclear diffusion limit in mammalian cells, including primary neurons and neuroblastoma cells, established that large molecules (molecular masses above 70 kDa) cannot freely diffuse into nuclei of intact, healthy cells [66]. It is unlikely that high molecular mass ubiquitin conjugates, most with molecular masses above 75 kDa as judged by Western blot analysis, passively diffuse from the cytoplasm into the nucleus and vice versa. In the cell, the size of the aggregates may be even larger as they may be bound to proteins/complexes with UBA domains. The large aggregates could only passively enter the nucleus if the nuclear membrane was disrupted. Accordingly, nuclear migration of full-length mutant huntingtin can only occur upon deterioration of the nuclear membrane [66]. Wild type huntingtin is a cytoplasmic protein with a molecular mass of ~350 kDa. Neither wild type nor mutant huntingtin have a nuclear targeting signal and thus cannot be actively transported across the nuclear membrane [66]. On the other hand, the subcellular distribution of transfected GFP (~30 kDa) fused to full length or truncated forms of wild type or mutant huntingtin is not homogeneous. These fusion proteins can accumulate in the cytoplasm, nucleus or both, depending on the size of the fusion protein, on the nuclear diffusion limit of the specific transfected cells and on their nuclear membrane integrity [66]. We conclude that aggregate size, nuclear diffusion limit and nuclear membrane integrity are some of the factors dictating the subcellular distribution of protein aggregates.
Host-to-graft aggregate transmission: “prion-like” properties or toxic microenvironment?
A new premise has emerged for a “prion-like” neuropathological propagation based on recent data involving host-to-graft studies in PD [67–69] and HD [70] patients receiving neural transplants. In the PD patients α-synuclein-positive Lewy-like inclusions were detected in long term mesencephalon transplants. In the HD cases the grafts suffered disease-like neuronal degeneration. These studies raise concerns that genetically and immunologically unrelated grafts are susceptible to the disease processes as well. To explain this host-to-graft transmission phenomenon, neuronal cells were incubated with media containing polyQ amyloids [71], tau aggregates [72] or α-synuclein oligomers [73]. Entry of these proteins/peptides into cells most likely through endocytosis, was confirmed by immunofluorescence and/or western blot analyses. The data indicate that protein aggregates can propagate from the outside to the inside of a cell, a phenomenon potentially relevant to many neurodegenerative diseases associated with protein misfolding.
Another possible explanation for the host-to-graft transmission is the potentially toxic microenvironment into which the grafts are transplanted into. It is well established that neuroinflammation is a common denominator among diverse neurodegenerative diseases, such as AD, PD, HD and ALS [74–76] and that chronic neuroinflammation is a critical mechanism responsible for the progressive nature of neurodegeneration. Microglia, the resident innate immune cells in the CNS, produce many factors, such as interleukin-1, tumor necrosis factor α, nitric oxide, PGJ2, superoxide, which are toxic to neurons. The chronic and unregulated activation of microglia in the affected CNS areas into which the grafts are transplanted into, could be responsible for propagating the neuronal injury to the grafts.
Regardless of which mechanisms are responsible for the host-to-graft transmission, this phenomenon raises uncertainty about this potential therapeutical approach [73]. It is important to consider different underlying mechanisms and to explore additional therapeutic paradigms for these devastating disorders.
UPP dysfunction in neurodegenerative disorders
Most neurodegenerative disorders are associated with formation of protein aggregates, resulting ultimately in proteinaceous inclusions, such as neurofibrillary tangles in AD and Lewy bodies in PD [77, 78]. While the composition of these inclusion bodies varies with the disorder, a general feature is that these aggregates contain ubiquitinated proteins. These inclusions also contain components of the UPP, such as the ubiquitin carboxyl-terminal hydrolase-L1 (UCH-L1) and proteasome subunits. Proteins unrelated to the UPP are also found in these inclusions. For example, α-synuclein is a major component of PD Lewy bodies and the microtubule-associated tau protein is a major component of AD neurofibrillary tangles. Although selective sets of neurons are affected in these neurodegenerative disorders, they share the inability to degrade ubiquitinated proteins and thus UPP impairment is likely to contribute to the observed neuronal death. Support for this view comes from studies demonstrating that proteasome function is impaired in the affected brain areas of patients with AD [79] and PD [80] and declines with age [81] and oxidative stress [82]. Furthermore, mutations in ubiquitin were linked to AD [83] and in the ubiquitin ligase parkin were linked to familial PD [84] and to autosomal recessive juvenile parkinsonism [85]. A missense mutation (Ile93Met) in the uch-l1 gene was identified in a German family with PD [86]. UCH-L1 is very abundant in the brain and the Ile93Met mutation decreases its catalytic activity [86]. Collectively, these findings indicate that the UPP may be deficient in these disorders [87]. We discuss below in more detail some of the changes in UPP components directly associated with these chronic neurodegenerative disorders characterized by abnormal protein deposition.
Proteasome dysfunction
The most consistent risk factor for developing a neurodegenerative disorder, especially AD or PD, is increasing age [88]. One of the most accepted theories of aging is the loss of quality control in protein turnover with the concomitant build-up of oxidatively modified proteins (reviewed in [89]). Since proteasomes selectively degrade oxidatively damaged as well as ubiquitinated proteins it is postulated that proteasome activity declines with aging. A loss of proteasome activity with age is supported by decreased subunit expression, alterations and/or replacement of proteasome subunits and formation of inhibitory cross-linked proteins (reviewed in [81, 90]). Food restriction, which is one of the experimental paradigms that halts the aging process, prevents the age-dependent changes in proteasome function and structure in mice and rats, further supporting the notion that the proteasome plays a role in the aging process (reviewed in [91]). Several mechanisms explain the observed UPP changes with aging (reviewed in [92]). We recently identified a unique aging-dependent mechanism that contributes to proteasome dysfunction in Drosophila melanogaster [93]. We observed that the major proteasome form in old flies is the weakly active 20S proteasome, while in younger flies highly active 26S proteasomes are preponderant. Old flies also exhibited a decline in ATP levels, which is relevant to 26S proteasomes, as their assembly is ATP-dependent. The perturbation in proteasome activity in “old-age” flies most likely deprives them of the ability to effectively cope with proteotoxic damages caused by environmental and/or genetic factors.
UBB+1
There are at least three genes (A, B and C) encoding human ubiquitin and two of them the polyubiquitin B and C genes, contain heat-shock promoters [94]. A mutant form of the polyubiquitin B gene, known as UBB+1, was originally detected in brains of AD patients and not in age matched controls [83]. This aberrant form of ubiquitin was also detected in patients with Down syndrome, progressive supranuclear palsy, Pick’s disease, frontotemporal dementia, argyrophilic grain disease and HD but not in PD (reviewed in [95]).
UBB+1 is the product of a frameshift generated by a dinucleotide deletion in the polyubiquitin B mRNA resulting in a 19-amino acid extension at the C-terminus and the absence of a C-terminal Gly76, which is present in wild-type ubiquitin [66]. Without Gly76 at the C-terminus, UBB+1 fails to conjugate with other ubiquitin molecules and is deprived of the capacity to tag protein substrates. However, due to its unaffected Lys residues, UBB+1 by itself can form polyubiquitin chains and is recognized by substrate receptors on the proteasome. UBB+1-capped polyubiquitin chains are refractory to disassembly by de-ubiquitinating enzymes and potently inhibit proteasome degradation of a polyubiquitinated substrate in vitro [96] and in neuronal cells [97].
The toxic effects of UBB+1 expression were demonstrated in several model systems. In yeast, expression of a protein analogous to UBB+1 significantly enhanced cellular susceptibility to toxic protein aggregates [98]. In rodent primary neuronal cultures, transfection of UBB+1 impaired mitochondrial trafficking along neurites leading to activation of both the mitochondrial stress and p53-dependent cell death pathways [99]. Post-natal neuronal expression of UBB+1 in transgenic mice reduced proteasome activity and increased ubiquitinated protein levels in the cerebral cortex, and caused a deficit in contextual memory in both water maze and fear conditioning paradigms [100]. The inhibitory activity of UBB+1 may thus be an important determinant of neurotoxicity and contribute to an environment that favors the accumulation of misfolded proteins. In rodent primary neuronal cultures, suppressing UBB+1 expression through siRNA diminished the damage caused by UBB+1, suggesting a therapeutic approach for neurodegeneration associated with this aberrant form of ubiquitin [99].
Parkin
Mutations in the parkin gene (PARK2) have emerged as a major factor in familial PD [101]. The human parkin protein has a DNA binding domain, at least 15 suspected phosphorylation sites, and has homology to ubiquitin. Mutations in the exon regions of parkin are associated with an autosomal recessive juvenile parkinsonism [85] and with early-onset autosomal recessive familial PD [101].
The human parkin gene contains 12 exons and encodes a 465 amino acid protein that is abundant in the brain and belongs to a group of heterogeneous proteins collectively known as “ubiquitin-domain proteins or UDPs”. They bear a ubiquitin-like domain, usually at their N-terminus, but are mostly unrelated to each other in the remainder of their sequences. UDPs play an important role as adapter proteins, mediating the assembly of heteromeric complexes through their ubiquitin-like domains. Parkin binds the Rpn10 subunit of 26S proteasome through its UBL domain [102, 103].
Parkin also contains two RING-finger and one in between RING finger (IBR) domains at its C-terminus. RING-finger containing proteins are ubiquitin ligases and recruit E2 ubiquitin-conjugating enzymes [103, 104]. Parkin is an E3 ubiquitin ligase that recruits the ubiquitin-conjugating enzymes UbcH7 and Ubc8 via the RING fingers [105, 106]. The ubiquitin ligase activity of parkin facilitates the assembly of both K48- and K63-linked polyubiquitin chains [107]. The latter chains target misfolded proteins to dynein motor complexes and promote the sequestration of misfolded proteins to aggresomes with subsequent autophagic degradation [108].
In addition to mutations in the parkin gene, post-translational modifications are also involved in the inactivation of parkin in PD. Parkin was found to be S-nitrosylated in PD brains [109]. In vitro studies demonstrated that S-nitrosylation of parkin by nitric oxide reduces its E3 ligase activity and impairs its protective function [109]. Parkin has multiple consensus sequences for casein kinase I and II, protein kinase C (PKC), protein kinase A (PKA), and tyrosine kinase [84]. Phosphorylation of parkin by casein kinase I and cyclin-dependent kinase 5 (Cdk5) decreases parkin solubility resulting in its aggregation and inactivation [110]. It is likely that phosphorylation/dephosphorylation events regulate parkin activity or its targeting to cellular compartments other than the cytosol or the Golgi, where it has been frequently detected [111].
Many studies report an interaction between parkin and PTEN-induced kinase 1 (PINK1, also known as PARK6). Mutations in both genes are implicated in autosomal recessive PD. PINK1 is a serine/threonine kinase with a mitochondrial targeting sequence. Phosphorylation of parkin by PINK1 increases the ligase activity of parkin, and enhances parkin-mediated ubiquitin signaling through the NF-κB pathway [112]. While deletion of PINK1 in Drosophila causes mitochondrial morphological defects and loss of dopaminergic neurons, the PINK1 mutants are rescued by overexpressing parkin [113, 114]. Parkin is essential for mitochondrial function and is selectively recruited to impaired mitochondria to promote their engulfment by autophagosomes [115]. The ubiquitin ligase protects mitochondrial DNA from oxidative damage and stimulates its repair [116]. Moreover, parkin regulates cytochrome c release and apoptosis, which may be relevant to the selective vulnerability of certain neuronal populations in PD [117].
The neuroprotective role of parkin is supported by different kinds of evidence. For example, parkin promotes ubiquitination and proteasomal degradation of intracellular Aβ1–42 an effect that could prevent the formation of amyloid deposits in AD [118]. The E3 ligase was also shown to negatively regulate excitatory glutamatergic synapses thus reducing the excitotoxic vulnerability of dopaminergic neurons [119]. The type of RING finger architecture found in parkin, RING-IBR-RING, is also present in other proteins such as the mouse RBCK1, shown to participate in gene expression [120]. Notably, parkin is a p53 transcriptional repressor that interacts with the p53 promoter and reduces p53 expression and activity [121]. On the other hand, parkin depletion enhances p53 levels and activity, a phenomenon observed in autosomal recessive juvenile PD [121].
In conclusion, parkin is a multifunctional protein that contributes to several vital cellular functions, including ubiquitination, gene expression, and molecular scaffolding through protein-protein interactions. One or more of these interactions is important for neuronal survival.
UCH-L1 (ubiquitin carboxyl-terminal hydrolase-L1)
The gene uch-l1 (PARK5) encodes for PGP9.5, a de-ubiquitinating enzyme. UCH-L1 is one of the most abundant proteins in the CNS accounting for 1–2% of soluble neuronal protein [122]. This enzyme has at least four functions: removes ubiquitin from small or unfolded proteins, disassembles polyubiquitin chains for ubiquitin recycling, stabilizes monoubiquitin and was shown to exhibit ubiquitin ligase activity [123, 124]. Besides polyubiquitin chains, the UCH-L1 substrates remain unidentified.
An in-frame deletion including exons 7 and 8 of the uch-l1 gene was described as the cause of gracile axonal dystrophy (gad) in mice, which is a condition characterized by “dying back” type axonal degeneration [125]. This mutation results in a truncated protein lacking 42 amino acids including a possible histidine at the active site of the enzyme [125]. This genetic model is characterized by a retrograde accumulation of amyloid β-protein and ubiquitin-conjugates in sensory and motor neurons, as seen in certain inherited human neurodegenerative diseases [125, 126]. The gad mouse was the first mammalian model of a hereditary neurodegenerative disorder that resulted from a mutation in a component of the UPP.
The uch-l1 gene is also a susceptibility gene in PD. The two most studied mutations in uch-l1 in association with PD are I93M and S18Y. I93M was originally identified in two German siblings with PD [86]. This autosomal dominant mutation leads to a reduction in the in vitro hydrolytic activity of UCH-L1 [123]. An inverse association between the S18Y variant of the uch-l1 gene and the risk of developing PD was found in Asian and Caucasian samples in a recent Meta-analysis [127]. However, this protection mechanism is not well understood and was suggested to be due to its reduced ligase activity [123]. An antioxidant function of the S18Y variant in neurons was also suggested to mediate its protective effect [128].
In addition to mutations, UCH-L1 is a major contributor to idiopathic AD and PD [129]. UCH-L1 is required for normal synaptic and cognitive function, but its activity is down-regulated in APP/PS1 mice that overexpress Aβ [130]. Restoration of UCH-L1 levels rescues deficits in synaptic transmission in Aβ-treated hippocampal slices and in slices from APP/PS1 mice [130]. UCH-L1 is a major target for oxidative damage. Both the familial PD-associated UCH-L1 I93M mutant and the sporadic PD-associated carbonyl-modified UCH-L1, induce an increase in α-synuclein levels. Both forms of UCH-L1 enhance an aberrant interaction with LAMP-2A (lysosome associated membrane protein type 2A), Hsc70 and Hsp90, which are components of the chaperone-mediated autophagic pathway [131, 132]. Moreover, farnesylation of a membrane-associated form of UCH-L1 was shown to promote α-synuclein neurotoxicity [133].
UCH-L1 plays an important regulatory role in the pattern, activity and plasticity of synaptic connections. Initial data with Aplysia, later shown to be conserved in mammals [134], demonstrated that uch-l1 is one of the immediate early genes essential for long-term facilitation involved in long-term memory storage [135].
A recent study searched for alterations in gene expression in replaceable neurons in the high vocal center of male zebra finches and in granule neurons of mouse hippocampus and olfactory bulb, two well characterized replaceable neurons in mammals [136]. Notably, these studies established that uch-l1 was the most consistently underexpressed gene in the replaceable neurons as compared to non-replaceable neurons. Moreover, the levels of uch-l1 expression were increased by singing behavior in the male birds, a stimulus known to increase the survival of those specifically replaceable neurons [136].
Together, these studies highlight the notion that the neuronal-specific UCH-L1 plays an important role in neuronal fate: its reduced function jeopardizes survival of CNS neurons, while its up-regulation increases neuronal plasticity and survival. Based on these important findings we postulate that increasing UCH-L1 levels may prevent neurodegeneration.
Ataxin-3
Ataxin-3 is another de-ubiquitinating enzyme. This protein is a member of the Josephin family and contains an N-terminal catalytic Josephin domain, three C-terminal UIMs and an expandable polyQ stretch located between the second and the third UIM [137]. The normal polyQ stretch contains between 14 and 40 glutamines. Abnormal expansion to over 53 glutamines is pathological and causes Machado-Joseph disease (MJD), also known as spinocerebellar ataxia type 3 [138]. This neurodegenerative disorder is characterized by ubiquitinated intranuclear inclusions that also contain ataxin-3 [139].
Ataxin-3 is actively imported to and exported from the cell nucleus [140]. Heat shock and oxidative stress promote nuclear localization of this de-ubiquitinating enzyme [141]. Ataxin-3 interacts with the UBL domain of HHR23 proteins. The latter are human homologs of the yeast DNA repair protein Rad23 that shuttles substrates to the proteasome for degradation [142]. The ataxin-3 recruitment of HHR23A to intranuclear inclusions formed by mutant ataxin-3 could contribute to the neuropathological process.
The ubiquitin-specific cysteine protease ataxin-3 binds Lys63- and Lys48-linked polyubiquitin chains, but it preferentially cleaves Lys63-linkages [143]. The cellular functions of ataxin-3 in protein quality control are modulated through ubiquitination. On one hand, its hydrolytic activity is enhanced by ubiquitination [144], and on the other hand its ubiquitination by Gp78, which is an endoplasmic reticulum-associated E3 ligase, promotes its degradation [145].
PolyQ-expanded ataxin-3 is neurotoxic and induces neuronal apoptosis through the mitochondrial pathway [146]. Suppressing caspase cleavage of ataxin-3 by caspase inhibitors in cells, or by mutating the caspase recognition sites on ataxin-3 in a Drosophila model of MJD, slows down neurodegeneration [147]. These data suggest that targeting ataxin-3 cleavage by caspases might slow disease progression in MJD patients.
Sequestosome 1 (p62): overseeing substrate delivery to proteasomes or autophagosomes
Sequestosome 1, also known as p62 (p62/sqstm1), is a protein that is implicated in the UPP due to its ability to bind to proteasomes as well as to polyubiquitinated proteins. P62/sqstm1 has at its C-terminus a UBA domain that binds non-covalently to polyubiquitin chains. For example, this UBA domain interacts with K63-linked polyubiquitinated tau protein and delivers it to proteasomes for degradation [148]. Loss of p62/sqstm1 led to hyperphosphorylation of tau and accumulation of polyubiquitinated tau [149].
P62/sqstm1 contains additional structural motifs that promote its interaction with other proteins: a ZZ type zinc finger that binds the receptor interactive protein (RIP) involved in TNFα-induced apoptosis [150, 151], a binding site for the RING-finger protein tumor necrosis factor receptor-associated factor 6 (TRAF6) that is an E3 ubiquitin ligase [151, 152] and two PEST (proline, glutamic acid, serine, threonine) sequences (reviewed in [153]). An additional 22 amino acid structural motif known as LC3-interacting region (LIR), binds to LC3 that is an autophagosomal marker [154]. This LC3-interacting region is thought to target p62/sqstm1 to autophagosomes for lysosomal degradation.
At its N-terminus, p62/sqstm1 has a PB1 domain, which is a protein–protein interaction domain that binds the atypical PKC ζ [155]. The PB1 domain assumes a ubiquitin-like folding and can directly bind to proteasomes and other PB1-containing proteins including itself [156]. The PB1 domain also interacts with a mutant form of superoxide dismutase 1 (SOD1) and delivers it to autophagosomes for degradation [157].
Its ability to bind proteasomes as well as autophagasomes supports the notion that p62/sqstm1 selectively sequesters polyubiquitinated proteins and decides their proteolytic fate. P62/sqstm1 could target the polyubiquitinated proteins to autophagy and, ultimately, to degradation by the lysosome when proteasomes are impaired or overwhelmed [154, 158]. P62/sqstm1 may thus be a candidate for the missing link between the UPP and the autophagy-lysosome pathway.
P62/sqstm1 was detected in ubiquitin-containing intraneuronal and intraglial inclusions in a variety of neurodegenerative disorders [159–162]. For example, increased p62 immunoreactivity was detected early during neurofibrillary pathogenesis and was consistently present in neurofibrillary tangles but absent in neuropil threads and senile plaques in AD patients [163]. Furthermore, p62/sqstm1 expression in neuronal cells is induced by serum withdrawal conditions that trigger apoptosis and by proteasome inhibitors [159, 164] as well as by products of inflammation, such as PGJ2 [46] and by expression of expanded pathologic polyglutamine repeats [165]. These findings suggest that p62/sqstm1 is involved in the cellular defense mechanism triggered by the accumulation of misfolded and/or ubiquitinated proteins to enhance their degradation and/or aggregation.
The role played by p62/sqstm1 in the aggregation of ubiquitinated proteins was addressed by in vitro studies in which expression of this protein was inhibited by specific antisense oligonucleotides or by siRNA [46, 164, 166]. These studies established that abolishing p62/sqstm1 up-regulation prevents aggregation of ubiquitinated proteins thus indicating that p62/sqstm1 promotes protein aggregation. Notably, increased oxidative damage to the p62/sqstm1 promoter was found to correlate with a decline in protein level observed in many neurodegenerative disorders such as AD, PD and HD brains [167]. It was proposed that pharmacological means that increase p62/sqstm1 expression could be beneficial in delaying the onset of neurodegeneration. The reasoning is that p62/sqstm1 up-regulation would increase trafficking of polyubiquitinated proteins for proteasomal degradation and/or their sequestration into aggregates for autophagic removal. However, whether protein aggregation is beneficial or detrimental to cells is a highly controversial issue as discussed above.
Therapeutic approaches targeting the UPP
For the prevention/treatment of neurodegenerative disorders it is of therapeutic interest to find strategies to activate the UPP in order to avoid the accumulation/aggregation of ubiquitinated proteins. The challenge rests on developing strategies that will enhance the degradation of misfolded and aggregation-prone proteins without compromising the normal function of the UPP. We discuss below some potential therapeutic strategies that aim to activate proteasome activity, ubiquitination or SUMOylation to prevent neurodegeneration associated with protein misfolding, accumulation and aggregation.
Activate proteasomes
One of the most appealing targets for therapeutic intervention is the proteasome. In the case of cancer, the aim is to inhibit the proteasome. However, for neurodegenerative diseases the emphasis is to activate not inhibit the proteasome. Notably, ~36% of cancer patients treated with the proteasome inhibitor bortezomib develop peripheral neuropathy [168], implicating proteasome dysfunction in neurological impairment. We will discuss endogenous, genetic and pharmacological approaches to increasing proteasome activity.
Endogenous activators of the proteasome
There are three endogenous activators of the 20S proteasome: PA700, PA200 and PA28. The PA700 is also known as the 19S regulatory particle, a multimeric complex comprising 9 subunits in the lid and 10 subunits in the base. The 19S regulatory particle activates 20S proteasomes in a ubiquitin- and ATP-dependent manner. We discussed its properties above under “26S proteasome and substrate recognition”.
PA200 is a large nuclear protein that binds to 20S proteasomes as a 200 kDa monomer [169]. It activates proteasomal hydrolysis of peptides rather than proteins, by opening the axial gate of the α-rings [170]. PA200 forms hybrid proteasomes with the 19S regulatory particle and 20S proteasomes. Upon exposure of cells to ionizing radiation PA200-20S-19S hybrid proteasomes are recruited to chromatin with enhanced proteolytic activity to participate in DNA repair [171].
Like PA200, PA28 activates peptide hydrolysis by the 20S proteasome by associating with its α rings [18]. This activation is independent of ATP and ubiquitin, therefore it is not involved in degrading ubiquitinated proteins. PA28, also known as 11S regulatory particle (REG), can be either a heteroheptamer composed of PA28α and PA28β subunits primarily localized in the cytoplasm, or a homoheptamer composed of PA28γ predominantly in the nucleus [172]. PA28α/β is upregulated by interferon-γ and is required for the assembly of immunoproteasomes [173]. Upregulation of immunoproteasome was detected in injured [174], AD [175] and HD brains [176], suggesting that it plays a role in neuronal protection and damage repair. Notably, PA28γ overexpression recovered proteasome activity in skin fibroblasts from HD patients, and improved the viability of striatal neurons expressing mutant huntingtin [177].
The most successful therapeutical approach would be to target the PA700 (19S particle) in order to increase the degradation of polyubiquitinated proteins. Molecules known as ubistatins block proteasome-polyubiquitin chain interaction [178]. A search for molecules that have the opposite effect and enhance this interaction could stimulate substrate degradation and prevent protein aggregation [179].
Genetic activation of the proteasome
It is known from studies in yeast that the cellular abundance of proteasomes is controlled by the zinc finger transcription factor Rpn4 which allows the concerted induction of all proteasome subunits via interaction with PACE (proteasome associated control elements) sequences (reviewed in [180]). In humans a similar coordinated regulation of proteasome subunits must exist. Accordingly, stable overexpression of the β5 subunit in primary human fibroblasts resulted in elevated levels of other β subunits, and increased the levels of all three proteasome activities [181]. This genetic manipulation resulted in increased survival against oxidants and a delay in senescence. In a follow up study proteasomal up-regulation was achieved via overexpression of the proteasome maturation protein (POMP), the accessory factor for proteasome assembly in humans [182]. POMP overexpression in fibroblasts led to increased levels of assembled and functional proteasomes, and enhanced the capacity to effectively cope with various oxidative stressors.
An alternate therapeutic approach for maintenance of proteasome function would be to overexpress the arsenite-inducible RNA-associated protein (AIRAP) as it dramatically stabilizes proteasome activity in the absence of ATP [183]. In addition, AIRAP containing proteasomes show a higher rate of hydrolysis using model substrates. Together, these data strengthen the therapeutic prospect of genetic manipulation of the proteasomal system.
Natural and synthetic activators
The detergent SDS and some fatty acids such as oleic, linoleic and linolenic acids stimulate proteasome activity in vitro by favoring the open conformation of the proteasome [184, 185]. Some synthetic compounds such as peptidyl alcohols, esters, p-nitroaniliades and nitriles reversibly stimulate proteasome activity probably by binding to the same site as PA28 [186]. In addition, some proteasome-activating hydrophobic peptides bind as modifiers at noncatalytic sites, thus mimicking the effect of the PA28 complex by opening the gate of the α-rings [187]. Oleuropein, the most abundant phenolic compound in Olea europaea leaf extract, olive oil, and olives has a stimulatory impact on proteasome activity in vitro, probably acting through conformational changes of the gate of 20S α-rings [188]. Some natural antioxidants such as dithiolethione and sulforaphane, were also shown to enhance mammalian proteasome expression through the Keap1/Nrf2 (Kelch-like ECH-associated protein 1/nuclear factor-erythroid 2-related factor 2) signaling pathway, resulting in increased protection against various oxidants [189, 190]. Some synthetic triterpenoid derivatives, such as 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), its methyl ester (CDDO-Me) and imidazolide (CDDO-Im) derivatives, are Nrf2 activators [191] and could also be considered as proteasome activators. Betulinic acid, a lupene-type pentacyclic triterpene derived from many plants, such as birch trees, is a potent activator of the chymotrypsin-like activity of the proteasome [192]. Notably, C-3 modifications on betulinic acid change it from a proteasome activator into a proteasome inhibitor [192].
As a group these natural and synthetic proteasome activators might be important to slow down the progression of neurodegenerative diseases by enhancing proteasome activity and preventing the abnormal accumulation/aggregation of ubiquitinated proteins.
Concern
Proteasome inhibitors are currently used as anti-cancer drugs mostly because they induce apoptosis preferentially in transformed cells (reviewed in [193]). Based on the anti-carcinogenic effect of proteasome inhibition, one could argue that increasing proteasome function to prevent/treat neurodegenerative diseases could have the opposite effect and thus induce carcinogenesis [179]. However, studies with human senescent fibroblasts strongly oppose this view. Overexpressing the β5 subunit of the 20S proteasome [181] or its assembly chaperone POMP [182] in the fibroblasts enhanced proteasome activity without increasing cell proliferation. Interestingly, fibroblasts from healthy centenarians exhibited proteasome characteristics (expression levels and activity) similar to younger rather than elderly individuals [194]. These studies strengthen the prospect that increasing/maintaining proteasome function as we age is a promising therapeutical strategy to prevent neurodegeneration.
Enhance ubiquitination
“Chemical knockdown” of specific proteins by PROTACS
The development of PROteolysis TArgeting Chimera moleculeS (PROTACS) is a strategy with potential for selectively inducing ubiquitination and proteasomal degradation of specific substrates. Basically, PROTACS are heterobifunctional molecules comprising a ligand for the target protein, a linker moiety, and a ligand for an E3 ubiquitin ligase [195]. PROTACS function as a bridge between the target protein and an E3 ubiquitin ligase. Upon binding to a PROTAC, the E3 ubiquitin ligase promotes the synthesis/attachment of a polyubiquitin chain to the target protein, followed by its recognition and degradation by the 26S proteasome [196]. Potential advantages of PROTACS are (a) their selectivity based on a unique site on the target protein and recruitment of the E3 ubiquitin ligase with subsequent enhanced ubiquitination and proteasomal degradation of the target protein; (b) tissue-specific knockdown of a protein since some E3 ubiquitin ligases are expressed in a tissue-specific manner. PROTACS represent a novel approach for small molecule-induced targeted proteolysis through the UPP in intact cells. One could envision designing specific PROTACS for degradation of disease-promoting proteins, such as α-synuclein, truncated tau and huntingtin, in order to prevent their accumulation/aggregation and promote neuronal survival.
Parkin gene therapy
This genetic manipulation might be an effective approach for patients with loss-of-function mutations in parkin associated with the demise of dopaminergic neurons in autosomal recessive juvenile PD. Prior to clinical trials preliminary studies were conducted in primates. The serotype-1 recombinant adeno-associated virus (rAAV1) vector was used to deliver α-synuclein or α-synuclein plus parkin unilaterally into striatum of monkeys [197]. Overexpression of parkin diminished the accumulation of α-synuclein.
Modulation of carboxyl terminus of Hsc 70-interacting protein (CHIP)
Like parkin, the E3 ubiquitin ligase CHIP is a good candidate for enhancing ubiquitination. CHIP is also a co-chaperone with Hsp70 and Hsp90 and one of its roles is to ubiquitinate client proteins. CHIP is a dimeric protein containing a U-box domain at the C-terminus as its ubiquitin ligase domain, and a tetratricopeptide repeat (TPR) domain at the N-terminus as its chaperone binding domain [198].
Overexpression of CHIP is neuroprotective, while its deletion is neurotoxic in cell and animal models of a variety of neurodegenerative diseases. In a cell model of PD, overexpression of CHIP reduced α-synuclein levels by promoting its proteasomal and lysosomal degradation via the TPR and U-box domains respectively, and suppressed α-synuclein aggregation [199]. Furthermore, CHIP up-regulation protects against mutant leucine-rich repeat kinase-2 (LRRK2)-induced toxicity, which is one of the most common causes of autosomal dominant PD [200]. In vivo studies of AD brains demonstrate that higher CHIP levels correlate with less tau aggregation and fewer neurofibrillary tangles in early stages of AD [201]. Restoring CHIP levels in a transgenic mouse model of AD rescued Aβ-induced effects on tau pathology [202]. On the other hand, CHIP deletion in transgenic mice induced the accumulation of non-aggregated, ubiquitin-negative, hyperphosphorylated tau species [203]. In cell models of HD and MJD, CHIP overexpression promoted ubiquitination and degradation of the polyglutamine-expanded proteins huntingtin and ataxin-3, and suppressed their aggregation as well as cell death [204].
Besides its roles as a chaperone and E3 ubiquitin ligase, CHIP was proposed to play a role in the aggresome pathway [205]. As such, CHIP interacts with the inducible form of nitric oxide synthase, promotes its ubiquitination and degradation by the proteasome as well as its sequestration into aggresomes, and enhances its interaction with histone deacetylase 6, a linker between ubiquitinated proteins and the dynein motor.
Collectively these studies suggest that CHIP is an important regulator for ubiquitination of multiple substrates. Enhancing CHIP levels may thus be a promising therapeutic approach to facilitate ubiquitination and degradation of misfolded proteins, prevent their aggregation, and slow the neurodegenerative process.
Promote SUMO conjugation
SUMO (small ubiquitin-like modifier) conjugation is a reversible pathway that provides a rapid and effective manner for regulating subcellular localization, activity and stability of many substrates (reviewed in [206, 207]). Several proteins implicated in neurodegenerative disorders such as tau in AD, α-synuclein in PD, and huntingtin in HD are SUMOylated suggesting that this post-translational modification is involved in the neurodegenerative process (reviewed in [207]). Two recent studies demonstrate that SUMOylation attenuates aggregation and cell toxicity of proteins that contain expanded polyQs, such as ataxin 7 [208] and the androgen receptor [209]. Apparently SUMOylation functions at least in part as a steric impediment to the formation of higher order polyglutamine β-sheet structures thus preventing misfolding of these proteins. It is conceivable that enhancing SUMOylation could be of clinical value for preventing/treating neurodegenerative disorders associated with high order protein aggregation.
Conclusions
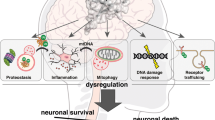
In order to survive under various environmental stress conditions or those induced by mutations, cells have a repertoire of mechanisms that they can activate or inhibit according to their needs. The UPP is the ultimate mechanism that cells use to ensure the selective destruction of misfolded or damaged proteins [210]. We reviewed a large body of evidence linking UPP impairment with neuronal loss in most chronic neurodegenerative diseases such as AD, PD, HD and ALS. When functional, the UPP provides neurons with the fundamental ability to protect themselves from a certain amount of damage before there is a severe disruption of function and viability (Fig. 1). The life or death of individual neuronal populations depends on the overall intracellular burden of accumulated misfolded or aggregated proteins (reviewed in [211]). Neuronal survival demands a dynamic, effective and safe capacity to deal with this abnormal protein burden.

Time line for neurodegeneration. The following sequence of events is proposed for neurodegeneration associated with protein misfolding. The CNS is subjected to all kinds of injury, such as infectious, chemical and physical stimuli. In many instances, these insults induce inflammation resulting in astrocytes and microglia activation. Activated glia release both protective and toxic factors. Protective factors, such as BDGF, GDNF, IGF-1 and VEGF, act in concert to repair the damage. However, if the toxic factors, such as nitric oxide, IL6, TNFα, reactive oxygen species and PGJ2, prevail they generate a toxic microenvironment that damages neurons by causing protein misfolding. In addition, mutations and the initial insults themselves can act in/on neurons and also promote protein misfolding. Ubiquitination of these damaged proteins and removal by the UPP is essential for cell survival. However, if due to mitochondrial dysfunction ATP levels decline and the UPP and chaperones activities also deteriorate, the ubiquitinated proteins accumulate, the cytoskeleton collapses and apoptosis is activated. All of these events contribute to protein aggregation. Autophagy activation is an attempt to remove the aggregates, but at this point the cells may have reached a point of no return leading to neurodegeneration. Abolishing/diminishing the toxic effects associated with inflammation and/or activating the UPP early in the process seem to be attractive approaches to prevent the neurodegenerative cascade before the damage is too far advanced for the therapy to be effective and the cells are committed to die resulting in neurodegeneration
It is critical to understand the causes and establish tools for early pre-symptomatic diagnosis to slowdown or halt the underlying neurodegenerative process. Current therapies for neurodegenerative diseases treat only the symptoms and not the causes of these disorders. Among potential strategies, preventing apoptosis with caspase inhibitors failed to prevent neurodegeneration [212]. Likewise, it is not clear if targeting the removal of protein aggregates will promote neuronal survival. It is possible that like for apoptosis, when cells develop protein aggregates the damage is too far advanced for the therapy to be effective. When protein aggregates develop cells may be already at a point of no return and committed to the death pathway.
Strategies that focus on enhancing proteasome activity in general, and ubiquitination and/or SUMOylation of particular proteins may prove to be a worthwhile endeavor. The genetic and pharmacological therapeutical approaches targeting the UPP and relevant to neurodegeneration that we discussed in this review focus on modulating different aspects of the UPP. A critical requirement for any of these degradation-enhancing therapies is the ability to cross the blood brain barrier. The latter is particularly relevant to diseases of the CNS.
Neuroinflammation is likely to be one of the mechanisms that contributes to the cascade of events leading to the accumulation of ubiquitinated proteins and UPP impairment in the neurodegenerative process (reviewed in [213]). Neuroinflammation is viewed as a process that occurs in the CNS and that involves primarily non-cell-autonomous pathological mechanisms mediated by activated glia leading to progressive neurodegeneration [214]. During neuroinflammation activated microglia migrate to sites of neuronal injury, phagocytose neighboring cells, and produce large amounts of oxygen free radicals and other toxic products that elicit protein misfolding and impair the UPP. This process of microglia activation involving cell-to-cell interaction may explain the host-to-graft transmission phenomenon observed in patients receiving neural transplants. The host-to-graft transmission phenomenon is reviewed in [215]. Interestingly, one of the mechanisms used by microglia to withstand such oxidative challenges is to up-regulate the proteasome through PARP activation [216]. Microglia can elicit pathological cell–cell interactions causing vulnerable neurons to become dysfunctional and at risk for degeneration. Therapeutic strategies aimed at down-regulating these inflammatory processes might help to slow down the progression of neurodegeneration. However, a better understanding of the complex function of microglia (reviewed in [217]) is required in order to provide a basis for therapeutically targeting neuroinflammation associated with neurodegeneration.
Abbreviations
- AD:
-
Alzheimer’s disease
- AIRAP:
-
Arsenite-inducible RNA-associated protein
- ALS:
-
Amyotrophic lateral sclerosis
- CDDO:
-
2-Cyano-3,12-dioxooleana-1,9-dien-28-oic acid
- CHIP:
-
Carboxyl terminus of Hsc 70-interacting protein
- CNS:
-
Central nervous system
- DALIS:
-
Dendritic cell aggresome-like induced structures
- DriPs:
-
Defective ribosomal products
- E1:
-
Ubiquitin-activating enzyme
- E2:
-
Ubiquitin-conjugating enzyme
- E3:
-
Ubiquitin ligase
- Gad:
-
Gracile axonal dystrophy
- GFP:
-
Green fluorescent protein
- HD:
-
Huntington’s disease
- Hsp:
-
Heat shock protein
- LC3:
-
Light chain 3
- MJD:
-
Machado-Joseph disease
- MTOC:
-
Microtubule organizing center
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Nrf2:
-
Nuclear factor-erythroid 2-related factor 2
- PA:
-
Proteasome activator
- PD:
-
Parkinson’s disease
- PGJ2:
-
Prostaglandin J2
- PINK1:
-
PTEN-induced kinase 1
- PolyQ:
-
Polyglutamine
- POMP:
-
Proteasome maturation protein
- PROTACS:
-
Proteolysis targeting chimera molecules
- Rpn:
-
19S Regulatory particle, non ATP-dependent
- UBA:
-
Ubiquitin-associated domain
- UBL:
-
Ubiquitin-like domain
- UCH-L1:
-
Ubiquitin carboxyl-terminal hydrolase-L1
- UDP:
-
Ubiquitin-domain proteins
- UIM:
-
Ubiquitin-interacting motif
- UPP:
-
Ubiquitin/proteasome pathway
References
Ciechanover A (2005) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ 12:1178–1190
Lee DH, Goldberg AL (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8:397–403
Yewdell JW (2001) Not such a dismal science: the economics of protein synthesis, folding, degradation and antigen processing. Trends Cell Biol 11:294–297
Haas AL, Bright PM (1985) The immunochemical detection and quantitation of intracellular ubiquitin-protein conjugates. J Biol Chem 260:12464–12473
Gronostajski RM, Pardee AB, Goldberg AL (1985) The ATP dependence of the degradation of short- and long-lived proteins in growing fibroblasts. J Biol Chem 260:3344–3349
Hendil KB (1988) The 19 S multicatalytic “prosome” proteinase is a constitutive enzyme in HeLa cells. Biochem Int 17:471–477
Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW (2003) Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18:343–354
Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME (1998) Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 21:516–520
Jung T, Catalgol B, Grune T (2009) The proteasomal system. Mol Aspects Med 30:191–296
Segref A, Hoppe T (2009) Think locally: control of ubiquitin-dependent protein degradation in neurons. EMBO Rep 10:44–50
Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C (1996) Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci USA 93:861–866
Hochstrasser M (2009) Origin and function of ubiquitin-like proteins. Nature 458:422–429
Deshaies RJ, Joazeiro CA (2009) RING domain E3 ubiquitin ligases. Annu Rev Biochem 78:399–434
Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96:635–644
Reyes-Turcu FE, Ventii KH, Wilkinson KD (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78:363–397
Tanaka K (2009) The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85:12–36
Finley D (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78:477–513
Rechsteiner M, Realini C, Ustrell V (2000) The proteasome activator 11 S REG (PA28) and class I antigen presentation. Biochem J 345(Pt 1):1–15
Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ (2009) Catalytic mechanism and assembly of the proteasome. Chem Rev 109:1509–1536
Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–471
Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL (2003) ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell 11:69–78
Bech-Otschir D, Helfrich A, Enenkel C, Consiglieri G, Seeger M, Holzhutter HG, Dahlmann B, Kloetzel PM (2009) Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat Struct Mol Biol 16:219–225
Babbitt SE, Kiss A, Deffenbaugh AE, Chang YH, Bailly E, Erdjument-Bromage H, Tempst P, Buranda T, Sklar LA, Baumler J, Gogol E, Skowyra D (2005) ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell 121:553–565
Orlowski M, Wilk S (2003) Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys 415:1–5
Grune T, Davies KJ (2003) The proteasomal system and HNE-modified proteins. Mol Aspects Med 24:195–204
Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19:94–102
Young P, Deveraux Q, Beal RE, Pickart CM, Rechsteiner M (1998) Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J Biol Chem 273:5461–5467
Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898
Wojcik C, DeMartino GN (2003) Intracellular localization of proteasomes. Int J Biochem Cell Biol 35:579–589
Reits EAJ, Benham AM, Plougastel B, Neefjes J, Trowsdale J (1997) Dynamics of proteasome distribution in living cells. EMBO J 16:6087–6094
Welchman RL, Gordon C, Mayer RJ (2005) Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol 6:599–609
Elsasser S, Finley D (2005) Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 7:742–749
Mayer RJ (2003) From neurodegeneration to neurohomeostasis: the role of ubiquitin. Drug News Perspect 16:103–108
Medicherla B, Goldberg AL (2008) Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J Cell Biol 182:663–673
Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH (2006) ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2:189–199
Katayama H, Yamamoto A, Mizushima N, Yoshimori T, Miyawaki A (2008) GFP-like proteins stably accumulate in lysosomes. Cell Struct Funct 33:1–12
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552–1555
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–4170
Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004) Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem 279:4625–4631
Cookson MR, Lockhart PJ, McLendon C, O’Farrell C, Schlossmacher M, Farrer MJ (2003) RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet 12:2957–2965
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Mitra S, Tsvetkov AS, Finkbeiner S (2009) Protein turnover and inclusion body formation. Autophagy 5:1037–1038
Orr HT (2004) Neurodegenerative disease: neuron protection agency. Nature 431:747–748
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66
Wang Z, Figueiredo-Pereira ME (2005) Inhibition of sequestosome 1/p62 up-regulation prevents aggregation of ubiquitinated proteins induced by prostaglandin J2 without reducing its neurotoxicity. Mol Cell Neurosci 29:222–231
Arnaud LT, Myeku N, Figueiredo-Pereira ME (2009) Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem 110:328–342
Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ (1999) Dynamic association of proteasomal machinery with the centrosome. J Cell Biol 145:481–490
Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES (1999) Characterization and dynamics of aggresome formation by a cytosolic GFP- chimera. J Cell Biol 146:1239–1254
Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN (2000) Activity and regulation of the centrosome-associated proteasome. J Biol Chem 275:409–413
Ogburn KD, Figueiredo-Pereira ME (2006) Cytoskeleton/endoplasmic reticulum collapse induced by prostaglandin J2 parallels centrosomal deposition of ubiquitinated protein aggregates. J Biol Chem 281:23274–23284
Cappelletti G, Pedrotti B, Maggioni MG, Maci R (2001) Microtubule assembly is directly affected by MPP(+)in vitro. Cell Biol Int 25:981–984
Diaz-Corrales FJ, Asanuma M, Miyazaki I, Miyoshi K, Ogawa N (2005) Rotenone induces aggregation of gamma-tubulin protein and subsequent disorganization of the centrosome: relevance to formation of inclusion bodies and neurodegeneration. Neuroscience 133:117–135
Marshall LE, Himes RH (1978) Rotenone inhibition of tubulin self-assembly. Biochim Biophys Acta 543:590–594
Roy S, Zhang B, Lee VM, Trojanowski JQ (2005) Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol 109:5–13
Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, Gatti E, Pierre P (2004) Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 164:667–675
Ebstein F, Lange N, Urban S, Seifert U, Kruger E, Kloetzel PM (2009) Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int J Biochem Cell Biol 41:1205–1215
Hartmann-Petersen R, Semple CA, Ponting CP, Hendil KB, Gordon C (2003) UBA domain containing proteins in fission yeast. Int J Biochem Cell Biol 35:629–636
Su V, Lau AF (2009) Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci 66:2819–2833
Wilkinson CR, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, Gordon C (2001) Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol 3:939–943
Madura K (2002) The ubiquitin-associated (UBA) domain: on the path from prudence to prurience. Cell Cycle 1:235–244
Shin J (1998) P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res 21:629–633
Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI (2002) Polyglutamine protein aggregates are dynamic. Nat Cell Biol 4:826–831
Tran PB, Miller RJ (1999) Aggregates in neurodegenerative disease: crowds and power? Trends Neurosci 22:194–197
Sharma D, Sharma S, Pasha S, Brahmachari SK (1999) Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. FEBS Lett 456:181–185
Trushina E, Heldebrant MP, Perez-Terzic CM, Bortolon R, Kovtun IV, Badger JD, Terzic A, Estevez A, Windebank AJ, Dyer RB, Yao J, McMurray CT (2003) Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington’s disease. Proc Natl Acad Sci USA 100:12171–12176
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506
Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB (2008) Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 23:2303–2306
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503
Cicchetti F, Saporta S, Hauser RA, Parent M, Saint-Pierre M, Sanberg PR, Li XJ, Parker JR, Chu Y, Mufson EJ, Kordower JH, Freeman TB (2009) Neural transplants in patients with Huntington’s disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci USA 106:12483–12488
Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11:219–225
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106:13010–13015
McGeer EG, Klegeris A, McGeer PL (2005) Inflammation, the complement system and the diseases of aging. Neurobiol Aging 26(Suppl 1):94–97
Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ (2008) A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 205:1869–1877
Boillee S, Vande VC, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59
Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ (1988) Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol 155:9–15
Lowe J, Lennox G, Jefferson D, Morrell K, McQuire D, Gray T, Landon M, Doherty FJ, Mayer RJ (1988) A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci Lett 94:203–210
Keller JN, Hanni KB, Markesbery WR (2000) Impaired proteasome function in Alzheimer’s disease. J Neurochem 75:436–439
McNaught KS, Jenner P (2001) Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett 297:191–194
Keller JN, Gee J, Ding Q (2002) The proteasome in brain aging. Ageing Res Rev 1:279–293
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–S36
van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RDJ, Salehi A, Martens GJM, Grosveld FG, Peter J, Burbach H, Hol EM (1998) Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 279:242–247
Kitada T, Asakawa S, Minoshima S, Mizuno Y, Shimizu N (2000) Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm Genome 11:417–421
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson’s disease. Nature 395:451–452
Moore DJ, Dawson VL, Dawson TM (2003) Role for the ubiquitin-proteasome system in Parkinson’s disease and other neurodegenerative brain amyloidoses. Neuromolecular Med 4:95–108
Przedborski S, Vila M, Jackson-Lewis V (2003) Neurodegeneration: what is it and where are we? J Clin Invest 111:3–10
Gray DA, Tsirigotis M and Woulfe J (2003) Ubiquitin, proteasomes, and the aging brain. Sci Aging Knowl Environ 2003:RE6
Carrard G, Bulteau A, Petropoulos I, Friguet B (2002) Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol 34:1461
Gaczynska M, Osmulski PA, Ward WF (2001) Caretaker or undertaker? The role of the proteasome in aging. Mech Ageing Dev 122:235–254
Vernace VA, Schmidt-Glenewinkel T, Figueiredo-Pereira ME (2007) Aging and regulated protein degradation: who has the UPPer hand? Aging Cell 6:599–606
Vernace VA, Arnaud L, Schmidt-Glenewinkel T, Figueiredo-Pereira ME (2007) Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J 21:2672–2682
Mayer RJ, Arnold J, Laszlo L, Landon M, Lowe J (1991) Ubiquitin in health and disease. Biochim Biophys Acta 1089:141–157
van Leeuwen FW, Hol EM, Fischer DF (2006) Frameshift proteins in Alzheimer’s disease and in other conformational disorders: time for the ubiquitin-proteasome system. J Alzheimers Dis 9:319–325
Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, Mayer RJ, Layfield R (2000) Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc Natl Acad Sci USA 97:9902–9906
Lindsten K, de Vrij FM, Verhoef LG, Fischer DF, van Leeuwen FW, Hol EM, Masucci MG, Dantuma NP (2002) Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol 157:417–427
Tank EM, True HL (2009) Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet 5:e1000382
Tan Z, Sun X, Hou FS, Oh HW, Hilgenberg LG, Hol EM, van Leeuwen FW, Smith MA, O’Dowd DK, Schreiber SS (2007) Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ 14:1721–1732
Fischer DF, van DR, van TP, Hobo B, Verhage MC, van der Schors RC, Li KW, van MJ, Hol EM and van Leeuwen FW (2009) Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol Aging 30:847–863
Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N Engl J Med 342:1560–1567
Sakata E, Yamaguchi Y, Kurimoto E, Kikuchi J, Yokoyama S, Yamada S, Kawahara H, Yokosawa H, Hattori N, Mizuno Y, Tanaka K, Kato K (2003) Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep 4:301–306
Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA 96:11364–11369
Ulrich HD, Jentsch S (2000) Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J 19:3388–3397
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305
Beasley SA, Hristova VA, Shaw GS (2007) Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson’s disease. Proc Natl Acad Sci USA 104:3095–3100
Doss-Pepe EW, Chen L, Madura K (2005) Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J Biol Chem 280:16619–16624
Olzmann JA, Chin LS (2008) Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 4:85–87
Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004) S-nitrosylation of parkin regulates ubiquitination and compromises Parkin’s protective function. Science 304:1328–1331
Rubio la de TE, Luzon-Toro B, Forte-Lago I, Minguez-Castellanos A, Ferrer I, Hilfiker S (2009) Combined kinase inhibition modulates parkin inactivation. Hum Mol Genet 18:809–823
Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y (1999) Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol 45:668–672
Sha D, Chin LS, Li L (2010) Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet 19:352–363
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, Patenge N (2009) Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 18:3832–3850
Berger AK, Cortese GP, Amodeo KD, Weihofen A, Letai A, LaVoie MJ (2009) Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum Mol Genet 18:4317–4328
Burns MP, Zhang L, Rebeck GW, Querfurth HW, Moussa CE (2009) Parkin promotes intracellular Abeta1–42 clearance. Hum Mol Genet 18:3206–3216
Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD (2008) Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci USA 105:19492–19497
Morett E, Bork P (1999) A novel transactivation domain in parkin. Trends Biochem Sci 24:229–231
da Costa CA, Sunyach C, Giaime E, West A, Corti O, Brice A, Safe S, bou-Sleiman PM, Wood NW, Takahashi H, Goldberg MS, Shen J, Checler F (2009) Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat Cell Biol 11:1370–1375
Wilkinson KD (1997) Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J 11:1245–1256
Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 111:209–218
Setsuie R, Wada K (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51:105–111
Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K (1999) Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet 23:47–51
MacDonald ME (1999) Gadzooks!. Nat Genet 23:10–11
Ragland M, Hutter C, Zabetian C, Edwards K (2009) Association between the ubiquitin carboxyl-terminal esterase L1 gene (UCHL1) S18Y variant and Parkinson’s disease: a HuGE review and meta-analysis. Am J Epidemiol 170:1344–1357
Kyratzi E, Pavlaki M, Stefanis L (2008) The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum Mol Genet 17:2160–2171
Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L (2004) Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 279:13256–13264
Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O (2006) Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126:775–788
Kabuta T, Furuta A, Aoki S, Furuta K, Wada K (2008) Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 283:23731–23738
Kabuta T, Wada K (2008) Insights into links between familial and sporadic Parkinson’s disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 4:827–829
Liu Z, Meray RK, Grammatopoulos TN, Fredenburg RA, Cookson MR, Liu Y, Logan T, Lansbury PT Jr (2009) Membrane-associated farnesylated UCH-L1 promotes alpha-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc Natl Acad Sci USA 106:4635–4640
Lopez-Salon M, Alonso M, Vianna MR, Viola H, Mello e Souza T, Izquierdo I, Pasquini JM, Medina JH (2001) The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur J Neurosci 14:1820–1826
Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH (1997) Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89:115–126
Lombardino AJ, Li XC, Hertel M, Nottebohm F (2005) Replaceable neurons and neurodegenerative disease share depressed UCHL1 levels. Proc Natl Acad Sci USA 102:8036–8041
Masino L, Musi V, Menon RP, Fusi P, Kelly G, Frenkiel TA, Trottier Y, Pastore A (2003) Domain architecture of the polyglutamine protein ataxin-3: a globular domain followed by a flexible tail. FEBS Lett 549:21–25
Maciel P, Costa MC, Ferro A, Rousseau M, Santos CS, Gaspar C, Barros J, Rouleau GA, Coutinho P, Sequeiros J (2001) Improvement in the molecular diagnosis of Machado-Joseph disease. Arch Neurol 58:1821–1827
Lieberman AP, Trojanowski JQ, Leonard DG, Chen KL, Barnett JL, Leverenz JB, Bird TD, Robitaille Y, Malandrini A, Fischbeck KH (1999) Ataxin 1 and ataxin 3 in neuronal intranuclear inclusion disease. Ann Neurol 46:271–273
Macedo-Ribeiro S, Cortes L, Maciel P, Carvalho AL (2009) Nucleocytoplasmic shuttling activity of ataxin-3. PLoS One 4:e5834
Reina CP, Zhong X, Pittman RN (2010) Proteotoxic stress increases nuclear localization of ataxin-3. Hum Mol Genet 19:235–249
Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N (2000) Ataxin-3, the MJD1 gene product, interacts with the two human homologs of yeast DNA repair protein RAD23, HHR23A and HHR23B. Hum Mol Genet 9:1795–1803
Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ, Cohen RE, Peng J, Paulson HL (2008) The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 283:26436–26443
Todi SV, Winborn BJ, Scaglione KM, Blount JR, Travis SM, Paulson HL (2009) Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J 28:372–382
Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, Wang G (2009) Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 18:4268–4281
Chou AH, Yeh TH, Kuo YL, Kao YC, Jou MJ, Hsu CY, Tsai SR, Kakizuka A, Wang HL (2006) Polyglutamine-expanded ataxin-3 activates mitochondrial apoptotic pathway by upregulating Bax and downregulating Bcl-xL. Neurobiol Dis 21:333–345
Jung J, Xu K, Lessing D, Bonini NM (2009) Preventing Ataxin-3 protein cleavage mitigates degeneration in a Drosophila model of SCA3. Hum Mol Genet 18:4843–4852
Babu JR, Geetha T, Wooten MW (2005) Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 94:192–203
Ramesh BJ, Lamar SM, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106:107–120
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J (1999) The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J 18:3044–3053
Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J (2001) The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem 276:7709–7712
Sanz L, Diaz-Meco MT, Nakano H, Moscat J (2000) The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J 19:1576–1586
Geetha T, Wooten MW (2002) Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett 512:19–24
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145
Puls A, Schmidt S, Grawe F, Stabel S (1997) Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci USA 94:6191–6196
Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC (2006) Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62’s role in neurodegenerative disease. J Biomed Biotechnol 2006:62079
Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H (2009) Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 111:1062–1073
Seibenhener ML, Geetha T, Wooten MW (2007) Sequestosome 1/p62–more than just a scaffold. FEBS Lett 581:175–179
Kuusisto E, Salminen A, Alafuzoff I (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12:2085–2090
Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160:255–263
Furukawa Y, Iseki E, Hino H, Kanai A, Odawara T, Kosaka K (2004) Ubiquitin and ubiquitin-related proteins in neurons and dendrites of brains of atypical Pick’s disease without Pick bodies. Neuropathology 24:38–45
Nakano T, Nakaso K, Nakashima K, Ohama E (2004) Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta Neuropathol (Berl) 107:359–364
Kuusisto E, Salminen A, Alafuzoff I (2002) Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol 28:228–237
Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K, Araga S, Yanagawa T, Ishii T, Nakashima K (2004) Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson’s disease. Brain Res 1012:42–51
Nagaoka U, Kim K, Jana NR, Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N (2004) Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J Neurochem 91:57–68
Nan L, Wu Y, Bardag-Gorce F, Li J, French BA, Fu AN, Francis T, Vu J, French SW (2004) p62 is involved in the mechanism of Mallory body formation. Exp Mol Pathol 77:168–175
Du Y, Wooten MC, Wooten MW (2009) Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis 35:302–310
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352:2487–2498
Ustrell V, Hoffman L, Pratt G, Rechsteiner M (2002) PA200, a nuclear proteasome activator involved in DNA repair. EMBO J 21:3516–3525
Ortega J, Heymann JB, Kajava AV, Ustrell V, Rechsteiner M, Steven AC (2005) The axial channel of the 20S proteasome opens upon binding of the PA200 activator. J Mol Biol 346:1221–1227
Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, Pandita TK, Bangia N (2008) Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA 105:16165–16170
Mao I, Liu J, Li X, Luo H (2008) REGgamma, a proteasome activator and beyond? Cell Mol Life Sci 65:3971–3980
Schwarz K, Eggers M, Soza A, Koszinowski UH, Kloetzel PM, Groettrup M (2000) The proteasome regulator PA28alpha/beta can enhance antigen presentation without affecting 20S proteasome subunit composition. Eur J Immunol 30:3672–3679
Ferrington DA, Hussong SA, Roehrich H, Kapphahn RJ, Kavanaugh SM, Heuss ND, Gregerson DS (2008) Immunoproteasome responds to injury in the retina and brain. J Neurochem 106:158–169
Mishto M, Bellavista E, Santoro A, Stolzing A, Ligorio C, Nacmias B, Spazzafumo L, Chiappelli M, Licastro F, Sorbi S, Pession A, Ohm T, Grune T, Franceschi C (2006) Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains. Neurobiol Aging 27:54–66
Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J, Lucas JJ (2003) Neuronal induction of the immunoproteasome in Huntington’s disease. J Neurosci 23:11653–11661
Seo H, Sonntag KC, Kim W, Cattaneo E, Isacson O (2007) Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS One 2:e238
Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW (2004) Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306:117–120
Hol EM, Fischer DF, Ovaa H, Scheper W (2006) Ubiquitin proteasome system as a pharmacological target in neurodegeneration. Expert Rev Neurother 6:1337–1347
Hanna J, Finley D (2007) A proteasome for all occasions. FEBS Lett 581:2854–2861
Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES (2005) Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem 280:11840–11850
Chondrogianni N, Gonos ES (2007) Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp Gerontol 42:899–903
Stanhill A, Haynes CM, Zhang Y, Min G, Steele MC, Kalinina J, Martinez E, Pickart CM, Kong XP, Ron D (2006) An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol Cell 23:875–885
Dahlmann B, Rutschmann M, Kuehn L, Reinauer H (1985) Activation of the multicatalytic proteinase from rat skeletal muscle by fatty acids or sodium dodecyl sulphate. Biochem J 228:171–177
Watanabe N, Yamada S (1996) Activation of 20S proteasomes from spinach leaves by fatty acids. Plant Cell Physiol 37:147–151
Wilk S, Chen WE (1997) Synthetic peptide-based activators of the proteasome. Mol Biol Rep 24:119–124
Kisselev AF, Kaganovich D, Goldberg AL (2002) Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J Biol Chem 277:22260–22270
Katsiki M, Chondrogianni N, Chinou I, Rivett AJ, Gonos ES (2007) The olive constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life span extension of human embryonic fibroblasts. Rejuvenation Res 10:157–172
Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW (2003) Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol 23:8786–8794
Kwak MK, Cho JM, Huang B, Shin S, Kensler TW (2007) Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic Biol Med 43:809–817
Thimmulappa RK, Fuchs RJ, Malhotra D, Scollick C, Traore K, Bream JH, Trush MA, Liby KT, Sporn MB, Kensler TW, Biswal S (2007) Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid Redox Signal 9:1963–1970
Huang L, Ho P, Chen CH (2007) Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett 581:4955–4959
Orlowski RZ, Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14:1649–1657
Chondrogianni N, Petropoulos I, Franceschi C, Friguet B, Gonos ES (2000) Fibroblast cultures from healthy centenarians have an active proteasome. Exp Gerontol 35:721–728
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ (2001) Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA 98:8554–8559
Schneekloth AR, Pucheault M, Tae HS, Crews CM (2008) Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett 18:5904–5908
Yasuda T, Miyachi S, Kitagawa R, Wada K, Nihira T, Ren YR, Hirai Y, Ageyama N, Terao K, Shimada T, Takada M, Mizuno Y, Mochizuki H (2007) Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience 144:743–753
Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C, Pearl LH (2005) Chaperoned ubiquitylation–crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell 20:525–538
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ (2005) The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280:23727–23734
Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, Zhang YJ, Jiang H, Ross CA, Moore DJ, Patterson C, Petrucelli L, Dawson TM, Dawson VL (2009) CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci USA 106:2897–2902
Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A (2005) In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem 94:1254–1263
Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, LaFerla FM (2008) Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci 28:12163–12175
Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci 26:6985–6996
Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, Nukina N (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 280:11635–11640
Sha Y, Pandit L, Zeng S, Eissa NT (2009) A critical role for CHIP in the aggresome pathway. Mol Cell Biol 29:116–128
Matunis MJ, Pickart CM (2005) Beginning at the end with SUMO. Nat Struct Mol Biol 12:565–566
Dorval V, Fraser PE (2007) SUMO on the road to neurodegeneration. Biochim Biophys Acta 1773:694–706
Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, Duyckaerts C, Brice A, Dejean A, Sittler A (2010) SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet 19:181–195
Mukherjee S, Thomas M, Dadgar N, Lieberman AP, Iniguez-Lluhi JA (2009) Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J Biol Chem 284:21296–21306
Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899
Gorman AM (2008) Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med 12:2263–2280
Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL (2006) Recent clinical failures in Parkinson’s disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol 72:1197–1206
Schmidt-Glenewinkel T, Figueiredo-Pereira ME (2006) Inflammation as a mediator of oxidative stress and UPS dysfunction. In: Stefanis L, Keller JN (eds) The proteasome in neurodegeneration. Springer, New York, pp 105–131
Streit WJ, Miller KR, Lopes KO, Njie E (2008) Microglial degeneration in the aging brain–bad news for neurons? Front Biosci 13:3423–3438
Brundin P, Li JY, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson’s disease pathology spread. Nat Rev Neurosci 9:741–745
Ullrich O, Diestel A, Bechmann I, Homberg M, Grune T, Hass R, Nitsch R (2001) Turnover of oxidatively damaged nuclear proteins in BV-2 microglial cells is linked to their activation state by poly-ADP-ribose polymerase. FASEB J 15:1460–1462
Lucin KM, Wyss-Coray T (2009) Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64:110–122
Acknowledgments
Please note that this review is not intended to be comprehensive and we apologize to the authors whose work is not mentioned. Supported by National Institutes of Health (NIH) [AG028847 to M.F.-P. from National Institute of Aging; NS41073 (Specialized Neuroscience Research Programs) to M.F.-P. (head of subproject) from National Institute of Neurological Disorders and Stroke; RR03037 to Hunter College (infrastructure) from National Institute of General Medical Sciences (NIGMS)/RCMI (Research Centers in Minority Institutions)].
Conflict of interest statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huang, Q., Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis 15, 1292–1311 (2010). https://doi.org/10.1007/s10495-010-0466-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-010-0466-z