
Abstract
Cryopreservation, a widely utilized technique for the long-term preservation of in vitro cultures, effectively arrests metabolic processes, obviating the need for frequent subcultures and mitigating the risk of somaclonal variation. In this study, we applied cryopreservation methods to intact rice (Oryza sativa L.) calli to determine the optimal age for cryopreservation, investigating the timelines for greening and shoot initiation in R0 plants. Results revealed that three-month-old calli exhibited the highest regeneration percentage, with greening observed within twelve days and shoot initiation within fifteen days. Using 3% mannitol in the callus culture medium provided osmotic stress, aiding in the formation of compact calli masses suitable for regeneration. Vitrification with cryoprotectants (DMSO, PEG, and glucose) and gradual dehydration improved cell survival. Thawing and post-thaw damage were minimized using rapid thawing, fast cryoprotectant removal, and gradual rehydration. We assessed the phenotypic variations in R1 and R2 generation and genotypic fidelity of regenerants in R1. Phenotypic variations from seed-derived plants were observed in seed characters both in vitrified and cryopreserved calli-derived plants. However, these variations were unstable and disappeared in the R2. SSR markers were used to detect genetic variations in R1, with results showing a 3.6% change in vitrified calli-derived plants and an 8.61% change in cryopreservation-derived plants, likely due to reversible DNA methylation or SNPs in non-coding region. Our study confirms the feasibility of cryopreservation for rice calli, ensuring high regeneration rates and minimal long-term genetic variations.
Key message
Three-month-old rice calli maintained in callus proliferation medium with 3% mannitol and vitrified in a combination of cryoprotectants ensure high regeneration rates post-cryopreservation, maintaining phenotypic stability and minimal genetic variation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is a crucial global food crop. It primarily caters to over half of the global population in terms of their main nutritional intake. It serves as a model organism in molecular biology and tissue culture studies due to its small genome size, genome sequence availability (IRGSP 2005), and ease of regeneration (Hiei et al. 1994). However, the long-term maintenance of rice cultures faces challenges as morphogenic competence declines over time (Lynch and Benson 1990). Cryopreservation emerges as a vital technique for preserving plant cell cultures, offering advantages like low maintenance costs and reduced risk of somaclonal variation (Helliot et al. 2002). Genetic and agronomic stability of plants regenerated from frozen somatic embryos is crucial, ensuring viability and maintaining phenotype and genotype integrity (Bomal et al. 2000; Ren et al. 2021; Ichikawa et al. 2023).
Cryopreservation is a multi-step procedure that starts with the selection of appropriate tissue, followed by cooling, thawing, and reestablishment of plants. Attention to cryopreservation was first drawn by the report of Sakai (1956), who stressed the dehydration and precooling of the tissue. In subsequent phases of research, in vitro cultured cells were tried. These cells posed the challenge of cooling such highly hydrated cells without the formation of damaging ice crystals within cells (Panis 2019). This led to the development of the two-step protocol, wherein precooling in a vitrification solution effectively removed water from cells (Benson 2008). Although this proved to be effective for unorganized tissues like callus (Zeliang et al. 2010) and suspension cells (Lynch et al. 1995), the efficiency of organized tissue (like shoot tips) cryopreservation improved with the development of fast freezing methods like droplet vitrification and freezing, encapsulation dehydration, and V-cryoplate procedures (Panis et al. 2019; Banelli et al. 2021).
The process of cryopreservation of rice cell suspension was initially documented by Sala et al. (1979), and Finkle and Ulrich’s study (1982) came next. In a thorough investigation on the cryopreservation of Taipei 309 suspension cells, Lynch and Benson (1990) proposed that the composition of the pre- and post-freezing culture media, as well as cryogenic procedures, are critical to the success of cryopreservation. A two-step cooling procedure where protected cells were first cooled to -25 °C and then stored in liquid nitrogen (LN) (Jain et al. 1996), vitrification and cooling (Wang et al. 1998), rapid freezing (Wang et al. 2001), pre-growth desiccation and subsequent freezing in LN (Zhang et al. 2001), direct immersion in LN (Moukadiri et al. 2002), and encapsulation dehydration (Zeng et al. 2009) are some of the various cryopreservation techniques that have been successfully used for rice. All these studies were carried out with O. sativa. Also, there are two reports of cryopreservation of wild rice, O. meyeriana Baill (He et al. 1998)and O. rufipogon (Zeliang et al. 2010). In case of O. rufipogon, which was reported by our group, a slow cooling of mature embryo-derived calli in vitrification solution preceded freezing in liquid nitrogen.
The two-step cooling protocol with vitrification has been widely used in the cryopreservation of rice callus and cell suspensions. In this protocol, the choice of the vitrification solution is crucial. Dimethyl sulfoxide (DMSO), ethylene glycol (such as PEG), glycerol, and sugars (sucrose, glucose) and sugar alcohols (mannitol, sorbitol) have been used as cryoprotectants in vitrification solutions. Using rice in vitro cells and Coherent Anti-Stokes Raman Scattering (CARS) microscopy, it has been demonstrated that the distribution of DMSO, ethylene glycol, and glycerol is not uniform within the cell mass (Samuels et al. 2021, 2023). Although cryoprotectants enter cells within minutes of application, visible cell response may be delayed. They suggested the use of a combination of cryoprotectants rather than any single one (Samuels et al. 2023). This study also made two interesting observations. First, the concentrations of cryoprotectants inside cells never exceed the outside concentration. Second, the concentration of glycerol inside cells may be lower than that of DMSO or ethylene glycol, although it is used at a higher concentration (30% and 15% w/v, respectively). Therefore, it appears that glycerol is not as efficient as DMSO/ethylene glycol as a cryoprotectant for plant cells. Cincotti and Fada (2013), using a mathematical model, concluded that in a population of varying cell sizes, intracellular ice formation may be most damaging for large size cells, may not seriously damage medium size cells, while it may not happen at all in smaller cells. Samuels et al. (2023), in their study using CARS microscopy, have shown that cryoprotectant concentration is the highest in starch granules of rice cells. These two studies together probably explain why suspensions of embryogenic rice cells, which are smaller in size with dense cytoplasm and rich in starch granules, showed better regrowth after cryopreservation (Lynch et al. 1994; Benson et al. 1995).
Controlled, slow cooling in cryopreservation aims to facilitate osmotic water loss, preventing ice crystal formation in cells at the final cryogen temperature (Grout 1995). Freeze tolerance is higher in smaller cells, tissues, or organs. The cells exhibited optimal regrowth during the late lag, early exponential, or early stationary phase and the cells in the late lag or early exponential phase exhibit the lowest survival rate (Ishikawa et al. 1996). Callus age influences chromosomal aberrations, with older callus exhibiting increased chromosomal instability (Jain and Maluszynski 2004). A variety of procedures have been used for thawing, the most common being rapid warming in a water bath (40–45oC) followed by removal of vitrification solution and gradual rehydration with culture medium (Popova et al. 2023).
A great deal of study has been done on the medium used for the recovery of calli after cryopreservation. Generally, the callus proliferation medium has been used. However, additives like antifreeze proteins (Pe et al. 2021), iron-chelating agents (Benson et al. 1995), antioxidants (Diengdoh et al. 2019), vitamins (Uchendu et al. 2010), ABA (Moukadiri et al. 2002), and ammonium-free medium (Kuriyama et al. 1989; Lynch et al. 1994) have been shown to improve recovery and regrowth after cryopreservation. Recently, the use of gold and carbon nanoparticles in the pre-culture and recovery medium has been reported (Kulus and Tymoszuk 2021; Ren et al. 2020). A semisolid medium has been widely preferred for recovery (Popova et al. 2023).
Plant vitrification solutions (PVS) can induce complicated challenges as osmotic damage and dehydration stress, even though they are crucial for cell survival (Uchendu et al. 2010). Previous studies show that during cryopreservation, a few genes and proteins linked to stress are activated. Using comparative transcriptomic technology, Ren et al. (2013) discovered that the application of cryoprotectants causes the generation of reactive oxygen species (ROS), which can lead to oxidative stress and processes akin to apoptosis. In plant cells, oxidative stress typically stimulates the genes for peroxidation and anti-oxidation, where anti-oxidation is a beneficial element and peroxidation is a detrimental one for cell survival. The most significant ROS molecule causing oxidative damage and impacting cell viability is H2O2 (Zhang et al. 2015; Wei et al. 2023), which is scavenged by catalase, peroxidase, superoxide dismutase to shield cells from damage (Wei et al. 2023).
Since cryopreservation and the ensuing regeneration steps have the potential to induce somaclonal variation, which can manifest itself as changes in morphology, chromosome number, gene expression, protein profiles, or DNA sequences, the extent of variation in the regenerated plants is an important factor in in vitro conservation (Harding 2004). A variety of molecular and biochemical markers have been used for assessing genetic fidelity of cryopreservation-derived plants (for detailed review please see Martinez 2018). According to Cornejo et al. (1995), cryopreservation has no effect on rice cells’ morphogenic potential, which allows them to express and incorporate foreign genes. Transgenic rice suspension cells have been shown to maintain the transgene functioning after recovery from cryopreservation (Cho et al. 2007). It is significant to remember that plants of the R1 generation may not have the original aberrant features, and that phenotypic variation displayed by the original (R0) plantlets is said to be infrequently inherited in the seed offspring (Monk 1990). Reviewing investigations pertaining to morphology, cytology, histology, and biochemistry, Harding (2004) concluded minimal indication of instability in plants that had been cryopreserved. In plants grown in fields, Kaity et al. (2009) found 0.45–4.25% DNA modification and 0–1.42% methylation modification in a study using cryopreserved papaya clones. Research conducted by Zhang and Hu (1999) and by Moukadiri et al. (1999a, b) revealed that tissue culture-induced variations, as opposed to the effects of cold storage, were the primary cause of phenotypic variations observed in some plants regenerated following cryopreservation in cultivated rice. In our study with O. rufipogon, (Zeliang et al. 2010) in the R1 generation, phenotypic variations were seen in the seed physical characters like length, breadth and pericarp colour while SSR band variations ranged from 4.78 to 7.25%.
Recent advances in DNA sequencing technology have made it possible to study genetic variation in regenerants from cell cultures using whole-genome sequencing. Oryza sativa L. is one of the plant species that is studied the most. Using whole-genome sequencing, somaclonal variation in rice plants, including transgenic and modified plants, has been examined (Park et al. 2019; Qin et al. 2018; Zhang et al. 2014; Ichikawa et al. 2023). Numerous mutations brought on by cell culture were found during these investigations. However, the ratio of single nucleotide polymorphism between regenerants and their source seed-derived plants were comparable (Ichikawa et al. 2023). Wang et al. (2021) did a comprehensive review of published reports on epigenetic, genetic and biochemical integrity of cryopreserved plants. They found that in some plant species, regenerants obtained through cryopreservation did show certain alterations in DNA methylation. Specifically, with regard to embryogenic tissues, several of these modifications were ascribed to in vitro cultivation techniques. However, when in vitro cryopreserved plants are cultivated in field, DNA methylation was found transient and plants returned to their original DNA state. Thus, in cryopreserved plants, DNA methylation was not significantly correlated with genetic differences. While several plant species showed evidence of genetic diversity in their regenerants retrieved after cryopreservation, these differences were tiny in locus and low in frequency, with some of the variations being ascribed to in vitro growth procedures.
Analyses by flow cytometry (Kulus et al. 2019) and molecular markers (Harding 2004; Zeliang 2010) proved that overall genetic stability was maintained in the regenerants recovered after cryopreservation in many of the plant species. Therefore, cryopreservation can maximally maintain genetic stability of cryo-derived plants compared to other traditional methods. Cryopreservation of transgenes provides a safe and reliable strategy for the long-term preservation of transgenic plant materials. The biochemical profiles of plants originating from cryogenic sources are preserved in their regenerated progeny, and the metabolic stability of these plants is not negatively impacted by cryopreservation procedures. Transgenes in the altered materials can also be preserved via cryopreservation.
In this study, an effort was made to determine the optimum age of the rice callus for cryopreservation, days taken for the calli to turn green, and subsequent shoot formation in the R0 generation and comparison of the phenotypic and genotypic effect of cryopreservation with that of cryoprotectant treatment in the R1generation. We also assessed the stability of phenotypic variations in the R2 generation.
Materials and methods
Plant material
Seeds from a single seed-derived line of Oryza sativa L., cv. Lamphnah, were utilized for the experiment, which included controls for both phenotypic and genotypic evaluations. At each stage of seed multiplication, panicles were bagged to ensure selfing.
Callus induction
Ten dehusked and surface-sterilized seeds per 90-mm Petri dish were inoculated on MS medium (Murashige and Skoog 1962) supplemented with 11.31 µM 2,4-D, 300 mg/l casein hydrolysate, 4.34 mM proline, and 0.088 M sucrose, solidified with 0.3% Phytagel (pH 5.8), and incubated in the dark at 27 ± 2 °C. After 2 weeks, swollen scutella were separated from the endosperm and roots and plated onto callus maintenance medium N6M (Bhattacharjee et al. 1998) containing N6 salts and vitamins, 11.31 µM 2,4-D, 2% (w/v) sucrose, and 3% (w/v) mannitol, solidified with 0.3% Phytagel. At all steps where N6M was used, it was solidified with 0.3% Phytagel.
Callus maintenance
At every 15-day intervals, nodular embryogenic calli were separated from loose non-embryogenic calli and subcultured onto fresh N6M medium. Calli were maintained in the dark.
Cryopreservation and recovery of the calli
Pretreatment of calli
About 3.0 g of calli (of various ages) were first washed with liquid MS medium, then suspended in 25 ml of filter-sterilized vitrification (cryoprotectant) solution PGD (Finkle and Ulrich 1983) containing 10% (w/v) polyethylene glycol (PEG) 6000, 0.44 M glucose, and 10% (w/v) dimethyl sulfoxide (DMSO) in a 100-ml conical flask and chilled on ice for 1 h while shaking at 50 rpm. About 15 ml of the vitrification solution was then discarded. Vitrified (cryoprotectant-treated) calli (VF calli) were retained from each batch before proceeding to the freezing treatment which were then processed in the same way as described for the thawing of cryopreserved calli starting from dipping in 40oC water bath.
Freezing of calli
For freezing the calli, a slow cooling method (Zeliang et al. 2010) was followed. Approximately 1.5 ml of calli (around 250 mg) in vitrification solution was placed in 2 ml round-bottom microcentrifuge tubes and cooled to -23 °C in a cryocooler bath (Cryo1C cooler, Tarsons, India) filled with cold (4 °C) isopropyl alcohol (propan-2-ol, BDH, Aristar grade). The tubes were then kept inside a -70 °C freezer for 1 h. After the initial cooling, the microcentrifuge tubes containing the calli were plunged into liquid nitrogen (LN) and kept frozen for one week.
Thawing
Microcentrifuge tubes containing calli were taken out of liquid nitrogen (LN), placed on a float, and immediately dipped in a 40 °C water bath to melt the ice crystals (approximately 1 min), and then transferred to crushed ice (for five minutes). Vitrification solution was removed followed by three washes with liquid MS medium (two of them with 0.2 M glucose) without hormones. After air drying for three hours on sterile filter paper in a laminar hood, the calli were plated on N6M for recovery and stored for ten days in the dark. Using fluorescein diacetate (FDA) labeling (Widholm 1972), cell survival following freezing/vitrification treatment (from three hours post-washing up to five days at 24-hour intervals) was evaluated and compared with untreated calli of the same age.
Assessment of effect of age of calli on regeneration after cryopreservation
Calli were regularly subcultured every fifteen days onto fresh N6M medium, selecting for nodular embryogenic calli at each step. These calli were used to test regeneration capability after vitrification / cryogenic treatment at different ages, ranging from one month to nine months of subculture. The days taken for callus greening, days to shoot formation, and the number of regenerated plants were studied. Data were collected from three batches of 30 calli each.
Plant regeneration
For regeneration of plantlets, nodular and healthy calli from both the treatments, after five subcultures on N6M medium, were transferred to four modified MS medium as listed below.
-
(i)
MSA - MS medium + 2.22µM BAP + 2.69µM NAA (Gupta and Pattanayak 1993).
-
(ii)
MSB– MS medium + 13.9µM kinetin + 2.69µM NAA (Gupta and Pattanayak 1993).
-
(iii)
MSBT - MS medium + 13.9µM kinetin + 2.69µM NAA + 1µM TDZ.
-
(iv)
MST- MS medium + 1µMTDZ.
All media were solidified with 0.3% Phytagel.
The culture tubes containing the calli were first kept in the dark for one week and then transferred to a 16/8-hour light/dark period (illumination 35–40 µmol quanta/m²/sec) at 27 ± 2 °C and maintained for 3–4 weeks. The percentage of transferred calli exhibiting shoot and root regeneration after four weeks under the 16/8-hour light/dark regime was used to measure regeneration efficiency. Plantlets with roots were transferred to half-strength MS medium for further root development and hardening, and after 21 days, they were transplanted into pots containing an equal mixture of soil and compost. Potted plants were kept within a polyethylene cover for seven days in the greenhouse with natural light under 27/21°C day/night temperature conditions, after which the cover was removed. Plants were grown to maturity in the greenhouse. Clones of each plant that regenerated from a single cluster of calli were kept intact. All panicles were bagged, and the seeds were collected when the plants reached maturity.
Assessment of phenotypic variation in R1 and R2 plants
Seeds from R0 plants derived from both VF and cryopreserved calli, along with those from normal seed-derived plants, were cultivated in the subsequent years. In the R0 generation, seeds from each tiller were individually harvested, and R1 plants were grown as panicle-to-row progeny. Each panicle from R1 plants was harvested separately. For R2, five seeds from each panicle in a progeny row were pooled and grown as a block of 3 m². Thirty progeny rows (in R1)/blocks (in R2) from cryopreservation/VF calli were selected randomly for data collection. In R1 and R2, seed-derived plants grown in three replications were used as controls. Data were collected from ten randomly chosen plants in each progeny/block/replication, focusing on twelve agrobotanic traits: plant height (cm), leaf length (cm), number of ear-bearing tillers, number of days to 50% flowering, number of days to maturity, panicle length (cm), number of spikelets per panicle, spikelet fertility (as percentage of fertile spikelets in a panicle), 100-seed weight (g), seed girth (mm), seed length (mm), and seed breadth (mm). Data were analyzed using the balanced ANOVA routine of Crop Stat 7.2 software. Duncan’s multiple range test with the least significant difference (LSD) was used to compare mean trait values across treatments.
Assessment of genotypic variation in R1 plants
Eight progeny rows each from cryopreservation-derived and VF calli, and thirty seed-derived plants, selected randomly, were included in the study. Leaf samples from all plants in a progeny row were bulked before DNA isolation. Similarly, leaf samples from all thirty seed-derived plants were bulked. DNA extraction followed the Qiagen DNeasy Miniprep Kit protocol. PCR was conducted in a 15 µl reaction mixture, comprising 50 ng of template DNA, 5 pmol each of forward and reverse primers, 7.5 µl Qiagen PCR 2X Master Mix, and nuclease-free water. The mixture was then amplified in an ABI GeneAmp 9700 thermal cycler, involving a 4-minute initial denaturation at 94 °C, followed by 35 cycles of 94 °C for 1 min, primer-specific annealing for 1 min, and extension at 72 °C for 2 min. A final extension at 72 °C for 5 min concluded the process. The PCR products were separated in a 2% ISSR agarose gel at 7 V/cm for 1 h. A gel documentation system (Alpha Innotech, USA) was used to visualize and capture gel pictures. Using Alpha Imager FC software, bands were adjusted for the smiling effect, and genotype presence or absence was assessed. The amplification patterns of alleles (bands) in cryopreserved, and VF calli-derived plants were compared with those of seed-derived plants, and the percentage variation resulting from changes in band size and lack of amplification was computed for all markers.
Results
Callus induction, maintenance and cryopreservation
Seeds cultured on callus induction medium produced swelled scutellum. After separation and subculture on N6M medium, they showed good proliferation. Continuous selection for dry and nodular calli resulted in healthy and compact calli. These calli did not show any disintegration during washing, pre-cooling, cryo-treatment, or thawing.
Recovery and viability of the calli
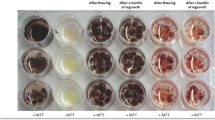
After thawing treatment and performing the FDA test, the initial viability (after three hours of thawing) of VF calli was higher (56–63%, Fig. 1a) than that of cryopreserved calli (28–31%, Fig. 1b). A careful observation of the cryopreserved calli (Fig. 1b) indicated that almost all cells on the outer layer lost viability (single arrow), while viability was maintained in the inner layers (double arrow). However, by the third day, both cryopreserved calli and VF calli showed recovery, with 42–48% and 70–77% viability, respectively, reaching up to 80% within 12 days with nodular growth and embryoid formation (Fig. 1c).

Plant regeneration from mature embryo-derived cryopreserved calli. (a and b) Three-month calli stained with FDA 3 h after thawing (Scale 300 μm). a VF calli, (b) cryopreserved calli. In the cryopreserved calli, the cells in the outer layer (single arrow) lost viability while cells in the inner layer were viable (double arrow). (c) Cryopreserved calli after 3 days of recovery (Scale 100 μm). (d) Formation of shoot from cryopreserved calli after recovery (scale 50 μm). (e) Plantlet with leaves and roots developed from cryopreserved calli. Occasionally, albino shoots were seen along with green shoots in the same calli mass. These plants although produced root, did not survive in pots. (f) Cryopreserved calli-derived plants established in pots inside a greenhouse
Plant regeneration
Upon exposure to light, cryopreserved calli exhibited green pigmentation and initiated shoot and root development within 12–14 days (Fig. 1d). Some albino plantlets were occasionally observed in the same calli mass from which green plants were regenerated (Fig. 1e). Among the media tested, MSB proved most effective for callus regeneration, with a regeneration rate of 67.8% compared to 32.3% in MSA medium. In comparison, calli treated only with cryoprotectant (VF) achieved an 80.2% regeneration rate in MSB compared to 40.3% in MSA. Significant growth of shoots and roots was observed in both cryopreserved (Fig. 1e) and VF calli after three weeks. There was no regeneration in MSBT or MST medium in either type of calli. 98% of the plantlets transferred to pots following a 21-day hardening period on half-strength MS medium successfully survived in the greenhouse.
Effect of callus age on regeneration
In one-month-old callus, both cryopreserved and VF calli samples required 20 days for greening, and 30 and 35 days for shoot formation in VF calli and cryopreserved calli, respectively. The percentage of regeneration was 22.22% in cryopreserved calli and 30.33% in VF calli (Table 1). For two-month-old callus, the respective days for greening were 14, and shoot formation took 30 days in cryopreserved calli and 17 days in VF calli, yielding 50.33% and 67.77% regenerated plants, respectively. Three-month-old calli required 12 days and 13 days for greening and 15 days for shoot formation. Plantlet regeneration was 75.55% from cryopreserved calli and 81.11% from VF calli. Four-month-old calli took 13 days for greening, and shoot formation was observed within 15 days in both cryopreserved and VF calli, resulting in 68.88% and 74.44% plantlet regeneration, respectively. For five-month-old callus, greening took 15 and 14 days, and shoot formation took 19 days in both cryopreserved and VF calli, yielding 43.77% and 55.55% plantlets, respectively. Calli beyond five months of age did not exhibit greening or shoot formation after cryopreservation, while VF calli could regenerate shoots and plantlets up to nine months, although with a reduced percentage (Table 1).
Assessment of variation in R1 and R2 plants
To assess variations induced by vitrification and cryopreservation, data on quantitative and qualitative traits were gathered from R1 and R2 plants. Twelve traits were assessed in cryopreserved calli, VF calli, and seed-derived plants, all planted simultaneously. Molecular analysis for variation was also conducted in the R1 plants.
Phenotypic variation
In R1 plants, out of the twelve agrobotanic characters compared, according to Duncan’s multiple range test, there was no statistically significant difference among the three sources of plants in height, leaf length, number of ear-bearing tillers per plant, panicle length, spikelets per plant, and spikelet fertility percentage. For days to 50% flowering, cryopreserved calli-derived plants showed significantly higher values than seed-derived plants but they were not significantly different from VF calli-derived plants. For days to maturity, 100-seed weight, and seed length, VF calli-derived plants did not show a significant difference from seed-derived plants, although seed breadth and seed girth were lower (Table 2). Cryopreserved calli-derived plants showed significant difference from seed-derived plants in all seed characters viz. 100 seed weight, seed girth, seed breadth and seed length.
For R2 plants, height, leaf length, effective tillers, days to 50% flowering, days to maturity, panicle length, spikelets per panicle, spikelet fertility, and 100-seed weight were comparable across all three groups. Leaf length remained longer in seed-derived plants compared to VF and cryopreserved calli-derived plants. The seed-derived plants also matured earlier than VF and cryopreserved calli-derived plants. Seed girth, seed breadth, and seed length were slightly greater in seed-derived plants, but all these differences were statistically not significant (Table 2).
Genotypic variation
For genotype analysis in the R1 plants, 30 rice SSR primers distributed across all 12 rice chromosomes were tested, but six markers (OSR13, RM338, RM507, RM44, RM271, and RM118) with non-reproducible bands were excluded. The remaining 24 markers successfully amplified 30 alleles, and some markers amplified two alleles each, while others amplified one allele each within the expected range (Table 3). A representative picture of band variations is presented in Fig. 2. Two different alleles were seen in markers RM237, RM161, RM125, RM484, RM316, and RM495 located on chromosomes 1, 5, 7, 9, and 11. RM484 amplified a band of 220 bases, which was shorter than the expected size (Table 3). Null alleles (no amplification) in different groups of plants were observed in RM125, RM161, RM237, RM316, RM413, RM484, and RM495 located on chromosomes 1, 5, 7, 9, and 10 (Table 4). Variations (band size and null allele combined) from seed-derived plants were highest in cryopreserved calli-derived plants (8.6%) compared to VF calli-derived plants (3.6%, Table 5). Variation attributed to altered amplification from seed-derived plants (Fig. 2a, b, c, e) was greater compared to no amplification (Fig. 2c, d).

SSR amplification profiles of five RM primers in R1 plants. A second allele (RM316, RM237, RM125 and RM161), altered amplification (RM484) and no amplification (RM215 and RM484) were observed. (Lane M (Marker) Lane 1–8 (Cryopreserved), Lane 9–12 (VF) and Lane 13–16 (Seed))
Discussion
Cryopreservation of plant cells is difficult compared to animal cells because of the presence of large water-filled vacuoles (Schillberg et al. 2019; Zhang et al. 2024). Although cryopreservation techniques for various plant cell cultures have been developed (Hakkinen et al. 2018; Kurki et al. 2021), the management of ice formation inside cells, as well as osmotic, temperature, mechanical, and oxidative stresses, are important aspects of successful recovery of cryopreserved cells (Zhang et al. 2015; Bradï et al. 2023). Preculture of cells on a high osmoticum medium, either liquid or semisolid (Meijer et al. 1991; Lambardi et al. 2008; Bradï et al. 2023), has been shown to improve recovery. Effects of inoculum size and density have also been reported, although mostly with cell suspension cultures (Marum et al. 2004; Nausch and Buyel 2021). In light of the above reports, mannitol in our callus culture medium N6M appears to have played a critical role, as has been reported for sorbitol by Ma et al. (2023). The presence of 3% mannitol in the culture medium provided sufficient osmotic stress to keep the cell moisture content at a lower level and made the callus friable, resulting in small but compact masses of globular calli, each capable of plant regeneration. The small and compact nature also reduced mechanical damage during handling.
Vitrification is an important step in a cryopreservation protocol that reduces cell water content before freezing, thereby preventing ice crystallization. Samuels et al. (2023) suggested the use of a combination of cryoprotectants based on their study of cell penetration of cryoprotectants vis-à-vis cell response time. This finding supports our choice of vitrification solution, which contained DMSO, PEG, and glucose. Zhang et al. (2024) evaluated several aspects of cryopreservation of a petunia hybrid callus culture and concluded that gradual and gentle dehydration improves cell survival. Our results also support this view, as we maintained the callus under moderate osmotic stress and used a longer vitrification time of one hour. Shaking was used to facilitate uniform penetration of cryoprotectants, which also helped in the removal of loose cells from the surface of compact calli.
Both direct freezing in liquid nitrogen (Moukadiri et al. 2002; Jain et al. 1996) and slow cooling before freezing (Moukadiri et al. 2002) have been used with rice callus, and it was shown that direct freezing is better. However, we did not observe a similar result neither in O. rufipogon (Zeliang et al. 2010) nor in the present experiment. Reports suggest that cryoprotectants enter rice cells within minutes of application (Samuels et al. 2021, 2023). However, the study was performed at room temperature. Since precooling is done at a much lower temperature, it appeared safe to us to use a longer incubation time. Slow cooling was also suggested as an ideal method for cells with higher water content (Wesley-Smith et al. 2004). The cryocooler used in our experiment has been shown to be an effective replacement for a programmable freezer (Martinez-Montero et al. 1998; Zeliang et al. 2010). Thawing and post-thaw culture are two very important steps in a cryopreservation protocol. Rapid thawing, fast removal of cryoprotectants, and gradual rehydration, as used in this study, have been shown to minimize physical and chemical damage (Lynch et al. 1994; Panis 2019; Ma et al. 2023; Popova et al. 2023). The use of an iron-chelating agent (Benson et al. 1995) and ammonium-free medium (Kuriyama et al. 1989; Lynch et al. 1994) has been reported to improve post-thaw recovery of rice suspension cells. However, in our experiment, the regeneration percentage (Table 1, rows 2–4) was comparable to their results, even though ammonium was not completely removed. It should be noted here that survival (callus growth) was reported in their experiment, which is usually higher than regeneration (Martinez-Montero and Harding 2015).
Lynch et al. (1994), in a comprehensive study with embryogenic and non-embryogenic rice cell suspensions, showed significantly higher post-thaw viability and callus growth in embryogenic cell lines. The cell line that was embryogenic prior to the experiment showed lower viability than embryogenic cell lines but higher viability than non-embryogenic ones. Although there may be several reasons for non-embryogenicity in rice cell lines, age (duration of subculture) is an important factor. This finding indirectly supports the result of our study, where the regeneration percentage from cryopreserved calli is significantly higher in 3–4 month-old calli (6–8 subcultures) than in 5–9 month-old calli (11–18 subcultures, Table 1).
Phenotypic and genotypic fidelity of recovered/regenerated plants are crucial to the routine use of cryopreservation techniques. We evaluated the regenerated plants in the R1 and R2 generations to avoid the unstable variations usually observed in R0 plants (Meijer et al. 1991). In the R1 generation, variations were seen in the seed physical characteristics (Table 2) of cryopreservation-derived and VF calli-derived plants when compared with seed-derived plants. The extent of these variations is comparable to previous reports of somaclonal variations in rice (Sun et al. 1994; Zeliang et al. 2010). Studies with cryopreserved seeds of sorghum (Villalobos et al. 2019) and maize (Arguedas et al. 2018) also reported non-significant variations in the regenerated plants. There is no report in rice where cryopreservation-derived plants were followed up to the R2 generation. We assessed plants in the R2 generation to ensure that recessive mutations, if any, would become homozygous and express. However, in the R2 generation, the variations observed in the R1 also disappeared, suggesting that the variations were unstable. A similar observation was made by Martinez-Montero et al. (2002), who reported the disappearance (at the late growth stage) of initial variations observed at the early growth stage of Saccharum officinalis cryopreservation-derived plants. Our results also suggest that somaclonal variations in rice regenerants, if observed, should be followed up to the R2 generation for confirmation.
Studying the genetic fidelity of tissue culture-derived plants has become feasible due to the availability of numerous marker systems (Martinez 2018) and advancements in DNA sequencing technology (Ichikawa et al. 2023). Gupta and Varsney (1999) suggested that DNA markers suitable for fingerprinting can be used for assessing genetic fidelity. We used 30 SSR markers recommended for diversity analysis of O. sativa (AA genome) and shown to detect variations in cryopreservation-derived plants (Zeliang et al. 2010). The cost and complexity involved in whole genome sequencing of the number of individual plants handled also influenced our choice of SSR markers. The percent change in banding pattern, compared to seed-derived plants, of 3.6% in the VF calli-derived plants is lower compared to previous reports of tissue culture-derived (Yang et al. 1999; Gao et al. 2009) or cryopreservation-derived (Zeliang et al. 2010) rice plants. The 8.61% variation in SSR banding patterns observed in the cryopreservation-derived plants is higher than the above reports but similar to the RAPD variations observed by Moukadiri et al. (1999b), who suggested the variations were either physiological or epigenetic. Wang et al. (2021) reviewed epigenetic and genetic changes in cryopreservation-derived plants of several species and concluded that the epigenetic changes are reversible and plants can revert back to their normal state. Whole genome sequencing of rice somaclones has shown that single nucleotide polymorphisms (Ichikawa et al. 2023) as well as DNA methylation-related epigenetic changes (Wang et al. 2022) do occur (which may be the reason for banding pattern variations in our study). Both studies suggested that such changes do not result in clear morphological changes, which supports our observation as well.
DNA banding pattern variations observed in our case can come from the seed source (Ichikawa et al. 2023), the tissue culture process, or cryogenic treatment. To avoid seed-derived variation, a single seed-derived starting material was used, and panicles of R0, R1, and R2 were bagged. To avoid overestimation of variations due to cryogenic treatment, a cryoprotectant-treated (VF) control group was also kept apart from the seed-derived control. Thus, it appears that the additional 5% change in banding pattern in cryopreservation-derived plants is probably caused by freezing. Such changes were either due to reversible DNA methylation or SNPs in the non-coding region, as no phenotypic effect was seen in the R2 generation. To conclude, we presented a protocol for the cryopreservation of rice mature embryo-derived calli and determined the optimal calli age for the highest plant regeneration. We have also demonstrated that the phenotypic variations observed in the progeny of regenerants are not stable over generations. Genotypic variations observed in the progeny are either very small or reversible.
Data availability
All data are included in the manuscript and tables.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- ABA:
-
Abscicic Acid
- ANOVA:
-
Analysis of variance
- BAP:
-
Benzyl amino purine
- CD:
-
Critical difference
- LSD:
-
Least significant difference
- NAA:
-
α-Napthaleneacetic acid
- PCR:
-
Polymerase chain reaction
- PGR:
-
Plant growth regulator
- TDZ:
-
Thiadizuron
References
Arguedas M, Villalobos A, Gómez D, Hernández L, Zevallos BE, Cejas I, Yabor L, Martínez-Montero ME, Lorenzo JC (2018) Field performance of Cryopreserved seed-derived Maize plants. CryoLetters 39:366–370
Benelli C (2021) Plant cryopreservation: a look at the present and the future. Plants 10:2744. https://doi.org/10.3390/plants10122744
Benson EE (2008) Cryopreservation of phytodiversity: a critical appraisal of theory & practice. Crit Rev Plant Sci 27:141–219. https://doi.org/10.1080/07352680802202034
Benson EE, Lynch PT, Jones J (1995) The use of the iron chelating agent desferrioxamine in rice cell cryopreservation: a novel approach for improving recovery. Plant Sci 110:249–258. https://doi.org/10.1016/0168-9452(95)04201-5
Bhattacharjee B, Pattanayak A, Gupta HS (1998) Fertile plant regeneration from suspension culture derived protoplasts of two indica rice lines and field evaluation of the seed progeny. J Genet Breed 52:135–141
Bomal C, Tremblay FM (2000) Dried cryopreserved somatic embryos of two picea species provide suitable material for direct plantlet regeneration and germplasm storage. Annals Bot 86:177–183. https://doi.org/10.1006/anbo.2000.1176
Bradaï F, Almagro-Bastante J, Sánchez-Romero C (2023) Effect of sucrose preculture and culture inoculum density on cryopreservation of olive somatic embryos. Sci Hort 322:112385. https://doi.org/10.1016/j.scienta.2023.112385
Cho JS, Hong SM, Sung-Yeon Joo SY, Yoo JS, Kim DI (2007) Cryopreservation of transgenic rice suspension cells producing recombinant hCTLA4Ig. Appl Microbiol Biotechnol 73:1470–1476. https://doi.org/10.1007/s00253-006-0627-8
Cincotti A, Fadda S (2012) Modelling the cryopreservation process of a suspension of cells: the effect of a size-distributed cell population. In: Geris L (ed) Computational modeling in tissue engineering. Studies in mechanobiology, tissue engineering and biomaterials, vol 10. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8415_2012_134
Cornejo MJ, Wong VL, Blechl AE (1995) Cryopreserved callus: a source of protoplasts for rice transformation. Plant Cell Rep 14:210–214. https://doi.org/10.1007/bf00233635
Diengdoh RV, Kumaria S, Das MC (2019) Antioxidants and improved regrowth procedure facilitated cryoconservation of Paphiopedilum Insigne Wall. Ex. Lindl —An Endanger Slipper Orchid Cryobiology 87:60–67. https://doi.org/10.1016/j.cryobiol.2019.02.003
Finkle BJ, Ulrich JM (1982) Cryoprotectant removal temperature as a factor in the survival of frozen rice and sugarcane cells. Cryobiol 19:329–335. https://doi.org/10.1016/0011-2240(82)90161-4
Finkle BJ, Ulrich JM (1983) Protocols of cryopreservation. In: Evans DA (ed) Hand book of plant cell culture, vol I. Macmillan Publishing Co, New York, pp 806–815
Gao DY, Vallejo VA, He B, Gai YC, Sun LH (2009) Detection of DNA changes in somaclonal mutants of rice using SSR markers and transposon display. Plant Cell Tiss Organ Cult 98:187–196. https://doi.org/10.1007/s11240-009-9551-9
Grout BWW (1995) Introduction to the in vitro preservation of plant cells, tissues and organs. In: Grout B (ed) Genetic preservation of plant cells in vitro, Springer Lab manuals. Springer, Berlin, Heidelberg, pp 1–20. https://doi.org/10.1007/978-3-642-78661-7_1
Gupta HS, Pattanayak A (1993) Plant regeneration from mesophyll protoplasts of rice (Oryza sativa L). Bio/Technology 11:90–94. https://doi.org/10.1038/nbt0193-90
Gupta PK, Varsney RK (1999) Molecular markers for genetic fidelity during micropropagation and germplasm conservation. Curr Sci 76:1308–1310
Hakkinen ST, Reuter L, Nuorti N, Joensuu JJ, Rischer H, Ritala A (2018) Tobacco BY-2 media component optimization for a cost-efficient recombinant protein production. Front Plant Sci 9:45. https://doi.org/10.3389/fpls.2018.00045
Harding K (2004) Genetic integrity of cryopreserved plant cells: a review. CryoLetters 25:3–22
He G, Shu L, Liao L, Yin X, Sheng L, Wang X (1998) Somatic cell preservation and protoplast regeneration of important disease resistant wild rice Oryza Meyeriana Baill. Sci China 41:393–399. https://doi.org/10.1007/bf02882739
Helliot B, Madur D, Dirlewanger E, de Boucaud MT (2002) Evaluation of genetic stability in cryopreserved Prunus. Vitro Cell Dev Biol Plant 38:493–500. https://doi.org/10.1079/ivp2002325
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282. https://doi.org/10.1046/j.1365-313X.1994.6020271
Ichikawa M, Kato N, Toda E, Kashihara M, Ishida Y, Hiei Y, Isobe SN, Shirasawa K, Hirakawa H, Okamoto T, Komari T (2023) Whole-genome sequence analysis of mutations in rice plants regenerated from zygotes, mature embryos, and immature embryos. Breed Sci 73:349–353. https://doi.org/10.1270/jsbbs.22100
IRGSP (2005) International Rice genome sequencing project. The map-based sequence of the rice genome. Nature 436:793–800. https://doi.org/10.1038/nature03895
Ishikawa M, Tandon P, Suzuki M, Yamaguishi-Ciampi A (1996) Cryopreservation of bromegrass (Bromus inermis Leyss) suspension cultured cells using slow prefreezing and vitrification procedures. Plant Sci 120:81–88. https://doi.org/10.1016/s0168-9452(96)04474-3
Jain SM, Maluszynski M (2004) Induced mutations and biotechnology in improving crops. In: Mujib A (ed) In vitro application in crop improvement. CRC, pp 187–220. https://doi.org/10.1201/9781482280111
Jain S, Jain RK, Wu R (1996) A simple and efficient procedure for cryopreservation of embryonic cells of aromatic indica rice varieties. Plant Cell Rep 15:712–717. https://doi.org/10.1007/BF00231931
Kaity A, Ashmore Se, Drew RA (2009) Field performance evaluation and genetic integrity assessment of cryopreserved papaya clones. Plant Cell Rep 28:1421–1430. https://doi.org/10.1007/s00299-009-0742-y
Karki U, Fang H, Guo W, Cofre CU, Xu J (2021) Cellular engineering of plant cells for improved therapeutic protein production. Plant Cell Rep 40:1087–1099. https://doi.org/10.1007/s00299-021-02693-6
Kulus D, Tymoszuk A (2021) Gold nanoparticles affect the cryopreservation efficiency of in vitro-derived shoot tips of bleeding heart. Plant Cell Tiss Organ Cult 146:297–311. https://doi.org/10.1007/s11240-021-02069-4
Kulus D, Rewers M, Serocka M, Mikuta A (2019) Cryopreservation by encapsulation-dehydration affects the vegetative growth of chrysanthemum but does not disturb its chimeric structure. Plant Cell Tiss Organ Cult 138:153–166. https://doi.org/10.1007/s11240-019-01614-6
Kuriyama A, Watanabe K, Ueno S, Mitsuda H (1989) Inhibitory effects of ammonium ion on recovery of cryopreserved rice cells. Plant Sci 64:231–235. https://doi.org/10.1016/0168-9452(89)90028-9
Lambardi M, Ozudogru EA, Benelli C (2008) Cryopreservation of embryogenic cultures. In: Reed BM (ed) Plant Cryopreservation: a practical guide. Springer Science + Business Media, New York, pp 177–210. https://doi.org/10.1007/978-0-387-72276-4_9
Lynch PT, Benson EE (1990) Cryopreservation: a method for maintaining plant regeneration capability of rice cell suspension cultures. Rice Gen II. IRRI, Manila, Philippines, pp 321–332 https://doi.org/10.1142/9789812814272_0034
Lynch PT, Benson EE, Jones J, Cocking EC, Power JB, Davey MR (1994) Rice cell cryopreservation: the influence of culture methods and the embryogenic potential of cell suspensions on post-thaw recovery. Plant Sci 98:185–192. https://doi.org/10.1016/0168-9452(94)90008-6
Lynch PT, Benson EE, Jones J, Cocking EC, Power JB, Davy MR (1995) The embryogenic potential of rice cell suspensions affects their recovery following cryogenic storage. Euphytica 4:1–3. https://doi.org/10.1007/bf00023966
Ma M, Wang X, Zhang C, Pak S, Wu H, Yang J, Li C (2023) Enhancing the cryopreservation system of Larch embryogenic culture by optimizing pre-culture, osmoprotectants, and rapid thawing. Forests 14:1621. https://doi.org/10.3390/f14081621
Martínez O (2018) Selection of molecular markers for the estimation of somaclonal variation. In: Loyola-Vargas V, Ochoa-Alejo N (eds) Plant Cell Culture protocols. Methods in Molecular Biology: 1815. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-8594-4_6
Martinez-Montero ME, Harding K (2015) Cryobionomics: Evaluating the concept in plant cryopreservation. In: Barh D, Khan M, Davies E (eds) Plant Omics: The Omics of Plant Science, Springer, New Delhi, India, pp. 655–682 https://doi.org/10.1007/978-81-322-2172-2_23
Martínez-Montero ME, González-Arnao MT, Borroto-Nordelol C, Puentes-Díaz C, Engelmann F (1998) Cryopreservation of sugarcane embryogenic callus using a simplified freezing process. CryoLetters 19:171–176
Martínez-Montero ME, Mora N, Quiñones J, GonzáLez-arnao MT, enGeLMann F, Lorenzo JC (2002) Effect of cryopreservation on the structural and functional integrity of cell membranes of sugarcane (Saccharum sp.) embryogenic calluses. CryoLetters 23:237–244
Marum L, Estêvão C, Oliveira MM, Amâncio S, Rodrigues L, Miguel C (2004) Recovery of cryopreserved embryogenic cultures of maritime pine - effect of cryoprotectant and suspension density. CryoLetters 25:363–374
Meijer EGM, Iren Ev, Schrijnemakers E, Hensgens LAM, Zijderveld Mv, Schilperoort RA (1991) Retention of the capacity to produce plants from protoplasts in cryopreserved cell lines of rice (Oryza sativa L). Plant Cell Rep 10:171–174. https://doi.org/10.1007/bf00234288
Monk M (1990) Variation in epigenetic inheritance. Trends Genet 6:110–114. https://doi.org/10.1016/0168-9525(90)90124-O
Moukadiri O, Deming J, O’Connor JE, Cornejo MJ (1999a) Phenotypic characterization of progenies of rice plants derived from cryopreserved calli. Plant Cell Rep 18:625–632. https://doi.org/10.1007/s002990050633
Moukadiri O, Lopes CR, Cornejo MJ (1999b) Physiological and genomic variations in rice cells recovered from direct immersion and storage in liquid nitrogen. Physiol Plant 105:442–449. https://doi.org/10.1034/j.1399-3054.1999.105308.x
Moukadiri O, O´Connor JE, Cornejo MJ (2002) Effect of the cryopreservation procedures on recovered rice cell populations. CryoLetters 23:11–20
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 15:473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Nausch H, Buyel JF (2021) Cryopreservation of plant cell cultures– diverse practices. New BIOTECH 62:86–95. https://doi.org/10.1016/j.nbt.2021.02.002
Panis B (2019) Sixty years of plant cryopreservation: from freezing hardy mulberry twigs to establishing reference crop collections for future generations. Acta Hortic 1234:1–8. https://doi.org/10.17660/ActaHortic.2019.1234.1
Park D, Park SH, Kim YS, Choi BS, Kim JK, Kim NS, Choi IK (2019) NGS sequencing reveals that many of the genetic variations in transgenic rice plants match the variations found in natural rice population. Genes Genomics 41:213–222. https://doi.org/10.1007/s13258-018-0754-5
Pe PPW, Naing AH, Kim CK, Park KI (2021) Antifreeze protein improves the cryopreservation efficiency of Hosta capitata by regulating the genes involved in the low-temperature tolerance mechanism. Horticulturae 7:82. https://doi.org/10.3390/horticulturae7040082
Popova E, Kulichenko I, Kim HH (2023) Critical role of regrowth conditions in post-cryopreservation of in vitro plant germplasm. Biology 12:542. https://doi.org/10.3390/biology12040542
Qin Y, Shin KS, Woo HJ, Lim MH (2018) Genomic variations of rice regenerants from tissue culture revealed by whole genome re-sequencing. Plant Breed Biotech 6:426–433. https://doi.org/10.9787/pbb.2018.6.4.426
Ren L, Zhang D, Jiang XN, Gai Y, Wang WM, Reed BM, Shen XH (2013) Peroxidation due to cryoprotectant treatment is a vital factor for cell survival in Arabidopsis cryopreservation. Plant Sci 212:37–47. https://doi.org/10.1016/j.plantsci.2013.07.011
Ren L, Deng S, Chu Y, Zhang Y, Zhao H, Hairong Chen, Zhang D (2020) Single-wall carbon nanotubes improve cell survival rate and reduce oxidative injury in cryopreservation of Agapanthus praecox embryogenic callus. Plant Methods 16:130. https://doi.org/10.1186/s13007-020-00674-6
Ren L, Wang MR, Wang QC (2021) ROS-induced oxidative stress in plant cryopreservation: occurrence and alleviation. Planta 254:124. https://doi.org/10.1007/s00425-021-03784-0
Sakai A (1956) Survival of plant tissue of super-low temperatures. Low Temp Sci B 1956(14):17–23
Sala F, Cella R, Rollo F (1979) Freeze preservation of rice cell grown in suspension culture. Plant Physiol 45:170–176. https://doi.org/10.1111/j.1399-3054.1979.tb01682.x
Samuels FMD, Stich DG, Bonnart R, Volk GM, Levinger NE (2021) Non-uniform distribution of cryoprotecting agents in rice culture cells measured by CARS microscopy. Plants 10:589. https://doi.org/10.3390/plants10030589
Samuels FMD, Pearce KC, Soderlund S, Stich DG, Bonnart R, Volk GM, Levinger NE (2023) Direct observation of common cryoprotectant permeation into rice callus by CARS microscopy. Cell Rep Phy Sci 4:101469. https://doi.org/10.1016/j.xcrp.2023.101469
Schillberg S, Raven N, Spiegel H, Rasche S, Buntru M (2019) Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front Plant Sci 10:720. https://doi.org/10.3389/fpls.2019.00720
Sun LH, Wang YF, Jiang N, Li HB (1994) A recessive tall culm somatic mutant with wide compatibility in rice (Oryza sativa L). Acta Genet Sin 21:67–73
Uchendu EE, Leonard SW, Traber MG, Reed BM (2010) Vitamins C and E improve regrowth and reduce lipid peroxidation of blackberry shoot tips following cryopreservation. Plant Cell Rep 29:25–35. https://doi.org/10.1007/s00299-009-0795-y
Villalobos A, Arguedas M, Escalante D, Martínez J, Zevallos BE, Cejas I, Yabor L, Martínez-Montero ME, Sershen, Lorenz JC (2019) Cryopreservation of sorghum seeds modifies germination and seedling growth but not field performance of adult plants. J Appl Bot Food Qual 92:94–99. https://doi.org/10.5073/JABFQ.2019.092.013
Wang J, Liu F, Huang C, Yan Q, Zhang X (1998) Observation on ultrastructural change of embryonic suspension cells cryopreserved by vitrification in rice (O. sativa L). Chin Rice Res Newslett 6:5–6
Wang JH, Bian HW, Zhang YX, Cheng HP (2001) The dual effect of antifreeze protein on cryopreservation of rice (Oryza sativa L.) embryogenic suspension cells. CryoLetters 22:175–182
Wang MR, Bi W, Shukla MR, Ren L, Hamborg Z, Blystad DR, Saxena PK, Wang QC (2021) Epigenetic and genetic integrity, metabolic stability, and field performance of cryopreserved plants. Plants 10:1889. https://doi.org/10.3390/plants10091889
Wang N, Yu Y, Zhang D, Zhang Z, Wang Z, Xun H, Li G, Liu B, Zhang J (2022) Modification of gene expression, DNA methylation and small RNAs expression in rice plants under in vitro culture. Agronomy 12:1675. https://doi.org/10.3390/agronomy12071675
Wei Q, Shi P, Khan FS, Htwe YM, Zhang D, Li Z, Wei X, Yu Q, Zhou K, Wang Y (2023) Cryopreservation and cryotolerance mechanism in zygotic embryo and embryogenic callus of oil palm. Forests 14:966. https://doi.org/10.3390/f14050966
Wesley-Smith J, Walters C, Berjak P, Pammenter NW (2004) The influence of water content, cooling and warming rate upon survival of embryonic axes of Poncirus trifolia (L). CryoLetters 25:129–138
Widholm JM (1972) The use of Fluorescein Diacetate and Phenosafranine for determining viability of cultured plant cells. Stain Technol 47:189–194
Yang H, Tabei Y, Kamad H, Kayano T, Takaiwa F (1999) Detection of somaclonal variation in cultured rice cells using digoxigenin based random amplified polymorphic DNA. Plant Cell Rep 18:520–526. https://doi.org/10.1007/s002990050615
Zeliang PK, Pattanayak A, Iangrai B, Khongwir E, Sarma BK (2010) Fertile plant regeneration from cryopreserved calli of Oryza rufipogon Griff. And assessment of variation in the progeny of regenerated plants. Plant Cell Rep 29:1423–1433. https://doi.org/10.1007/s00299-010-0932-7
Zeng BY, Wang Z, Zhang YF, Yang Q, Lu W (2009) Cryopreservation of rice (Oryza sativa L.) embryonic cell suspensions by encapsulation-dehydration. Plant Physiol Commun 45:603–606
Zhang ZH, Hu ZL (1999) Regenerating plants from cryopreserved adventitious buds of haploids in rice. Wuhan Univ J Nat Sci 4:115–117. https://doi.org/10.1007/bf02827638
Zhang YX, Wang JH, Bian HW, Zhu M (2001) Pregrowth–desiccation: a simple and efficient procedure for the cryopreservation of rice (Oryza sativa L.) embryogenic suspension cells. CryoLetters 22:221–229
Zhang D, Wang Z, Wang N, Gao Y, Liu Y, Wu Y, Bai Y, Zhang Z, Lin X, Dong Y et al (2014) Tissue culture-induced heritable genomic variation in rice, and their phenotypic implications. PLoS ONE 9:e96879. https://doi.org/10.1371/journal.pone.0096879
Zhang D, Ren L, Chen G-Q, Zhang J, Reed BM, Shen X-H (2015) ROS-induced oxidative stress and apoptosis-like event directly affect the cell viability of cryopreserved embryogenic callus in Agapanthus praecox. Plant Cell Rep 34:1499–1513. https://doi.org/10.1007/s00299-015-1802-0
Zhang Y, Guo X, Song J, Chen G, Shen X (2024) Enhancing cryopreservation survival in Petunia × Calibrachoa ‘Light yellow’ callus: insights into material characteristics and genetic integrity. Cryobiol 114:104846. https://doi.org/10.1016/j.cryobiol.2024.104846
Acknowledgements
The authors sincerely thank the Director, ICAR Research Complex for NEH Region for providing facilities. The work was funded by grant no. BT/PR2768/AGR/142/2001 of Department of Biotechnology, Government of India to AP. We thank the anonymous reviewers whose suggestions have helped to improve the manuscript to the present form.
Author information
Authors and Affiliations
Contributions
Both PKZ and AP contributed equally at all stages of planning, research, data collection, data analysis and manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Qiao-Chun Wang.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zeliang, P.K., Pattanayak, A. Cryopreservation of embryogenic callus in Oryza sativa L.: Assessment of impact of callus age on regeneration; morphological and genetic stability regenerants. Plant Cell Tiss Organ Cult 158, 23 (2024). https://doi.org/10.1007/s11240-024-02821-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11240-024-02821-6