
Abstract
Cryopreservation is a reliable technique for the long-term storage and preservation of embryogenic cells, maintaining their viability without loss of their embryogenic capacity. However, the large-scale conservation of grapevine embryogenic lines in cryobanks remains limited. A significant challenge is understanding somatic cell rejuvenation. Here, we investigate the encapsulation/dehydration and encapsulation/vitrification for cryopreserving embryogenic material. Cell rejuvenation and enhanced embryogenic competence were observed after cryopreservation, as evidenced through structural cellular changes observed by histology and electron scanning microscopy. Results showed that cryopreserved samples of 110-Richter, Riesling, and Tempranillo using encapsulation/dehydration had better survival rates, averaging 81%, 62%, and 48%, respectively, while encapsulation/vitrification yielded lower survival rates, averaging 58%, 42%, and 32%, respectively. Cryopreservation also improved post-thaw recovery and regeneration efficiency assessed through regrowth of proembryogenic masses and somatic embryo conversion reaching 54–72% against 11–17% in control samples. Cryopreservation triggered changes in gene expression patterns and exhibited considerable increase at genotype-specific basis of 1.5- to 4.5-fold in SERK1, BBM, and WOX associated to embryogenic competence as well as in ChitIV and LEA involved in stress response. Membrane stability index, hydrogen peroxide, and proline contents were used as indicators of oxidative stress uncovering a key role of an osmotic trans-priming effect leading to cryotolerance. Our finding highlighted that cryopreservation enhances embryogenic capacity in senescent callus and probably acts as a screening process allowing safe maintenance of proembryogenic cells and promoting their recovery. This study provides a high throughput innovation to set up cryolines for cell rejuvenation of grapevine and other important plant species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cryopreservation, the preservation of biological material at ultra-low temperatures, is a valuable tool for conserving field collections of vegetatively propagated plants requiring minimal space and maintenance (Engelmann 2004). To conserve these genetic resources, many gene banks rely on cryopreservation for long-term storage of biological material in liquid nitrogen (LN) at − 196 °C, since it is believed that all metabolic, cellular, and physiological processes are arrested under these conditions. This method has been particularly successful in maintaining the samples’ viability through osmotic dehydration followed by ultra-rapid freezing, thereby preventing the formation of cellular damaging ice crystals (Benelli 2021). Significant progress has been made over the past decades in cryopreserving numerous plant species, ensuring their germplasm preservation with respect of epigenetic integrity, genetic fidelity, and metabolic stability (Wang et al. 2021).
Grapevines (Vitis spp.) are among the most important fruit crops worldwide with an average of cultivated areas that reached about 7.4 million ha in 2019. In addition, grape production attained 77.8 million tons in 2018 (57% of wine grapes, 36% of table grapes, and 7% of dried grapes) with a world wine consumption of 246 million hl (OIV 2020). Grapevine breeding programs require access to hundreds of Vitis cultivars that could be maintained within clonal germplasm repository gene banks to develop and implement in vitro backup collections. In this context, grapevine has also benefited from cryopreservation techniques. Cryopreserved grapevine shoot tips were used to maintain genetic diversity and for cryotherapy-based sanitation to eliminate pathogens including viruses, phytoplasmas, and bacteria (Bettoni et al. 2019; Bettoni et al. 2021; Bi et al. 2018; Markovic et al. 2015; Marković et al. 2018; Wang et al. 2003; Wang and Valkonen 2009).
Somatic embryogenesis (SE) remains the preferred regeneration procedure for grapevine breeding programs, where the source and quality of embryogenic cultures are key factors for successful genetic transformation and plant regeneration (Ben-Amar et al. 2022b; Bouamama et al. 2007). During the last decades, pro-embryogenic masses (PEMs) are raised as the most appropriate material for crop improvement and genetic engineering with new breeding technologies including the genome editing tool (Campos et al. 2021). Cryopreservation of embryogenic cell suspension (ECS) cultures derived from PEMs has been achieved using both encapsulation-dehydration (Ben-Amar et al. 2013; González-Benito et al. 2009; Vasanth and Vivier 2011; Wang et al. 2002) and vitrification-based protocols (Miaja et al. 2004; Wang et al. 2004; Wang and Perl 2006). Such cryogenic technology is often based on tissue culture practice and requires specific handling of explant tissue, osmoprotection, loading/unloading, freeze/thawing, and secure recovery of produced plants. However, the cryopreservation process, along with the inherent stress of in vitro culture (Ben-Amar et al. 2022a), can induce oxidative stress during dehydration and freezing due to the generation of reactive oxygen species (ROS) (Ren et al. 2021). Maintenance of a basal level of ROS as signaling molecules is required for cellular proliferation and physiological functions. Conversely, excessive ROS production results in membrane peroxidation and theatrical cell injuries involving major structural damages and functional alterations (Mittler 2017). During cryopreservation, the generation of ROS can occur through the various steps involved in this process including tissue culture-mediated photo-oxidative stress, osmotic injury, and desiccation by cryoprotective agents (CPAs), as well as during cryostorage and rewarming (Benson and Bremner 2004). Thus, cryogenic-mediated stress during freeze–thaw cycles causes several cellular injuries resulting in structural damages, necrosis, loss of embryogenic capacity, decrease of sample survival, and failure of plant recovery. Advanced attempts for successful cryopreservation were made to overcome stress injury by using a wide range of CPAs for safe storage of embryogenic material leading to stress mitigation. As the priming occurs when a plant tissue’s exposure to a certain stimulus influences its response to a subsequent one, stress-priming studies differentiate (i) cis-priming when both stresses are physically and chemically identical, and (ii) trans-priming when they differ from each other in nature, duration, or strength, resulting in cross-tolerance (Baier et al. 2019). In this purpose, priming was achieved during cryopreservation to induce cryotolerance, in a way that exposure to one stimulus may influence a response to a subsequent stimulus. The loading of plant cells in high sucrose concentrations during 3–4 days (Wang et al. 2002) refers to an osmotic priming that can alleviate cryo-injuries imposed by freezing in extreme low temperatures.
Cryopreservation of embryogenic lines derived from woody species like grapevines offers several advantages, including reduced cost maintenance, minimized risk of somaclonal variation and contamination during regular subcultures (Ballesteros et al. 2024). It has been also known that the age of stock cultures from which explants were used considerably affected the success of cryopreservation (Wang and Perl 2006). Ren et al. (2013) found that survival of cryopreserved seedlings significantly declined when increasing the age of stock seedlings in Arabidopsis. Most interestingly, few previous reports noted an exceptional increase of embryogenic capacity in Vitis vinifera and Gentiana cruciata when PEMs were subjected to cryogenic treatment, probably by cell rejuvenation (Ben-Amar et al. 2013; Mikuła et al. 2011). However, the process by which cryopreserved cells acquire increased embryogenic capacity is still unknown. To our knowledge, the cryopreservation-mediated rejuvenation of ECS in grapevine and other plant species has not been documented, suggesting that cryopreservation may hold the key to keeping cells highly totipotent and suitable for various manipulations. In the present work, we provide evidence that cryopreservation promotes somatic cell rejuvenation and increases embryogenic potential, opening new avenues for grapevine breeding.
Materiel and methods
Plant material, induction of somatic embryogenesis, and establishment of cell suspensions
This study used the grapevine (Vitis vinifera) cultivars Tempranillo (from Spain) and Riesling (from Germany), as well as the rootstock 110 Richter (Vitis berlandieri × Vitis rupestris) (from the USA), were used in this study. Somatic embryogenesis (SE) was induced from anther culture, and the resulting yellow friable pro-embryogenic masses (PEMs) were maintained by secondary embryogenesis via monthly subcultures on solid medium. Explants were surface sterilized under aseptic conditions with ethanol 70% for 1 min, followed by washing in distilled water before being immersed for 20 min in a sodium hypochlorite solution NaOCl at 6%. The explants were rinsed twice with sterile distilled water before being plated on 9 cm Petri dishes containing MS basal medium (Murashige and Skoog 1962) enriched with 30 g/l sucrose and solidified with 3 g/l gelrite. The pH was adjusted to 5.8 before autoclaving at 120 °C, pressure 1.05 kg/cm2 for 20 min. The medium was supplemented with growth regulators 1 mg/l 2,4-dichlorophenoxyacetic acid (2,4-D) and 0.25 mg/l of benzyl amino-purine (BAP), filter sterilized on 0.22 µm millipore as previously reported (Ben-Amar et al. 2007). The cultures were incubated in a climate-controlled growth chamber with a photoperiod of 16 h/8 h light/darkness under white fluorescent light (Mazdafluor 7D TF 36 w/LJ) at a photonic flux density of 100 μmol m−2 s−1 at 24 ± 1 °C in light and 20 ± 1 °C in darkness. Embryogenic cell suspension (ECS) cultures were initiated and established by transferring embryogenic callus into Nitsch and Nitsch (NN, Nitsch and Nitsch 1969) liquid culture containing 20 g/l maltose, 4.6 g/l glycerol, 0.5 g/l 2-N-morpholino-ethane sulfonic acid (MES) and supplemented with 1 mg/l naphtoxy-acetic acid (NOA) and 0.25 mg/l BAP as growth regulators. The cultures were then maintained by weekly subcultures using the conditioned medium procedure according to Ben-Amar et al. (2007).
Cryogenic procedures, thawing, and recovery of cryopreserved material
Six years old embryogenic cultures were used for cryopreservation. Cells harvested 3 days after subculturing from exponentially established ECS, were subjected to these cryogenic procedures. Two cryogenic techniques were tested to cryopreserve embryogenic material in liquid nitrogen (LN): encapsulation/dehydration (Encap/deh) and encapsulation/vitrification (Encap/vit).
-
(i)
Encapsulation-dehydration (Encap/deh) was achieved as described previously (Ben-Amar et al. 2013). Briefly, PEMs were precultured in NN liquid medium with increasing sucrose concentrations (0.25 M; 0.5 M; 0.75 M and 1 M sucrose) for dehydration. Then, the cells were harvested and encapsulated within alginate beads in Na-alginate solution in presence of 0.1 M CaCl2 solution. The latter induces crosslinking of the alginate monomers resulting in the formation of alginate beads (~ 4 mm in diameter and PCV 30% (m/v)) that were subjected to air-flow desiccation for 5 h until the moisture content drops to 20–30%, and then placed in 2 ml sterile cryovials (10 beads per vial) before being plunged rapidly in LN at − 196 °C.
-
(ii)
Encapsulation-vitrification (Encap/vit) was carried out following the protocol of Wang et al. (2004) with some modifications. ECS were precultured with increasing sucrose concentrations (0.25 M; 0.5 M; 0.75 M and 1 M sucrose), each concentration for 1 day, before being suspended in 2.5% (w/v) Na-alginate and 0.4 M sucrose. Cells were encapsulated into beads (4 mm in diameter) by dripping in 0.1 M CaCl2 solution with 0.4 M sucrose. The beads were incubated in loading solution (NN, 2 M glycerol, 0.4 M sucrose) for 20 min followed by exposure to plant vitrification solution 2 (PVS2), which consists of 30% (w/v) glycerol, 15% (w/v) ethylene glycol, and 15% (w/v) DMSO in basal culture medium containing 0.4 M sucrose (Sakai et al. 1990), for 120 min at 0 °C, prior to direct immersion in LN.
Here, the loading in cryoprotectant agents (CPAs) is intended to act as an osmotic priming prior to immersion in LN using both approaches. Once thawed in a water bath at 40 °C for 5 min, beads are often crushed to release the cells after being in culture for several days. The recovery of cryopreserved samples was achieved during 8 weeks by transferring cryopreserved beads and embryo clusters to solid MS basal medium supplemented with 30 g/l sucrose, 3 g/l gelrite, 2.5 g/l activated charcoal, and 1 mg/l 2,4-D/0.25 mg/l BAP according to Wang and Perl (2006). Each treatment was replicated six times (6 biological replicates) and repeated twice (2 technical replicates).
Induction of plant regeneration in suspension culture
Cryopreserved embryogenic calli were transferred into NN liquid culture at a rate of 1 g fresh weight per 50 ml medium in 250 ml flasks. Liquid cultures were initiated based on conditioned medium procedure as described previously (Ben-Amar et al. 2007) to establish suspensions with efficient cell proliferation. Somatic embryo differentiation and conversion into plantlets were carried out in liquid culture by daily removal of the entire culture medium according to Ben-Amar et al. (2022b).
MTT cell viability assay and survival assessment
The diphenyl tetrazolium bromide (MTT)-based assay relies mainly on dehydrogenase conversion of the dye into insoluble formazan by living cells. ECS seeded to 24-well plates (1 ml/well) in presence of MTT solution at a final concentration of 0.45 mg/ml were incubated at 37 °C for 2 h following the protocol of Kamiloglu et al. (2020). Then, the formazan crystals were dissolved in 1 ml of 40% (v/v) dimethylformamide containing 2% (v/v) glacial acetic acid and the colored product was quantitatively measured calorimetrically at 570 nm with a multiplate reader as mentioned by Mosmann (1983). The intensity of colored product was directly proportional to the level of cell viability that was calculated according to Kamiloglu et al. (2020) using the following equation: % viability = (mean ODsample / mean ODblank) × 100.
Survival rate was expressed by the percentage of developing cryopreserved samples showing cell mass growth 2 months post-freezing according to Ben-Amar et al. (2013). Non-cryopreserved samples (− LN) were taken as control. Each experiment was carried out with six replicates.
Histological sections
SE developmental stages were studied histologically. PEMs as well as somatic embryos at different developmental stages were fixed in formol for more than 24 h, dehydrated in serial ethanol grades [70° (3 × 10 min), 95° (3 × 10 min), and 99° (2 × 25 min)] and then embedded in paraffin wax. Serial sections of 0.7–1.2 μm thickness were cut with a microtome and stained with hematoxylin. Sections were observed and photographed under appropriate magnifications (× 4, × 10, and × 20) using a Leica 3125 microscope (Leica GmbH, Germany) as indicated by Bouamama et al. (2011). Observations were performed on control and cryopreserved PEMs with encapsulation/dehydration-based cryopreservation procedure.
Environmental scanning electron microscopy (ESEM)
Samples of somatic embryos (0.5 cm diameter) were mounted on aluminum stubs with double sided adhesive tape and directly observed using an ESEM (Fei company, Oregon, USA) suitable for biological observation, without prior chemical fixation. Secondary electron images were taken at an accelerating voltage of 10 to 15 kV and a vapor pressure of 0.7–0.8 Torr according to Bouamama-Gzara et al. (2019). Observations were performed on control and cryopreserved PEMs with encapsulation/dehydration procedure.
RNA extraction and differential gene expression analysis
Total RNAs from in vitro plant material at 2 weeks post-recovery from cryopreservation were extracted using Trizol® reagent (Invitrogen™, CA, USA) following the manufacturer’s instructions. RNA concentration, purity, and integrity were determined. RNA preparation, DNase treatment, and cDNA synthesis were performed according to Ben-Amar et al. (2022a). PCR amplifications were carried out in GeneAmp9700® Thermal Cycler (Applied Biosystems, CA, USA) using 10 µl of RT-Mix buffer (2 ×), 0.4 μl of each of primer (10 μM), and 0.4 μl of SuperScript-III One-Step RT-PCR Taq DNA Polymerase (Invitrogen™, CA, USA) with 1 µl of template RNA (500 ng/µl) in a final volume of 20 µl. The reaction conditions were as follows: Reverse transcription at 50 °C for 30 min, denaturation at 94 °C for 2 min followed by 32 cycles of 94 °C for 15 s, 55 °C for 30 s, and 68 °C for 45 s, and final extension at 68 °C for 4 min. At the end of the PCR cycles, the amplified product was run on a 1.2% agarose gel with a 1 Kb DNA ladder (Fermentas, Germany). Differential expression of selected genes was quantified against an internal reference gene, VvEF1 (Vitis vinifera elongation factor 1) through digital image densitometry analysis of electrophoretic gel using the ImageJ software (NIH, Bethesda, USA). The sequences of RT-PCR primers are listed in Table 1.
Estimation of hydrogen peroxide, proline content, and cell membrane stability index
Hydrogen peroxide (H2O2) assay was performed as described by Velikova et al. (2000). Briefly, 100 mg of plant material was frozen in LN and ground to powder with 1 ml 0.1% (w/v) thrichloroacetic acid (TCA). The mixture was centrifuged at 10 000 × g for 20 min at 4 °C and the supernatant was treated with 1 M potassium iodide in the presence of phosphate buffer (10 mM, pH 7.0). Absorbance was determined at 390 nm using UV–Vis spectrophotometer (Ultrasepec 2000, PharmaciaBiotech, Sweeden). H2O2 content was calculated by comparison with a standard calibration curve, previously plotted using different concentrations of H2O2.
Proline content was determined as described by Bates et al. (1973) and Abraham et al. (2010) after homogenizing ground plant material in cold 3% aqueous sulfo-salicylic acid and quantitatively measured by ninhydrin-based colorimetric assay at 520 nm using toluene as reference. The proline concentration was determined using a calibration curve and calculated on fresh weight basis (expressed as µmol/g fresh weight).
Cell membrane stability index (MSI) reflects the lipid peroxidation of cell membranes and therefore the cellular oxidative damage estimated by measuring the amount of malondialdehyde (MDA) following the method of Hodges et al. (1999). First, MDA was extracted from plant material in 0.1% TCA as described above. After centrifugation, the supernatant was added to 0.1% (w/v) thiobarbituric acid (TBA) in 20% (w/v) TCA and incubated in boiling water for 30 min. The mix was then centrifuged at 10 000 × g for 5 min, before reading the absorbance at 532 nm. The value for non-specific absorption at 600 nm was subtracted. The amount of MDA-TBA red complex was calculated from the extinction coefficient of 155 mM−1 cm−1 and expressed as nmol MDA/g fresh weight.
Data assessment and statistical analysis
Experiments were performed in three biological replicates with three technical repetitions for each sample and the control treatment. All experiments described here were repeated independently twice. Data were expressed as mean value ± SD and were compared by one-way analysis of variance (ANOVA). Significant differences between means for all treatments were assessed by Fisher’s test at 0.05. Histogram followed by different letters are significantly different at p ≤ 0.05. All analyses were performed using the XL-STAT software.
Results
Effectiveness of encapsulation/dehydration and vitrification-based cryopreservation in terms of cell viability, regrowth, and regeneration
To assess the most reliable freezing method intended for cryopreserving grapevine embryogenic material, we compared here the encapsulation/vitrification (Encap/vit) with the encapsulation/dehydration (Encap/deh). Our results, based on an MTT-based viability assay conducted immediately after thawing and again 2 months later on grapevine 110 Richter, demonstrated that Encap/deh was significantly more efficient than Encap/vit in terms of cell viability and post-thaw recovery (Fig. 1). Specifically, Encap/deh yielded a viability rate of 82.16% ± 6.62, against only 40.66% ± 3.00 for Encap/vit (Fig. 1). There was no significant difference in viability between the immediate post-thaw assessment and the 2-month follow-up. The success of cryopreservation relied basically on high post-thaw cell viability together with suitable regrowth and regeneration rates.

MTT-based viability assay of unfrozen (− LN) and cryopreserved (+ LN) grapevine embryogenic material (110-Richter) by encapsulation-dehydration or encapsulation-vitrification method. Viability assay was conducted immediately after thawing, and after 2 months of regrowth. Non-cryopreserved embryogenic material was taken as control. Three repetitions were presented here
While unfrozen control samples exhibited optimal survival (100%), callus regrowth of cryopreserved material was constantly lower, regardless of the method. However, the decrease was less pronounced with Encap/vit than Encap/deh. Regrowth rates for Tempranillo, Riesling, and 110 Richter exposed to Encap/deh were 48%, 62%, and 81% respectively, while Encap/vit resulted in lower rates of 31%, 38%, and 44%, respectively (Table 2). Interestingly, cryopreservation treatment enhanced the regeneration ability of embryogenic cultures, despite the low regeneration efficiency (17%, 9%, and 11% for 110 Richter, Riesling, and Tempranillo, respectively) observed in unfrozen controls due to the advanced age (6 years) of the embryogenic callus. Encap/deh consistently outperformed Encap/vit in terms of regeneration (72%, 60%, and 54% respectively for Encap/deh of 110 Richter, Riesling, and Tempranillo versus 62%, 49%, and 43%, respectively, for Encap/vit), with 110 Richter displaying the highest recovery and regeneration percentages across all cultivars (Table 2).
Based on these findings, the Encap/deh method was selected for cryopreserving grapevine embryogenic cultures. Figure 2 illustrates the complete process using the 110 Richter genotype, showcasing the sequence of the main steps from SE induction to suspension establishment, bead formation, freezing, thawing, recovery, regrowth, and subsequent plant regeneration (Fig. 2).

Encapsulation/dehydration-based cryopreservation of grapevine embryogenic tissues and subsequent plant regeneration. a Anther culture performed to induce somatic embryogenesis. b Embryogenic calli of grapevine rootstock 110 Richter obtained from anther culture and subcultured for multiplication on solid medium. c Cell suspensions established from embryogenic callus in liquid culture. d Culture of alginate encapsulated beads after immersion in LN. e Encapsulated bead in culture immediately after cryogenic procedure. f–i Crushed bead following 2, 4, 6, and 8 weeks of culture, respectively. j Transfer of cryopreserved material to establish embryogenic cell suspension. k–l Embryogenic masses undergoing regeneration process in liquid culture at the globular (k) and torpedo (l) stages. m Elongated mature somatic embryos transferred in solid media regenerating plantlets showing green shoots. n Developed rooted vitroplants from cryopreserved embryogenic material. Scale bar represents 5 mm
Cryopreservation-induced de novo embryogenic stem cell formation as revealed by microscopic and histological analyses
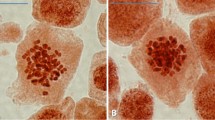
To investigate how cryopreservation enhances embryogenic potential, we examined the morphogenetic changes that increase cell competence through microscopic and histological analyses. We first observed the morphological features of a long-term maintained (6 years old) embryogenic callus of the rootstock 110 Richter, which displayed signs of senescence, before and after being subjected to cryopreservation procedure. ESEM micrographs revealed that non-cryopreserved cells started to lose their embryogenic potential (Fig. 3a). Epidermal cells lost rigidity, the tissue appeared completely collapsed, and embryogenic structures disappeared, leaving behind a disorganized material. In contrast, the same callus subjected to cryopreservation restarted the production of heart-stage and early cotyledonary somatic embryos, demonstrating renewed embryogenic ability (Fig. 3b). At this stage cells of the protodermic layer were well-aligned and organized. Histological staining carried out on the same samples confirmed huge morphogenetic changes. Non-cryopreserved callus showed large, vacuolated cells lightly stained (Fig. 3c), while cryopreserved callus exhibited darkly stained meristematic cells organized into well-shaped embryo structures (Fig. 3d).

Micro-morphological and histological analysis of non-cryopreserved vs. cryopreserved grapevine embryogenic material. a Environmental scanning electron microscopy (ESEM) micrography of long-term callus losing its embryogenic competence. b Histological analysis of long-term maintained callus showing the presence of large and highly vacuolated cells. c ESEM micrograph of grapevine callus revealing high embryogenic potential after cryopreservation. d Histological observation of cryopreserved embryogenic callus showing well-shaped somatic embryo structure with darkly stained meristematic cells
Figure 2 illustrates succession of early somatic embryo developmental stages following cryopreservation. ESEM micrographs revealed several globular and post-globular structures (Fig. 4a). The presence of a protodermic layer, a fibrillar extra-cellular matrix that coats the pro-embryogenic cells, serves as a key marker of the embryonic process. Histological study revealed continuous cell divisions, characterized by dense cytoplasm and prominent nucleus in pro-embryogenic cells (Fig. 4b). Numerous heart, torpedo, and early cotyledonary somatic embryo shapes were noticed (Fig. 4c). Clusters of single somatic embryos emerged from the border of the embryogenic tissue (Fig. 4d) and continued its development into late cotyledonary stage embryos (Fig. 4e) with clearly defined meristematic and radical poles. Further elongation and emergence of the apical meristem, surrounded by primordia, are illustrated in Fig. 4f.

Cryopreserved embryogenic material and sequence of somatic embryo developmental stages. a Embryogenic callus visualized by ESEM analysis. b ESEM micrograph showing a cluster of heart-stage somatic embryos. c Histological analysis of embryogenic tissue showing a cluster of stained meristematic cells with dense nucleus and cytoplasm. d Histological analysis showing several SE shapes. e Cluster of germinated somatic embryos. f Histological analysis showing the bipolar orientation of SE with shoots and root axis and the caulinar meristem
Efficient growth recovery after cryopreservation is associated with alleviation of oxidative stress by osmotic priming
Previous research has demonstrated that applying Encap/deh to cryopreserve grapevine embryogenic cultures resulted in the highest rates of survival and recovery, indicating that the vitrification process may be more aggressive for grapevine cells. Additionally, it was already known that similar environmental conditions, including freezing and osmotic stress, might cause ROS-related oxidative stress. Changes in hydrogen peroxide H2O2, MDA, and proline contents were monitored throughout the cryopreservation process involving (i) CPA loading/pre-freezing in LN, (ii) thawing/recovery, and (iii) 2-month regrowth phases, in order to assess the occurrence of such oxidative stress. The three studied cultivars that were either subjected to Encap/vit or Encap/deh were monitored in comparison to non-cryopreserved samples, which served as controls (Fig. 5). All cultivars demonstrated occurrence of oxidative stress during cryopreservation compared to controls (not subjected to cryoprotective solution). Encap/vit resulted in substantially more stress than Encap/deh, as evidenced by significant increases in MDA, H₂O₂, and proline. A dramatic increase in MDA was observed primarily during the loading phase before LN immersion, followed by a significant rise during the thawing, with no differences between cryopreserved and control samples during regrowth. When comparing the Encap/vit records to the control settings, the MDA increase was often larger (sevenfold increase) at CPA loading than via Encap/deh (fourfold increase) (Fig. 5a). The same behavior was observed for H2O2, which showed a significant increase in the first loading step when osmotic stress was intense, then decreased by roughly half, remaining slightly higher than controls (Fig. 5b). The Encap/deh-based cryopreservation induced less increase in H2O2 content than Encap/vit thereby alleviating cryogenic injury, despite the fact that all studied cultivars responded in the same way. This is possible thanks to proline synthesis that is a potent free amino-acid antioxidant, which makes this possible. In comparison to the untreated control material, the CPA-induced osmotic stress during loading step prior to LN immersion, greatly enhanced proline synthesis. The proline accumulation increased during pre-LN step in Encap/deh but not in Encap/vit (8 and 6 µmol/g FW, respectively) during pre-LN as the most stressful step, with a slight decrease at thawing while retaining the ability to support cryopreserved samples from oxidative injury (Fig. 5c). Such an alleviation of oxidative injury was associated to osmotic priming via CPA loading. Overall, Encap/deh outperformed Encap/vit in terms of reducing cryogenic-induced oxidative stress in the target embryogenic material. Cryopreservation performed efficiently via Encap/deh than by Encap/vit in terms of mitigating cryogenic-induced oxidative stress. Evidence of a priming effect of the osmotic treatment with CPAs was demonstrated as non-treated cultures cannot support freezing temperatures.

Oxidative status of grapevine embryogenic cultures (110 Richter, Tempranillo, and Riesling) during cryopreservation steps as revealed by a MDA content, b hydrogene peroxide (H2O2) content, and c proline content. MDA, H2O2, and proline contents were monitored throughout the three common steps of cryopreservation (i) PreLN osmotic treatment prior to LN immersion, (ii) thawing step following freezing and (iii) regrowth 2 months after thawing of the cryopreserved material. Investigation was carried out among encapsulation/vitrification and encapsulation/dehydration-based cryopreservation against non-cryopreserved material as control. Bars indicate the standard deviation of the mean. Lowercase letters represent significant differences according to the Turkey test (p < 0.05)
Rejuvenation of callus cell lines via cryopreservation with recovery of high embryogenic competence
After cryopreservation process, instead of noticing a tricky restart of callus development, we observed rather an active well-growing cryopreserved callus material with high embryogenic potential in the few weeks post-recovery, as revealed by regeneration rates (Table 2). To understand such results at the molecular basis, we examined the transcript level of some genes related to embryogenic competence (SERK1, SERK2, BBM, WOX, LEA, and ChitIV). Compared to the internal reference gene EF1α, data showed differential expression patterns of these target genes between the controls and the cryopreserved material among the three tested cultivars (“110R” for 110 Richter, “Temp” for Tempranillo, and “Ries” for Riesling) as presented in Fig. 6a. Relative gene expression highlighted a significant increase of 1.5- to twofold for VvSERK1 in cryopreserved material compared to control. Indeed, a raise of 1.5-fold in VvSERK2 was noticed only in cryopreserved 110R, without significant difference for the other cultivars (Fig. 6b). VvBBM and VvWOX were observed to be highly expressed in all cryopreserved material, regardless of the grapevine cultivar, while the expression was markedly amplified in 110R which recorded a considerable increase of 2.7-fold and 4.5-fold for BBM and WOX genes, respectively. Significant boosts of gene expression of VvChitIV involved in stress defense, and VvLEA associated with abiotic stress response, were observed in 110R and Temp (Fig. 6b).

Expression analysis of target genes associated with stem cell maintenance in control (C) and cryopreserved (Cryo) embryogenic calli of three grapevine cultivars: 110-Richter (110R), Tempranillo (Temp), and Riesling (Ries). a RT-PCR analysis of selected target genes against VvEF1α as an internal reference gene in cryopreserved (Cryo) and unfrozen control (C) embryogenic callus material. b Relative expression of VvSERK1, VvSERK2, VvBBM, VvWOX, VvLEA, and VvChitIV gene transcript levels compared with VvEF1α reference gene. Intensity of bands was quantified by image densitometry analysis. The dashed line on the histograms represents the expression level of VvEF1α internal control gene. Asterisks indicate statistically significant differences (p < 0.05) between control (C) and cryopreserved (Cryo) material
Discussion
Cryopreservation of old embryogenic material triggers de novo generation of stem cells with enhanced recovery and regeneration
Cryopreservation is a favored long-term storage method of valuable embryogenic lines already known for their potential in regeneration and genetic manipulation. However, inducing and maintaining these lines can be challenging. Our previous work reported successful cryopreservation of grapevine embryogenic cultures (Ben-Amar et al. 2013), noting a significant regrowth of cryopreserved samples, that has suggested a stimulating effect of cryogenic treatment. Similarly, Mikuła et al. (2011) observed enhanced embryogenic capacity in cryopreserved Gentiana cruciata cell suspension cultures. However, the exact mechanism behind this beneficial effect of freezing on the growth potential of embryogenic material is still unclear. This study investigates the cellular and molecular basis underlying cryopreservation-induced cell rejuvenation. Histological and scanning electron microscopy observations revealed profound cellular and morphogenetic changes, including de novo generation of meristematic cells with prominent nucleus and dense cytoplasm that are able to restart the embryogenic program. Our data showed that callus material was successfully cryopreserved enabling the recovery of "revitalized" cells with high embryogenic potential. This could occur through either the conversion of non-embryogenic cells to PEMs under cryogenic stress, or the selection and promotion of remaining PEMs over non-embryogenic cells. Our findings suggests more plausibly that cryopreservation may act as a selection or screening tool, preserving and promoting PEMs, while discarding damaged non-embryogenic cells. This hypothesis stipulating that cryopreservation differentially affects some subpopulations of cells, was more founded according to earlier studies indicating that the freezing/thawing cycles inflicted cryo-injuries on the parenchymatic regions (non-meristematic cells), while only embryogenic cells survived the treatment (Poobathy et al. 2013). These authors showed that cryogenic treatments affect the survival of cryopreserved cells, while there were no physical damages caused by cryopreservation of protocorn-like bodies in the ornamental orchid Dendrobium.
The viability, regrowth, and regeneration of cryopreserved samples were assessed in comparison to unfrozen controls in both tested cryopreservation methods, namely Encap/deh and Encap/vit. A decrease in survival percentages within Encap/vit may be linked to the chemical toxicity of PVS2 used in vitrification-based method. Previous studies have indicated that dimethyl sulfoxide (DMSO), an aggressive penetrating cryoprotectant CPA used to prevent freezing damage, can negatively impact cell viability (Kobayashi et al. 2006). Despite this, PVS2 remains necessary for some recalcitrant plant species in Encap/vit and droplet vitrification (Bettoni et al. 2019). Exhaustive CPA treatment can cause irreversible cell injury with low recovery. For instance, in the vitrification method, intensive CPA concentrations (8 M) led to reduced cell regrowth after thawing (59% vs. 74% in Encap/deh) as documented by Nausch and Buyel (2021). Current findings also highlight the importance of limiting PVS2 exposure time, as extended incubation significantly reduces survival and viability (van der Walt et al. 2023; Wei et al. 2023). In fact, exposure time is crucial, as PVS2 penetration into plant tissues was directly proportional to exposure time as reported for mint and garlic shoot tips (Volk and Walters 2006). Others authors showed that the regeneration of cryopreserved meristems exposed to PVS3 for more than 10 min was stunted and they died after being severely damaged (El Merzougui et al. 2023). In grapevine, extended exposure to PVS2 significantly reduced cell viability. Wang et al. (2002) showed that optimal viability of cryopreserved cell suspensions by Encap/deh was achieved within 2–4 days of preculture on 1 M sucrose. Improved Encap/deh protocols have achieved up to 50% viability of embryogenic cell suspensions (Ben-Amar et al. 2013; González-Benito et al. 2009).
Osmotic priming induces acquisition of freeze/thaw tolerance and alleviates oxidative stress in embryogenic cell lines during cryopreservation
Osmotic treatment achieved by loading cells in a cryoprotectant agent (CPA) solution, appears to be instrumental in maintaining membrane integrity for cryogenic protection. This process may induce the generation of ROS leading to oxidative stress and stimulating the biosynthesis of proline and other signaling metabolites. Preconditioning with non penetrating CPAs, such as sucrose, mannitol, or sorbitol below 1 M for up to 1–7 days can help acclimate cells to dehydration stress before exposure to higher CPA concentrations (Nausch and Buyel 2021). In the Encap/deh method, osmotic loading, which requires several days for full acclimation (Wang et al. 2002), acts as a priming stimulus to enhance plant tolerance to freezing during immersion in LN. Nonetheless, it is important to note that elevated sucrose concentrations during preculture and dehydration may alter the embryogenic competence in Gentiana cell suspensions (Markowski et al. 2024). Furthermore, embedding plant cells in a calcium alginate matrix can help to mitigate mechanical stress and osmotic pressure changes during freezing and thawing (Draget et al. 1988; Nausch and Buyel 2021). This process enhances plant cell adaptation and promotes resilience to further environmental stresses, such as chilling temperatures, highlighting the pivotal role of osmotic treatment during cryopreservation.
By themselves, extreme cold temperatures have been reported to affect protein structure, arrest plant growth, and activate defense mechanisms protection against a broad spectrum of biotic and abiotic stresses (Baier et al. 2019). This process, known as cold acclimation, can lead to increased concentrations of soluble sugars and α-amylase, enhanced membrane stabilization, and lower H2O2 levels. However, maintaining cold tolerance through these metabolic adjustments is cost-intensive. Consequently, cryopreserved plant material starts to de-acclimate rapidly during thawing phase as temperatures rise (Baier et al. 2019; Cvetkovic et al. 2017).
Osmotic priming, utilizing either a sucrose/glycerol solution for Encap/deh or PVS2 for Encap/vit, is crucial for establishing cross-tolerance in plant cell cultures and enhancing their resilience to freezing. ROS-induced oxidative stress can significantly hinder or even prevent successful cryopreservation (Ren et al. 2021). Cells exhibit two distinct responses to ROS-induced oxidative stress: resistance or sensitivity. Resistant cells mitigate oxidative stress and survive cryopreservation, while sensitive cells succumb to membrane lipid peroxidation, protein oxidation, DNA damage, and ultimately induced programmed cell death. Our study suggests that cryopreservation acts as a stress-based screening process, differentiating between resistant and sensitive cell populations, with resistant cells surviving and sensitive cells perishing after cryogenic treatment. Figure 7 outlines a flowchart of the major steps involved in Encap/deh and Encap/vit cryopreservation, along with the primary stresses causing cryoinjuries in plant cells exposed to low temperatures. Our work elucidates the enhancement of cryotolerance through osmotic pre-treatment based on trans-priming involving different types of stresses. A recent study in grapevine suspension cultures, showed enhanced tolerance to in vitro stress through ROS signaling and amplified endochitinase expression, leading to increased H2O2 and proline levels while maintaining membrane stability (Ben-Amar et al. 2022a). Notably, the amino acid proline is known to confer freezing tolerance in a wide variety of plant cells and is often incorporated into cryoprotective solutions or used for preconditioning plants or isolated cells or tissues prior to cryopreservation (Burritt 2008). Polesi et al. (2023) highlighted the importance of activating the enzymatic antioxidant system for the viability of Guadua chacoensis embryogenic cultures during cryopreservation. In our study, osmotic loading likely activates H2O2 signaling and ROS-mediated oxidative stress, enhancing freezing resilience during immersion into LN and improving survival and post-thaw recovery of grapevine callus material.

Flowchart illustrating stress-induced cryo-tolerance during cryopreservation. Major steps that relay on desiccation or vitrification protocols were described. Cryopreservation procedure involves several stresses (osmotic, freezing, and oxidative stress) that could trigger a rejuvenation of senescent embryogenic material into cryopreserved cells with greater embryogenic competence enabling high regrowth recovery and improved plant regeneration. Evidence of a stress-based cell screening differentiate resistant cells that will survive and sensitive cells that will dead following cryopreservation. This involves the upregulation of genes and transcription factors associated with embryogenic potential, as well as the accumulation of proline and raffinose oligosaccharides (RFOs), described previously (Ben-Amar et al. 2022a), that could be active with their osmolyte, antioxidant, and signaling functions. Recovering cryopreserved samples was made possible by the cells’ ability to overcome cryoinjuries thanks to the mitigation of oxidative stress. These embryogenic masses underwent several morphogenetic alterations when they were cryogenically treated; only resistant cells survived the treatment and gave rise to revitalized callus cell line with high embryogenic competence through rejuvenation
Evidence of cryopreservation-mediated cell rejuvenation highlighted promotion of embryogenic competence
While cryopreservation has been primarily used for the cold storage of embryogenic material, its potential to promote embryogenic capacity through cryogenic treatment is less understood. To our knowledge, this is the first report addressing this finding by investigating cryogenic-based cell rejuvenation at the cellular, biochemical, and molecular levels. Overall, our data revealed a substantial increase in the transcript levels of VvSERK1, VvBBM, and VvWOX genes associated with embryogenic competence and stem cell maintenance, following cryopreservation. Additionally, VvChitIV and VvLEA, related to plant stress defense and environmental response, were also upregulated. These genes may form a complex which is required for the maintenance of undifferentiated stem cells, similar to embryogenic cells. Notably, we found a remarkable increase in relative gene expression levels of SERK1, BBM, and WOX transcription factors (TFs), in most cryopreserved grapevine cultivars, activating the embryogenic pathway and modulating developmental change and cellular reprogramming. Plant growth regulators like auxins and cytokinins are known to inhibit senescence by downregulating TFs such as MYB, ERF, WRKY, and SAP involved in crosstalk during plant stress and ABA pathway (Hallmark and Rashotte 2020). Similar high expression levels of SERK, BBM, and WOX boosting embryogenic competence in early-stage somatic embryo development have been reported in Vitis vinifera (Martinez et al. 2021), and Thuja koraiensis (Ahn et al. 2023), suggesting a key role in inducing and maintaining embryogenic callus in an immature and juvenile state. These genes, identified as positive regulators of SE, have shown increased embryogenic competence after ectopic expression, even without external hormones. SERK1 was associated with new callus formation and cell reprogramming in Medicago truncatula (Nolan et al. 2009) and showed high expression 4 weeks after callus transfer onto induction medium at early pro-embryogenic stage before the visible appearance of somatic embryos in Vitis vinifera (Schellenbaum et al. 2008). On the other hand, the expression of VvSERK2 was relatively stable during culture as noticed by Schellenbaum et al. (2008). Similarly, in our experiments, VvSERK2 did not show a significant change. Our study also demonstrated an enhanced transcript levels of VvChitIV and VvLEA, both involved in stress response. Upregulation of VvChitIV under abiotic stress has been shown to alleviate in vitro stress and enabled the efficient establishment of well-growing homogenous cell suspensions with improved embryogenic competence (Ben-Amar et al. 2022a). The upregulation of these genes under cryogenic treatment compared to controls suggests their involvement in boosting embryogenic competence in cryopreserved samples. Taken together, these genes may act independently as positive regulators of cell reprogramming for the rejuvenation of embryogenic stem cells.
Additional research has revealed stress-induced increase in embryogenic potential in other single-cell studies. For example, the stress caused by enzymatic digestion of the donor plant tissue's cell wall stimulates protoplast division (Xu et al. 2021). Moreover, stress-induced microspores serve as the starting material for microspore-derived embryogenesis (Bouamama-Gzara et al. 2019; Maraschin et al. 2005). Such finding could be applied to other plant systems, such as promoting PEMs callus cultures in spruce for boosting SE, or for cryopreservation-mediated cell rejuvenation.
Conclusion
Cryopreservation of embryogenic cultures is a valuable tool for the secure storage of grapevine genetic resources. High regrowth rates are crucial for establishing a reliable cryopreservation workflow in applied plant cryobiology. In this study, we compared the efficiency of encapsulation/dehydration (Encap/deh) and encapsulation/vitrification (Encap/vit) methods in terms of survival, post-thaw recovery, and regeneration rates. Encap/deh emerged as a selected method for cryopreserving embryogenic cell lines. Beyond its established ability to preserve these lines, Encap/deh induced notable morphogenetic changes, significantly enhancing their embryogenic competence. This was further supported by a marked increase in transcript levels of several genes associated to embryogenic potential, suggesting that cryopreservation acts as a selection process triggering cell rejuvenation and enabling the safe preservation of embryogenic masses. Osmotic priming induced by cryoprotective agents (CPAs) also plays a vital role in acquiring cryotolerance and mitigating oxidative stress, thereby safeguarding cell membrane integrity from freeze–thaw damage. Further research is needed to explore additional mechanisms conferring cryotolerance, such as the ABA pathway, changes in enzyme activity, calcium-dependent signaling among other stress response networks regulating plant cell freezing tolerance.
References
Abraham E, Hourton-Cabassa C, Erdei L, Szabados L (2010) Methods for determination of proline in plants. Methods Mol Biol 639:317–331. https://doi.org/10.1007/978-1-60761-702-0_20
Ahn CH, Han JY, Park HS, Yoon HW, Shin JW et al (2023) Isolation and expression of transcription factors involved in somatic embryo development by transcriptome analysis of embryogenic callus of Thuja koraiensis. Horticulturae 9:131. https://doi.org/10.3390/horticulturae9020131
Baier M, Bittner A, Prescher A, van Buer J (2019) Preparing plants for improved cold tolerance by priming. Plant Cell Environ 42(3):782–800. https://doi.org/10.1111/pce.13394
Ballesteros D, Martinez MT, Sanchez-Romero C, Montalban IA, Sales E, Moncalean P, Arrillaga I, Corredoira E (2024) Current status of the cryopreservation of embryogenic material of woody species. Front Plant Sci 14:1337152. https://doi.org/10.3389/fpls.2023.1337152
Bates LS, Waldren RP, Teare ID (1973) Rapid determination of free proline for water-stress studies. Plant Soil 39:205–207. https://doi.org/10.1007/BF00018060
Ben-Amar A, Cobanov P, Boonrod K, Bouzid S, Ghorbel A, Krczal G, Reustle G (2007) Efficient procedure for grape embryogenic suspensions establishment and plant regeneration: role of conditioned medium in cell proliferation. Plant Cell Rep 26:1439–1447. https://doi.org/10.1007/s00299-007-0341-8
Ben-Amar A, Daldoul S, Allel D, Reustle G, Mliki A (2013) Reliable encapsulation-based cryopreservation protocol for safe storage and recovery of grapevine embryogenic cell cultures. Sci Hort 157:32–38. https://doi.org/10.1016/j.scienta.2013.04.005
Ben-Amar A, Allel D, Mliki A (2022a) Up-regulation of a stress-responsive endochitinase Vv.Chit-IV in grapevine cell cultures improves in vitro stress tolerance. Protoplasma 259:1189–1203. https://doi.org/10.1007/s00709-021-01733-y
Ben-Amar A, Boonrod K, Krczal G, Ghorbel A, Reustle G (2022b) Unravelling the role of arabinogalactan proteins in promoting embryogenic cultures and enhancing transient gene expression in grapevine. Plant Cell Tiss Org Cult 151(3):579–591. https://doi.org/10.1007/s11240-022-02373-7
Benelli C (2021) Plant cryopreservation: a look at the present and the future. Plants (Basel) 10(12):2744. https://doi.org/10.3390/plants10122744
Benson EE, Bremner D (2004) Oxidative stress in the frozen plant: a free radical point of view. In: Fuller BJ, Lane N, Benson EE (eds) Life in the frozen state, pp 206– 241, CRC Press, Boca Raton, Florida
Bettoni JC, Bonnart R, Shepherd A, Kretzschmar AA, Volk GM (2019) Cryopreservation of grapevine (Vitis spp.) shoot tips from growth chamber-sourced plants and histological observations. Vitis 58:71–78. https://doi.org/10.5073/vitis.2019.58:71-78
Bettoni JC, Marković Z, Bi W, Volk GM, Matsumoto T, Wang QC (2021) Grapevine shoot tip cryopreservation and cryotherapy: secure storage of disease-free plants. Plants 10(10):2190. https://doi.org/10.3390/plants10102190
Bi WL, Hao XY, Cui ZH, Volk GM, Wang QC (2018) Droplet-vitrification cryopreservation of in vitro-grown shoot tips of grapevine (Vitis spp.). In Vitro Cell Dev Biol-Plant 54:590–599. https://doi.org/10.1007/s11627-018-9931-0
Bouamama B, Ben Salem A, Ben Jouira H, Ghorbel A, Mliki A (2007) Influence of the flower stage and culture medium on the induction of somatic embryogenesis from anther culture in Tunisian grapevine cultivars. J Int Sci Vigne Vin 41(4):185–192. https://doi.org/10.20870/oeno-one.2007.41.4.835
Bouamama B, Ben Salem A, Ben Youssef F, Chaieb S, Jaafoura MH, Mliki A, Ghorbel A (2011) Somatic embryogenesis and organogenesis from mature caryopses of North African barley accession “Kerkena” (Hordeum vulgare L.). In Vitro Cell Dev Biol-Plant 47:321–327. https://doi.org/10.1007/s11627-011-9357-4
Bouamama-Gzara B, Zemni H, Zoghlami N, Gandoura S, Mliki A, Arnold M, Ghorbel A (2019) Behavior of Opuntia ficus-indica (L.) Mill. heat-stressed microspores under in vitro culture conditions as evidenced by microscopic analysis. In Vitro Cell Dev Biol- Plant 56:122–133
Burritt DJ (2008) Efficient cryopreservation of adventitious shoots of Begonia x erythrophylla using encapsulation-dehydration requires pretreatment with both ABA and proline. Plant Cell Tiss Organ Cult 95:209–215
Campos G, Chialva C, Miras S, Lijavetzky D (2021) New technologies and strategies for grapevine breeding through genetic transformation. Front Plant Sci 12:767522. https://doi.org/10.3389/fpls.2021.767522
Cvetkovic J, Müller K, Baier M (2017) The effect of cold priming on the fitness of Arabidopsis thaliana accessions under natural and controlled conditions. Sci Rep 7:4405
Draget KI, Myhre S, Skjåk-Bræk G, Østgaard K (1988) Regeneration, cultivation and differentiation of plant protoplasts immobilized in Ca-alginate beads. J Plant Physiol 132(5):552–556. https://doi.org/10.1016/S0176-1617(88)80252-9
El Merzougui S, Benelli C, El Boullani R, Serghini MA (2023) The cryopreservation of medicinal and ornamental geophytes: application and challenges. Plants 12(11):2143. https://doi.org/10.3390/plants12112143
Engelmann F (2004) Plant cryopreservation: progress and prospects. In Vitro Cell Dev Biol-Plant 40:427–433
González-Benito ME, Martín C, Vidal JR (2009) Cryopreservation of embryogenic cell suspensions of the Spanish grapevine cultivars Albarino and Tempranillo. Vitis 48:131–136
Hallmark HT, Rashotte AM (2020) Cytokinin isopentenyladenine and its glucoside isopentenyladenine-9G delay leaf senescence through activation of cytokinin-associated genes. Plant Direct 4:e00292. https://doi.org/10.1002/pld3.292
Hodges DM, Delong JM, Forney CF, Prange RK (1999) Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta 207:604–611. https://doi.org/10.1007/s00425-017-2699-3
Kamiloglu S, Sari G, Ozdal T, Capanoglu E (2020) Guidelines for cell viability assays. Food Front 1:332–349. https://doi.org/10.1002/fft2.44
Kobayashi T, Niino T, Kobayashi M (2006) Cryopreservation of tobacco BY-2 suspension cell cultures by vitrification with encapsulation. Plant Biotechnol 23(3):333–337. https://doi.org/10.5511/plantbiotechnology.23.333
Maraschin SF, de Priester W, Spaink HP, Wang M (2005) Androgenic switch: an example of plant embryogenesis from the male gametophyte perspective. J Exp Bot 56(417):1711–1726. https://doi.org/10.1093/jxb/eri190
Markovic Z, Preiner D, Stupic D, Andabaka Z, Simon S et al (2015) Cryopreservation and cryotherapy of grapevine (Vitis vinifera L.). Vitis 54:247–251. https://doi.org/10.5073/vitis.2015.54.special-issue.247-251
Marković Z, Preiner D, Stupić D, Andabaka Z, Šikuten I, Kontić JK, Maletić E, Štambuk P (2018) Cryopreservation protocols for grapevine shoot tips. https://doi.org/10.5772/intechopen.81802
Markowski M, Czarnomska Z, Tomiczak K, Mikula A, Granica S, Podwyszynska M, Szypula WJ (2024) The influence of cryopreservation via encapsulation-dehydration on growth kinetics, embryogenic potential and secondary metabolite production of cell suspension cultures of Gentiana capitata Buch.-Ham. ex D.Don and Gentiana decumbens L.f. Indus Crops Prod 212:118349. https://doi.org/10.1016/j.indcrop.2024.118349
Martinez O, Arjones V, Gonzalez MV, Rey M (2021) Histone deacetylase inhibitors increase the embryogenic potential and alter the expression of embryogenesis-related and HDAC-encoding genes in grapevine (Vitis vinifera L., cv. Mencia). Plants 10:1164. https://doi.org/10.3390/plants10061164
Miaja ML, Gambino G, Vallania R, Gribaudo I (2004) Cryopreservation of Vitis vinifera L. somatic embryos by vitrification or encapsulation-dehydration. Acta Hortic 663:599–604
Mikuła A, Tomiczak K, Rybczyński JJ (2011) Cryopreservation enhances embryogenic capacity of Gentiana cruciata (L.) suspension culture and maintains epigenetic uniformity of regenerants. Plant Cell Rep 30:565–574
Mittler R (2017) ROS are good. Trend Plant Sci 22:11–19
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immun Meth 65(1–2):55–63
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–479. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Nausch H, Buyel JF (2021) Cryopreservation of plant cell cultures - diverse practices and protocols. Nat Biotechnol 62:86–95. https://doi.org/10.1016/j.nbt.2021.02.002
Nitsch JP, Nitsch C (1969) Haploid plants from pollen grains. Science 163:85–87
Nolan KE, Kurdyukov S, Rose RJ (2009) Expression of the somatic embryogenesis receptor-like kinase (SERK1) gene is associated with developmental change in the life cycle of the model legume Medicago truncatula. J Exp Bot 60(6):1759–1771. https://doi.org/10.1093/jxb/erp046
OIV (2020) State of the world viti-vinicultural sector in 2019; International Organisation of Vine and Wine: Paris, France. Available online: https://www.oiv.int/public/medias/7298/oiv-state-of-the-vitiviniculturalsector-in-2019.pdf
Polesi LG, Goeten D, Fraga HPdF, Steiner N, Guerra MP (2023) Enzymatic antioxidant system activation assures the viability of Guadua chacoensis (Bambusoideae, Poaceae) embryogenic cultures during cryopreservation. Plants 12(3):673. https://doi.org/10.3390/plants12030673
Poobathy R, Uma Rani S, Xavier R, Sreeramanan S (2013) Histology and scanning electron microscopy observations of cryopreserved protocorm-like bodies of Dendrobium sonia-28. Turk J Biol 37(2):10. https://doi.org/10.3906/biy-1206-22
Ren L, Zhang D, Jiang X, Gai Y, Wang W, Reed BM, Shen X (2013) Peroxidation due to cryoprotectant step is a vital factor for cell survival in Arabidopsis cryopreservation. Plant Sci 212:37–47
Ren L, Wang MR, Wang QC (2021) ROS-induced oxidative stress in plant cryopreservation: occurrence and alleviation. Planta 254(6):124. https://doi.org/10.1007/s00425-021-03784-0
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Schellenbaum P, Jacques A, Maillot P, Bertsch C, Mazet F, Farine S, Walter B (2008) Characterization of VvSERK1, VvSERK2, VvSERK3 and VvL1L genes and their expression during somatic embryogenesis of grapevine (Vitis vinifera L.). Plant Cell Rep 27:1799–1809
van der Walt K, Nadarajan J, Mathew L, Bettoni JC, Scouza JA (2023) Advances in cryopreservation of Syzygium maire (swamp maire, maire tawake) zygotic embryos, a critically endangered species endemic to new Zealand. Front Conserv Sci 4:1269881. https://doi.org/10.3389/fcosc.2023.1269881
Vasanth K, Vivier MA (2011) Improved cryopreservation for long term storage of synchronized culture of grapevine. Biol Plant 55:365–369
Velikova V, Yordanov I, Edreva A (2000) Oxidative stress and some antioxidant systems in acid rain-treated bean plants: protective role of exogenous polyamines. Plant Sci 151:59–66. https://doi.org/10.1016/S0168-9452(99)00197-1
Volk GM, Walters C (2006) Plant vitrification solution 2 lowers water content and alters freezing behavior in shoot tips during cryoprotection. Cryobiol 52:48–61
Wang Q, Valkonen JPT (2009) Cryotherapy of shoot tips: novel pathogen eradication method. Trends Plant Sci 14(3):119–122. https://doi.org/10.1016/j.tplants.2008.11.010. ISSN 1360-1385
Wang Q, Gafny R, Sahar N, Sela I, Mawassi M, Tanne E, Perl A (2002) Cryopreservation of grapevine (Vitis vinifera L.) embryogenic cell suspensions by encapsulation-dehydration and subsequent plant regeneration. Plant Sci 162:551–558
Wang Q, Mawassi M, Li P, Gafny R, Sela I, Tanne E (2003) Elimination of grapevine virus A (GVA) by cryopreservation of in vitro-grown shoot tips of Vitis vinifera L. Plant Sci 165:321–327. https://doi.org/10.1016/S0168-9452(03)00091-8. ISSN 0168-9452
Wang W, Mawassi M, Sahar N, Li P, Violeta CT, Gafny R, Sela I, Tanne E, Perl A (2004) Cryopreservation of grapevine (Vitis spp.) embryogenic cell suspensions by encapsulation-vitrification. Plant Cell Tiss Org Cul 77:267–275
Wang MR, Bi W, Shukla MR, Ren L, Hamborg Z, Blystad DR, Saxena PK, Wang QC (2021) Epigenetic and genetic integrity, metabolic stability, and field performance of cryopreserved plants. Plants (Basel) 10(9):1889. https://doi.org/10.3390/plants10091889
Wang QC, Perl A (2006) Cryopreservation of embryogenic cell suspensions by encapsulation-vitrification. In: Víctor M, Loyola-Vargas, Felipe Vázquez-Flota (eds) Plant cell culture protocols, methods in molecular biology, Humana Press, USA, Chapter 17, pp 77–86
Wanner LA, Junttila O (1999) Cold-induced freezing tolerance in Arabidopsis. Plant Physiol 120(2):391–400. https://doi.org/10.1104/pp.120.2.391
Wei Q, Shi P, Khan FS, Htwe YM, Zhang D, Li Z, Wei X, Yu Q, Zhou K, Wang Y (2023) Cryopreservation and cryotolerance mechanism in zygotic embryo and embryogenic callus of oil palm. Forests 14(5):966. https://doi.org/10.3390/f14050966
Xu M, Du Q, Tian C, Wang Y, Jiao Y (2021) Stochastic gene expression drives mesophyll protoplast regeneration. Sci Adv 7(33):eabg8466. https://doi.org/10.1126/sciadv.abg8466
Acknowledgements
We are grateful to Dr. Goetz Reustle (RLP-AgroScience, Germany) for kindly providing embryogenic material and to Dr. Pascal Cobanov (AlPlanta-Institute for Plant Research, Germany) for technical advice.
Funding
The work was supported by Tunisian Ministry of Higher Education and Scientific Research.
Author information
Authors and Affiliations
Contributions
AB conceived and designed the study, conducted the cryopreservation experiment, and wrote the manuscript. DA performed biochemical assays, data collection, and statistical analysis. BB carried out the histological sections and ESEM observations. AB, DA, and BB contributed in reviewing, editing, and approving the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Peter Nick.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ben-Amar, A., Allel, D. & Bouamama-Gzara, B. Osmotic priming-induced cryotolerance uncovers rejuvenation of grapevine cell cultures: morphogenetic changes and gene expression pattern highlighting enhanced embryogenic potential. Protoplasma (2024). https://doi.org/10.1007/s00709-024-01968-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00709-024-01968-5