
Abstract
Key message
Oxidative stress and apoptosis-like programmed cell death, induced in part by H 2 O 2 , are two key factors that damage cells during plant cryopreservation. Their inhibition can improve cell viability.
Abstract
We hypothesized that oxidative stress and apoptosis-like event induced by ROS seriously impact plant cell viability during cryopreservation. This study documented changes in cell morphology and ultrastructure, and detected dynamic changes in ROS components (O ·−2 , H2O2 and OH·), antioxidant systems, and programmed cell death (PCD) events during embryonic callus cryopreservation of Agapanthus praecox. Plasmolysis, organelle ultrastructure changes, and increases in malondialdehyde (a membrane lipid peroxidation product) suggested that oxidative damage and PCD events occurred at several early cryopreservation steps. PCD events including autophagy, apoptosis-like, and necrosis also occurred at later stages of cryopreservation, and most were apoptosis. H2O2 is the most important ROS molecule mediating oxidative damage and affecting cell viability, and catalase and AsA–GSH cycle are involved in scavenging the intracellular H2O2 and protecting the cells against stress damage in the whole process. Gene expression studies verified changes of antioxidant system and PCD-related genes at the main steps of the cryopreservation process that correlated with improved cell viability. Reducing oxidative stress or inhibition of apoptosis-like event by deactivating proteases improved cryopreserved cell viability from 49.14 to 86.85 % and 89.91 %, respectively. These results verify our model of ROS-induced oxidative stress and apoptosis-like event in plant cryopreservation. This study provided a novel insight into cell stress response mechanisms in cryopreservation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cryopreservation, the storage of viable cells, tissues, organs, and organisms at ultralow temperatures, usually in liquid nitrogen (LN), has successfully preserved various plant species (Bajaj 1995; Engelmann 2004; Benson 2008; Reed 2008). At this temperature, biochemical metabolic and cell division activities are arrested, allowing for long-term storage (Benelli et al. 2013; Kulus and Zalewska 2014). The primary goal of any cryopreservation procedure is to prevent lethal ice crystals forming within the cell and achieve intracellular vitrification. Once ice forms inside cells, it can do mechanical damage to intracellular structures. Crystallization can be avoided altogether by lowering the freezing point through the addition of highly concentrated solutes. The liquid solution becomes increasingly viscous as temperatures decrease, and at a low enough temperature and high enough solute concentration, becomes a glassy solid (vitrified) (Meryman 2007). Over the last 20 years, vitrification-based cryopreservation technology was developed and applied to long-term storage of plant species (Sakai et al. 2008). Although plant vitrification solutions (PVS) are important for cell survival, they can also cause complex stresses, such as osmotic injury and dehydration stress (Uchendu et al. 2010a). A relevant hypothesis is that the process of freezing and thawing increases the production of reactive oxygen species (ROS), which in turn will alter the redox state of the cells, eventually leading to stress responses (Dowling and Simmons 2009). Prior research provides evidence that some stress-related genes and proteins are induced during cryopreservation. Some genes were specifically up-regulated in response to cryoprotectant treatment in Arabidopsis shoot tips, mainly involved in dehydration responses (Volk et al. 2011). Ren et al. (2013) using comparative transcriptomic technology revealed that cryoprotectant treatment induces the production of ROS that may cause oxidative stress and apoptosis-like events. Oxidative stress usually induces genes for anti-oxidation and peroxidation in plant cells; anti-oxidation is a positive factor and peroxidation is a negative agent for cell survival.
Biosynthesis and accumulation of ROS, such as the superoxide anion radical (O ·−2 ), hydrogen peroxide (H2O2), hydroxyl radical (OH·), and singlet oxygen (1O2), which are highly reactive and toxic chemical species, are central to oxidative stress-related metabolisms (Gill and Tuteja 2010). The effects of ROS are dose-dependent, and at low levels, ROS act as signaling molecules to regulate cellular functions. However, they exert oxidative stress at high levels (Tatone et al. 2010). Skyba et al. (2012) observed increased production of ROS from cryopreserved Hypericum perforatum shoot tip meristems; and Xu et al. (2014) found that generation of ROS may improve Lilium × siberia pollen viability during cryopreservation; however, Whitaker et al. (2010) suggested that excessive O ·−2 formation results in poor survival of cryopreservation in Trichilia dregeana. Fang et al. (2008) confirmed that OH· induced oxidative stress and lipid peroxidation was the main cause of the viability loss in Theobroma cacao somatic embryo cryopreservation. Thus, the ROS molecule is a double-bladed sword, and each ROS component possibly plays a different role during cryopreservation.
Plants have complex antioxidant systems to resist external stress. The antioxidant system includes both enzymatic antioxidants, e.g., superoxide dismutase (SOD), catalase (CAT), ascorbate peroxidase (APX), monodehydroascorbate reductase (MDHAR), dehydroascorbate reductase (DHAR) and glutathione reductase (GR), and non-enzymatic antioxidants, such as ascorbic acid (AsA) and glutathione (GSH) (Gill and Tuteja 2010). Among these enzymatic antioxidants, SOD and CAT represent the first line of antioxidant defense (Van Breusegem et al. 2001), whereas SOD is responsible for the removal of O ·−2 by dismutation to form H2O2, and CAT metabolizes H2O2 into H2O and O2 (Scandalios 1997). Among non-enzymatic antioxidants, AsA is considered a powerful ROS scavenger because of its ability to donate electrons in a number of enzymatic and non-enzymatic reactions (Gill and Tuteja 2010). GSH is necessary to maintain the normal reduced state of cells so as to counteract the inhibitory effects of ROS-induced oxidative stress (Meyer 2008), and plays a key role in the antioxidative defense system by regenerating AsA, via the AsA–GSH cycle (Foyer and Halliwell 1976).
Programmed cell death (PCD) in plants is a crucial component of development and defense mechanisms. PCD is largely used to describe the processes of autophagy and apoptosis-like PCD, while necrosis is generally described as a chaotic and uncontrolled mode of death (Reape et al. 2008). Autophagy is a relatively slow process, with cell viability not being altered within the first 24 h, and cells displaying an enlarged vacuole and decreased cytoplasmic width (Reape et al. 2008). It is very different compared to the relatively rapid apoptosis-like PCD that occurs within 6 h after heat shock or other forms of abiotic stress in Arabidopsis cells (McCabe and Leaver 2000). Several studies have demonstrated that ROS production and mitochondrial dysfunction are important early indicators and necessary components of cell death in response to various stimuli (Vacca et al. 2004, 2006; Gao et al. 2008; Zhang and Xing 2008). In various plant systems, the release of cytochrome c from plant mitochondria as caused by ROS, elevated calcium levels, or inhibition of electron transport has been postulated to be a common means for integrating cellular stress and activating plant PCD (Balk et al. 1999; Sun et al. 1999; Hansen 2000; Jones 2000).
Generally, seeds, with characteristic dense cytoplasm and low water content, are easily cryopreserved by vitrification. However, callus cultures, suspension cultures, shoot tips, and recalcitrant seeds are more difficult to cryopreserve due to their higher water content, vigorous metabolic activities, complicated biological processes, and sensitive stress responses (Ren et al. 2013). Agapanthus praecox (Agapanthaceae) is a monocotyledonous, herbaceous, perennial plant (Zhang et al. 2014), which grows in tropical or subtropical regions and is sensitive to cold stress. Recently, we established a somatic embryogenesis system and successfully cryopreserved the embryonic callus (EC) of A. praecox. This study aims to determine the dynamic processes of ROS, PCD, and stress responses of plant cells during cryopreservation in order to improve cryobiological theory and the practical application of plant cryopreservation.
Materials and methods
Preparation of plant materials
EC of A. praecox was induced from pedicel tissue (Wang et al. 2012) and is under continuous subculture on MS medium supplemented with 1.5 mg L−1 picloram at 25 °C.
Cryopreservation procedure
Agapanthus praecox EC cells were cryopreserved by vitrification as described by Ren et al. (2013) with some modifications as detailed below:
-
1.
Pre-culture (PC): EC cells were cultured on MS medium with 0.5 M sucrose at 4 °C for 48 h.
-
2.
Osmoprotection (OP): EC cells (0.2 g) were immersed in 1.0 mL loading solution (MS liquid medium + 2.0 M glycerol + 0.4 M sucrose) at 0 °C for 60 min.
-
3.
Dehydration (DH): the loading solution was replaced by the vitrification solution PVS2 (30 % w/v glycerol, 15 % w/v ethylene glycol, and 15 % w/v dimethyl sulfoxide in MS liquid medium with 0.4 M sucrose) at 0 °C for 40 min.
-
4.
Rapid cooling and warming (RW): EC cells after dehydration in cryovials were rapidly plunged into LN, held for 1 h (cryogenic treatment) or long term (cryopreservation), and rewarmed in 40 °C water bath for 1 min.
-
5.
Dilution (DL): PVS2 was replaced by the unloading solution (MS liquid medium + 1.2 M sucrose) for 40 min, and the solution was replaced with fresh unloading solution every 10 min.
-
6.
Recovery (RC): the EC cells were cultured on MS medium under the same conditions as for subculture.
Application of chemical compounds
This study evaluated the effects of GSH (BBI, Markham, Ontario, Canada) at 0.08 mM, AsA (BBI) at 1 mM, cinnamtannin B-1 (a potent antioxidant; Enzo Life Sciences) at 50 μg mL−1, 2-methoxy-antimycin A3 (BCL-2 inhibitor; Enzo Life Sciences) at 3.8 μM, and Ac-AAVALLPAVLLALLAPDEVD-CHO (Caspase inhibitor, cell permeable inhibitor of caspase-3, -6, -7, -8, and -10; Enzo Life Sciences) at 1.0 μM. These compounds were added to the cryoprotectant PVS2 during the cryopreservation process. Experiments were run individually 3 times.
Electron and light microscopy
The samples were pre-fixed in 3 % glutaraldehyde, rinsed with phosphate buffer saline (PBS), and then post-fixed with 2 % osmic acid for 24 h. The fixed tissues were dehydrated in an ethanol series, and infiltrated and embedded with epoxy resin. Material sections were cut to a thickness of 50–70 nm and double-stained with uranyl acetate and lead citrate. Sections were examined using a transmission electron microscope (FEI Tecnai G2 Spirit Biotwin, USA) at 80 kV.
Sections were double-stained with the periodic acid–Schiff and toluidine blue O reagents to observe polysaccharide arrangement and cell morphology. Digital images were captured by using an Olympus camera system.
Determination of O ·−2 , H2O2, and OH·
The determination of O ·−2 inhibition activity was performed by using an inhibition and production superoxide anion assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. The absorbance of reaction solution was measured at 550 nm. The difference between the sample and the standards (0.15 mg/mL vitamin C) was expressed as O ·−2 inhibition activity.
H2O2 content was assayed according to Peng et al. (2009) method. The 0.2 g EC cells were ground in 1.8 mL of 100 mM cool PBS (pH 7.4) and centrifuged at 5000g at 4 °C for 10 min. Add 1.0 mL of 16 % H2SO4, 0.2 mL of 20 % KI, and 60 μL of 50 mM molybdic acid to 1.0 mL supernatant for a reaction time of 5 min. The absorbance of the solution was measured at 405 nm. Standards were prepared by known amounts of H2O2 in the same manner as the tested samples.
Hydroxyl radical generation activity was assayed by its capacity to oxidize bromopyrogallol red (BPR) as described by Wu et al. (2001). EC (0.2 g) cells were ground in 1.8 mL of distilled water on ice. Extracts were then centrifuged at 5000g at 4 °C for 10 min. Add 0.3 mL of 0.2 mM FeSO4, 0.3 mL of 1.0 mM BPR, and 0.3 mL of 1 % (v/v) H2O2 solutions to 1.0 mL of the supernatant (sample) while 1.0 mL of distilled water served as the CK. After reaction for 1 min, the solutions were measured at 550 nm. The difference between the sample and the CK was expressed as hydroxyl radical generation activity. Each detection of O ·−2 , H2O2, and OH· conducted 3 biological repeats.
Antioxidant enzymes activities
Extract preparation: About 0.2 g of EC was ground in 1.8 mL of 100 mM cool PBS (pH 7.4) and centrifuged at 5000g at 4 °C for 10 min; the supernatant was used for assay of the enzyme activities of SOD, CAT, and peroxidase (POD). The protein concentration of the supernatant was measured according to Lowry et al. (1951). Each experiment included 3 biological repeats.
The SOD activity was measured at 550 nm. Production of superoxide was measured by conversion of xanthine catalyzed by xanthine oxidase in the reagent solution containing 50 mM Na2CO3 (pH 10.2), 0.1 mM EDTA, 0.1 mM xanthine, 0.025 mM sodium benzenesulfonic acid hydrate, and 9 U L−1 xanthine oxidase. One unit of the SOD activity was defined as the amount of the enzyme required for 50 % inhibition of reaction rate of benzenesulfonic acid hydrate as a detection molecule reduced by superoxide.
The CAT activity was measured in a reaction mixture containing 0.85 mL of 50 mM PBS (pH 7.4), 0.5 mL of 30 mM H2O2, and 0.15 mL of the extract using a method described by Fowler and Aebi (1983). The CAT activity was determined following the decomposition of H2O2 at 240 nm.
The POD activity was measured according to Erdei et al. (2002). A 3 mL of the assay mixture contained 20 mM guaiacol, 25 mL PBS (pH 6.8), 40 mM H2O2, and 10 μL enzyme extracts. The POD activity was read at 470 nm.
Quantification of ascorbic acid and glutathione
Extraction of AsA and GSH was accomplished as described by Queval et al. (2007). EC (0.2 g) cells were ground in LN and then extracted into 2 mL of 0.2 M HCl. The homogenate was centrifuged at 5000g at 4 °C for 15 min. Then 0.5 mL supernatant was neutralized with 50 μL of 0.2 M NaH2PO4 and 0.4 mL of 0.2 M NaOH. To measure the AsA content, the initial absorbance of 30 μL of supernatant was measured at 265 nm in NaH2PO4, and then re-measured over 3 min following the addition of ascorbate oxidase (0.5 U). The measurement of GSH content was based on the GR-dependent reduction of 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) at 412 nm. The assay mixture contains 100 mM NaH2PO4 (pH 7.8), 0.6 mM DTNB, 6 mM EDTA, 0.1 mM NADPH, 25 μL extract, and 0.6 U GR.
Determination of malondialdehyde (MDA) content
Each sample (0.2 g) was homogenized in 5.0 mL of 10 % (w/v) trichloroacetic acid (TCA) on ice. The homogenate was centrifuged at 5000g at 4 °C for 10 min and then 2.0 mL of supernatant was mixed with 2 mL of 0.6 % thiobarbituric acid (TBA) (dissolved in 10 % TCA). The mixture was incubated at 100 °C for 30 min and cooled rapidly with flowing water, and then centrifuged at 5000g for 10 min. The MDA content was measured at 532, 600, and 450 nm, where MDA (μmol L−1) = 6.45 (A 532 − A 600) − 0.56A 450.
Viability detection
Survival assessment of cryopreserved cells was detected by the 2,3,5-triphenyltetrazolium chloride (TTC) method. EC tissue (0.2 g) was placed into 2 mL of TTC buffer (0.8 % w/v TTC in 0.05 M PBS) and incubated in the dark at 25 °C for 20 h. Three sterile water rinses were performed, and cells were incubated in 95 % ethanol at 85 °C for 1 h. The cells were centrifuged at 5000 rpm for 2 min and the supernatant was measured at 485 nm. Each sample was repeated 3 times.
Autophagy detection
Autophagy status of each sample was detected using a Cyto-ID® Autophagy detection kit (Enzo Life Sciences, PA, USA) according to the manufacturer’s instructions. This kit measured autophagic vacuoles and monitors autophagic flux in live cells using a novel dye that selectively labeled autophagic vacuoles. Cells were collected by centrifugation (5 min, 1000 rpm at room temperature) and washed twice with 1× assay buffer. Positive control cells were pretreated with the Autophagy Inducer (500 nM rapamycin) for 16–18 h. The supernatant was removed and 100 μL of Microscopy Dual Detection Reagent solution (every 1 mL of 1× assay buffer, add 2 μL of Cyto-ID® Green Detection Reagent and 1 μL of Hoechst 33342 Nuclear Stain) was added to cover the cell pellet. The pellet was re-suspended and incubated for 30 min at 37 °C. Cells were washed twice with 1× assay buffer and placed on a glass slide. The stained cells were analyzed by wide-field fluorescence microscopy, using a standard FITC (488 nm) and DAPI (550 nm) filter set for imaging the autophagic and nuclear signals.
Apoptosis and necrosis detection
Apoptosis and necrosis of each sample were detected using GFP-Certified® Apoptosis/Necrosis detection kit (Enzo Life Sciences, PA, USA) according to the manufacturer’s instructions. An Annexin V-EnzoGold (enhanced Cyanine-3; Ex/Em: 550/570 nm) conjugate enabled detection of apoptosis. The Necrosis Detection Reagent (Red) similar to the red-emitting dye 7-AAD (Ex/Em: 546/647 nm) facilitated late apoptosis and necrosis detection. Cells were collected by centrifugation (5 min, 1000 rpm at room temperature) and washed twice with 1× assay buffer. Positive control cells were pretreated with the apoptosis inducer (2 μM staurosporine) for 6–8 h. The supernatant was removed and 100 μL of Microscopy Dual Detection Reagent solution (Table 1) dispensed to cover the cell pellet. The pellet was re-suspended and incubated for 15 min at 37 °C, washed twice with 1× assay buffer, and placed onto a glass slide. The stained cells were analyzed by wide-field fluorescence microscopy, using Cyanine-3 (550 nm) filter set for imaging the apoptosis and necrosis signal simultaneously.
TUNEL assay
Apoptosis can be analyzed by detection of DNA fragmentation via a fluorescence assay based on terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) technique (Beyotime Institute of Biotechnology, China). This method takes advantage of DNA fragmentation, characteristic of apoptosis. The DNA breaking points (nicks) expose the 3′ OH ends of DNA, which are labeled, thus allowing the identification of apoptotic cells. Briefly, terminal deoxynucleotidyl transferase was used to incorporate residues of digoxigenin nucleotide into the 3′ OH ends of DNA fragments (Zhang et al. 2009). Cells were collected by centrifugation and washed twice with PBS buffer. Supernatant was removed and 100 μL of TUNEL Detection solution dispensed to cover the cell pellet. The pellet was re-suspended and incubated for 1 h at 37 °C, washed twice with PBS buffer, and placed onto a glass slide. The cells were imaged under a fluorescent microscope (488 nm).
qRT-PCR analysis
The method was as described by Zhang et al. (2013). About 0.2 g EC of each sample were collected for total RNA extraction using RNAiso Plus (TaKaRa, Otsu, Shiga, Japan) according to the manufacturer’s instructions. The total RNA was purified using the DNase I and RNase inhibitors (TaKaRa). cDNA was synthesized using SYBR PrimeScript RT-PCR Kit II (TaKaRa), according to the manufacturer’s instructions. Real-Time qPCR was performed according to SYBR Premix EX Taq II Kit (TaKaRa) on the Bio-Rad CHROMO4 Gradient cycler system. The data were analyzed using Opticon Monitor software (Version 3.1, Bio-Rad); actin 2 was used as an internal control parameter for normalization, and all primers for qRT-PCR are shown in Table S1. Gene expression quantification was calculated by \(2^{{ - \Delta \Delta C_{\text{t}} }} (\Delta \Delta C_{\text{t}} = \Delta \, [(C_{{t_{\text{targe\,gene}} }} - C_{{{\text{t}}_{\text{actin}} }} )_{\text{treated}} - (C_{{{\text{t}}_{\text{targe\,gene}} }} - C_{{{\text{t}}_{\text{actin}} }} )_{\text{control}} ])\).
Statistical analysis
Statistical analysis was performed using SAS 9.1.3 software. For all quantitative data, a one-way ANOVA was used, followed by a LSD multiple range test when significance differences were detected (P < 0.05). Correlation analysis used SAS 9.1.3 software; P values <0.05 were considered significant.
Results
Cell morphological characteristics
In the process of cryopreservation, the EC cells underwent a serious plasmolysis response during the steps of the cryopreservation protocol. Generally, the EC cells have a dense cytoplasm, containing some starch grains and the cell membrane is close to the cell wall (Fig. 1a). The cells exhibited large vacuole, more starch grain, and obvious plasmolysis after hypertonic pre-culture treatment (Fig. 1b). However, plasmolysis was reversed after osmoprotection and dehydration treatments (Fig. 1c); cellular organelles and starch grains mainly accumulated in the center of the cell, and the large vacuole appeared in the cell (Fig. 1c). Plasmolysis occurred again during dilution process; cytoplasm and starch grain were uniformly distributed in the cell, and the vacuole volume was obviously decreased at this stage (Fig. 1d).

Cell morphological characteristics of Agapanthus praecox EC during cryopreservation. a CK, b pre-culture stage, c dehydration stage, d dilution stage. CM cell membrane, CW cell wall, Cy cytoplasm, Nu nucleus, SG starch grain, Va vacuole, scale bars 20 μm
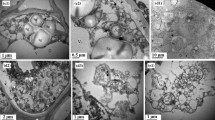
Cell ultrastructure observation indicated that oxidative damage and PCD event occurred in the EC cells during cryopreservation. Many starch grains, mitochondria, Golgi apparatus, and endoplasmic reticulum (ER) distributed in the control cell (Fig. 2a). After pre-culture, mitochondria, Golgi apparatus, and endoplasmic reticulum exhibited slight contraction, and many autophagic vesicles appeared in the cell (Fig. 2b). Additionally, autophagy, organelle degradation, and some lipid bodies appeared in the EC cells during the dehydration treatment (Fig. 2c); and these events were further exacerbated at dilution stage (Fig. 2d).

Cell ultrastructure observation of Agapanthus praecox during cryopreservation. a CK, b pre-culture stage, c dehydration stage, d dilution stage. AVi autophagic vesicle, CM cell membrane, CW cell wall, ER endoplasmic reticulum, G golgi apparatus, LB lipid body, M mitochondria, SG starch grain
Determination of cell viability, lipid peroxidation, ROS, and antioxidant system
Cell relative viability detected by TTC showed that the viability of EC cells continuously decreased in all steps as well as the recovery stage. The minimum value of TTC occurred at rapid warming (13.6 %) and dilution (12.7 %); however, after 24 h of recovery culture, the cell relative viability reached 49.1 % (Fig. 3). MDA, the product of membrane lipid peroxidation, also changed obviously in EC cells during cryopreservation. The MDA concentrations were highest at the dehydration, rewarming, and dilution steps, and the level was 2.5 times higher than the CK cells (Fig. 3). This result indicated that the cells underwent serious lipid peroxidation during cryopreservation. The H2O2 production trend was very similar to that of MDA. The H2O2 content significantly increased at the dehydration and rewarming steps and reached a peak of 110.91 μmol g−1 after rapid warming 218 % higher than the CK cells, and significantly decreased to 73.76 μmol g−1 after 24-h recovery. OH· generation activity was significantly enhanced at dehydration, dilution, and recovery and O ·−2 inhibition at the rapid warming stage (Fig. 3).

Quantitative analysis of MDA, ROS, and antioxidant system of A. praecox EC during cryopreservation. Bars represent means and standard errors over triplicate detections, and columns with different lowercase letters were significantly different (P < 0.05, least significant difference test). CK control, PC pre-culture, DH dehydration, RW rapid cooling and warming, DL dilution, RC recovery
The antioxidant systems showed significant changes during cryopreservation. The enzymatic antioxidants exhibited first decreased and then increased levels; however, non-enzymatic antioxidants presented the opposite trend to enzymatic antioxidants (Fig. 3). SOD and POD activities decreased at the dehydration stage and the maximum appeared in the recovery process. CAT activity remained steady until significantly increasing at last two stages (Fig. 3). AsA and GSH levels were significantly higher at dilution and rewarming, and the contents at those steps increased to 299 % for AsA and 173 % for GSH compared to the CK cells.
Correlation analysis indicated that cell relative viability had a significant negative correlation to MDA (P = 0.0141), H2O2 (P = 0.0468), AsA (P = 0.0077), and GSH (P = 0.0228) contents; and MDA content had a significant positive correlation to H2O2 (P = 0.0021), OH· (P = 0.0474), and GSH (P = 0.0306) contents; additionally, the GSH content had a significant positive correlation to H2O2 (P = 0.0414) and AsA (P = 0.0441) (Table 2). This result suggested that oxidative stress was mainly induced by H2O2 and OH·, and membrane lipid peroxidation mediated by H2O2 is a major factor leading cell viability decreases in A. praecox EC cells during cryopreservation. GSH and AsA were the main antioxidants to scavenge H2O2 in this system. Furthermore, there was a significant positive correlation between SOD and CAT (P = 0.0182) suggesting that these two enzymes have cooperative action to scavenge ROS components during EC cell cryopreservation. These data demonstrated that CAT, SOD, AsA, and GSH had a close relationship in the antioxidant system.
Detection of PCD
Programmed cell death detection showed that some EC cells exhibited autophagy and apoptosis-like events and a few cells underwent necrosis during cryopreservation. Positive controls of autophagy detection exhibited obvious bright green and apoptosis yellow-green fluorescence (Fig. 4a). The control cells showed faint fluorescent signals (Fig. 4b). Autophagy fluorescence signals were enhanced at the dehydration and dilution stage (Fig. 4d, e). Apoptosis-like events began at the pre-culture and continued through the recovery stage (Fig. 4c–e). Some cells became necrotic at the pre-culture and dilution stages (Fig. 4c, e). All PCD events were reduced at recovery (Fig. 4f). Nuclear integrity and TUNEL detection confirmed that apoptosis-like event occurred through the entire cryopreservation procedure (Fig. 5), and was most serious at the dehydration and dilution stages (Fig. 5d, e). Hoechst staining showed that the nucleus began to concentrate at pre-culture (Fig. 5c), and decomposed during dehydration and dilution treatments (Fig. 5d, e). TUNEL detection also exhibited an obvious nuclear fluorescence signal at the dehydration and dilution stages, indicating that DNA was highly fragmented in apoptotic cells.

Autophagy, apoptosis and necrosis detection of A. praecox EC cells during cryopreservation. a Positive control cells, b the EC cells; c pre-culture stage, d dehydration stage, e dilution stage, f 24-h recovery stage. 1 and 3 fluorescence microscopy bright field images, 2 and 4 fluorescence microscopy dark field images, 2 green fluorescence represents autophagy, 4 green fluorescence represents apoptosis, and red fluorescence indicates necrosis (white arrows). Scale bars 500 μm (color figure online)

Hoechst and TUNEL detection reveal apoptosis occurrence in A. praecox EC cells during cryopreservation. Hoechst staining indicates the integrity of the nucleus via blue fluorescence, and TUNEL reactions detect the DNA fragmentation via green fluorescence. a The CK cells, b pre-culture stage, c osmoprotection stage, d dehydration stage, e dilution stage, f 24-h recovery stage. Scale bars a1–f1, a3–f3 100 μm, a2–f2, a4–f4 50 μm (color figure online)
Gene quantitative analysis by qRT-PCR
The expression levels of antioxidant system-related genes indicated that the EC cells underwent serious oxidative damage during the dilution and recovery stages, and PCD-related genes testified that apoptosis-like event also occurred at these steps. In plant cells, NADPH oxidase catalyzes the conversion of O2 to O ·−2 , and O ·−2 can be continuously catalyzed by SOD forming H2O2 and CAT, forming H2O. NADPH oxidase was up-regulated at recovery and down-regulated at the other stages; SOD was up-regulated and CAT was down-regulated during all stages of cryopreservation (Fig. 6). The above results suggest that the cells produce large amounts of ROS molecules during recovery, and SOD and CAT gene expression levels indicate that the cell accumulated more H2O2 from pre-culture through the dilution stage. Furthermore, in addition to CAT, the AsA–GSH cycle and glutathione peroxidase (GPX) cycle are involved in scavenging intracellular H2O2. APX, MDHAR, and GR drive the AsA–GSH cycle, and GPX and GR regulate the GPX cycle. The expression patterns of APX, MDHAR, GR, and peroxiredoxin had the same trend; they were only significantly up-regulated at recovery. However, GPX was down-regulated at this stage (Fig. 6). This result indicated that AsA–GSH cycle plays an important role in scavenging excess H2O2 during recovery. Thus, AsA and GSH as exogenous antioxidants should be able to relieve oxidative damage of the cells in cryopreservation.

Real-time PCR quantitative analysis of several differentially expressed genes during cryopreservation in Agapanthus praecox. Bars show means of gene expression level and standard errors over triplicate detections; columns with different lowercase letters were significantly different (P < 0.05, least significant difference test). APX, ascorbate peroxidase; ATG5, autophagy protein 5; BAG, BCL-2-associated athanogene; CAT, catalase; CLPs, metacaspase-like proteins; DAD1, defender against apoptotic cell death 1; GPX, glutathione peroxidase; GR, glutathione reductase; MDHAR, monodehydroascorbate reductase; SOD, superoxide dismutase
PCD protein 5-like and defender against apoptotic cell death 1 (DAD1) were up-regulated in EC cells at recovery stage and were down-regulated at pre-culture and dehydration stage (Fig. 6). DAD1 is a universal negative regulator of PCD, and the gene expression levels indicated that the PCD event arising from oxidative stress has happened in EC cells during cryopreservation and DAD1 is inhibiting this event during recovery. The expression level of autophagy protein 5 (ATG5) has no significant differences between the control cells and the other treated samples, but it is significantly down-regulated at the dehydration and dilution stages compared to recovery. Apoptosis-related genes including BCL-2-associated athanogene (BAG) and Cytochrome c biogenesis C have a similar expression pattern which is specifically up-regulated expression at pre-culture and recovery, and these genes play important regulatory role in caspase-like enzymes activation. The RNA levels of metacaspase-like proteins (CLPs) and apoptosis inhibitory protein were significantly up-regulated at the recovery stage (Fig. 6). These results suggested that the EC cells experience apoptosis-like event after the cryopreservation process is completed. Therefore, inhibition of the upstream event (Cyt c release) or downstream event (caspase-like enzymes activity) of apoptosis-like PCD may effectively improve the cell viability in cryopreservation.
From relative viability analysis, EC cells exhibited differences in cell viability that were improved by GSH, AsA, cinnamtannin B-1, BCL-2 inhibitor, and caspase inhibitor after cryopreservation. Of these compounds, caspase inhibitor and cinnamtannin B-1 produced the greatest improvements during EC cryopreservation, and cell viability was significantly increased from 49.14 % (CK) to 89.91 % for caspase inhibitor and 86.85 for cinnamtannin B-1 (Fig. 7). AsA, GSH, and the BCL-2 inhibitor also increased the cell viability by 10–25 % and were significantly better than the control cells (Fig. 7). Antioxidants, the anti-stress compound, and apoptosis-like inhibitors significantly increased cell viability after cryopreservation, which also suggested that reducing the oxidative damage or apoptosis-like event is an effective way to improve recovery after cryopreservation.

Cell viability of EC cryopreservation as affected by antioxidants (cinnamtannin B-1), the anti-stress compound and apoptosis inhibitors. Bars represent means and standard errors over triplicate detections; columns with different lowercase letters were significantly different (P < 0.05, least significant difference test)
Discussion
Osmotic and oxidative damage occurred in the EC cells during cryopreservation
Plants are highly sensitive to low temperatures and require cryogenic protective treatments to ensure conservation of their structural integrity (Engelmann 2000). Low temperature or hypertonic pre-culture is required for plants to improve and induce dehydration or cold tolerance. Cryoprotectants are important for cell survival during cryopreservation; however, they can also cause some complex stresses, such as osmotic injury and dehydration stress (Uchendu et al. 2010a). In this study, cell morphological observation showed that the EC cells underwent serious plasmolysis and expansion during cryopreservation. Cell ultrastructure observation showed that autophagy and organelle degradation appeared in the EC cells during this process. MDA as the product of membrane lipid peroxidation continuously increased from pre-culture to the dilution stage, and the relative cell viability decreased concurrently during this process. These results indicated that the pre-culture and cryoprotectant treatments resulted in osmotic and oxidative damage to the cells. Thus, adding some antioxidants into pre-culture medium and/or cryoprotectant should improve the cell viability as suggested by Uchendu et al. (2010a, b). Wen et al. (2010, 2012) investigated cytological changes of Livistona chinensis and maize embryos during cryopreservation, and found that both dehydration and cryopreservation caused numerous ultrastructural alterations. Dehydration seriously impaired plasma membrane integrity, while cooling caused a further increase in electrolyte leakage. In A. praecox EC cells, plasma membrane damage, autophagy, and some apoptosis-like PCD characteristics appeared during dehydration treatment and these events were further exacerbated at the dilution stage. Our previous study also found that oxidative stress and apoptosis-like events were involved in response to cryogenic treatment in Arabidopsis seedlings (Ren et al. 2013). Furthermore, some reports demonstrated that oxidative stress is a mediator of apoptosis (Buttke and Sandstrom 1994; Chakraborti et al. 1999), and apoptosis can be initiated by ROS (Gechev et al. 2006). Therefore, this study provides evidence that osmotic and oxidative damage occurred in the EC cells during cryopreservation, and that ROS were involved in mediating this process.
ROS and antioxidant system
In plants, various abiotic stresses lead to the overproduction of ROS which are highly reactive, toxic, and ultimately result in oxidative stress (Gill and Tuteja 2010). ROS-induced oxidative stress is thought to be a fundamental cause of cell death in plant cryopreservation (Benson 1990). Membrane lipids are primary targets for oxidative stress (Benson and Bremner 2004). ROS are localized in several cellular compartments including the chloroplasts, mitochondria, and peroxisomes (del Rio et al. 2006; Navrot et al. 2007). Atmospheric oxygen (O2) is relatively non-reactive, but it can give rise to ROS which include O ·−2 , H2O2, OH·, and 1O2. A single electron reduction of O2 results in the generation of the O ·−2 ; SOD led dismutation of O ·−2 will produce H2O2; O ·−2 and H2O2 can be further catalyzed by the Haber–Weiss cycle and the Fenton reaction to generate OH· (Fig. 8). 1O2 can be formed by photo excitation of chlorophyll and its reaction with O2. In this study, EC cells contained only undifferentiated chloroplasts; thus, ROS in the EC tissue mainly included O ·−2 , H2O2, and OH·. ROS detection and correlation analysis showed that H2O2 and OH· generation activity had a significant positive correlation to MDA content. Therefore, H2O2 and OH· play important roles in mediating osmotic and oxidative damage in EC cells during cryopreservation. Furthermore, the cell relative viability had a significant negative correlation to MDA and H2O2 content. Consequently, H2O2 is the most important ROS molecule for mediating stress damage and affecting cell viability in the whole cryopreservation process.

Overall schematic of ROS inducing oxidative stress and apoptosis events in A. praecox EC during cryopreservation. The gray dashed line boxes represent each step of A. praecox EC cryopreservation. The black dashed line box represents EC cells' responses to low temperature and osmotic treatments from pre-culture and cryoprotectants. The red dashed line box represents ROS synthesis and metabolism; H2O2 with the orange stellate background indicates H2O2 is the most important ROS molecule mediating oxidative stress and apoptosis. The green dashed line box represents the antioxidant system, blue dotted line boxes denote AsA–GSH cycle and GPX cycle, red font with green oval background indicates enzymes involved in the antioxidant system; pink dashed line box represents apoptosis process; orange dashed line box represents oxidative stress; purple dashed line box represents membrane lipid peroxidation; blue dashed line box represents cell viability. Various color dashed lines represent the timelines of corresponding event occurrence in cryopreservation. APIs apoptosis inhibitory proteins, APX ascorbate peroxidase, AsA ascorbate acid, CAT catalase, cyt c cytochrome c, DHA dehydroascorbate, DHAR dehydroascorbate reductase, GPX glutathione peroxidase, GR glutathione reductase, GSH reduced glutathione, GSSG oxidized glutathione, MDHA monodehydroascorbate, SOD superoxide dismutase (color figure online)
Antioxidant systems work to control cascades of uncontrolled oxidation and protect cells from oxidative damage by scavenging of ROS (Gill and Tuteja 2010). O ·−2 can be catalyzed by SOD forming H2O2; H2O2 can be further transformed into H2O by CAT, the AsA–GSH cycle, and the GPX cycle (Fig. 8). OH· is thought to be largely responsible for mediating oxygen toxicity in vivo. OH· can potentially react with all of the biological molecules in cells and there are no enzymatic mechanisms for the elimination of this ROS (Vranová et al. 2002). Thus, scavenging excess H2O2 is a feasible method to alleviate abiotic stress damage during plant cryopreservation. CAT and the AsA–GSH cycle are involved in scavenging the intracellular H2O2 in this study. Adding AsA and GSH during the cryopreservation process increased cell viability 11.4% with AsA and 25.1 % with GSH. Poobathy et al. (2013) found that cryopreserved protocorm-like bodies (PLBs) of Dendrobium ‘Sonia-28’ underwent excessive oxidative stress due to decreased levels of CAT at the recovery stage, which led to poor survival. Fleck et al. (2003) suggested that higher tolerance in Haematococcus pluvialis cryopreservation was associated with high CAT and low SOD activities. Quan et al. (2008) indicated that H2O2 plays a dual role in plants; at low concentrations, it acts as a signal molecule triggering acclimation and resulting in tolerance to various biotic and abiotic stresses, and at high concentrations, it leads to PCD.
PCD events produce dynamic changes in the EC cells during cryopreservation
Programmed cell death is a process aimed at the removal of redundant, misplaced, or damaged cells. It is a crucial component of development and defense mechanisms in plants. Post cryopreservation apoptosis and necrosis are normally observed within 6–24 h after post-thaw culture, and result in a massive loss of cell viability and cellular function after cryopreservation (Bissoyi et al. 2014). Several morphological and biochemical features of plant PCD include DNA laddering, caspase-like proteolytic activity, cytochrome c release from mitochondria, and fragmentation of the cell into membrane-defined vesicles. These cell death processes are called apoptosis-like PCD (Hoeberichts and Woltering 2002). ROS and cellular stresses have emerged as important signals in the activation of plant PCD; they can trigger caspase-like proteins activation mediated by cytochrome c release from the mitochondria. Alternatively, endoplasmic reticulum stress can directly induce caspase-like enzymes activity. Once activated, caspase-like enzymes cleave numerous cellular proteins, eventually leading to dismantling of the cell (Fig. 8). The ROS and cellular redox environment play important regulatory role in caspase-like enzymes activation through effects on mitochondrial permeability. A second family of apoptosis-like PCD regulators is apoptosis inhibitory proteins (APIs), believed to suppress apoptosis-like event. DAD1 is a universal negative regulator of PCD, and the DAD1 encodes the protein involved in apoptosis-like PCD (Gallois et al. 1997). Our previous study found that DAD1 gene is up-regulated and can inhibited apoptosis-like event initiation during Arabidopsis seedling cryopreservation (Ren et al. 2013). Baust (2002) and Harding et al. (2009) also found that cryopreservation-induced cell death was a significant contributory factor in storage failure, and oxidative stress and apoptosis-like PCD may become the main factors in immediate and delayed-onset cryoinjury. The intrinsic ability to overcome oxidative stress and the apoptosis-like cascade may well be genetically determined (Johnston et al. 2009). To date, evidence for the existence of caspase-like proteins in plants is still indirect and mainly based on the inhibitory effects of caspase-specific inhibitors in plant cells. Such caspase-specific inhibitors can abolish bacteria-induced PCD in tobacco (del Pozo and Lam 1998). Caspase-like activity was also demonstrated in barley cell extracts and could only be inhibited by a specific caspase 3 inhibitor (Korthout et al. 2000). Recent studies have demonstrated that loss of seed viability may correspond to PCD, as shown by increased DNA fragmentation, caspase activity and Cyt c leakage in Pisum sativum, sunflower, and elm seeds (Kranner et al. 2006; El-Maarouf-Bouteau et al. 2011; Hu et al. 2012). The release of Cyt c and caspase-3-like/DEVDase activity increases during elm seed deterioration, and those increases can be suppressed by treatment with AsA and the caspase-3 inhibitor (Ac-DEVD-CHO), respectively (Hu et al. 2012). In this study, caspase inhibitor and cinnamtannin B-1 (a potent antioxidant) greatly improved EC cell viability after cryopreservation (Fig. 7). Caspase inhibitor is a cell permeable inhibitor of caspase-3, -6, -7, -8, and -10; and cinnamtannin B-1 effectively inhibits lipid peroxidation. Therefore, reducing oxidative stress or inhibiting apoptosis-like PCD by deactivating caspase-like proteins is a possible way to improve cell survival after cryopreservation.
Conclusions
This study analyzed the ROS, antioxidant system, and PCD events in A. praecox EC during cryopreservation. Successful cryopreservation depends on protective effects of pre-culture and cryoprotectant treatments; however, these treatments cause complex stresses from osmotic injury, dehydration, and low temperatures. These factors induce the production of ROS that may cause oxidative stress and apoptosis-like PCD, and H2O2 is the most important ROS molecule mediating stress damage and affecting cell viability in the whole cryopreservation process. Oxidative stress usually induces anti-oxidation and peroxidation in plant cells. Anti-oxidation is a positive and peroxidation a negative factor for cell survival. Furthermore, excess ROS and serious oxidative stress due to cryopreservation can lead to apoptosis-like PCD. Based on these results, we established a schematic of ROS induction of oxidative stress and apoptosis-like event affecting cell viability in plant cryopreservation, and indicated the timelines of occurrence of each biological process during cryopreservation (Fig. 8). Overall, H2O2-induced peroxidation and apoptosis-like PCD are the most important agents for reducing cell survival; reducing injury from these factors is the key to successful cryopreservation.
Author contribution statement
Di Zhang and Li Ren were responsible for conception and design of experiments, data analysis, and drafting of the manuscript. Guan-qun Chen and Jie Zhang completed part of experiment or offered some help. Barbara M. Reed and Xiao-hui Shen took care of study conception and design, and edited the manuscript.
References
Bajaj YPS (1995) Cryopreservation of plant cell, tissue and organ culture for the conservation of germplasm and biodiversity. In: Bajaj YPS (ed) Biotechnology in agriculture and forestry. Cryopreservation of plant germplasm I. Springer, USA, pp 3–18
Balk J, Leaver CJ, McCabe PF (1999) Translocation of cytochrome c from the mitochondria to the cytosol occurs during heat-induced programmed cell death in cucumber plants. FEBS Lett 463:151–154
Baust JM (2002) Molecular mechanisms of cellular demise associated with cryopreservation failure. Cell Preserv Technol 1:17–31
Benelli C, De Carlo A, Engelmann F (2013) Recent advances in the cryopreservation of shoot-derived germplasm of economically important fruit trees of Actinidia, Diospyros, Malus, Olea, Prunus, Pyrus and Vitis. Biotechnol Adv 31:175–185
Benson EE (1990) Free radical damage in stored plant germplasm. International Board for Plant Genetic Resources, Italy
Benson EE (2008) Cryopreservation of phytodiversity: a critical appraisal of theory and practice. Crit Rev Plant Sci 27:141–219
Benson EE, Bremner D (2004) Oxidative stress in the frozen plant: a free radical point of view. In: Fuller BJ, Lane N, Benson EE (eds) Life in the frozen state. CRC Press, USA, pp 205–241
Bissoyi A, Nayak B, Pramanik K, Sarangi SK (2014) Targeting cryopreservation-induced cell death: a review. Biopreserv Biobank 12:23–34
Buttke TM, Sandstrom PA (1994) Oxidative stress as a mediator of apoptosis. Immunol Today 15:7–10
Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S (1999) Oxidant, mitochondria and calcium: an overview. Cell Signal 11:77–85
del Pozo O, Lam E (1998) Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr Biol 8:1129–1132
del Rio LA, Sandalio LM, Corpas FJ, Palma JM, Barroso JB (2006) Reactive oxygen species and reactive nitrogen species in peroxisomes. Production, scavenging, and role in cell signaling. Plant Physiol 141:330–335
Dowling DK, Simmons LW (2009) Reactive oxygen species as universal constraints in life-history evolution. Proc R Soc B 276:1737–1745
El-Maarouf-Bouteau H, Mazuy C, Corbineau F, Bailly C (2011) DNA alteration and programmed cell death during ageing of sunflower seed. J Exp Bot 62:5003–5011
Engelmann F (2000) Importance of cryopreservation for the conservation of plant genetic resources. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm: current research progress and application. JIRCAS, Tsukuba/IPGRI, Rome, pp 8–20
Engelmann F (2004) Cryopreservation: progress and prospects. In Vitro Cell Dev Plant 40:427–433
Erdei L, Tari I, Csiszár J, Pécsváradi A, Horváth F, Szabó M, Ördög M, Cseuz L, Zhiponova M, Szilák L, Györgyey J (2002) Osmotic stress responses of wheat species and cultivars differing in drought tolerance: some interesting genes (advices for gene hunting). Acta Biol Szeged 46:63–65
Fang JY, Wetten A, Johnston J (2008) Headspace volatile markers for sensitivity of cocoa (Theobroma cacao L) somatic embryos to cryopreservation. Plant Cell Rep 27:453–461
Fleck RA, Benson EE, Bremner DH, Day JG (2003) A comparative study of antioxidant protection in cryopreserved unicellular algae Euglena gracilis and Haematococcus pluvialis. CryoLett 24:213–228
Fowler WE, Aebi U (1983) Preparation of single molecules and supramolecular complexes for high-resolution metal shadowing. J Ultrastruct Res 83:319–334
Foyer CH, Halliwell B (1976) The presence of glutathione and glutathione reductase in chloroplasts: a proposed role in ascorbic acid metabolism. Planta 133:21–25
Gallois P, Makishima T, Hecht V, Despres B, LaudiéM Nishimoto T, Cooke R (1997) An Arabidopsis thaliana cDNA complementing a hamster apoptosis suppressor mutant. Plant J 11:1325–1331
Gao C, Xing D, Li L, Zhang L (2008) Implication of reactive oxygen species and mitochondrial dysfunction in the early stages of plant programmed cell death induced by ultraviolet-C overexposure. Planta 227:755–767
Gechev TS, van Breusegem F, Stone JM, Denev I, Laloi C (2006) Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. BioEssays 28:1091–1101
Gill SS, Tuteja N (2010) Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem 48:909–930
Hansen G (2000) Evidence for Agrobacterium-induced apoptosis in maize cells. Mol Plant Microbe Interact 13:649–657
Harding K, Johnston JW, Benson EE (2009) Exploring the physiological basis of cryopreservation success and failure in clonally propagated in vitro crop plant germplasm. Agric Food Sci 18:103–116
Hoeberichts FA, Woltering EJ (2002) Multiple mediators of plant programmed cell death: interplay of conserved cell death mechanisms and plant-specific regulators. BioEssays 25:47–57
Hu D, Ma G, Wang Q, Yao J, Wang Y, Pritchard HW, Wang X (2012) Spatial and temporal nature of reactive oxygen species production and programmed cell death in elm (Ulmus pumila L.) seeds during controlled deterioration. Plant Cell Environ 35:2045–2059
Johnston JW, Benson EE, Harding K (2009) Cryopreservation induces temporal DNA methylation epigenetic changes and differential transcriptional activity in Ribes germplasm. Plant Physiol Biochem 47:123–131
Jones A (2000) Does the plant mitochondrion integrate cellular stress and regulate programmed cell death? Trends Plant Sci 5:225–230
Korthout HA, Berecki G, Bruin W, Van Duijn B, Wang M (2000) The presence and subcellular localization of caspase 3-like proteinases in plant cells. FEBS Lett 475:139–144
Kranner I, Birtic S, Anderson KM, Pritchard HW (2006) Glutathione half-cell reduction potential: a universal stress marker and modulator of programmed cell death? Free Radic Biol Med 40:2155–2165
Kulus D, Zalewska M (2014) Cryopreservation as a tool used in long-term storage of ornamental species—a review. Sci Hortic 168:88–107
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
McCabe PF, Leaver CJ (2000) Programmed cell death in cell cultures. Plant Mol Biol 44:359–368
Meryman HT (2007) Cryopreservation of living cells: principles and practice. Transfusion 47:935–945
Meyer AJ (2008) The integration of glutathione homeostasis and redox signaling. J Plant Physiol 165:1390–1403
Navrot N, Rouhier N, Gelhaye E, Jaquot JP (2007) Reactive oxygen species generation and antioxidant systems in plant mitochondria. Physiol Plant 129:185–195
Peng J, Wang D, Xu C, Chen L, Deng F, Zhang X (2009) Ammonium molybdate method for detecting the activities of rice catalase. Chin Agric Sci Bull 25:61–64
Poobathy R, Sinniah UR, Xavier R, Subramaniam S (2013) Catalase and superoxide dismutase activities and the total protein content of protocorm-like bodies of Dendrobium Sonia-28 subjected to vitrification. Appl Biochem Biotechnol 170:1066–1079
Quan LJ, Zhang B, Shi WW, Li HY (2008) Hydrogen peroxide in plants: a versatile molecule of the reactive oxygen species network. J Integr Plant Biol 50:2–18
Queval G, Issakidis-Bourguet E, Hoeberichts FA, Vandorpe M, Gakière B, Vanacker H, Miginiac-Maslow M, Breusegem FV, Noctor G (2007) Conditional oxidative stress responses in the Arabidopsis photorespiratory mutant cat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of H2O2-induced cell death. Plant J 52:640–657
Reape TJ, Molony EM, McCabe PF (2008) Programmed cell death in plants: distinguishing between different modes. J Exp Bot 59:435–444
Reed BM (2008) Cryopreservation—practical considerations. In: Reed BM (ed) Plant cryopreservation: a practical guide. Springer, USA, pp 3–13
Ren L, Zhang D, Jiang XN, Gai Y, Wang WM, Reed BM, Shen XH (2013) Peroxidation due to cryoprotectant step is a vital factor for cell survival in Arabidopsis cryopreservation. Plant Sci 212:37–47
Sakai A, Hirai D, Niino T (2008) Development of PVS-based vitrification and encapsulation-vitrification protocols. In: Reed BM (ed) Plant cryopreservation: a practical guide. Springer, USA, pp 33–57
Scandalios JG (1997) Molecular genetics of SOD in plants. In: Scandalios JG (ed) Oxidative stress and the molecular biology of antioxidant defense. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 527–568
Skyba M, Petijová L, Košuth J, Koleva DP, Ganeva TG, Kapchina-Toteva VM, Cellárová E (2012) Oxidative stress and antioxidant response in Hypericum perforatum L. plants subjected to low temperature step. J Plant Physiol 169:955–964
Sun YL, Zhao Y, Hong X, Zhai ZH (1999) Cytochrome c release and caspase activation during menadione-induced apoptosis in plants. FEBS Lett 462:317–321
Tatone C, Emidio GD, Ventol M, Ciriminna R, Artini PG (2010) Cryopreservation and oxidative stress in reproductive cells. Gynecol Endocrinol 26:563–567
Uchendu EE, Leonard SW, Traber MG, Reed BM (2010a) Vitamins C and E improve regrowth and reduce lipid peroxidation of blackberry shoot tips following cryopreservation. Plant Cell Rep 29:25–35
Uchendu EE, Muminova M, Gupta S, Reed BM (2010b) Antioxidant and anti-stress compounds improve regrowth of cryopreserved Rubus shoot tips. In Vitro Cell Dev-Pl 46:386–393
Vacca RA, de Pinto MC, Valenti D, Passarella S, Marra E, De Gara L (2004) Production of reactive oxygen species, alteration of cytosolic ascorbate peroxidase, and impairment of mitochondrial metabolism are early events in heat shock-induced programmed cell death in tobacco Bright-Yellow 2 cells. Plant Physiol 134:1100–1112
Vacca RA, Valenti D, Bobba A, MeraWna RS, Passarella S, Marra E (2006) Cytochrome c is released in a reactive oxygen species-dependent manner and is degraded via caspase-like proteases in tobacco Bright-Yellow 2 cells en route to heat shock-induced cell death. Plant Physiol 141:208–219
Van Breusegem F, Vranová E, Dat JF, Inzé D (2001) The role of active oxygen species in plant signal transduction. Plant Sci 161:405–414
Volk GM, Henk A, Basu C (2011) Gene expression in response to cryoprotectant and liquid nitrogen exposure in Arabidopsis shoot tips. Acta Hortic 908:55–66
Vranová E, Atichartpongkul S, Villarroel R, Van Montagu M, Inzé D, Van Camp W (2002) Comprehensive analysis of gene expression in Nicotiana tabacum leaves acclimated to oxidative stress. Proc Natl Acad Sci USA 99:870–875
Wang Y, Fan X, Zhang D, Shen X (2012) Regeneration of Agapanthus praecox ssp. orientalis ‘big blue’ via somatic embryogenesis. Propag Ornam Plants 12:148–154
Wen B, Wang RL, Cheng HY, Song SQ (2010) Cytological and physiological changes in orthodox maize embryos during cryopreservation. Protoplasma 239:57–67
Wen B, Cai C, Wang R, Song S, Song J (2012) Cytological and physiological changes in recalcitrant Chinese fan palm (Livistona chinensis) embryos during cryopreservation. Protoplasma 249:323–335
Whitaker C, Beckett RP, Minibayeva FV, Kranner I (2010) Production of reactive oxygen species in excised, desiccated and cryopreserved explants of Trichilia dregeana Sond. S Afr J Bot 76:112–118
Wu N, Ren F, Wu X (2001) Assay of hydroxyl radical produced in Co2+-H2O2 reaction system by spectrophotometry with bromopyrogallol red. J Instrum Anal 20:52–54
Xu J, Liu Q, Jia M, Liu Y, Li B, Shi Y (2014) Generation of reactive oxygen species during cryopreservation may improve Lilium × siberia pollen viability. In Vitro Cell Dev Plant 50:369–375
Zhang LR, Xing D (2008) Methyl jasmonate induces production of reactive oxygen species and alterations in mitochondrial dynamics that precede photosynthetic dysfunction and subsequent cell death. Plant Cell Physiol 49:1092–1111
Zhang Y, Wang J, Tang J, Guo L, Yang J, Huang Y, Tan Y, Fu S, Kong X, Zheng F (2009) In vivo protein transduction: delivery of PEP-1-SOD1 fusion protein into Myocardium efficiently protects against Ischemic Insult. Mol Cells 27:159–166
Zhang D, Ren L, Yue JH, Wang L, Zhuo LH, Shen XH (2013) A comprehensive analysis of flowering transition in Agapanthus praecox ssp. orientalis (Leighton) Leighton by using transcriptomic and proteomic techniques. J Proteomics 80:1–25
Zhang D, Ren L, Yue J, Wang L, Zhuo L, Shen X (2014) GA4 and IAA were involved in the morphogenesis and development of flowers in Agapanthus praecox ssp. orientalis. J Plant Physiol 171:966–976
Acknowledgments
This study received support from the National Natural Science Foundation of China (No. 31300580 and No. 31170655), Special Financial Grant from the China Postdoctoral Science Foundation (2013T60451), and Shanghai Graduate Education and Innovation Program (Horticulture).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. S. Judelson.
D. Zhang and L. Ren contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
299_2015_1802_MOESM1_ESM.xlsx
Supplementary material 1 (XLSX 10 kb) Supporting Information Table S1 the sequence and annealing temperature of all primers for qRT-PCR
Rights and permissions
About this article

Cite this article
Zhang, D., Ren, L., Chen, Gq. et al. ROS-induced oxidative stress and apoptosis-like event directly affect the cell viability of cryopreserved embryogenic callus in Agapanthus praecox . Plant Cell Rep 34, 1499–1513 (2015). https://doi.org/10.1007/s00299-015-1802-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-015-1802-0