
Abstract
In the past three decades, fungal respiratory colonization and fungal respiratory infections increasingly raised concern in cystic fibrosis (CF). Reasons for this are a better knowledge of the pathogenicity of fungi, whereby detection is sought in more and more CF centers, but also improvement of detection methods. However, differences in fungal detection rates within and between geographical regions exist and indicate the need for standardization of mycological examination of respiratory secretions. The still existing lack of standardization also complicates the assessment of fungal pathogenicity, relevance of fungal detection and risk factors for fungal infections. Nevertheless, numerous studies have now been conducted on differences in detection methods, epidemiology, risk factors, pathogenicity and therapy of fungal diseases in CF. Meanwhile, some research groups now have classified fungal disease entities in CF and developed diagnostic criteria as well as therapeutic guidelines.The following review presents an overview on fungal species relevant in CF. Cultural detection methods with their respective success rates as well as susceptibility testing will be presented, and the problem of increasing azole resistance in Aspergillus fumigatus will be highlighted. Next, current data and conflicting evidence on the epidemiology and risk factors for fungal diseases in patients with CF will be discussed. Finally, an overview of fungal disease entities in CF with their current definitions, diagnostic criteria and therapeutic options will be presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past three decades, several studies have been conducted regarding fungal colonization of the airways and fungal respiratory infections in the context of cystic fibrosis (CF). Although Aspergillus fumigatus for filamentous fungi and Candida albicans for yeasts remain by far the most common fungal species in patients with CF, the pattern of fungal species associated with CF considerably diversified since the beginning of this century [1,2,3,4,5,6]. Thus, beside A. fumigatus, some Scedosporium species (Scedosporium boydii, as well as Scedosporium ellipsoideum, Scedosporium apiospermum, Scedosporium aurantiacum and to a lesser extent Scedosporium minutisporum) and the closely related species Lomentospora prolificans are worldwide recognized today as significant fungal pathogens in CF [6, 7]. Likewise, other Aspergillus species belonging to the sections Fumigati, Terrei, Nigri or Flavi and some species of the Rasamsonia argillacea complex or the black yeast Exophiala dermatitidis for molds, as well as Candida dubliniensis, Blastobotrys adeninivorans/Blastobotrys raffinosifermentans or the Basidiomycete Apiotrichum mycotoxinovorans (formerly Trichosporon mycotoxinivorans) for yeasts, are increasingly reported in CF patients as responsible for chronic colonization of the airways and sometimes causing respiratory infections or severe disseminated infections from a primary pulmonary site [6]. In addition, some atypical yeast species like Candida blankii or the basidiomycete Cutaneotrichosporon cyanovorans (formerly Cryptococcus cyanovorans) that have been recently reported in some CF patients [8, 9] might be on the rise in a next future because of repeated cures of azole antifungals.
Moreover, several epidemiological studies have been performed recently in different countries in order to determine the frequency of fungal pathogens and their clinical significance, or to identify risk factors for airway colonization or respiratory infection. Likewise, particular attention has been paid to understanding the pathogenic mechanisms of these fungi and to evaluate new therapeutic approaches. Nevertheless, our knowledge on fungal infections in CF is hampered by the lack of standardization of the biological diagnosis, leading to individual diagnostic approaches and interpretation of clinical parameters. This may result in underestimation of the real frequency of fungi in this context and in false conclusions regarding geographic differences or influence of lifestyle on their frequency. Also, the clinical significance of isolating fungi and risk factors could be misinterpreted due to the possible lack of detection of fungi in some individuals included in control groups. In this review, we will present limitations of the routine procedures used for biological diagnosis in clinical laboratories and improvements made recently on this topic. We will also summarize progress made in the epidemiology of fungal respiratory infections in CF, as well as in the definition of different clinical entities, risk factors for these infections and their treatment.
Pitfalls in the Biological Diagnosis
Culture-Based Detection Methods
Comparison of the data from one study to another is hampered by the lack of standardization of mycological examination of respiratory secretions that has been pointed out since the beginning of the past decade [10, 11]. Some laboratories inoculate the samples on agar slants which offer a more limited surface for fungal growth compared to agar plates. Some fungal species may therefore be missed in case of mixed fungal populations which are common in the CF airways. Likewise, great variations may be seen from one laboratory to another in the volume of the sample being analyzed, as well as in the number of culture media inoculated and in incubation time and temperature [10]. Moreover, in most countries, mycological analysis of clinical samples is specifically performed only for immunocompromised patients with clinical signs and symptoms suggestive of an invasive fungal infection. The microbiological follow-up of CF patients is usually limited to a bacteriological analysis of respiratory secretions and samples are inoculated on Sabouraud dextrose agar supplemented with antibiotics (SDA) only when the presence of a fungus is suspected at direct microscopic examination of the sample.
In addition, even when microbiological surveillance comprises a specific search for fungi, there may be differences in the periodicity of the analysis. For example, although a mycological analysis is now recommended on all sputum samples from CF patients in France, it is still limited to the routine annual visit of the patient in many CF-care centers. Such differences unable consensus definitions of chronic colonization of the airways.
Differences in the population studied, in environmental exposure and in lifestyle of the patients possibly account for part of the differences that can be seen in the prevalence of filamentous fungi from one study to another. Nevertheless, limitations in the biological diagnosis need to be considered as attested by the recent publication from Reece et al. [12] or the comparison of current practices in clinical laboratories in the UK [13, 14].
This was perfectly illustrated by Hong et al. [15] in Baltimore (MA, USA). During one year, consecutive sputa (n = 487) collected from outpatients were inoculated in parallel on bacteriological agar plates, which were incubated for three days at 37 °C, or on SDA, Brain–Heart infusion (BHI) agar plates supplemented with antibiotics and inhibitory mold agar (IMA) plates which were incubated for two weeks at 30 °C. Only 48 of the samples (9.8%) revealed to be positive for fungi using bacteriological media, whereas they were about 120 (from 23.8% to 24.8%) on each of the three mycological media incubated at 30 °C. Interestingly, 32% of the samples revealed the presence of fungi when combining the results obtained on SDA, BHI agar and IMA and the prevalences of Aspergillus and Scedosporium/Lomentospora species were found to be twofold to threefold higher than their reported prevalences in the USA (40.8% vs. 20.4% and 5.2% vs. 1.9%, respectively). Likewise, only 3 out of the 20 Scedosporium/Lomentospora and 1 out of the 17 Trichosporon positive samples were detected using bacteriological media and none of the Exophiala positive samples. Of note, samples were not digested with a mucolyticum nor sonicated or diluted before inoculation of the plates and no Scedosporium-selective culture media were used [15].
Many countries currently have their own recommendations. Nevertheless, these need to be updated to allow a correct overview of the fungal biota colonizing the CF lungs. For example, the UK CF Trust Laboratory Standards for Processing Microbiological Samples from People with Cystic Fibrosis recommend the use of SDA with incubation for up to seven days at 35–37 °C, whereas inoculation of the clinical specimens on mycological culture media with antibiotics is only required when search for fungi is specifically requested by clinicians according to the US Cumulative Techniques and Procedures in Clinical Microbiology (Cumitech) Guidelines. Thus, fungal surveillance is not routinely performed in most clinical centers in the USA [15]. Improved guidelines for mycological examination of sputum samples from CF patients, therefore, are urgently needed [16].
In this aim, two multicenter studies were conducted recently within the Fri-CF (Fungal respiratory infections in Cystic Fibrosis) working group launched by both the European Confederation of Medical Mycology (ECMM) and the International Society for Human and Animal Mycology (ISHAM). The studies encompassed 7 CF-care centers from France for the MucoFong project [17] and 19 laboratories (9 from France and 4 from Italy, as well as laboratories from Spain, UK, Belgium, Austria, Greece and Australia, one each) for the MFIP project [18]. All laboratories agreed to use the same procedure for mycological examination of samples, including inoculation on a wide range of culture media and incubation of agar plates at two different temperatures. From the analysis of the obtained data, a combination of three to four culture media was proposed [17, 18]. Single-center studies were also conducted. For example, the added value of a new protocol including prior digestion of the sample with a mucolyticum, increased inoculum size, additional culture media (SDA, B + medium, Sce-Sel + and dichloran-glycerol agar) and longer incubation time (3 weeks) was evaluated in Nijmegen (The Netherlands) [19]. Based on this new protocol, a marked increase was noted in both the frequency and the diversity of molds.
Nevertheless, some semi-selective culture media were not evaluated in these studies, such as dichloran-rose bengal-chloramphenicol agar, the inhibitory mold agar used by Hong et al. [15] and others [20, 21] or the Scedo-Select III culture medium developed by Pham et al. [22] for environmental studies and used recently in clinical practice [21]. Likewise, the Burkholderia cepacia selective agar was recently reported to enhance the recovery of Exophiala dermatitidis from sputum samples [23]. In addition, all growing fungi should be identified, which is not the case presently, for example, because of a lack of consideration of some fungi as potential pathogens, misidentifications using conventional methods or limits of commercially available databases for MALDI-TOF mass spectrometry. For example, in a questionnaire survey of current practices in UK CF centers, only 7 out of the 11 respondents performed species identification for Exophiala species and only 2 for non-Candida yeasts [13]. Likewise, some molds colonizing the CF airways may be misidentified on the basis of the sole macroscopic and microscopic morphology, or when using only commercially available databases for MALDI-TOF mass spectrometry. This is particularly true for Rasamsonia species which are frequently misidentified as Penicillium or Paecilomyces species [24,25,26].
Further multicenter studies comparing these "new" culture media with those previously selected [17, 18] and using gold-standard procedures for species identification should be conducted in order to provide evidence-based guidelines for an optimal service for the patients.
Antifungal Susceptibility Testing
In vitro antifungal susceptibility testing of mold isolates is mandatory because of primary resistance of some fungal species and increasing occurrence of acquired resistance in other species. To do this, well-standardized methods are available such as the microbroth reference method in RPMI culture medium and the Etest procedure on RPMI agar plates. Nevertheless, the results may be disappointing with discrepancies between in vitro susceptibility to triazole antifungals and lack of efficacy of these drugs in vivo.
Analysis of transcriptomic changes induced in S. apiospermum in response to the particular physicochemical environment encountered by the fungus in the CF airways revealed reprogramming of genes involved in the synthesis of some important cell wall or membrane components. This includes genes encoding the glycosylphosphatidylinositol anchor and sphingolipids, but also ergosterol with the down-regulation of five of the genes involved in ergosterol biosynthesis pathway [27]. These metabolic changes could explain the discrepancies mentioned above regarding triazole drugs, since environmental conditions in CF lungs may lead to a drastic reduction in the ergosterol content of the plasma membrane, maintaining fungal growth despite the inhibition of the azole target, the 14 alpha-demethylase encoded by the gene Cyp51A (also called ERG11, especially in yeasts).
In agreement with these data, Mello et al. [28] reported that cultivation of some Scedosporium species under 5% carbon dioxide in a medium mimicking the biochemical composition and viscosity of the CF bronchial mucus resulted in increased resistance to triazole drugs compared to reference growth conditions. By contrast, opposite results were reported on A. fumigatus clinical isolates collected from CF patients [29]: The use of a synthetic medium mimicking the CF bronchial mucus resulted for 8 out the 10 isolates studied in a downshift of the set toward greater susceptibility to voriconazole compared to data obtained in RMPI culture broth (results unchanged for the two other isolates). However, all isolates failed to grow sufficiently in the synthetic culture medium and further studies are needed to decide on the best culture conditions for antifungal susceptibility testing of clinical isolates recovered from CF patients.
Spread of Azole Resistance in Aspergillus Fumigatus
Azoles are used intensively to treat invasive and chronic aspergillosis. Therefore, the likelihood for selection of resistant isolates is high. To address this concern, particular attention has been paid in recent years investigating the susceptibility to azole drugs of A. fumigatus isolates using the microbroth dilution method and, whenever necessary, cyp51A gene sequencing.
Analysis of isolates collected from respiratory samples of CF patients revealed resistance rates ranging from 5.3% to 13.2%. For example, a resistance rate of up to 8.2% was found for A. fumigatus isolates collected from CF patients in Italy [30]. In Germany, 2888 A. fumigatus isolates from 961 CF patients were screened prospectively and 101 isolates from 51 of these patients (5.3% of the patients) were found to be azole resistant [31]. Likewise, in the UK, 167 A. fumigatus isolates collected from 135 patients with CF were investigated; resistance to at least one azole drug was confirmed in 22 out of these isolates (13.2%) [32]. Finally, similar results were reported in France and in the Netherlands. During a one-year period, all A. fumigatus isolates collected from CF patients in a French reference center for CF were investigated prospectively; 23 out of the 355 isolates studied (6.5%) were found to be resistant to at least one triazole drug, leading to a prevalence of 6.8% (6/88 patients) [33]. In the Netherlands, azole-resistant A. fumigatus isolates were cultured from 7.3% (10/137) of the patients with CF [34].
These prevalence rates, sometimes alarmingly high, are of clinical relevance and should lead clinicians and microbiologists to perform susceptibility testing in patients with CF [35].
In patients with CF, azole-resistant A. fumigatus strains may be selected during the course of azole therapy, but they may also be recovered from azole-naïve CF patients, in relation with the extensive use of triazole fungicides in agriculture. Contrasting with the acquired resistance to azole drugs in the course of azole therapy which is usually due to point mutations in the cyp51A gene sequence, analysis of resistant isolates from environmental origin revealed one or two point mutations in the coding sequence of this gene associated with the insertion of a 34-bp or a 46-bp tandem repeat in the promoter region of the gene. Only some TR46/Y121F/T289A azole-resistant isolates have been reported so far in CF [31, 36,37,38], yet the environmentally driven mutation TR34/L98H is now distributed worldwide. For instance, it has been reported during the past decade from CF patients in France [33, 39,40,41], Germany [31, 37, 42], the Netherlands [43], Denmark [34, 36, 44], Italy [30], UK [32] and Argentina [45], with a markedly high rate in the most recent studies. However, no mutations in the cyp51A gene or its promoter were found in some azole-resistant isolates from CF patients that were investigated in Argentina [45] or Turkey [46]. Therefore, other mechanisms independent of this gene should be looked for to fully understand the acquired resistance to triazole drugs.
Prevalence rates of and Risk factors for Fungal Respiratory infections in Cystic Fibrosis
Epidemiology of Fungal Respiratory Infections
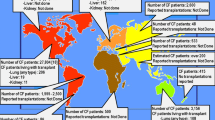
Epidemiological studies on fungal colonization of the respiratory tract in CF reveal a high proportion of filamentous fungi and yeasts. Among filamentous fungi, A. fumigatus is the most frequently detected species (up to 50% of the patients in some studies) and C. albicans among yeasts (up to 50%) [4,5,6, 21, 47,48,49,50,51,52,53,54]. However, various other fungal species are also reported in patients with CF, such as E. dermatitidis, A. mycotoxinivorans, or species of the R. argillacea complex [5, 6]. In a recent work on the epidemiology of fungal colonization of the airways in German CF patients (Fig. 1), 4153 patients were studied and mycological examination of their respiratory secretions revealed, as expected, C. albicans and the filamentous fungus A. fumigatus as predominant fungal species. Scedosporium/Lomentospora species, which rank the second among the filamentous fungi associated with CF in most epidemiological studies, were less frequent, as well as Exophiala species [48]. Analysis of the data from the 2017 CF registry cohort in Germany also showed that, apart from Candida yeasts which may be detected very early in the evolution of the CF disease, colonization of the airways by filamentous fungi is very rare in young children and its frequency progressively increases since the age of six years, reaching a plateau in young adults.

Prevalence of fungi in German CF patients according to the national 2017 CF registry [46]
Conversely to Aspergillus species which exhibit rapid and extensive growth on SDA plates, other mold species colonizing CF airways may be missed due to a short incubation period of the agar plates, or in case of co-colonization with an Aspergillus species. As some of these fungi such as Scedosporium and Lomentospora species may also cause severe and sometimes fatal disseminated infections in immunosuppressed patients, like patients undergoing lung transplantation, they are therefore considered as a contraindication to lung transplantation in some centers [55]. Considering this, all efforts should be made to detect these fungi as early as possible, at least before registration on the lung transplantation waiting list. However, as abovementioned, detection of these fungi from bronchial secretions is much less successful if semi-selective culture media are not used [1, 6, 16, 19,20,21, 49].
The reasons for the increasingly frequent detection of fungi in the respiratory tract of patients with CF are still unclear. Different hypotheses have been discussed, including the frequent use of oral, intravenous or inhaled antibiotics [56, 57] and of inhaled corticosteroids [58], the increase in life expectancy [59] and the significant improvement of fungal detection methods [6, 16], which has already been highlighted in the first section of this review. In addition, the extent to which the aforementioned fungi have clinical relevance in patients with CF is only partially known. But in recent years, relevant scientific results have been added in the context of clinical trials that shed more light on the relevance of their detection.
Interestingly, during the past few years, preliminary data on respiratory infections in CF patients were reported from China [52], as well as the first epidemiological data from Poland [53], Argentina [20] and Iran [21, 54] on the prevalence of fungi in the CF lungs. These studies highlight some geographical variations in the prevalence of some fungal species. For instance, up to 90% of the patients were found to be colonized by Aspergillus species in Argentina [20]. In this study, which used a variety of mycological culture media (including erythritol agar, IMA and Sce-Sel +), E. dermatitidis was recovered from 14% of the patients and Scedosporium species from 10%. However, A. fumigatus was not the predominant species in Iranian CF patients. For instance, by the follow-up of 40 CF patients, Aspergillus terreus was identified as the predominant filamentous fungus followed by A. fumigatus, Aspergillus oryzae and Aspergillus flavus [54]. Likewise, a highly rigorous study was published by Hedayati et al. [21]. During one year (2017/02 to 2018/02), sputum samples from all patients attending a pulmonary reference center in Tehran (n = 90) were analyzed for fungi by parallel inoculation on Malt extract agar, IMA, BHI agar and Scedo-Select III, with incubation for 7 days at 37 °C. As expected, Scedosporium species ranked second among the filamentous fungi (3.3% of the samples), after Aspergillus species, but compared to A. fumigatus (14.5% of the samples), higher frequencies were found for other Aspergillus species, A. flavus (29.4%), A. tubingensis (24.7%) and A. niger (17.0%).
Identification of Risk Factors for Fungal Diseases in Cystic Fibrosis
As fungal colonization in the respiratory tract is a frequent finding and different fungal disease entities can occur such as bronchitis, pneumonia or allergic bronchopulmonary aspergillosis (ABPA) that influence patient’s outcome, identification of risk factors is of paramount importance.
Several risk factors for A. fumigatus colonization, like continuous antibiotic therapy or chronic lung inflammation as well as older age, are currently being discussed [60], but patient-to-patient transmission has been recently reported as a potential risk factor [61]. Likewise, intermittent co-colonization of the airways with Pseudomonas aeruginosa and A. fumigatus seems to lead to more frequent hospitalizations and lower lung function like chronic P. aeruginosa infection [62, 63]. Numerous studies have been conducted on interkingdom interactions in CF. However, whether the interactions between P. aeruginosa and A. fumigatus result in antagonistic or synergistic effects on the lung inflammation remains to be elucidated [62, 64,65,66,67].
The most recent publication in this field was from Germany. A total of 5665 CF patients were followed up during two different years [48]. In order to investigate specifically risk factors for A. fumigatus colonization (at least two positive cultures in 12 months), exclusion criteria included medical history of lung transplantation and colonization of the airways by P. aeruginosa as it has the strongest impact on the clinical outcome. Main risk factors for colonization with A. fumigatus were age, continuous antibiotic treatment, CFTR modulators and number of pulmonary exacerbations (Table 1). Conflicting results, however, were reported by Baxter et al. [68] who found that intravenous antibiotics reduce the occurrence of A. fumigatus in sputum samples. But as patients usually have less abundant respiratory secretions after intravenous therapy, the amount and quality of sputum specimens might have influenced the results of this study.
Interestingly, we recently also identified pet ownership [69] or frequent pet contacts [70] as a risk factor for ABPA, which may have important implications for clinicians since they should pay greater attention to patients that are in contact with pets.
Regarding Scedosporium and Lomentospora species, a registry analysis revealed the use of inhaled antibiotics as a risk factor for detection of these fungi from CF respiratory secretions [71]. In a German prospective study, adjusted multivariate regression analysis showed that colonization of the airways by these fungi was associated with younger age (OR 0.8684, 95% CI 0.7955–0.9480, p < 0.005) and less frequent colonization with Haemophilus influenzae (OR 0.0118, 95% CI 0.0009–0.1585, p < 0.001). In addition, patients colonized by these fungi more often suffered from ABPA (OR 14.6663, 95% CI 2.1873–98.3403, p < 0.01) and have been more frequently colonized by the mucoid phenotype of P. aeruginosa (OR 9.8941, 95% CI 1.0518–93.0705, p < 0.05) [72].
Aspergillus Fumigatus Clinical Entities
Only few publications are available regarding the clinical significance of fungal detection from respiratory secretions. However, using registry data from the German Cystic Fibrosis Registry, an impact on lung function was demonstrated for the first time on a large cohort of patients [73]. Of the 2599 patients, 32.5% were colonized with Aspergillus species. These patients had significantly worse FEV1 percent predicted values with a median of 64.5 ± 24.6 compared to 75.7 ± 27.3 (p < 0.0001) in the control group. A limitation of this study was the failure to differentiate patients with positive cultures for Aspergillus species into their respective disease entity. Thus, patients with Aspergillus species detection may have harmless colonization, sensitization, or clinically relevant ABPA and there is current evidence in the literature for new aspergillosis disease entities [73,74,75]. Indeed, individual patient cases have been described as Aspergillus bronchitis and, currently, Aspergillus pneumonia [76].
ABPA is recognized as a disease entity in CF since a long time and is well described in terms of diagnosis and therapy [76,77,78,79,80,81,82,83]. However, in the setting of CF, diagnosis of ABPA can be very difficult because its symptoms often cannot be separated from those of bronchopulmonary exacerbation. For this reason, a consensus conference has defined diagnostic criteria to facilitate diagnosis in the context of CF [84]. Nevertheless, even with this recommendation, some cases of ABPA are diagnosed lately or even overlooked, so there is a high need for an improved diagnosis, especially with regard to ABPA. Defining the different Aspergillus clinical entities is a prerequisite for comparing patients’ data, especially in relation to therapeutic indications.
Definition of Aspergillus Colonization
Up to now, there is no clear, consensual definition regarding chronic colonization of the airways with Aspergillus or any other fungal species. Nevertheless, there is now an increasing body of evidence that chronic colonization of the airways by A. fumigatus or even Candida yeasts is associated with lung function decline [48, 59, 85,86,87,88,89,90,91]. However, even intermittent co-colonization with P. aeruginosa and A. fumigatus also seems to lead to more frequent hospitalizations and lower lung function like chronic P. aeruginosa infections [62, 92]. To increase evidence about impact of A. fumigatus colonization on CF patients’ lung disease, data from the German CF Registry were correlated with lung function, chronic P. aeruginosa infection and treatment for CF lung disease. CF patients without chronic P. aeruginosa infection, however, had a significantly lower FEV1 in case of only one or at least two positive A. fumigatus cultures per year (p < 0.0001). Pulmonary exacerbations requiring antibiotic treatment were experienced by significant more CF patients with at least two positive A. fumigatus cultures (53.9% vs. 39.5% without A. fumigatus detection in 2016 and 73.9% vs. 59.2% in 2017; p < 0.0001 in both cases) [48]. Different statuses were defined for this study: patients with no A. fumigatus-positive culture from respiratory samples, with only one A. fumigatus-positive culture per year or with at least two A. fumigatus-positive cultures per year, defined as no A. fumigatus colonization, transient colonization and chronic colonization. Nevertheless, the quality of respiratory samples is crucial for defining any of the statuses.
Definition of Aspergillus spp. Bronchitis
Aspergillus bronchitis was first described in 2006 [75]. It is defined as by Aspergillus-positive cultures from sputum, respiratory exacerbations unresponsive to antibiotic treatment, successful antifungal treatment and exclusion of ABPA [75]. Further specifications and a modified definition of Aspergillus bronchitis were published in 2013 by Baxter et al. [74] including a cutoff index of > 0.5 for sputum galactomannan, positive A. fumigatus-specific PCR and elevated levels for specific serum IgG antibodies (> 75 mg/l) in combination with a total serum IgE level < 500 IU/ml (whereas a total serum IgE level > 500 IU/ml indicates an ABPA) and the lack of specific serum IgE. The latest publication on Aspergillus bronchitis, using the aforementioned definition, could demonstrate a significant increase in FEV1 and decrease in cough and sputum after antifungal therapy with a duration of up to 6 weeks. The impact of Aspergillus species on the clinical course of patients with CF has been shown in several studies and is not, therefore, a matter of discussion anymore [2, 48, 59, 75,76,77, 79,80,81,82,83, 85,86,87, 93, 94]. But which patient groups would profit from antifungal or even anti-inflammatory therapy is hard to define as randomized clinical trials are not available to answer this question. Thus, the recommendation would be to screen for Aspergillus bronchitis when patients’ status is instable and signs of exacerbations occur without responding to antibiotics.
Definition of Aspergillus Pneumonia
Severe invasive lung infections caused by fungal species are well known in immunosuppressed patients [6, 7, 26, 95, 96], but their occurrence in non-immunocompromised CF patients has been described only recently [6, 7, 94, 97]. As many patients with CF are colonized with A. fumigatus, the bottleneck is to distinguish between harmless colonization and clinically relevant infections. To overcome this issue, international expert groups proposed a definition for pulmonary infections caused by fungi to guide clinicians [94, 97, 98]. These fungal infections are defined as follows:
-
Increased sputum production;
-
Multiple isolation of the same fungal species from sputum or bronchoalveolar lavage (≥ twice over a 6-month period);
-
Pulmonary infiltrate(s) on chest CT scan, MRI or X-ray;
-
Treatment failure with antibiotic therapy (≥ 2 x antibiotic treatment, duration ≥ two weeks);
-
Unclear lung function decline (exclusion of new CF-related diseases, e.g., diabetes mellitus);
-
Exclusion of new/other bacteria (e.g., non-tuberculous mycobacteria or P. aeruginosa);
-
And exclusion of ABPA.
Although this definition may help to identify patients with fungal pneumonia, it can be still difficult to confirm this diagnosis. CT scans might be a very helpful tool in diagnosing fungal pneumonia. Typical findings for this specific diagnosis may be ground-glass density surrounding a nodule, also known as “halo sign,” or consolidation with internal ground-class density. But other findings like semi-solid and ground-glass nodules and especially peripheral well-circumscribed nodules also might be specific hints for severe pulmonary fungal infections [94, 99, 100].
A novel diagnostic tool has been proposed in recent years relying on the measurement of A. fumigatus-specific T cell response during the onset of fungal infections [101,102,103]. This method represents the direct host–pathogen reaction and could also identify the right pathogen despite negativity of culture-based detection methods or PCR-based methods.
Allergic Bronchopulmonary Aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) is known as a hypersensitivity reaction to A. fumigatus. The prevalence of ABPA is estimated to be 10% in patients with CF [104]. Patients with ABPA are likely to have lower lung function and are less likely to fully recover following treatment of pulmonary exacerbations [76, 105].
Given that CF and ABPA have overlapping clinical, radiographic and immunological features, the diagnosis of ABPA remains challenging and sometimes very difficult. Therefore, guidelines have been published to standardize the diagnosis and management of ABPA in patients with CF [84].
The ISHAM ABPA working group proposed the following criteria to diagnose ABPA [106]:
-
Aspergillus fumigatus-positive skin prick test or presence of serum specific IgE associated with an elevated total serum IgE level (> 1000 kU/L);
-
And two of the following criteria:
-
Presence of specific anti-A. fumigatus IgG antibodies;
-
Eosinophilia of more than 500 cells/µL (in steroid-naïve patients);
-
Radiographic pulmonary opacities consistent with ABPA.
A recently published systemic review and meta-analysis on biomarkers for diagnosing ABPA demonstrated that serum IgE directed toward the recombinant proteins rAsp f4 and rAsp f 6 and hyperattenuating mucus on CT scan complement current diagnostic criteria to improve diagnostic specificity [107]. Another very promising tool for diagnosis of ABPA again is the Aspergillus-specific T cell response. In a recent pilot study in patients with CF and ABPA, we could demonstrate that Aspergillus-specific TH17 cells confirmed the diagnosis in patients with acute ABPA without specific treatment. The assay could also discriminate patients with acute ABPA from patients after treatment, so that this biomarker might not only be helpful for defining acute ABPA but also to prove the clinical response to therapy [108]. Nevertheless, randomized clinical trials are needed in order to guide clinicians in diagnosing and treating ABPA in an evidence-based manner.
Antifungal Therapy
The current antifungal armamentarium is divided into four chemical classes: azole dugs, polyenes, echinocandins and flucytosine. Being able to draw on a wide range of antifungals is crucial to select the best therapeutic option for each patient. It is not uncommon for the clinician to be confronted with a situation where antifungals in combination with other medication induce drug–drug interactions [109]. In addition, acquired or primary resistance of some fungal species needs to be taken into account.
ABPA was the first A. fumigatus-related clinical entity recognized in CF [77, 110]. Thus, therapy recommendations exist over a long time already [84]. Nevertheless, a recent survey in the UK on clinicians’ behavior in terms of treatment options of ABPA demonstrated the need for establishing evidence-based guidelines [14]. 66.7% of the respondent clinicians were using prednisolone alongside itraconazole for treatment of first diagnosis of ABPA, whereas 35.1% reported using prednisolone alone and 3.5% prednisolone with voriconazole. This survey also demonstrated a significant difference in treatment choices between adult and pediatric physicians. Prednisolone therapy alone was chosen for a first diagnosis of ABPA by 47% of adult clinicians, vs. 20% of pediatricians (p < 0.0001). Finally, no adult physicians reported the use of voriconazole, but 8% of pediatricians would use prednisolone with voriconazole.
Fortunately, progress has been made in the past few years for treatment of Aspergillus infections in CF patients, with the availability of some new antifungal drugs (isavuconazole and echinocandins, caspofungin, micafungin and anidulafungin). Recently, the Pulmatrix company (Lexington, MA) started the development of a new formulation combining itraconazole with the dry powder iSPERSE™ technology which allows delivering high doses of drug directly into the lungs by inhalation while minimizing side effects [111]. This formulation called PUR1900 has been designated by the US Food and Drug Administration a Qualified Infectious Disease Product (QIDP) for treatment of ABPA in patients with CF.
Treatment of A. fumigatus bronchitis is recommended, but as abovementioned this clinical entity is not always easy to identify. The recommended treatment relies on the use of a single azole drug orally, with a duration of 2 to 6 weeks [73,74,75]. An interesting alternative would be the inhalation of an antifungal drug such as amphotericin B, but unfortunately it is not approved for inhalation. Therefore, good news is the start of clinical trials with inhaled itraconazole [111].
Aspergillus fumigatus or non-Aspergillus species pneumonia is a new disease entity in patients with CF [6, 7, 94] which needs attention from the radiologist and the clinician as well. Aspergillus fumigatus infections are usually treated with monotherapy such as voriconazole, posaconazole, caspofungin, micafungin or amphotericin B [94, 112].
Nevertheless, treatment of fungal respiratory infections in CF remains challenging when they are caused by multi-resistant fungi like Scedosporium species and Lomentospora prolificans or the emerging Rasamsonia species. In vitro studies showed that these fungi exhibit a primary resistance or low susceptibility to most available antifungals [6, 26]. However, several bodies of work performed in recent years highlight the potential of combination therapy for Scedosporium/Lomentospora infections. In those cases, a combination of two or even three different antifungals is recommended [6, 7, 26, 94, 97, 113], comprising usually an echinocandin, particularly micafungin which may also be used for treatment of Rasamsonia infections [6, 94, 97]. For all treatment recommendations, performing susceptibility testing is mandatory as resistance can occur [6, 7, 35].
Finally, another important question for clinicians is whether or not colonization without clinical impact should be treated. Randomized trials are needed to answer this question. Nevertheless, there is now increasing evidence that chronic colonization of the airways by Candida yeasts or A. fumigatus results in diminished pulmonary function [48, 57, 60, 85,86,87,88,89,90,91]. In addition, colonization of the airways with resistant fungi such as Lomentospora prolificans should lead to at least think about eradication therapy since these fungi can impact the clinical course with causing severe infections [7, 94, 97] and may be considered a contraindication to lung transplantation in some centers.
Conclusion
Although significant progress has been made regarding fungal colonization of the airways and fungal respiratory infections, many challenges remain to be addressed by the CF community (Fig. 2). The newly described selective culture media and the various procedures for mycological examination of respiratory secretions from CF patients should be evaluated in multicenter studies, in order to propose guidelines to the European Cystic Fibrosis Society and relevant national societies. Culture methods mimicking the particular physicochemical conditions encountered in the CF bronchial mucus also need to be evaluated for determination of azole susceptibility of fungal isolates recovered from sputum samples. Then, large multicenter epidemiological studies using well-standardized procedures for both bacteriological and mycological examinations of sputum samples could be conducted in order to determine the real frequency of fungal species associated with CF, the clinical relevance of fungal colonization of the airways and the risk factors for fungal diseases in CF. In addition, clinical and radiological criteria supporting the different clinical entities that may be caused by fungal respiratory pathogens should be validated by the CF community, as well as some recently proposed biological markers. Finally, treatment strategies should be defined taking into account the new formulations and new antifungals in the pipeline.

Key actions to be addressed by the CF community for a better understanding of fungal infections in CF and improvement of patient management. ECFS: European Cystic Fibrosis Society
References
Pihet M, Carrère J, Cimon B, Chabasse D, Delhaes L, Symoens F, et al. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis – a review. Med Mycol. 2009;47(4):387–97.
Middleton PG, Chen SCA, Meyer W. Fungal infections and treatment in cystic fibrosis. Curr Opin Pulm Med. 2013;19(6):670–5.
Chotirmall SH, McElvaney NG. Fungi in the cystic fibrosis lung: Bystanders or pathogens? Int J Biochem Cell Biol. 2014;52:161–73.
Bouchara JP, Symoens F, Schwarz C, Chaturvedi V. Fungal respiratory infections in cystic fibrosis (CF): Recent progress and future research agenda. Mycopathologia. 2018;183(1):1–5.
Schwarz C, Bouchara JP, Buzina W, Chrenkova V, Dmeńska H, de la Pedrosa EGG, et al. Organization of patient management and fungal epidemiology in cystic fibrosis. Mycopathologia. 2018;183(1):7–19.
Schwarz C, Vandeputte P, Rougeron A, Giraud S, Dugé De Bernonville T, Duvaux L, et al. Developing collaborative works for faster progress on fungal respiratory infections in cystic fibrosis. Med Mycol. 2018;56:S42–59.
Ramirez-Garcia A, Pellon A, Rementeria A, Buldain I, Barreto-Bergter E, Rollin-Pinheiro R, et al. Scedosporium and Lomentospora: An updated overview of underrated opportunists. Med Mycol. 2018;56(suppl_1):S102–25.
Zaragoza S, Galanternik L, Vazquez M, Teper A, Córdoba S, Finquelievich J. 318 Candida blankii: New agent in cystic fibrosis airways? J Cyst Fibros. 2015;14(Suppl 1):S140.
van der Bruggen T, Kolecka A, Theelen B, Kwakkel-van Erp JM, Arets B, Boekhout T. Cutaneotrichosporon (Cryptococcus) cyanovorans, a basidiomycetous yeast, isolated from the airways of cystic fibrosis patients. Med Mycol Case Rep. 2018;22:18–20.
Borman AM, Palmer MD, Delhaes L, Carrère J, Favennec L, Ranque S, et al. Lack of standardization in the procedures for mycological examination of sputum samples from CF patients: a possible cause for variations in the prevalence of filamentous fungi. Med Mycol. 2010;48(Suppl 1):S88-97.
Pashley CH, Fairs A, Morley JP, Tailor S, Agbetile J, Bafadhel M, et al. Routine processing procedures for isolating filamentous fungi from respiratory sputum samples may underestimate fungal prevalence. Med Mycol. 2012;50(4):433–8.
Reece E, McClean S, Greally P, Renwick J. The prevalence of Aspergillus fumigatus in early cystic fibrosis disease is underestimated by culture-based diagnostic methods. J Microbiol Methods. 2019;164:105683.
Boyle M, Moore JE, Whitehouse JL, Bilton D, Downey DG. Laboratory diagnosis and characterization of fungal disease in patients with cystic fibrosis (CF): A survey of current UK practice in a cohort of clinical microbiology laboratories. Mycopathologia. 2018;183(4):723–9.
Boyle M, Moore JE, Whitehouse JL, Bilton D, Downey DG. The diagnosis and management of respiratory tract fungal infection in cystic fibrosis: A UK survey of current practice. Med Mycol. 2019;57(2):155–60.
Hong G, Miller HB, Allgood S, Lee R, Lechtzin N, Zhang SX. Use of selective fungal culture media increases rates of detection of fungi in the respiratory tract of cystic fibrosis patients. J Clin Microbiol. 2017;55(4):1122–30.
Chen SC, Meyer W, Pashley CH. Challenges in laboratory detection of fungal pathogens in the airways of cystic fibrosis patients. Mycopathologia. 2018;183(1):89–100.
Coron N, Pihet M, Fréalle E, Lemeille Y, Pinel C, Pelloux H, et al. Toward the standardization of mycological examination of sputum samples in cystic fibrosis: Results from a French multicenter prospective study. Mycopathologia. 2018;183(1):101–17.
Delhaes L, Touati K, Faure-Cognet O, Cornet M, Botterel F, Dannaoui E, et al. Prevalence, geographic risk factor and development of a standardized protocol for fungal isolation in cystic fibrosis: Results from the international prospective study “MFIP.” J Cyst Fibros. 2019;18(2):212–20.
Engel TGP, Tehupeiory-Kooreman M, Melchers WJG, Reijers MH, Merkus P, Verweij PE. Evaluation of a new culture protocol for enhancing fungal detection rates in respiratory samples of cystic fibrosis patients. J Fungi (Basel). 2020;6(2):82.
Brito Devoto T, Alava KSH, Pola SJ, Pereda R, Rubeglio E, Finquelievich JL, et al. Molecular epidemiology of Aspergillus species and other moulds in respiratory samples from Argentinean patients with cystic fibrosis. Med Mycol. 2020;58(7):867–73.
Hedayati MT, Tavakoli M, Maleki M, Heidari S, Mortezaee V, Gheisari M, et al. Fungal epidemiology in cystic fibrosis patients with a special focus on Scedosporium species complex. Microb Pathog. 2019;129:168–75.
Pham T, Giraud S, Schuliar G, Rougeron A, Bouchara JP. Scedo-Select III: a new semi-selective culture medium for detection of the Scedosporium apiospermum species complex. Med Mycol. 2015;53(5):512–9.
Raclavsky V, Novotny R. Burkholderia cepacia selective agar can be useful for recovery of Exophiala dermatitidis from sputum samples of cystic fibrosis patients. J Cyst Fibros. 2016;15(2):e19.
Giraud S, Pihet M, Razafimandimby B, Carrère J, Degand N, Mely L, et al. Geosmithia argillacea: an emerging pathogen in patients with cystic fibrosis. J Clin Microbiol. 2010;48(7):2381–6.
Giraud S, Favennec L, Bougnoux ME, Bouchara JP. Rasamsonia argillacea species complex: taxonomy, pathogenesis and clinical relevance. Future Microbiol. 2013;8(8):967–78.
Hong G, White M, Lechtzin N, West NE, Avery R, Miller H, et al. Fatal disseminated Rasamsonia infection in cystic fibrosis post-lung transplantation. J Cyst Fibros. 2017;16(2):e3-7.
Vandeputte P, Dugé de Bernonville T, Le Govic Y, Le Gal S, Nevez G, Papon N, et al. Comparative transcriptome analysis unveils the adaptative mechanisms of Scedosporium apiospermum to the microenvironment encountered in the lungs of patients with cystic fibrosis. Comput Struct Biotechnol J. 2020;18:3468–83.
Mello TP, Lackner M, Branquinha MH, Santos ALS. Impact of biofilm formation and azoles’ susceptibility in Scedosporium/Lomentospora species using an in vitro model that mimics the cystic fibrosis patients’ airway environment. J Cyst Fibros. 2020;S1569–1993(20):30933–4.
Stevens DA, Moss RB, Hernandez C, Clemons KV, Martinez M. Effect of media modified to mimic cystic fibrosis sputum on the susceptibility of Aspergillus fumigatus and the frequency of resistance at one center. Antimicrob Agents Chemother. 2016;60:2180–4.
Prigitano A, Esposto MC, Biffi A, De Lorenzis G, Favuzzi V, Koncan R, et al. Triazole resistance in Aspergillus fumigatus isolates from patients with cystic fibrosis in Italy. J Cyst Fibros. 2017;16(1):64–9.
Seufert R, Sedlacek L, Kahl B, Hogardt M, Hamprecht A, Haase G, et al. Prevalence and characterization of azole-resistant Aspergillus fumigatus in patients with cystic fibrosis: a prospective multicentre study in Germany. J Antimicrob Chemother. 2018;73(8):2047–53.
Abdolrasouli A, Scourfield A, Rhodes J, Shah A, Elborn JS, Fisher MC, et al. High prevalence of triazole resistance in clinical Aspergillus fumigatus isolates in a specialist cardiothoracic centre. Int J Antimicrob Agents. 2018;52(5):637–42.
Lavergne RA, Morio F, Danner-Boucher I, Horeau-Langlard D, David V, Hagen F, et al. One year prospective survey of azole resistance in Aspergillus fumigatus at a French cystic fibrosis reference centre: prevalence and mechanisms of resistance. J Antimicrob Chemother. 2019;74(7):1884–9.
Risum M, Hare RK, Gertsen JB, Kristensen L, Johansen HK, Helweg-Larsen J, et al. Azole-resistant Aspergillus fumigatus among Danish cystic fibrosis patients: Increasing prevalence and dominance of TR34/L98H. Front Microbiol. 2020;11:1850.
Hamprecht A, Morio F, Bader O, Le Pape P, Steinmann J, Dannaoui E. Azole resistance in Aspergillus fumigatus in patients with cystic fibrosis: A matter of concern? Mycopathologia. 2018;183(1):151–60.
Astvad KM, Jensen RH, Hassan TM, Mathiasen EG, Thomsen GM, Pedersen UG, et al. First detection of TR46/Y121F/T289A and TR34/L98H alterations in Aspergillus fumigatus isolates from azole-naive patients in Denmark despite negative findings in the environment. Antimicrob Agents Chemother. 2014;58(9):5096–101.
Fischer J, van Koningsbruggen-Rietschel S, Rietschel E, Vehreschild MJ, Wisplinghoff H, Krönke M, et al. Prevalence and molecular characterization of azole resistance in Aspergillus spp. isolates from German cystic fibrosis patients. J Antimicrob Chemother. 2014;69(6):1533–6.
Lavergne RA, Morio F, Favennec L, Dominique S, Meis JF, Gargala G, et al. First description of azole-resistant Aspergillus fumigatus due to TR46/Y121F/T289A mutation in France. Antimicrob Agents Chemother. 2015;59(7):4331–5.
Morio F, Aubin GG, Danner-Boucher I, Haloun A, Sacchetto E, Garcia-Hermoso D, et al. High prevalence of triazole resistance in Aspergillus fumigatus, especially mediated by TR/L98H, in a French cohort of patients with cystic fibrosis. J Antimicrob Chemother. 2012;67(8):1870–3.
Burgel PR, Baixench MT, Amsellem M, Audureau E, Chapron J, Kanaan R, et al. High prevalence of azole-resistant Aspergillus fumigatus in adults with cystic fibrosis exposed to itraconazole. Antimicrob Agents Chemother. 2012;56(2):869–74.
Guégan H, Chevrier S, Belleguic C, Deneuville E, Robert-Gangneux F, Gangneux JP. Performance of molecular approaches for Aspergillus detection and azole resistance surveillance in cystic fibrosis. Front Microbiol. 2018;9:531.
Bader O, Weig M, Reichard U, Lugert R, Kuhns M, Christner M, et al. cyp51A-Based mechanisms of Aspergillus fumigatus azole drug resistance present in clinical samples from Germany. Antimicrob Agents Chemother. 2013;57(8):3513–7.
Engel TGP, Slabbers L, de Jong C, Melchers WJG, Hagen F, Verweij PE, et al. Prevalence and diversity of filamentous fungi in the airways of cystic fibrosis patients - A Dutch, multicentre study. J Cyst Fibros. 2019;18(2):221–6.
Mortensen KL, Jensen RH, Johansen HK, Skov M, Pressler T, Howard SJ, et al. Aspergillus species and other molds in respiratory samples from patients with cystic fibrosis: a laboratory-based study with focus on Aspergillus fumigatus azole resistance. J Clin Microbiol. 2011;49(6):2243–51.
Brito Devoto T, Hermida-Alva K, Posse G, Finquelievich JL, García-Effrón G, Cuestas ML. High prevalence of triazole-resistant Aspergillus fumigatus sensu stricto in an Argentinean cohort of patients with cystic fibrosis. Mycoses. 2020;63(9):937–41.
Güngör Ö, Sampaio-Maia B, Amorim A, Araujo R, Erturan Z. Determination of azole resistance and TR(34)/L98H mutations in isolates of Aspergillus section Fumigati from Turkish cystic fibrosis patients. Mycopathologia. 2018;183(6):913–20.
Sudfeld CR, Dasenbrook EC, Merz WG, Carroll KC, Boyle MP. Prevalence and risk factors for recovery of filamentous fungi in individuals with cystic fibrosis. J Cyst Fibros. 2010;9(2):110–6.
Düesberg U, Wosniok J, Naehrlich L, Eschenhagen P, Schwarz C. Risk factors for respiratory Aspergillus fumigatus in German cystic fibrosis patients and impact on lung function. Sci Rep. 2020;10(1):18999.
Sedlacek L, Graf B, Schwarz C, Albert F, Peter S, Würstl B, et al. Prevalence of Scedosporium species and Lomentospora prolificans in patients with cystic fibrosis in a multicenter trial by use of a selective medium. J Cyst Fibros. 2015;14(2):237–41.
Valenza G, Tappe D, Turnwald D, Frosch M, König C, Hebestreit H, et al. Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J Cyst Fibros. 2008;7(2):123–7.
Paugam A, Baixench M-T, Demazes-Dufeu N, Burgel P-R, Sauter E, Kanaan R, et al. Characteristics and consequences of airway colonization by filamentous fungi in 201 adult patients with cystic fibrosis in France. Med Mycol. 2010;48(O1):S32–6.
Shen Y, Liu J, Zhong L, Mogayzel PJ Jr, Zeitlin PL, Sosnay PR, Zhao S. Clinical phenotypes and genotypic spectrum of cystic fibrosis in Chinese children. J Pediatr. 2016;171:269-76.e1.
Garczewska B, Jarzynka S, Kuś J, Skorupa W, Augustynowicz-Kopeć E. Fungal infection of cystic fibrosis patients - single center experience. Pneumonol Alergol Pol. 2016;84(3):151–9.
Nasri E, Fakhim H, Vaezi A, Khalilzadeh S, Ahangarkani F, Laal Kargar M, et al. Airway colonisation by Candida and Aspergillus species in Iranian cystic fibrosis patients. Mycoses. 2019;62(5):434–40.
Renner S, Nachbaur E, Jaksch P, Dehlink E. Update on respiratory fungal infections in cystic fibrosis lung disease and after lung transplantation. J Fungi (Basel). 2020;6(4):381.
Burns JL, Van Dalfsen JM, Shawar RM, Otto KL, Garber RL, Quan JM, et al. Effect of chronic intermittent administration of inhaled tobramycin on respiratory microbial flora in patients with cystic fibrosis. J Infect Dis. 1999;179(5):1190–6.
Bargon J, Dauletbaev N, Köhler B, Wolf M, Posselt H-G, Wagner TOF. Prophylactic antibiotic therapy is associated with an increased prevalence of Aspergillus colonization in adult cystic fibrosis patients. Respir Med. 1999;93(11):835–8.
Noni M, Katelari A, Dimopoulos G, Kourlaba G, Spoulou V, Alexandrou-Athanassoulis H, et al. Inhaled corticosteroids and Aspergillus fumigatus isolation in cystic fibrosis. Med Mycol. 2014;52(7):715–22.
Saunders RV, Modha DE, Claydon A, Gaillard EA. Chronic Aspergillus fumigatus colonization of the pediatric cystic fibrosis airway is common and may be associated with a more rapid decline in lung function. Med Mycol. 2016;54(5):537–43.
Hong G, Psoter KJ, Jennings MT, Merlo CA, Boyle MP, Hadjiliadis D, et al. Risk factors for persistent Aspergillus respiratory isolation in cystic fibrosis. J Cyst Fibros. 2018;17(5):624–30.
Engel TGP, Erren E, Vanden Driessche KSJ, Melchers WJG, Reijers MH, Merkus P, et al. Aerosol transmission of Aspergillus fumigatus in cystic fibrosis patients in the Netherlands. Emerg Infect Dis. 2019;25(4):797–9.
Reece E, Segurado R, Jackson A, McClean S, Renwick J, Greally P. Co-colonisation with Aspergillus fumigatus and Pseudomonas aeruginosa is associated with poorer health in cystic fibrosis patients: an Irish registry analysis. BMC Pulm Med. 2017;17(1):70.
Hector A, Kirn T, Ralhan A, Graepler-Mainka U, Berenbrinker S, Riethmueller J, et al. Microbial colonization and lung function in adolescents with cystic fibrosis. J Cyst Fibros. 2016;15(3):340–9.
Mowat E, Rajendran R, Williams C, McCulloch E, Jones B, Lang S, et al. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol Lett. 2010;313(2):96–102.
Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, et al. The airway microbiota in cystic fibrosis: A complex fungal and bacterial community—Implications for therapeutic management. PLoS One. 2012;7(4):e36313.
Shirazi F, Ferreira JAG, Stevens DA, Clemons KV, Kontoyiannis DP. Biofilm filtrates of Pseudomonas aeruginosa strains isolated from cystic fibrosis patients inhibit preformed Aspergillus fumigatus biofilms via apoptosis. PLoS One. 2016;11(3):e0150155.
Briard B, Mislin GLA, Latgé JP, Beauvais A. Interactions between Aspergillus fumigatus and pulmonary bacteria: Current state of the field, new data and future perspective. J Fungi (Basel). 2019;5(2):48.
Baxter CG, Rautemaa R, Jones AM, Webb AK, Bull M, Mahenthiralingam E, et al. Intravenous antibiotics reduce the presence of Aspergillus in adult cystic fibrosis sputum. Thorax. 2013;68(7):652–7.
Thronicke A, Heger N, Antweiler E, Krannich A, Roehmel J, Brandt C, et al. Allergic bronchopulmonary aspergillosis is associated with pet ownership in cystic fibrosis. Pediatr Allergy Immunol. 2016;27(6):597–603.
Grehn C, Eschenhagen P, Temming S, Düesberg U, Neumann K, Schwarz C. Frequent pet contact as risk factor for allergic bronchopulmonary aspergillosis in cystic fibrosis. Front Cell Infect Microbiol. 2021;10:601821.
Hong G, Lechtzin N, Hadjiliadis D, Kawut SM. Inhaled antibiotic use is associated with Scedosporium/Lomentospora species isolation in cystic fibrosis. Pediatr Pulmonol. 2018;54(2):133–40.
Schwarz C, Brandt C, Antweiler E, Krannich A, Staab D, Schmitt-Grohé S, et al. Prospective multicenter German study on pulmonary colonization with Scedosporium /Lomentospora species in cystic fibrosis: Epidemiology and new association factors. PLoS One. 2017;12(2):e0171485.
Brandt C, Roehmel J, Rickerts V, Melichar V, Niemann N, Schwarz C. Aspergillus bronchitis in patients with cystic fibrosis. Mycopathologia. 2018;183(1):61–9.
Baxter CG, Dunn G, Jones AM, Webb K, Gore R, Richardson MD, et al. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol. 2013;132(3):560-6.e10.
Shoseyov D, Brownlee KG, Conway SP, Kerem E. Aspergillus bronchitis in cystic fibrosis. Chest. 2006;130(1):222–6.
Mastella G, Rainisio M, Harms H., Hodson M., Koch C, Navarro J, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. A European epidemiological study. Eur Respir J. 2000;16(3):464.
Cohen-Cymberknoh M. Fungal infection and ABPA in CF. Paediatr Respir Rev. 2013;14:S34–5.
Tanou K, Zintzaras E, Kaditis AG. Omalizumab therapy for allergic bronchopulmonary aspergillosis in children with cystic fibrosis: A synthesis of published evidence. Pediatr Pulmonol. 2014;49(5):503–7.
Elphick HE, Southern KW. Antifungal therapies for allergic bronchopulmonary aspergillosis in people with cystic fibrosis. Cochrane Database Syst Rev. 2016;11(11):CD002204.
Janahi IA, Rehman A, Al-Naimi AR. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann Thorac Med. 2017;12(2):74–82.
Patel G, Greenberger PA. Allergic bronchopulmonary aspergillosis. Allergy Asthma Proc. 2019;40(6):421–4.
Sunman B, Ademhan Tural D, Ozsezen B, Emiralioglu N, Yalcin E, Özçelik U. Current approach in the diagnosis and management of allergic bronchopulmonary aspergillosis in children with cystic fibrosis. Front Pediatr. 2020;8:582964.
Lattanzi C, Messina G, Fainardi V, Tripodi MC, Pisi G, Esposito S. Allergic bronchopulmonary aspergillosis in children with cystic fibrosis: An update on the newest diagnostic tools and therapeutic approaches. Pathogens. 2020;9(9):716.
Stevens DA, Moss RB, Kurup VP, Knutsen AP, Greenberger P, Judson MA, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis—State of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis. 2003;37(s3):S225–64.
Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest. 2010;137(1):171–6.
Fillaux J, Brémont F, Murris M, Cassaing S, Rittié JL, Tétu L, et al. Assessment of Aspergillus sensitization or persistent carriage as a factor in lung function impairment in cystic fibrosis patients. Scand J Infect Dis. 2012;44(11):842–7.
Noni M, Katelari A, Dimopoulos G, Doudounakis SE, Tzoumaka-Bakoula C, Spoulou V. Aspergillus fumigatus chronic colonization and lung function decline in cystic fibrosis may have a two-way relationship. Eur J Clin Microbiol Infect Dis. 2015;34(11):2235–41.
Chotirmall SH, O’Donoghue E, Bennett K, Gunaratnam C, O’Neill SJ, McElvaney NG. Sputum Candida albicans presages FEV1 decline and hospital-treated exacerbations in cystic fibrosis. Chest. 2010;138(5):1186–95.
Gileles-Hillel A, Shoseyov D, Polacheck I, Korem M, Kerem E, Cohen-Cymberknoh M. Association of chronic Candida albicans respiratory infection with a more severe lung disease in patients with cystic fibrosis. Pediatr Pulmonol. 2015;50(11):1082–9.
AbdulWahab A, Salah H, Chandra P, Taj-Aldeen SJ. Persistence of Candida dubliniensis and lung function in patients with cystic fibrosis. BMC Res Notes. 2017;10(1):326.
Al Shakirchi M, Klingspor L, Bergman P, Hjelte L, de Monestrol I. A 16-year retrospective study on fungal prevalence and diversity in patients with cystic fibrosis: Candida dubliniensis was associated with a decline in lung function. Int J Infect Dis. 2020;96:663–70.
Hector A, Frey N, Hartl D. Update on host-pathogen interactions in cystic fibrosis lung disease. Mol Cell Pediatr. 2016;3(1):12.
King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J Infect. 2016;72:S50–5.
Schwarz C, Brandt C, Whitaker P, Sutharsan S, Skopnik H, Gartner S, et al. Invasive pulmonary fungal infections in cystic fibrosis. Mycopathologia. 2018;183(1):33–43.
Iversen M, Burton CM, Vand S, Skovfoged L, Carlsen J, Milman N, et al. Aspergillus infection in lung transplant patients: Incidence and prognosis. Eur J Clin Microbiol Infect Dis. 2007;26(12):879–86.
Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. Hidden killers: Human fungal infections. Sci Transl Med. 2012;4(165):1–10.
Schwarz C, Brandt C, Melichar V, Runge C, Heuer E, Sahly H, et al. Combined antifungal therapy is superior to monotherapy in pulmonary scedosporiosis in cystic fibrosis. J Cyst Fibros. 2019;18(2):227–32.
Schwarz C, Hartl D, Eickmeier O, Hector A, Benden C, Durieu I, et al. Progress in definition, prevention and treatment of fungal infections in cystic fibrosis. Mycopathologia. 2018;183(1):21–32.
Marchiori E, Zanetti G, Hochhegger B, Irion KL, Carvalho ACP, Godoy MCB. Reversed halo sign on computed tomography: State-of-the-art review. Lung. 2012;190(4):389–94.
Hussien A, Lin CT. CT findings of fungal pneumonia with emphasis on aspergillosis. Emerg Radiol. 2018;25(6):685–9.
Bacher P, Scheffold A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A. 2013;83(8):692–701.
Bacher P, Schink C, Teutschbein J, Kniemeyer O, Assenmacher M, Brakhage AA, et al. Antigen-reactive T cell enrichment for direct, high-resolution analysis of the human naive and memory Th cell repertoire. J Immunol. 2013;190(8):3967–76.
Scheffold A, Schwarz C, Bacher P. Fungus-specific CD4 T cells as specific sensors for identification of pulmonary fungal infections. Mycopathologia. 2018;183(1):213–26.
Maturu VN, Agarwal R. Prevalence of Aspergillus sensitization and allergic bronchopulmonary aspergillosis in cystic fibrosis: Systematic review and meta-analysis. Clin Exp Allergy. 2015;45(12):1765–78.
Sanders DB, Bittner RCL, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med. 2010;182(5):627–32.
Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, Guleria R, et al. Allergic bronchopulmonary aspergillosis: Review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. 2013;43(8):850–73.
Li BCM, Huh SM, Prieto MD, Hong G, Schwarz C, Moss RB, et al. Biomarkers for the diagnosis of allergic bronchopulmonary aspergillosis in cystic fibrosis: A systematic review and meta-analysis. J Allergy Clin Immunol Pract. 2021;S2213–2198(21):00056–8.
Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell. 2019;176(6):1340-55.e15.
Van Daele R, Spriet I, Wauters J, Maertens J, Mercier T, Van Hecke S, et al. Antifungal drugs: What brings the future? Med Mycol. 2019;57(Suppl 3):S328–43.
Cohen-Cymberknoh M, Blau H, Shoseyov D, Mei-Zahav M, Efrati O, Armoni S, et al. Intravenous monthly pulse methylprednisolone treatment for ABPA in patients with cystic fibrosis. J Cyst Fibros. 2009;8(4):253–7.
Hava DL, Tan L, Johnson P, Curran AK, Perry J, Kramer S, et al. A phase 1/1b study of PUR1900, an inhaled formulation of itraconazole, in healthy volunteers and asthmatics to study safety, tolerability and pharmacokinetics. Br J Clin Pharmacol. 2020;86(4):723–33.
Ruhnke M, Cornely OA, Schmidt-Hieber M, Alakel N, Boell B, Buchheidt D, et al. Treatment of invasive fungal diseases in cancer patients—Revised 2019 recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Oncology (DGHO). Mycoses. 2020;63(7):653–82.
Bentley S, Davies JC, Carr SB, Balfour-Lynn IM. Combination antifungal therapy for Scedosporium species in cystic fibrosis. Pediatr Pulmonol. 2020;55(8):1993–5.
Author information
Authors and Affiliations
Contributions
CS, PE and JPB equally contributed to this review. CS and JPB are members of the ECMM/ISHAM (European Confederation of Medical Mycology; International Society for Human and Animal Mycology) working group Fungal Respiratory Infections in Cystic Fibrosis (Fri-CF).
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Additional information
Handling Editor: Sanjay Haresh Chotirmall
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Schwarz, C., Eschenhagen, P. & Bouchara, J.P. Emerging Fungal Threats in Cystic Fibrosis. Mycopathologia 186, 639–653 (2021). https://doi.org/10.1007/s11046-021-00574-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-021-00574-w