
Abstract
Purpose of Review
Cystic fibrosis (CF) is an inherited disease that can progressively affect multiple organs including respiratory tract. CF has been believed to occur infrequently in the Middle East (ME). However, it seems that CF maybe more common in ME countries than expected before. Despite the fact that surveillance of fungal infections is essential, data in the ME region remain scarce. This narrative review aims to evaluate the previously published data on the prevalence of fungal respiratory colonization and infections, spectrum of isolated fungi from the respiratory tract, and antifungal treatment in CF patients across ME countries.
Recent Findings
Among different opportunistic fungal pathogens, Aspergillus fumigatus and Candida species were reported as the most prevalent mold and yeast like fungi isolated from respiratory tract of CF patients from ME countries. A. fumigatus was reported as the common filamentous colonizing fungi of the respiratory tract of CF patients from Turkey in the range 10.4–76.3%. Colonization due to Aspergillus also showed the highest frequency for A. fumigatus (50.0%) in CF patients from Qatar. However, in a more comprehensive study from Iran, A. flavus was the most common. Scedosporium spp. has not been reported in CF patients from ME countries except for one study in Iran.
Summary
Future efforts should focus on improving laboratory capacities in ME for detection of common and rare fungal pathogens. National CF registries could help to collect more systematic data on fungal colonization and infection in patients in the ME.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic fibrosis (CF) is a fatal autosomal recessive genetic disease that systemically affects multiple organs and affects patients from different aspects of life throughout their lives [1]. Although the disease is considered a multi-organ disorder, respiratory involvement represents the most important cause of morbidity and mortality in CF patients [2]. Mutation in cystic fibrosis transmembrane conductance regulator (CFTR) gene which encodes CFTR protein, leads to dysfunction of the chloride ion (Cl−) transporter, followed by dehydration of the organs and production of thick and sticky mucus secretions, and susceptibility to pulmonary infections [3, 4]. CFTR mutations at position of ΔF508, N1303K, G542X, 1717-1G > A, R553X, W1282X, G551D, 621 + 1G > T, ΔI507, and R560 becoming important as clinical aspect [1]. European Cystic Fibrosis Society Patient Registry reports F508del substitution accounts for the most frequent mutation in CF patients, more than 80% of all mutations occurred [5]. Mutation at F508del was also reported in the Lebanese (35%), Turkish (24%), and Iranian (17.5–23%) populations [6,7,8,9,10]. In Qatar substitution at the pIle1234Val (I1234V) position is known as the most prevalent mutation in CFTR gene [11]. Traditionally, CF has been known as a prevalent disease in Europe, North America, and Australasia affecting the Caucasian population, exclusively [12, 13]. However, recent reports show that the disorder is also present in other areas such as the Middle East (ME), Asia, and Latin America at lower rates [14, 15]. Although actual incidence rates of CF may be lower in these regions, the overall disease burden can be substantial given the high population density. Measures such as national CF registries or proper reporting systems, newborn screening programs, and accuracy and timeliness in diagnosis of CF contribute to a higher incidence of diagnosed disease in high-income countries [16,17,18]. For the ME, registry data are only available for Iran [19], Turkey [20], and Israel [21] as ME countries. Estimates for the ME indicate a CF incidence of one in every 2560 to 15,876 live births [22]. Epidemiological studies have shown considerable variation in the incidence of CF in different ME countries, with only one CF patient in Bahrain within the last 15 years, compared to 11 CF infants in 1 year in Lebanon [23, 24]. The reported rate for Iran was 1 in 100,000 [25]. According to the report of Health Ministry’s Management center for Transplantation and Special Diseases of Iran in 2020, there were 1100 CF patients in Iran [26]. Nevertheless, in a recent study, Guo et al. [17] estimated 162,428 individuals with CF across 94 countries, with 47,650 of these cases located in 40 European countries, 37,002 in seven North American countries, 10,034 in eight South American countries, 3652 in two Australasian countries, 5349 in 28 Asian countries, and 1665 in nine African countries. However, despite different studies, the exact number of CF patients worldwide is unclear [16, 17].
People with CF (pwCF) exhibit significant variability in the clinical course of their disease. Disparities of race, ethnicity, socioeconomic status (SES), and gender likely contribute to this variability due to increased family stress, exposure to pollution, and tobacco smoke, differences in medication beliefs, and illness management. Studies have shown individuals from racial and ethnic minorities, low SES, and women all face worse health outcomes when diagnosed with CF [27, 28]. PwCF are at high risk for developing severe pulmonary infections caused by a variety of microorganisms including bacteria, viruses and fungi [29]. Bacterial pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus are the most common cause of respiratory infection in CF patients [30,31,32]. Besides bacteria, fungi are often isolated from airway samples [33]. Different fungal species are able to colonize and develop an acute or chronic disease in the respiratory tract of CF patients depending on fungal characteristics and host immune response [34, 35]. Frequent bacterial infections and as a result long-term use of antibacterial agents in CF patients may also predispose to fungal colonization and fungal infection in the lungs. Studies have shown that fungal colonization is associated with worse clinical outcomes in some CF patients; however, whether chronic fungal colonization independently leads to poor outcomes or reflects decreased lung function is not clear [36]. Airways fungal colonization and/or infection in CF patients is commonly diagnosed with broad opportunistic fungal pathogens including Aspergillus, Candida, Scedosporium species, Exophiala dermatitidis, Rasamsonia argillacea complex, and Lomentospora prolificans [37,38,39,40,41]. On the other hand, the spread of resistance to currently available antifungals challenges therapeutic options against these opportunistic pathogens and consequently endangers the lives of CF patients [40, 42,43,44,45,46].
Fungal respiratory infections in pwCF represent a wide spectrum of clinical presentations including sensitization (an immune-mediated response to a fungus that is clinically documented by elevated titers of fungal-specific immunoglobulin E (IgE) or a positive skin test) and allergic bronchopulmonary mycosis, bronchitis and invasive pulmonary form, mostly caused by Aspergillus spp. [33, 47,48,49,50]. Studies have demonstrated that pwCF are prone to sensitivity to several environmental fungal allergens and represent higher rates of atopy and asthma [51,52,53]. Allergic bronchopulmonary aspergillosis (ABPA) is a severe disease affecting 8.9% of the CF population which causes mucus impaction and blockage of the airways [47, 54]. However, it is not completely understood how ABPA is developed and who is exactly susceptible [55].
Based on the different definition for classifications of ME countries, we considered the countries that are classified as always ME countries including Iran, Cyprus, Lebanon, Syria, Iraq, Israel, Jordan, Saudi Arabia, Kuwait, Qatar, Bahrain, United Arab Emirates, Oman, and Yemen (Fig. 1) [26].

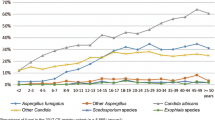
(a) Overall incidence of allergic bronchopulmonary aspergillosis in cystic fibrosis patients in Middle East countries. (b) Distribution of isolated Aspergillus species in cystic fibrosis with allergic bronchopulmonary aspergillosis who screened for mycological culture findings according to the extraction of relevant results obtained from the studies conducted in Iran [56, 57, 63] and Turkey [64, 65]
According to these realities, we aimed to evaluate the previously published data between 1990 and 2022 on the prevalence of fungal respiratory colonization and infections, spectrum of isolated fungi from the respiratory tract, and antifungal treatment in pwCF across ME countries.
Search Strategy
Data were collected using available sources including PubMed, Google Scholar, Web of Science, Scopus, and several Persian databases (Iran Medex, Iran Doc, and Scientific Information Database) to find all published papers from 1990 to 2022 using the following terms: (Cystic fibrosis OR CF) AND (Fungal infection OR Aspergill* OR Candida OR candidemia OR candidiasis OR mucormyc* OR Rhizopus OR Scedosporiosis* OR Scedosporium) AND (invasive OR putative OR probable OR infection OR case OR patient OR report) AND (ME countries OR Middle East OR Cyprus OR Lebanon OR Syria OR Iraq OR Iran OR Jordan OR Saudi Arabia OR Kuwait OR Qatar OR Bahrain OR United Arab Emirates OR Oman OR Yemen). Then, we specified all of the potentially relevant studies and included them if appropriate. In the last step, we used EndNote to search the currently selected references for duplicates.
Respiratory Clinical Entities
According to published data from ME countries, the main reported respiratory clinical entities associated with fungal pathogens were ABPA and fungal sensitization. However, it has been reported in several studies that respiratory fungal colonization is associated with different complications [56,57,58,59,60,61].
Allergic Bronchopulmonary Aspergillosis (ABPA)
ABPA is a non-invasive inflammatory lung disease as a common event in immunocompetent individuals, usually with a diagnosis of asthma or CF that manifests as an abnormal pulmonary hypersensitivity reaction, generally caused by Aspergillus fumigatus [62]. The prevalence rate of ABPA was reported between 2.1 and 13.6% in CF patients, globally [61]. There are limited published data from ME countries including Iran [57, 58, 63] and Turkey [64, 65] which reported the prevalence rate of ABPA in CF patients. In addition, there are also some case reports [60, 61, 66]. Two studies from Turkey showed differing prevalence rates of ABPA in CF patients as 9.6% [65] and 2.4% [64]. The reported rate from Iran was 7.1–10.5% according to different studies [56, 57, 63]. Overall, of the 404 and 269 CF patients screened for ABPA from Iran and Turkey, 103 (25.5%) and 46 (17.1%) had proven ABPA, respectively (Fig. 1).
The species of Aspergillus involved in CF patients with ABPA was considered only in one study from Iran [57] where A. flavus (37.5%) was the most common followed by A. fumigatus (25%), A. terreus (25%), and A. tubingensis (12.5%). Interestingly, Hassanzad et al. [56] from Iran reported a case of recurrent ABPA caused by two species of Aspergillus, A. terreus, and A. ochraceus. However, according to a study conducted by Emiralioğl et al. [65] in Turkey, A. fumigatus was the most common (76.3%) species from CF patients including those with ABPA (Fig. 1). It seems that the lack of CF registry in most ME countries and the need for a precise approach and following the different clinical and laboratory practices to identify ABPA are the main reasons for the absence ABPA reporting from most ME countries.
Fungal Sensitization
CF patients have been reported to have higher rates of atopy and asthma, indicating greater sensitivity to environmental allergens [60]. Among molds, Aspergillus fumigatus was the most commonly reported allergen in CF patients [57, 61]. Studies have also shown that adults with CF are more sensitive to several environmental fungal allergens, leading to asthma-like phenotypes, these fungi may not even have been isolated from the sputum of these people [67].
A study by Atay et al. [68] showed a higher rate of sensitivity (33.3%) to aeroallergens in Turkish CF children compared to healthy children. Fungi (19.6%) was identified as the most prevalent allergen followed by pollen (15.7%), animals (9.8%), and house dust mites (7.8%). Among fungi, Aspergillus fumigatus was the most frequent allergen with a rate of 17.6%. In another cohort study from Turkey [65], Aspergillus sensitization was reported in 3.6% of patients with CF.
Respiratory Colonization
Fungal colonization is commonly caused by Candida and Aspergillus species, and has shown some association with clinical presentation in patients with CF [11]. In some studies, from ME countries the correlation between colonization with fungi and respiratory function was considered [56, 60, 66, 68]. Three reports noted that fungal colonization in CF patients leads to a decrease in forced expiratory volume in the first second (FEV1), Forced vital capacity (FVC), and forced mid-expiratory flow (FEF) by 25–75% [15]. They also emphasized that Aspergillus colonization may contribute to lung failure and should be followed up in patients with CF. In addition, the association between fungal colonization and pulmonary exacerbations, asthma, and pancreatic insufficiency has been mentioned [11]. In two studies, persistent fungal colonization especially with Candida dubliniensis and A. fumigatus was significantly associated with body mass index (BMI) [11, 69].
In Irmak et al. [70] study from Turkey, three different groups of CF patients (fungal disease group, colonization group, and non-isolated group) were studied. History of pulmonary worsening symptoms, number of admissions to emergency unit, ABPA, and higher isolation rate of bacterial species from sputum were significantly higher in the fungal disease group. Parenteral administration of antibiotics in the fungal disease group was higher than in other groups whereas lung functions were similar. Potential risk factors for fungal isolation were the use of dietary supplements and intravenous antibacterial therapy. They have also shown that the association of increased fungal colonization with age may be due to frequent severe bacterial infections and long-term use of antibiotics, which lead to damage to the lung structure.
Although, there is no evidence of the significant relationship between fungal colonization and mutations in CFTR in ME, in a study from Qatar [11], the isolation of Aspergillus and Candida species in CF patients with CFTR I1234V mutation was reported. Interestingly, 62% of CF patients who had HLA-DRB1 alleles from Iran showed C. albicans colonization and 6% had ABPA [71].
Spectrum of Isolated Fungi From Respiratory Tract of CF Patients in ME
Candida albicans, Aspergillus fumigatus, Scedosporium/Lomentospora spp. and Exophilia dermatitidis were reported as the most frequently detected fungal species in respiratory tract of CF patients worldwide [15, 57, 68, 70, 71].
According to a recent review by Magee et al. [72], the prevalence rate of colonization and/or infection with Aspergillus fumigatus or Candida albicans in CF patients has been reported in the range of 5–78%. Data from a retrospective study in nine European CF centers showed that of 66,616 samples from 3235 CF patients [73], C. albicans is the most common yeast in each center (33.8% up to 77.9%) and A. fumigatus (3.9% up to 42.4%) is the most prevalent among mold fungi. Scedosporium spp. and Lomentospora prolificans, Exophiala dermatitidis, Trichosporon mycotoxinivorans, and Pneumocystis jirovecii have also been isolated [33].
Epidemiological studies on respiratory fungal infections or colonization in CF patients in ME countries are mostly limited to Iran, Turkey, and Qatar. In addition, it should be indicated that most of these studies were small single center studies. In a study by Hedayati et al. [67], sputum samples were collected from 90 CF patients and cultured in different culture media. Generally, Candida spp. (48.3%) were the most commonly isolated fungi followed by Aspergillus spp. (47.4%). In this study, among filamentous fungi, Aspergillus spp. (91.7%) was the most common followed by Penicillium spp., Alternaria alternata, Fusarium fujikuroi, and Scedosporium species. Among Aspergillus species, A. flavus (29.4%) was the most common followed by A. tubingensis (24.7%), A. niger (17.0%), and A. fumigatus (14.5%). Of three isolated species of Scedosporium, two isolates were S. boydii, and one S. ellipsoideum. A. flavus was also reported as the most common species causing invasive aspergillosis (IA) and allergic fungal rhinosinusitis (AFRS) from Iran [67, 74]. However, geographical variation in reported isolated species of Aspergillus may exist within the ME; as shown by Nasri et al. [63] in a cohort of 42 CF patients, where the most common filamentous fungi were Aspergillus terreus (7.14%) followed by A. fumigatus (4.76%), A. oryzae (2.38%), and A. flavus (2.38%). Hassanzad et al. [75] reported Candida albicans (9.6%) and Aspergillus fumigatus (8.8%) as the most common fungi in the lung sputum of 192 CF patients. In several studies from Turkey, C. albicans and A. fumigatus were the most commonly isolated fungi from CF patients [64, 65, 68, 70]. In Turkey, the reported rates of colonization with A. fumigatus as the most prevalent mold in the respiratory tract were 10.4% [76], 29.5% [70], and 76.3% [65].
In a study from Qatar [11], Candida and Aspergillus species were the most commonly colonizing species in CF patients with a prevalence of 81% and 45%, respectively. Among Candida species, 55.6% of CF patients were colonized by Candida dubliniensis, whereas Aspergillus fumigatus was the most common (50.0%) among Aspergillus colonized patients.
With the exception of the study conducted by Hedayati et al. [67] from Iran, there is no report on Scedosporium species isolation from ME countries, which may have to do with the fact that this pathogen can be identified in many settings. In fact, Hedayati et al. [67] used specific culture media targeting these rare molds, including Scedo-Select III, a semi-selective medium, which is not available in many other laboratories in ME. Table 1 shows detailed information on the obtained spectrum of fungal pathogens across different studies in CF patients in the ME countries during 1990–2022. Moreover, two case reports from Turkey [60, 61] and one from Qatar [66] reported the isolation of A. fumigatus, and also, Exophiala (Wangiella) dermatitidis and Candida krusei from Qatar [77] and A. ochraceus and A. terreus from Iran [56].
Geographical location, number of included patients in the study, type of clinical sample, type of collected samples, type of culture media and temperature and duration of incubation for isolation of fungi from respiratory samples and the method of identification of isolated fungi at species level may have been influenced on the distribution pattern of isolated fungi in CF patients [76]. For the proper recovery of fungi, particularly some molds, the use of selective media enriched with antimicrobial agents such as erythritol-chloramphenicol agar for Exophiala dermatitidis and Scedo-Select III medium for detecting Scedosporium in CF patient samples has been recommended [41, 63, 77].
Mixed Colonization with Fungal and Bacterial Pathogens
In some studies, co-colonization of bacterial and fungal pathogens in the respiratory tract of patients with CF was described [63, 78]. A study in Turkey [76] has shown mixed fungal cultures, yeast along with mold, which were detected in 31 (41.3%) of the 75 culture-positive samples. Analysis of fungal-bacterial showed in the mentioned study, bacterial co-colonization with A. fumigatus and C. albicans were observed in 100% and 98.2% of the fungal culture-positive samples, respectively. A. fumigatus and Pseudomonas aeruginosa or A. fumigatus and Staphylococcus aureus were detected together in 75% of A. fumigatus positive samples. Stenotrophomonas maltophilia was co-colonized with A. fumigatus in 16.60% of positive samples [76]. In a similar cohort study in Iran, mixed bacterial and fungal colonization was reported in 68.9% of patients with CF [63]. In both of these reports, no relationship was found between the specific fungal and bacterial agents grown on the culture medium in samples obtained from CF patients.
Antifungal Treatment
There are three main approaches to preventing and treating respiratory tract infections in pwCF: (a) continuous prophylaxis, (b) treatment based on respiratory symptoms only, and (c) treatment based on microbial detection of pathogens in the lung [78]. Based on new guidelines for children with CF [79], the treatment of ABPA focuses on downregulation of the inflammatory host response to Aspergillus antigens. For this reason, the use of corticosteroids (prednisolone) in conjunction with an oral antifungal agent posaconazole benefits treatment management. For this reason, the use of corticosteroids (prednisolone) in conjunction with an oral antifungal agent, posaconazole, benefits treatment management. Nebulised amphotericin B and IV caspofungin may be useful in difficult and refractory cases of ABPA, respectively. A newer azole anti-fungal, isavuconazole, is being used for ABPA treatment in CF adults. One month course of oral posaconazole shows promise for Aspergillus bronchitis treatment for children with positive growth Aspergillus in sputum but if Aspergillus is not eradicated, a 3-month course of posaconazole is needed. The incidence of invasive aspergillosis is lower but may occur in immunosuppressed, neutropenic, and post-transplant CF patients. In these cases, warrants treatment with IV caspofungin.
Posaconazole and combination posaconazole and terbinafine use for Scedosporium apiospermum and Lomentospora prolificans, highly drug-resistant and the second most common filamentous fungus recovered from CF respiratory secretions, respectively.
Dual therapy (terbinafine and an azole antifungal) is required for Exophiala dermatitidis. Finally, for Candida species, local treatment with nystatin is given; otherwise, oral fluconazole, if yeast is found in BAL, is needed.
Aaron et al. [80] conducted a randomized, double-blind, placebo-controlled trial to study the effect of itraconazole on CF patients with positive A. fumigatus pulmonary samples cultures. The analysis showed no obvious benefits in the frequency of exacerbation and poor lung function. By contrast, in a small study on CF patients (n = 13) with Aspergillus infection who were treated with 6 weeks of itraconazole, respiratory symptoms and pulmonary exacerbations were reduced [81]. Notably, in retrospective data from Sweden, the impact of antifungal therapy in 42 patients without clinical symptoms of infection with positive A. fumigatus cultures (19 treated with antifungals) was examined. The treated group had a more pronounced lung function decline during follow-up compared to the untreated group, suggesting no benefit from antifungal treatment in the setting of the positive culture without symptoms [82].
There are low data on the treatment of CF patients with aspergillosis in ME countries. High minimum inhibitory concentration (MIC) of all triazoles except voriconazole against Scedosporium isolates from CF patients were also reported in Iran [67]. Hasanzad et al. [56] reported an Iranian 12-year-old boy with CF who suffered from recurrent ABPA. Antifungal therapy started with itraconazole and corticosteroid therapy. Two months after discontinuing therapy, the patient was re-admitted to the hospital with progressive respiratory symptoms. Aspergillus terreus and A. ochraceus were identified from sputum using β-tubulin gene sequencing both of which were sensitive to itraconazole and voriconazole. The treatment was changed to voriconazole and the patient’s clinical symptoms improved.
The Role of Geoclimatic Factors in Global Distribution of Fungi
Several climatic factors including high temperature, water scarcity, relative humidity, and air quality were accounted for human health risks and also microbial viability and function [83].
Geographical and human-induced climate changes may alter the environmental reservoir of pathogenic bacteria and fungi and lead to widespread changes in fungal pathogenicity [84]. Significantly, higher stable temperatures in higher latitudes expand the geographical range of dimorphic fungi such as Coccidioides, Blastomyces, Histoplasma, Talaromyces marneffei, and Paracoccidioides [84]. In addition, recently due to global warming, species sensitive to climate change such as Trichphyton mentagrophytes have emerged in India [85]. It is also assumed that a higher stable environmental temperature in the future can lead to the emergence of less known and unknown pathogenic fungal species [84, 85]. Aspergillus fumigatus as the major causative agent of invasive aspergillosis is able to adapt to warmer climates compared with other Aspergillus spp. [86, 87]. Some studies suggest that constant temperatures lead to substitution of environmental species including A. flavus by A. fumigatus [88]. Although A. fumigatus is more thermotolerant, A. flavus is primarily involved in aspergillosis in Asia, the Middle East, and Africa [67, 74, 89, 90].
In the ME, warmer climates support fungal allergens survival in the environment. In addition, sand storms might be carrying fungal allergens and trigger allergic diseases in ME parts [83]. Interestingly, specific air pollutants may interact with A. fumigatus allergens and promote their allergenic potency [88, 91]. Several pollutants induce epithelial barrier damage and contribute to microbial or fungal infection [92]. In this situation, the fungal conidia have the opportunity for morphological alternation and germination and release allergenic proteins [93]. Recent studies show that higher ambient temperature, being close to the equator and also the summer season can be related to the increased prevalence of Pseudomonas aeruginosa in CF patients [94]. Emerging data suggest that environmental and climatic factors may influence the risk of fungal infections in people living in rural environments [94].
Climate change also impacts agricultural activities and can change microbial population [95]. For example, recent modeling-based studies show that climate change affects the ecology of Fusarium oxysporum [96]. Environmental factors may interact with plant pathogens and affect the effectiveness of pesticides, thereby increasing the use of fungicides [88, 97]. Therefore, it was hypothesized that the extensive use of fungicides has increased the trends of azole-resistant fungi [98]. Several factors such as climate change, human migration, transport of agricultural commodities, and migratory birds have increased the global extent of these isolates.
The ME is known as the most exposed region to climate change [99]. However, regarding the literature review, no data are available in the field of climate change and its role in ecology of fungi and the effect on CF patients throughout the ME.
Conclusion
A genetic predisposition in CF patients results in thick, sticky mucous secretions in the respiratory tract, which makes them significantly different from the normal population in terms of their susceptibility to bacterial and fungal respiratory infections and colonization. Chronic colonization of the airways due to a wide variety of opportunistic fungal pathogens including Aspergillus fumigatus, Candida spp. were reported in CF patients from ME countries. Some studies have also increasingly reported colonization with non-fumigatus species of Aspergillus including A. niger, A. flavus, A. nidulans, A. terreus, and other less common fungi such as Exophiala dermatitidis.
Although globally, Scedosporium spp. is the second most common fungal pathogen after A. fumigatus responsible for chronic airway colonization in CF patients, Scedosporium spp. have rarely been reported in CF patients from ME countries which may be driven in part by limited laboratory capacities to detect these organisms. Some other important fungal species such as the Rasamsonia argillacea complex and Lomentospora prolificans have not been reported in CF patients from ME so far, which may have again been driven also by limited laboratory capacities.
Future efforts should focus on improving laboratory capacities in ME for detection of common and rare fungal pathogens, including broad distribution of selective culture media and improved access to ITS sequencing and MALDI-TOF for species identification. National CF registries could help to collect more systematic data on fungal colonization and infection in patients in the ME.
Highly effective modulating therapy (HEMT) is an emerging and priority research topic for members of the CF community and clinicians [100]. The use of HEMT has profoundly influenced the care of many pwCF and could serve as considerations for ME countries regarding the future management of CF airway infections. Ongoing efforts to monitor and control infections with antifungal-resistant strains continue to be a cornerstone of care for clinicians in the care of pwCF.
Data Availability
No datasets were generated or analysed during the current study.
References
Mirtajani SB, Farnia P, Hassanzad M, Ghanavi J, Farnia P, Velayati AA. Geographical distribution of cystic fibrosis; the past 70 years of data analyzis. Biomed Biotechnol Res J. 2017;1(2):105–12. https://doi.org/10.4103/bbrj.bbrj_81_17.
Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662. https://doi.org/10.3389/fphar.2019.01662.
Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332(22):1475–80. https://doi.org/10.1056/NEJM199506013322204.
Page A, Goldenberg A, Matthews AL. Lived experiences of individuals with cystic fibrosis on CFTR-modulators. BMC Pulm Med. 2022;22(1):1–12. https://doi.org/10.1186/s12890-022-01825-2.
De Boeck K, Zolin A, Cuppens H, Olesen H, Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros. 2014;13(4):403–9. https://doi.org/10.1016/j.jcf.2013.12.003.
Bonyadi M, Omrani O, Rafeey M, Bilan N. Spectrum of CFTR gene mutations in Iranian Azeri Turkish patients with cystic fibrosis. Genet Test Mol Biomarkers. 2011;15(1–2):89–92. https://doi.org/10.1089/gtmb.2010.0091.
Zamani RAM. Mutation analysis of CFTR gene in 70 Iranian cystic fibrosis patients. Iran J Allergy Asthma Immunol. 2006:3–8
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. https://doi.org/10.1002/humu.10041.
Oskooei VK, Dooki MRE, Tabaripour R, Mirzajani S, Pourbagher R, Akhavan-Niaki H. CFTR haplotypes in northern Iranian population. Gene. 2013;512(1):55–60. https://doi.org/10.1016/j.gene.2012.09.096.
Hosseini Nami A, Kabiri M, Zafarghandi Motlagh F, Shirzadeh T, Fakhari N, Karimi A, et al. Genetic attributes of Iranian cystic fibrosis patients: the diagnostic efficiency of CFTR mutations in over a decade. Front Genet. 2023;14:1140034. https://doi.org/10.3389/fgene.2023.1140034.
Thomas M, Aboukhalaf S, Darwish T, Ali M, Elsaied O, Al Bakri M, et al. The spectrum of fungal colonization and their attributable effects on cystic fibrosis patients with rare CFTR genetic mutations. Microbiol Res. 2021;12(3):591–605. https://doi.org/10.3390/microbiolres12030042.
Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7(5):450–3. https://doi.org/10.1016/j.jcf.2008.03.007.
Silva Filho LVRF, Castaños C, Ruíz HH. Cystic fibrosis in Latin America—improving the awareness. J Cyst Fibros. 2016;15(6):791–3. https://doi.org/10.1016/j.jcf.2016.05.007.
Scotet V, Gutierrez H, Farrell PM. Newborn screening for CF across the globe—where is it worthwhile? Int J Neonatal Screen. 2020;6(1):18. https://doi.org/10.3390/ijns6010018.
Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet. 2021;397(10290):2195–211. https://doi.org/10.1016/s0140-6736(20)32542-3.
Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020;8(1):65–124. https://doi.org/10.1016/s2213-2600(19)30337-6.
Guo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J Cyst Fibros. 2022;21(3):456–62. https://doi.org/10.1016/j.jcf.2022.01.009.
da Silva LVRF, Zampoli M, Cohen-Cymberknoh M, Kabra SK. Cystic fibrosis in low and middle-income countries (LMIC): a view from four different regions of the world. Paediatr Respir Rev. 2021;38:37–44. https://doi.org/10.1016/j.prrv.2020.07.004.
Aghamohammadi A, Keivanfar M, Navaei S, Shirzadi R, Masiha F, Allameh Z, et al. First cystic fibrosis patient registry annual data report-cystic fibrosis foundation of Iran. Acta Med Iran. 2019:33–41.https://doi.org/10.18502/acta.v57i1.1751.
Dogru D, Çakır E, Şişmanlar T, Çobanoğlu N, Pekcan S, Cinel G, et al. Cystic fibrosis in Turkey: First data from the national registry. Pediatr Pulmonol. 2020;55(2):541–8. https://doi.org/10.1002/ppul.24561.
Stafler P, Mei-Zahav M, Wilschanski M, Mussaffi H, Efrati O, Lavie M, et al. The impact of a national population carrier screening program on cystic fibrosis birth rate and age at diagnosis: Implications for newborn screening. J Cyst Fibros. 2016;15(4):460–6. https://doi.org/10.1016/j.jcf.2015.08.007.
Cuppens H, Boulyjenkov V, Cassiman J, Cutting G, Dodge J, Des Georges M, et al. The molecular genetic epidemiology of cystic fibrosis: report of a joint meeting of WHO/ECFTN/ICF (M) A/ECFS. 2004
Desgeorges M, Mégarbané A, Guittard C, Carles S, Loiselet J, Demaille J, Claustres M. Cystic fibrosis in Lebanon: distribution of CFTR mutations among Arab communities. Hum Genet. 1997;100:279–83. https://doi.org/10.1007/s004390050505.
Al-Mahroos F. Cystic fibrosis in Bahrain incidence, phenotype, and outcome. J Trop Pediatr. 1998;44(1):35–9. https://doi.org/10.1093/tropej/44.1.35.
Havasian MR, Panahi J, Mahdieh N. Cystic fibrosis and distribution and mutation analysis of CFTR gene in Iranian patients. Faslnamahi Kumish. 2014;15(4):431–40.
https://worldpopulationreview.com/country-rankings/middle-east-countries. Accessed November 1, 2023.
Quittner AL, Schechter MS, Rasouliyan L, Haselkorn T, Pasta DJ, Wagener JS. Impact of socioeconomic status, race, and ethnicity on quality of life in patients with cystic fibrosis in the United States. Chest. 2010;137(3):642–50.
Palla JB. Disparities and therapeutic advances in cystic fibrosis. Pediatr Pulmonol. 2023. https://doi.org/10.1002/ppul.26445.
Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community—implications for therapeutic management. PLoS ONE. 2012;7(4):e36313. https://doi.org/10.1371/journal.pone.0036313.
Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34(2):91–100. https://doi.org/10.1002/ppul.10127.
Sagel SD, Gibson RL, Emerson J, McNamara S, Burns JL, Wagener JS, et al. Impact of Pseudomonas and Staphylococcus infection on inflammation and clinical status in young children with cystic fibrosis. J Pediatr. 2009;154(2):183–8. https://doi.org/10.1016/j.jpeds.2008.08.001.
Salsgiver EL, Fink AK, Knapp EA, LiPuma JJ, Olivier KN, Marshall BC, Saiman L. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest. 2016;149(2):390–400. https://doi.org/10.1378/chest.15-0676.
Tracy MC, Moss RB. The myriad challenges of respiratory fungal infection in cystic fibrosis. Pediatr Pulmonol. 2018;53(S3):S75–85. https://doi.org/10.1002/ppul.24126.
Sudfeld CR, Dasenbrook EC, Merz WG, Carroll KC, Boyle MP. Prevalence and risk factors for recovery of filamentous fungi in individuals with cystic fibrosis. J Cyst Fibros. 2010;9(2):110–6. https://doi.org/10.1016/j.jcf.2009.11.010.
Yan K, Yin H, Wang J, Cai Y. Subtle relationships between Pseudomonas aeruginosa and fungi in patients with cystic fibrosis. Acta Clin Belg. 2022;77(2):425–35. https://doi.org/10.1080/17843286.2020.1852850.
Chotirmall SH, McElvaney NG. Fungi in the cystic fibrosis lung: bystanders or pathogens? J Biochem Cell Biol. 2014;52:161–73. https://doi.org/10.1016/j.biocel.2014.03.001.
Touati K, Nguyen DNL, Delhaes L. The airway colonization by opportunistic filamentous fungi in patients with cystic fibrosis: recent updates. Curr Fungal Infect Rep. 2014;8:302–11. https://doi.org/10.1007/s12281-014-0197-7.
Gileles-Hillel A, Shoseyov D, Polacheck I, Korem M, Kerem E, Cohen-Cymberknoh M. Association of chronic Candida albicans respiratory infection with a more severe lung disease in patients with cystic fibrosis. Pediatr Pulmonol. 2015;50(11):1082–9. https://doi.org/10.1002/ppul.23302.
De Jong C, Slabbers L, Engel T, Yntema J, van Westreenen M, Croughs P, et al. Clinical relevance of Scedosporium spp. and Exophiala dermatitidis in patients with cystic fibrosis: A nationwide study. Med Mycol. 2020;58(7):859–66. https://doi.org/10.1093/mmy/myaa003.
Abdolrasouli A, Bercusson AC, Rhodes JL, Hagen F, Buil JB, Tang AY, et al. Airway persistence by the emerging multi-azole-resistant Rasamsonia argillacea complex in cystic fibrosis. Mycoses. 2018;61(9):665–73. https://doi.org/10.1111/myc.12789.
Hoenigl M, Salmanton-García J, Walsh TJ, Nucci M, Neoh CF, Jenks JD, et al. Global guideline for the diagnosis and management of rare mould infections: an initiative of the European Confederation of Medical Mycology in cooperation with the International Society for Human and Animal Mycology and the American Society for Microbiology. Lancet Infect Dis. 2021. https://doi.org/10.1016/s1473-3099(20)30784-2.
Sedlacek L, Graf B, Schwarz C, Albert F, Peter S, Würstl B, et al. Prevalence of Scedosporium species and Lomentospora prolificans in patients with cystic fibrosis in a multicenter trial by use of a selective medium. J Cyst Fibros. 2015;14(2):237–41. https://doi.org/10.1016/j.jcf.2014.12.014.
Seufert R, Sedlacek L, Kahl B, Hogardt M, Hamprecht A, Haase G, et al. Prevalence and characterization of azole-resistant Aspergillus fumigatus in patients with cystic fibrosis: a prospective multicentre study in Germany. J Antimicrob Chemother. 2018;73(8):2047–53. https://doi.org/10.1093/jac/dky147.
Stathi A, Loukou I, Kirikou H, Petrocheilou A, Moustaki M, Velegraki A, Zachariadou L. Isolation of Candida auris from cystic fibrosis patient, Greece, April 2019. Euro Surveill. 2019;24(29):1900400. https://doi.org/10.2807/1560-7917.ES.2019.24.29.1900400.
Jenks JD, Seidel D, Cornely OA, Chen S, van Hal S, Kauffman C, et al. Voriconazole plus terbinafine combination antifungal therapy for invasive Lomentospora prolificans infections: analysis of 41 patients from the FungiScope® registry 2008–2019. Clin Microbiol Infect. 2020:S1198–743X(20)30037–9.https://doi.org/10.1016/j.cmi.2020.01.012.
Boyer J, Feys S, Zsifkovits I, Hoenigl M, Egger M. Treatment of invasive Aspergillosis: how it’s going, where it’s heading. Mycopathologia. 2023. https://doi.org/10.1007/s11046-023-00727-z.
Janahi IA, Rehman A, Al-Naimi AR. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann Thorac Med. 2017;12(2):74. https://doi.org/10.4103/atm.ATM_231_16.
Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest. 2010;137(1):171–6. https://doi.org/10.1378/chest.09-1103.
Baxter CG, Dunn G, Jones AM, Webb K, Gore R, Richardson MD, Denning DW. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol. 2013;132(3):560–6. https://doi.org/10.1016/j.jaci.2013.04.007.
King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J Infect. 2016;72:S50–5.
Antunes J, Fernandes A, Borrego LM, Leiria-Pinto P, Cavaco J. Cystic fibrosis, atopy, asthma and ABPA. Allergol Immunopathol (Madr). 2010;38(5):278–84. https://doi.org/10.1016/j.aller.2010.06.002.
Henry M, Bennett D, Kiely J, Kelleher N, Bredin C. Fungal atopy in adult cystic fibrosis. Respir Med. 2000;94(11):1092–6. https://doi.org/10.1053/rmed.2000.0918.
Tobin M, Maguire O, Reen D, Tempany E, Fitzgerald M. Atopy and bronchial reactivity in older patients with cystic fibrosis. Thorax. 1980;35(11):807–13. https://doi.org/10.1136/thx.35.11.807.
Poore TS, Meier M, Towler E, Martiniano SL, Brinton JT, DeBoer EM, et al. Clinical characteristics of people with cystic fibrosis and frequent fungal infection. Pediatr Pulmonol. 2022;57(1):152–61. https://doi.org/10.1002/ppul.25741.
Poore TS, Hong G, Zemanick ET. Fungal infection and inflammation in cystic fibrosis. Pathogens. 2021;10(5):618. https://doi.org/10.3390/pathogens10050618.
Hassanzad M, Mortezaee V, Bongomin F, Poorabdollah M, Sharifynia S, Maleki M, et al. Successful control of exacerbation of allergic bronchopulmonary aspergillosis due to Aspergillus terreus in a cystic fibrosis patient with short-term adjunctive therapy with voriconazole: a case report. J Mycol Med. 2019;29(2):189–92. https://doi.org/10.1016/j.mycmed.2019.02.001.
Maleki M, Mortezaee V, Hassanzad M, Mahdaviani S, Poorabdollah M, Mehrian P, et al. Prevalence of allergic bronchopulmonary aspergillosis in cystic fibrosis patients using two different diagnostic criteria. Eur Ann Allergy Clin Immunol. 2020;52(3):104–11. https://doi.org/10.23822/eurannaci.1764-1489.121.
Alyasin S, Moghtaderi M, Farjadian S, Babaei M, Teshnizi SH. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis and non-cystic fibrosis bronchiectasis. Electron Physician. 2018;10(1):6273. https://doi.org/10.19082/6273.
Khalilzadeh S, Hassanzad M, Toutkaboni MP, Nejad ST, Sheikholeslami F-M, Velayati AA. Reappraisal of frequency of common cystic fibrosis transmembrane conductance regulator gene mutations in Iranian cystic fibrosis patients. Tanaffos. 2018;17(2):73.
Yakut N, Kadayifci EK, Eralp EE, Gokdemir Y. Successful treatment of allergic bronchopulmonary aspergillosis with posaconazole in a child with cystic fibrosis: case report and review of the literature. Lung India. 2020;37(2):161. https://doi.org/10.4103/lungindia.lungindia_288_19.
Cakir E, Uyan Z, Ersu RH, Karadag B, Karakoc F, Dagli E. Mucoid impaction: an unusual form of allergic bronchopulmonary aspergillosis in a patient with cystic fibrosis. Pediatr Pulmonol. 2006;41(11):1103–7. https://doi.org/10.1002/ppul.20499.
Emiralioglu N, Dogru D, Tugcu GD, Yalcin E, Kiper N, Ozcelik U. Omalizumab treatment for allergic bronchopulmonary aspergillosis in cystic fibrosis. Ann Pharmacother. 2016;50(3):188–93. https://doi.org/10.1177/1060028015624204.
Nasri E, Fakhim H, Vaezi A, Khalilzadeh S, Ahangarkani F, Laal Kargar M, et al. Airway colonisation by Candida and Aspergillus species in Iranian cystic fibrosis patients. Mycoses. 2019;62(5):434–40. https://doi.org/10.1111/myc.12898.
Kartal G. Asthma-like symptom or “cystic fibrosis asthma”? Tuberk Toraks. 2021;69(2):167–76. https://doi.org/10.5578/tt.20219806.
Emiralioğlu N, Dogru D, Dogan Ö, Gulmez D, Akdagli S, Polat S, et al. Diverse clinical characteristics of Aspergillus growth in patients with cystic fibrosis. Turk J Pediatr. 2020;62(4).https://doi.org/10.24953/turkjped.2020.04.005.
Hamad SG, Abu-Hasan M, AbdulWahab A. Use of intravenous pulse steroids to treat allergic bronchopulmonary aspergillosis in a non-compliant asthmatic adolescent. Children. 2022;9(2):252. https://doi.org/10.3390/children9020252.
Hedayati MT, Tavakoli M, Maleki M, Heidari S, Mortezaee V, Gheisari M, et al. Fungal epidemiology in cystic fibrosis patients with a special focus on Scedosporium species complex. Microb Pathog. 2019;129:168–75. https://doi.org/10.1016/j.micpath.2019.02.009.
Atay Ö, Asilsoy S, Köse S, Atakul G, Al S, Boyacioğlu ÖK, et al. The importance of aeroallergen sensitivity in children with cystic fibrosis. Allergol Immunopathol (Madr). 2022;50(3):1–9. https://doi.org/10.15586/aei.v50iSP3.764.
AbdulWahab A, Salah H, Chandra P, Taj-Aldeen SJ. Persistence of Candida dubliniensis and lung function in patients with cystic fibrosis. BMC Res Notes. 2017;10(1):1–5. https://doi.org/10.1186/s13104-017-2656-z.
Irmak I, Damadoğlu E, Güven DK, Huseynova X, İnkaya AÇ, Er B, et al. Clinical implications of fungal isolation from sputum in adult patients with cystic fibrosis. Turk J Med Sci. 2021;51(3):1191–200. https://doi.org/10.3906/sag-2006-94.
Asef A, Ghafaripour HA, Jamaati H, Varahram M, Adcock IM, Mortaz E. The role of HLA-DRB1 alleles in pulmonary cystic fibrosis. Iran J Allergy Asthma Immunol. 2022:1–8.https://doi.org/10.18502/ijaai.v21i2.9226.
Magee LC, Louis M, Khan V, Micalo L, Chaudary N. Managing fungal infections in cystic fibrosis patients: challenges in clinical practice. Infect Drug Resist. 2021:1141–53.https://doi.org/10.2147/IDR.S267219.
Schwarz C, Bouchara J-P, Buzina W, Chrenkova V, Dmeńska H, de La Pedrosa EGG, et al. Organization of patient management and fungal epidemiology in cystic fibrosis. Mycopathologia. 2018;183:7–19. https://doi.org/10.1007/s11046-017-0205-x.
Ghazanfari M, Arastehfar A, Davoodi L, Yazdani Charati J, Moazeni M, Abastabar M, et al. Pervasive but neglected: a perspective on COVID-19-associated pulmonary mold infections among mechanically ventilated COVID-19 patients. Front Med (Lausanne). 2021;8:649675. https://doi.org/10.3389/fmed.2021.649675.
Hassanzad M, Boloursaz MR, Darougar S, Nejad ST, Mohajerani SA, Baghaie N, et al. Long term outcome of cystic fibrosis patients with multisystem evaluation. Adv Respir Med. 2016;84(6):310–5. https://doi.org/10.5603/ARM.2016.0040.
Güngör Ö, Tamay Z, Güler N, Erturan Z. Frequency of fungi in respiratory samples from Turkish cystic fibrosis patients. Mycoses. 2013;56(2):123–9. https://doi.org/10.1111/j.1439-0507.2012.02221.x.
Taj-Aldeen S, El Shafie S, Alsoub H, Eldeeb Y, De Hoog G. Isolation of Exophiala dermatitidis from endotracheal aspirate of a cancer patient. Mycoses. 2006;49(6):504–9. https://doi.org/10.1111/j.1439-0507.2006.01280.x.
Koch C, Høiby N. Diagnosis and treatment of cystic fibrosis. Respiration. 2000;67(3):239–47. https://doi.org/10.1159/000029503.
Aidoo E, Alexander S,Alshafi K, Al-Yaghchi C, Anderson AK, Balfour-Lynn I, et al. Clinical guidelines: care of children with cystic fibrosis. 2023. Available on www.rbht.nhs.uk/childrencf.
Aaron SD, Vandemheen KL, Freitag A, Pedder L, Cameron W, Lavoie A, et al. Treatment of Aspergillus fumigatus in patients with cystic fibrosis: a randomized, placebo-controlled pilot study. PLoS ONE. 2012;7(4):e36077. https://doi.org/10.1371/journal.pone.0036077.
Coughlan CA, Chotirmall SH, Renwick J, Hassan T, Low TB, Bergsson G, et al. The effect of Aspergillus fumigatus infection on vitamin D receptor expression in cystic fibrosis. Am J Respir Crit Care Med. 2012;186(10):999–1007. https://doi.org/10.1164/rccm.201203-0478OC.
Blomquist A, Inghammar M, Al Shakirchi M, Ericson P, Krantz C, Svedberg M, et al. Persistent Aspergillus fumigatus infection in cystic fibrosis: impact on lung function and role of treatment of asymptomatic colonization-a registry-based case-control study. BMC Pulm Med. 2022;22(1):263. https://doi.org/10.1186/s12890-022-02054-3.
Soleimani Z, Teymouri P, Boloorani AD, Mesdaghinia A, Middleton N, Griffin DW. An overview of bioaerosol load and health impacts associated with dust storms: a focus on the Middle East. Atmos Environ. 2020;223:117187. https://doi.org/10.1016/j.atmosenv.2019.117187.
Nnadi NE, Carter DA. Climate change and the emergence of fungal pathogens. PLoS Pathog. 2021;17(4):e1009503. https://doi.org/10.1371/journal.ppat.1009503.
Gadre A, Enbiale W, Andersen LK, Coates SJ. The effects of climate change on fungal diseases with cutaneous manifestations: a report from the International Society of Dermatology Climate Change Committee. J Clim Chang Health. 2022:100156. https://doi.org/10.1016/j.joclim.2022.100156.
Chang YC, Tsai H-F, Karos M, Kwon-Chung K. THTA, a thermotolerance gene of Aspergillus fumigatus. Fungal Genet Biol. 2004;41(9):888–96. https://doi.org/10.1016/j.fgb.2004.06.004.
Kwon-Chung KJ, Sugui JA. Aspergillus fumigatus—what makes the species a ubiquitous human fungal pathogen? PLoS Pathog. 2013;9(12):e1003743. https://doi.org/10.1371/journal.ppat.1003743.
van Rhijn N, Bromley M. The consequences of our changing environment on life threatening and debilitating fungal diseases in humans. J Fungi (Basel). 2021;7(5):367. https://doi.org/10.3390/jof7050367.
Krishnan S, Manavathu EK, Chandrasekar PH. Aspergillus flavus: an emerging non-fumigatus Aspergillus species of significance. Mycoses. 2009;52(3):206–22. https://doi.org/10.1111/j.1439-0507.2008.01642.x.
Erami M, Hashemi SJ, Raiesi O, Fattahi M, Getso MI, Momen-Heravi M, et al. COVID-19-associated pulmonary aspergillosis (CAPA) in Iranian patients admitted with severe COVID-19 pneumonia. Infection. 2023;51(1):223–30. https://doi.org/10.1007/s15010-022-01907-7.
Lang-Yona N, Shuster-Meiseles T, Mazar Y, Yarden O, Rudich Y. Impact of urban air pollution on the allergenicity of Aspergillus fumigatus conidia: outdoor exposure study supported by laboratory experiments. Sci Total Environ. 2016;541:365–71. https://doi.org/10.1016/j.scitotenv.2015.09.058.
Lee P-H, Park S, Lee Y-G, Choi S-M, An M-H, Jang A-S. The impact of environmental pollutants on barrier dysfunction in respiratory disease. Allergy Asthma Immunol Res. 2021;13(6):850. https://doi.org/10.4168/aair.2021.13.6.850.
Bertuzzi M, Hayes GE, Icheoku UJ, Van Rhijn N, Denning DW, Osherov N, Bignell EM. Anti-Aspergillus activities of the respiratory epithelium in health and disease. J Fungi (Basel). 2018;4(1):8. https://doi.org/10.3390/jof4010008.
Ramsay K, Stockwell R, Bell S, Kidd T. Infection in cystic fibrosis: impact of the environment and climate. Expert Rev Respir Med. 2016;10(5):505–19. https://doi.org/10.1586/17476348.2016.1162715.
Lanz B, Dietz S, Swanson T. The expansion of modern agriculture and global biodiversity decline: an integrated assessment. Ecol Econ. 2018;144:260–77. https://doi.org/10.1016/j.ecolecon.2017.07.018.
Alkhalifah DHM, Damra E, Melhem MB, Hozzein WN. Fungus under a changing climate: modeling the current and future global distribution of Fusarium oxysporum using geographical information system data. Microorganisms. 2023;11(2):468. https://doi.org/10.3390/microorganisms11020468.
Garrett KA, Nita M, De Wolf E, Esker PD, Gomez-Montano L, Sparks AH. Plant pathogens as indicators of climate change. Climate change: Elsevier; 2021. 499–513.https://doi.org/10.1016/B978-0-12-821575-3.00024-4.
Snelders E, Huis in’t Veld RA, Rijs AJ, Kema GH, Melchers WJ, Verweij PE. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Appl Environ Microbiol. 2009;75(12):4053–7. https://doi.org/10.1128/AEM.00231-09.
Namdar R, Karami E, Keshavarz M. Climate change and vulnerability: the case of MENA countries. Int J Geo-Inf. 2021;10(11):794. https://doi.org/10.3390/ijgi10110794.
King JA, Nichols A-L, Bentley S, Carr SB, Davies JC. An update on CFTR modulators as new therapies for cystic fibrosis. Paediatr Drugs. 2022;24(4):321–33. https://doi.org/10.1007/s40272-022-00509-y.
Salah H, Lackner M, Houbraken J, Theelen B, Lass-Flörl C, Boekhout T, et al. The emergence of rare clinical Aspergillus species in Qatar: molecular characterization and antifungal susceptibility profiles. Front Microbiol. 2019;10:1677. https://doi.org/10.3389/fmicb.2019.01677.
Güngör Ö, Sampaio-Maia B, Amorim A, Araujo R, Erturan Z. Determination of azole resistance and TR 34/L98H mutations in isolates of Aspergillus section Fumigati from Turkish cystic fibrosis patients. Mycopathologia. 2018;183:913–20. https://doi.org/10.1007/s11046-018-0297-y.
Karaman M, Firinci F, Karaman O, Uzuner N, Hakki Bahar I. Long-term oropharyngeal colonization by C. albicans in children with cystic fibrosis. Yeast. 2013;30(11):429–36. https://doi.org/10.1002/yea.2977.
Wahab AA, Janahi I, Marafia M, El-Shafie S. Microbiological identification in cystic fibrosis patients with CFTR I1234V mutation. J Trop Pediatr. 2004;50(4):229–33. https://doi.org/10.1093/tropej/50.4.229.
Al Arrayed SS, Abdulla F. Incidence of cystic fibrosis in Bahrain. J Bahrain Med Soc. 1996;8:157–60.
Author information
Authors and Affiliations
Contributions
M.T.H., M.G., B.N.S., S.A.S., S.G., S.Y., S.P., S.H. and N.H. wrote the different part of the manuscript text and B.N.S. prepared figure and table. M.T.H. and M.H. conceptualized, supervised, reviewed and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hedayati, M.T., Ghazanfari, M., Shirvan, B.N. et al. Fungal Respiratory Colonization and Infections in Cystic Fibrosis Patients in the Middle East. Curr Fungal Infect Rep 18, 40–50 (2024). https://doi.org/10.1007/s12281-024-00486-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-024-00486-4