
Abstract
Wheat is one of the most important staple foods in the world. Genetic characterization of wheat agronomically important traits is crucial for yield improvement through molecular breeding. In this study, a recombinant inbred line (RIL) population was developed by crossing a local adapted high yield variety Jimai 22 (JM22) with an external variety Cunmai no.1 (CM1). A high-density genetic map containing 7,359 single nucleotide polymorphism (SNP) markers was constructed. Quantitative trait loci (QTL) mapping identified 61 QTL for eight yield-related traits under six environments (years). Among them, 17 QTL affecting spike number per plant, grain number per spike and thousand grain weight showed high predictability for theoretical yield per plant (TYP), of which, 12 QTL alleles positively contributed to TYP. Nine promising candidate genes for seven of the 12 QTL were identified including three known wheat genes and six rice orthologs. Four elite lines with TYP increased by 5.6%—15.2% were identified through genotype selection which carried 7—9 favorable alleles from JM22 and 2—3 favorable alleles from CM1 of the 12 QTL. Moreover, the linked SNPs of the 12 QTL were converted to high-throughput kompetitive allele-specific PCR (KASP) markers and validated in the population. The mapped QTL, identified promising candidate genes, developed elite lines and KASP markers are highly valuable in future genotype selection to improve wheat yield.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat is one of the most important cereal crops feeding about 40% of the world's population. With the rapid increase of global population, wheat production needs to increase by 60% in 2050 to eliminate hunger (Shiferaw et al. 2013), thus, improving wheat yield potential is the foremost task for wheat breeders. Conventional hybridization breeding has been widely adopted in wheat breeding which can combine favorable genes controlling different traits and accumulate multiple minor genes regulating the same trait by crossing two or more genetically diverse parents (Li et al. 2019). However, the genetic gain in yield appears to have reached a plateau in the past decade (Grassini et al. 2013). Breeding based solely on traditional phenotypic selection is becoming difficult to develop breakthrough high-yield varieties, hence, the genotyping-based selective breeding or molecular breeding is becoming increasingly crucial and essential (Rasheed and Xia 2019; Li et al. 2020).
Yield is a comprehensive trait that is difficult to be directly selected especially in early generations because of the low heritability (Jiang et al. 2017). Yield is multiplicatively determined by its three component traits including spike number (SN) per unit area, grain number per spike (GNS) and thousand grain weight (TGW). In addition, other agronomic traits such as heading date (HD), plant height (PH), spike length (SL), spikelet number per spike (SNS), grain length (GL) and grain width (GW), etc., are also important factors affecting yield (Tshikunde et al. 2019). As yield and its related traits are typically quantitatively inherited and controlled by multiple genes or quantitative trait loci (QTL) (Pang et al. 2020), understanding the genetic basis of these traits are essential to improve breeding efficiency.
QTL mapping is a widely used approach to analyze the genetic architecture of quantitative traits (Xu et al. 2017). Tremendous QTL mapping studies have been conducted for wheat agronomically important traits which usually use bi-parental populations derived from crosses between two inbred lines with contrasting performances (Singh et al. 2022). So far, at least 25,000 QTL have been reported for yield related traits, morphological and physiological traits, resistance to abiotic and biotic resistances, nutrient use efficiency traits, biofortification and end use quality traits in wheat (WheatQTLdb V2.0). Cao et al. (2020) summarized 253 environmental stable QTL for the three yield-component traits reported in the last decade including 139, 67 and 34 QTL for TGW, GNS and SN, respectively. Saini et al. (2022) conducted a meta-QTL analysis using 2,852 QTL for yield and yield related traits reported in the last two decades, and identified 63 stable and robust meta-QTL. However, the fine mapping and map-based cloning of QTL for agronomic traits are still challenging due to the large and complex genome of wheat (IWGSC 2018), which limited the application of marker assisted selection (MAS) in breeding (Song et al. 2023).
The recent advances in high-throughput genotyping platform and wheat functional genomics revolutionized the QTL mapping and cloning efficiency in wheat. Genome-wide single nucleotide polymorphisms (SNPs) can be easily obtained by DNA sequencing such as genotype-by-sequencing (GBS) (Spindel et al. 2013), targeted capture sequencing (Zhang et al. 2023a), and Wheat SNP arrays (Sun et al. 2020), which dramatically increased the marker density of genetic linkage map and improved the power, precision, and accuracy of QTL mapping. In a recombinant inbred line (RIL) population from the cross between Xiaoyan 54 and Jing 411, QTL mapping was conducted for 14 yield-related traits using a high-density genetic map constructed by the Wheat 660 K SNP array genotyping data, and 12 environmentally stable QTL were identified, with three of which were further validated in a panel of 190 wheat accessions (Ma et al. 2023). In a RIL population derived from the cross of TAM 112 and Duster, QTL mapping for 18 yield and kernel related traits was conducted using a genetic linkage map constructed by 5,081 SNPs generated by GBS, and 134 QTL were identified (Wang et al. 2022).
The advances in QTL mapping have accelerated the cloning of agronomically important QTL. In recent years, several important genes for yield and yield related traits have been cloned, such as WAPO1 (Kuzay et al. 2022) and TaCol-B5 (Zhang et al. 2022) for SNS and GN, TaD27-B (Zhao et al. 2020) and TN1 (Dong et al. 2023) for SN, TaDA1 (Liu et al. 2020) and TaD11-2A (Xu et al. 2022) for TGW, Rht8 (Xiong et al. 2022; Chai et al. 2022) and Rht25 (Zhang et al. 2023b) for PH, which can be applied in MAS to accelerate the breeding process and improve breeding efficiency. Therefore, more yield-related genes/QTL need to be identified to satisfy the demands of modern molecular design breeding.
In this study, a RIL population was developed by crossing an adapted local high yield variety Jimai 22 (JM22) with an external high yield variety Cunmai no.1 (CM1). The population was genotyped by the wheat 55 K SNP array and nine agronomic traits were evaluated under multiple environments. The objectives were to: 1) detect the favorable QTL/genes in JM22 and CM1 using a high-density genetic linkage map; 2) identify candidate genes of QTL for further gene function validation; 3) identify elite lines by combining the favorable alleles from both parents and develop kompetitive allele-specific PCR (KASP) markers for QTL genotype selection.
Materials and methods
Plant materials
One hundred and eighty-one RILs derived from the cross of JM22 and CM1 was constructed by single-seed descent. JM22 was a high yield commercial variety released in 2006 and has been widely grown for ~ 20 years in the Yellow and Huai River Valley Winter Wheat Region and the North Winter Wheat Region of China (Liu et al. 2022). Because of the outstanding comprehensive characters, JM22 has been used as the check variety in the regional test and an important backbone parent in wheat breeding (Yang et al. 2020). CM1 was a high yield variety as well released in 2011, and has been widely grown in the South part of Yellow and Huai River Valley Winter Wheat Region.
Field trials for trait evaluation
The RILs and the two parents were planted in six environments (locations by years) including the Experimental Station of Shandong Agricultural University, Taian, Shandong province in 2018–2019 (TA18), 2019–2020 (TA19), 2020–2021 (TA20) and 2021–2022 (TA21) wheat growing seasons, Yantai Academy of Agricultural Sciences, Yantai (YT20), and Henan Science and Technology University, Luoyang (LY20) in 2020–2021 wheat growing season. In each experiment, each RIL was space planted in a 3-m single-row plot with 5 cm between plants and 25 cm between rows. In each cropping season, fields were managed following the common practices of local wheat production.
Nine agronomic traits were evaluated including HD, PH, SN, SL, SNS, GNS, TGW, GL, and GW. HD was recorded in the TA18, TA20, and TA21 experiments. SL, was investigated in the TA19, TA20, TA21, YT20, and LY20 experiments. GL, GW, and TGW were investigated in the TA18, TA19, TA20, YT20, and LY20 experiments. The remaining traits, PH, SN, SNS, and GNS were evaluated across all six environments. HD was calculated at half heading in one plot as the days from sowing to heading. At maturity, PH was measured in centimeters (cm) by randomly selecting ten plants of each RIL. SN was recorded as mean of 10 independent plants of each RIL. SL, SNS, and GN were measured by means of 10 spikes of main tillers. GL, GW, and TGW were measured by a Crop Grain Appearance Quality Scanning Machine (SC-E, Wanshen Technology Company, Hangzhou, China) using dried seeds after harvesting. The theoretical yield per plant (TYP) across multiple environments was calculated using best linear unbiased predictions (BLUPs) of SN, GNS and TGW by the formula TYP = SN*GNS*TGW/1000.
Phenotypic data analysis
To estimate the variance components, multi-site analysis over environments was conducted by regarding genotype and environment as random effects. Based on the variance components, heritability (h2) was calculated by VG / (VG + Ve/s), where, VG, and Ve are the variance of genotype, and residual error, respectively, s is the number of environments. The BLUPs across environments were predicted. All analyses were conducted using the R package “lme4” (Bates et al. 2015). Histogram plots were depicted by the R package “ggplot2” (Wickham 2016) to show the phenotypic distributions of the nine traits using the BLUPs over environments. Pair-wise phenotypic correlations among agronomic traits were computed with the “rcorr” function implemented in the R package Hmisc (Harrell 2019) using the BLUPs. The significant threshold level was set to P = 0.05.
DNA extraction and genotyping
Genomic DNA was extracted from seedling leaves using a cetyl trimethyl ammonium bromide (CTAB) method with minor modifications (Stewart and Via 1993). Then, the DNA samples of the RILs and the two parents were genotyped using the wheat Axiom wTB55K SNP array.
Linkage map construction and QTL mapping
After genotyping, the SNP sits showing different genotyping between parents and among the population were picked out, and those with significant segregation distortion were removed. The remaining SNPs were used to construct genetic linkage map by R package “ASMap”. The linkage groups were visualized by “LinkageMapView” implemented in R software. The SNPs with identical genetic position were merged into one single bin marker. The Genome-Wide Composite Interval Mapping function in the software “QTL.gCIMapping” was used for single environment QTL analysis with the default sets (Zhou et al. 2022). The LOD threshold for significant QTL was 3.0, and the QTL with LOD scores ≥ 2.5 in at least two environments were also regarded as significant. Stable QTL were declared when QTL were identified in at least two environments. QTL for different traits with overlapped confidence intervals were regarded as a QTL cluster. The effects of QTL detected by single environmental analysis were further validated using the BLUPs across multiple environments. The SNPs linked with QTL were converted to KASP primers by PolyMarker (http://www.polymarker.info/).
Identification of rice orthologous genes in the QTL interval
According to the physical position of markers in the QTL confidence interval, the genes in the interval were extracted from the IWGSC reference genome v1.1 and annotated by IWGSC (https://wheat-urgi.versailles.inra.fr/). These sequences were used to search the 4,500 + cloned rice genes in funRiceGene (http://funricegenes.github.io/). The pairwise alignment of protein sequences between wheat and rice genes was conducted using the “pairwiseAlignment” function and the percentage of sequence identity was calculated using the “pid” function implemented in R package Biostrings (Pagès et al. 2019), and the genes with identity ≥ 70% was regarded as orthologs.
Results
Phenotype variation and trait correlations
JM22 and CM1 were broadly similar at most of agronomic traits such as HD, PH, SNS, SL, GL, and GW (Table S1). The three yield component traits SN, GNS, and TGW of JM22 was 8.1, 47.2 and 46.9 g, respectively, which were slightly higher than that of CM1 (7.5, 45.7 and 44.7 g) (Table 1). All nine traits showed continuous distributions with considerable variation and transgressive segregation in the population (Fig. 1A), indicating both parents contributed positive alleles for these agronomic traits. The coefficient of variation (CV) of these traits varied from 0.3% for HD to 7.1% for TGW, and the h2 ranged from 0.40 for SN to 0.87 for GL (Table 1).

The phenotype distribution and trait correlations. A The phenotype distribution of the RIL population. The values were the best liner unbiased predictions of across-site analysis, and arrows pointed to the two parents P1 (JM22) and P2 (CM1). B The correlations of the nine agronomic traits. The values in the low and upper triangular were correlation coefficients (r) and significant levels, respectively. HD, heading date; PH, plant height; SN, spike number per plant; SL, spike length; SNS, spikelet number per spike; GNS, grain number per spike; TGW, thousand grain weight; GL, grain length; GW, grain width
Significant correlations among these traits were identified with various correlation coefficients (r). The HD showed significantly negative correlations with most of agronomic traits except SN, SNS and GNS with the r varied from -0.14 for PH to -0.32 for GL, while PH showed significantly positive correlations with most of agronomic traits except HD, SN and SNS, with the r ranged from 0.23 for GNS to 0.51 for TGW. For three yield component traits, SN was significantly negatively correlated with TGW with the r of -0.16, while the negative correlations of GNS with SN and TGW were not significant. The spike morphological traits showed moderate positive correlation with the r ranged from 0.39 between SL and GNS to 0.44 between SNS and GNS. Similarly, the three grain related traits also showed positive correlation with the r ranged from 0.41 between GL and GW to 0.88 between TGW and GW (Fig. 1B).
Genetic linkage map
In total, 9,779 SNPs between the two parents were identified, and 9,104 SNPs were retained after removing SNPs with significant segregation distortion. A total of 8,238 SNPs showed reliable physical positions in the Chinese Spring reference genome V1.0 and were used to construct the linkage map. Totally, 7,359 SNPs in 1901 bins were mapped on 21 chromosomes with the genetic distance of 6,308.4 cM (Fig. 2A). The genetic distance of each chromosome ranged from 253.5 cM (1B) to 1043.6 cM (7D). The number of SNPs on each chromosome varied from 128 (2D) to 612 (2A) with marker space ranging from 4.2 cM (2A) to 11.9 cM (4D) (Fig. 2A). Except for chromosomes 2A, 2B and 7A, the genetic positions of markers showed good consistency with their physical location (Fig. 2B).

The summary statistics of genetic linkage map constructed using SNPs identified by wheat Axiom wTB55K SNP array. A The number of SNPs, number of bins, size of genetic distance and spacing of bins on each chromosome. B The consistency of genetic position with physical position for bins. The X-axis and Y-axis referred the bins order by genetic position and physical position, respectively
Identified QTL for measured traits
Sixty-one QTL for eight traits were detected except GW, including 1 for SN, 3 for GL, 3 for SNS, 6 for GNS, 9 for HD, 10 for TGW, 14 for PH and 15 for SL, which located on all chromosomes except 1D and 4D (Table S2 and Fig. 3). JM22 and CM1 contributed positive alleles for 28 and 33 QTL, respectively (Fig. 3). Among the detected QTL, 16 environmental stable QTL were identified, including qGL5D.1 and qGL7D.1 for GL, qHD1B.1 and qHD7A.1 for HD, qPH4A.1, qPH4A.3 and qPH5A.1 for PH, qSL2A.1, qSL5A.2 and qSL5D.1 for SL, qSNS7D.1 for SNS, qTGW4A.1, qTGW6D.1, qTGW6D.2, qTGW7B.2 and qTGW7D.1 for TGW (Fig. 3). Ten QTL clusters for 24 QTL were identified with each cluster harboring 2—4 QTL for different traits (Fig. 3).

The genetic position of QTL mapped in the RIL population derived from the cross of JM22/CM1. The QTL at the right and left side of chromosomes indicated the positive alleles from JM22 and CM1, respectively. The new QTL were indicated by blue color. The QTL with red point were detected under at least two environments. The red lines indicated the position of the QTL, and the blue rectangle indicated the region of QTL clusters
Predictability of QTL for yield
For the three yield-component traits SN, GNS and TGW, 1, 6 and 10 QTL were detected, respectively, of which, JM22 contributed favorable alleles for 10 and CM1 contributed favorable alleles for the remaining 7 QTL. The RILs carried 3 to 13 positive alleles totally with the positive alleles from JM22 varied from 1 to 10, and those from CM1 varied from 0 to 8.The TYP of RILs was significantly correlated with the number of positive alleles, with the coefficients of 0.19, 0.38 and 0.47 between TYP and the number of positive alleles from CM1, JM22 and the total positive alleles, respectively, indicating that the identified QTL were highly predictive for yield potential, and the JM22 positive alleles were much more predictive than those of CM1 alleles (Fig. 4A). Further correlation analysis of TYP with QTL for SN, GNS and TGW identified 12 QTL, including qGNS7B.2, qTGW7D.2, qTGW4A.1, qTGW6D.1, qTGW6D.2, qTGW7D.1, qGNS5A.1, qTGW5A.1, qTGW7A.1, qSN7A.1, qTGW7B.2 and qTGW7B.1 (Fig. 4B) that showed positive correlations with TYP, suggesting that these QTL are especially applicable for yield improvement, although they showed different or even opposite pleiotropic effects for the three yield-component traits.

The effects of yield-components related QTL on theoretical yield potential (TYP). A The correlations of the TYP with the number of positive alleles from CJ1, JM22, and the number of total positive alleles. The positive and negative alleles of QTL were indicated by 1 and 0, respectively. B The correlation coefficients of spike number per plant (SN), grain number per spike (GNS) and thousand grain weight (TGW) and TYP with QTL for the three yield-component traits. The X-axis showed QTL affecting SN, GNS and TGW, and the Y-axis showed correlation coefficients between phenotypes and genotypes of QTL
Candidate genes underlying the identified QTL
Among the 12 QTL positively contributing to TYP, three of them harbored previously cloned genes in wheat, including TaPIN1-6D for qTGW6D.1, TaIAA21-B for qTGW7B.2. and TaVRN3-7B for qTGW7B.1 (Table 2). Four QTL harbored one or two rice orthologs affecting relevant agronomic traits. qSN7A.1 harbored the ortholog of DLT affecting tiller number and grain size in rice. qTGW5A.1, qTGW6D.2 and qTGW7A.1 harbored orthologs of BSR2, OsTMN11 and BLS1, OsFD2 and BRD2, respectively, affecting grain size and grain weight in rice (Table 2). Hence, these orthologs are considered as the promising candidate genes underlying the QTL.
Selection of elite breeding lines and development of KASP assays
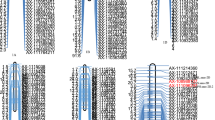
According to the performances of the RILs at TYP, four elite RILs CJ10, CJ58, CJ43 and CJ67 with TYP increased by 5.6% to 15.2% compared to JM22, the high value parent were identified (Fig. 5A). Further genotype fingerprinting of the four elite lines found that they carried 9 to 11 favorable alleles at the 12 QTL for SN, GNS and TGW that positively contributed to TYP (Fig. 5A). CJ10 performed better than JM22 at all of the three yield component traits, and carried eight favorable alleles from JM22 and two favorable alleles from CM1. CJ58, CJ43 and CJ67 showed similar SN and GNS with JM22, but obviously increased TGW. The three RILs carried 7, 8 and 9 favorable alleles from JM22, and 2, 3 and 2 favorable alleles from CM1, respectively (Fig. 5A).

QTL genotype of elite breeding lines and illustration of developed KASP assays in the RIL population. A The QTL genotype finger printing and traits performances of the two parents and the four elite RILs. The red and blue color indicated favorable and unfavorable alleles of the identified QTL for spike number per plant (SN), grain number per spike (GNS) and thousand grain weight (TGW) positively contributing to theoretical yield per plant (TYP). B The illustration of KASP assays for SNPs linked with the 12 QTL in the RIL population. The red, blue and green dots represent the HEX, FAM and heterozygote sits, respectively. The black dots represent negative controls (ddH2O)
To facilitate the employment in MAS of the 12 QTL, the linked SNPs were converted to KASP markers (Table S3) and were run in the RIL population (Fig. 5B). The identity between the KASP assay and the 55 K SNP array was ≥ 98.0%, suggesting these KASP assays can be used for genotype selection of the 12 QTL in MAS to further improve yield.
Discussion
In the current study, sixty-one QTL were identified for eight yield related traits. At least 12 QTL have been repeatedly detected in previous studies, and three QTL harbored cloned genes affecting target traits (Fig. 3 and Table 2). For instance, qPH4A.1 was overlapped with QPh.caas-4A.1 (Liu et al. 2022). In the interval of qTGW7A.1, several QTL for TGW have been reported including QTGW.cau-7A.2, QTGW.cau-7A.3, QTKW-7AL-AN, QTKW.ndsu.7A, QTKW.caas-7AL.1, TKW-IWB45030, and TKW-IWB34840 (Quarrie et al. 2005; Kumar et al. 2016; Li et al. 2018; Cao et al. 2020). qGL5D.1 was mapped in a 3.6 Mb interval on 5D harbored the known gene TaARF25-5D affecting grain size and grain weight (Jia et al. 2021). qTGW6D.1 harbored TaPIN1-6D that was reported affecting tiller number, grain number and weight (Yao et al. 2021). qTGW7B.2 harbored TaIAA21-B which has been determined to affect grain size and weight (Jia et al. 2021). The QTL cluster of qHD7B.1, qGNS7B.1 and qTGW7B.2 harbored the cloned gene TaVRN3-7B affecting vernalization and yield related traits (Yan et al. 2006; Xie and Sparkes 2021). These repeatable QTL in this study and other studies indicated that the variations affecting the same trait exist in different wheat germplasm lines.
The advanced high-throughput genotyping platform and advances in wheat functional genomics facilitated QTL mapping and candidate gene identification. In this study, a new high-density genetic linkage map containing 7,359 SNPs derived from 55 K SNP assay was constructed, which facilitated to map QTL to smaller chromosome intervals to identify candidate genes (Fig. 2A and Table S2). The cloned genes TaPIN1-6D, TaIAA21-B and TaVRN3-7B underlying the QTL for three yield-component traits were identified (Table 2). Interestingly, all these genes were rice orthologs, indicating the rice genes can provide valuable information for candidate gene identification of yield related traits in wheat. Here, six novel rice orthologs underlying four QTL for SN and TGW were further identified (Table 2). Among them, DLT encoding a GRAS family protein that has been determined to affect PH, tiller number and grain size in rice (Niu et al. 2022), and its orthologous TraesCS7A02G059000 was located in the region of qSN7A.1 affecting SN. BSR2 encoding a cytochrome p450 affected rice grain size (Maeda et al. 2019), and its orthologous TraesCS5A02G316600 was identified in the region of qTGW5A.1 for TGW. OsTMN11 belongs to transmembrane 9 superfamily member, and BLS1 is a DUF640 domain-like gene. Both of them have been identified affecting rice grain size and weight (Li et al. 2022; Ren et al. 2016), and their orthologous TraesCS6D02G344500 and TraesCS6D02G361900 were identified in the QTL region of qTGW6D.2 for TGW. OsFD2 is a bZIP transcription factor, and BRD2 is a brassinosteroid biosynthetic gene, both of which were involved in the regulation of rice grain size and weight (He et al. 2022; Qin et al. 2018), and their orthologs were located in the region of qTGW7A.1 affecting TGW. Hence, our results provided promising candidate genes for further gene cloning and functional characterization.
The three yield-component traits SN, GNS and TGW showed negative correlations with each other, although the correlations of GNS with SN and TGW didn’t reach the significant threshold (Fig. 1B), it still indicates a weak trade-off among yield related traits, which was consistent with previous studies (Xie and Sparkes 2021). The trade-off was partially genetically determined. For instance, four of the six QTL for GNS, qGNS3D.1, qGNS6D.1, qGNS7A.1 and qGNS7B.1 positively correlated with GNS, but showed negative correlations with at least one of the other two traits (Fig. 4B). For the 10 QTL for TGW, only the effects of qTGW6D.1 showed identical direction for all the three traits, while the other nine QTL showed negative correlations with at least one of the other two traits (Fig. 4B). Given the QTL’s contrary pleiotropic effects on SN, GNS and TGW, 12 of the 17 QTL for the three yield-component traits showed positive correlations with TYP, while the other five QTL showed negative correlations (Fig. 4B), hence, these QTL can be applied in wheat breeding to improve wheat yield by the simultaneous increase of the three yield component traits.
Because of the outstanding characters, many segregation populations have been constructed using JM22 as a parent, and lots of favorable QTL have been detected for yield related traits, abiotic and biotic resistances, such as QGI.caas-3A for pre-harvest sprouting (Sani Shawai et al. 2023), YrJ22 for stripe rust resistance (Chen et al. 2016, 2022), Pm52 for powdery mildew resistance (Qu et al. 2020). In a RIL population from Zhongmai 578 / JM22, 23 environmental stable QTL for six yield-related traits were detected, of which JM22 provided favorable alleles for 11 QTL (Liu et al. 2022). In a RIL population derived from the cross of landrace Banmangzi / JM22, 18 QTL for SL, SNS, TGW and PH were detected with seven favorable alleles from JM22 (Liu et al. 2022). In the current study, 61 QTL were identified for the eight agronomic traits using the RIL population derived from JM22 and CM1, and the positive alleles of 28 QTL were contributed by JM22 (Fig. 3 and Table S2). The correlation of TYP with the number of positive alleles of the 17 QTL for the three yield-component traits was 0.47 in the RIL population, indicating their high predictability for TYP (Fig. 4A). Interestingly, the correlation coefficient of TYP with the number of positive alleles from JM22 of the 10 QTL was 0.38 but 0.19 with that of positive alleles from CM1 of the seven QTL, suggesting keeping more alleles from JM22 was essential to gain high yield (Fig. 4A), which was consistent with a previous study that the rapid fixation of favorable haplotype of wheat backbone parents in its derivative lines are of importance for developing new varieties (Hao et al. 2020).
In this study, the external high yield variety, CM1, from the South part of Yellow and Huai River Valley Winter Wheat Region was used to cross with JM22. Although JM22 and CM1 were roughly similar in most of the agronomic traits, transgressive segregations were still observed for most traits in the derived RIL population (Fig. 1A), indicating different genes/alleles controlling the same trait between the two parents. By QTL mapping, 61 QTL were detected and both parents contributed positive QTL alleles (Fig. 3). Among the RIL population, four elite RILs with TYP increased by 5.6%—15.2% were identified. Genotype screening of the mapped 12 QTL for SN, GNS and TGW showed that all of these lines maintained most of the favorable alleles of JM22 and also added 2—3 favorable alleles from CM1 that controlled TGW (Fig. 5A), suggesting that by pyramiding favorable QTL alleles from both parents, we could increase yield considerably. Therefore, this study provided a valuable case of molecular breeding by mapping and genotype selection of agronomically important traits to further improve yield. The linked SNPs of the 12 QTL have been successfully converted to KASP primers (Fig. 5B) which are ready-to-use for MAS to develop elite wheat cultivars.
Conclusion
In summary, we mapped 12 QTL that can be used to enhance wheat yield potential, identified their promising candidate genes, developed and validated KASP markers of the SNPs linked with the QTL. Four TYP increased elite lines were identified through genotype selection, which carried 7- 9 favorable alleles from JM22 and 2—3 favorable alleles from CM1 of the 12 QTL. Our study provided a successful case of molecular breeding through QTL mapping and genotype selection to further improve wheat yield. The identified QTL, promising candidate genes, elite lines and high-throughput KASP markers are highly valuable in wheat molecular breeding.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67(1):1–48. https://doi.org/10.18637/jss.v067.i01
Cao S, Xu D, Hanif M, Xia X, He Z (2020) Genetic architecture underpinning yield component traits in wheat. Theor Appl Genet 133(6):1811–1823. https://doi.org/10.1007/s00122-020-03562-8
Chai L, Xin M, Dong C, Chen Z, Zhai H, Zhuang J, Cheng X, Wang N, Geng J, Wang X, Bian R, Yao Y, Guo W, Hu Z, Peng H, Bai G, Sun Q, Su Z, Liu J, Ni Z (2022) A natural variation in Ribonuclease H-like gene underlies Rht8 to confer “Green Revolution” trait in wheat. Mol Plant 15(3):377–380. https://doi.org/10.1016/j.molp.2022.01.013
Chen C, He Z, Lu J, Li J, Ren Y, Ma C, Xia X (2016) Molecular mapping of stripe rust resistance gene YrJ22 in Chinese wheat cultivar Jimai 22. Mol Breeding 36(8):118. https://doi.org/10.1007/s11032-016-0540-5
Chen C, Hao W, Wu J, Si H, Xia X, Ma C (2022) Fine mapping of stripe-rust-resistance gene YrJ22 in common wheat by bsr-seq and mutmap-based sequencing. Plants 11(23):3244
Dong C, Zhang L, Zhang Q, Yang Y, Li D, Xie Z, Cui G, Chen Y, Wu L, Li Z, Liu G, Zhang X, Liu C, Chu J, Zhao G, Xia C, Jia J, Sun J, Kong X, Liu X (2023) Tiller Number1 encodes an ankyrin repeat protein that controls tillering in bread wheat. Nat Commun 14(1):836. https://doi.org/10.1038/s41467-023-36271-z
Grassini P, Eskridge KM, Cassman KG (2013) Distinguishing between yield advances and yield plateaus in historical crop production trends. Nat Commun 4(1):2918. https://doi.org/10.1038/ncomms3918
Harrell FE Jr (2019) Hmisc: Harrell miscellaneous, R package version 4.2-0. https://CRAN.Rproject.org/package=Hmisc
Hao C, Jiao C, Hou J, Li T, Liu H, Wang Y, Zheng J, Liu H, Bi Z, Xu F, Zhao J, Ma L, Wang Y, Majeed U, Liu X, Appels R, Maccaferri M, Tuberosa R, Lu H, Zhang X (2020) Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol Plant 13(12):1733–1751. https://doi.org/10.1016/j.molp.2020.09.001
He Y, Li L, Shi W, Tan J, Luo X, Zheng S, Chen W, Li J, Zhuang C, Jiang D (2022) Florigen repression complexes involving rice CENTRORADIALIS2 regulate grain size. Plant Physiol 190(2):1260–1274. https://doi.org/10.1093/plphys/kiac338
IWGSC (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361(6403):eaar7191. https://doi.org/10.1126/science.aar7191
Jia M, Li Y, Wang Z, Tao S, Sun G, Kong X, Wang K, Ye X, Liu S, Geng S, Mao L, Li A (2021) TaIAA21 represses TaARF25-mediated expression of TaERFs required for grain size and weight development in wheat. Plant J 108(6):1754–1767. https://doi.org/10.1111/tpj.15541
Jiang Y, Schmidt RH, Zhao Y, Reif JC (2017) A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat Genet 49(12):1741–1746. https://doi.org/10.1038/ng.3974
Kumar A, Mantovani EE, Seetan R, Soltani A, Echeverry-Solarte M, Jain S, Simsek S, Doehlert D, Alamri MS, Elias EM, Kianian SF, Mergoum M (2016) Dissection of genetic factors underlying wheat kernel shape and size in an elite × nonadapted cross using a high density SNP linkage map. Plant Genome 9 (1):plantgenome2015.2009.0081. https://doi.org/10.3835/plantgenome2015.09.0081
Kuzay S, Lin H, Li C, Chen S, Woods DP, Zhang J, Lan T, von Korff M, Dubcovsky J (2022) WAPO-A1 is the causal gene of the 7AL QTL for spikelet number per spike in wheat. PLoS Genet 18(1):e1009747. https://doi.org/10.1371/journal.pgen.1009747
Li F, Wen W, He Z, Liu J, Jin H, Cao S, Geng H, Yan J, Zhang P, Wan Y, Xia X (2018) Genome-wide linkage mapping of yield-related traits in three Chinese bread wheat populations using high-density SNP markers. Theor Appl Genet 131(9):1903–1924. https://doi.org/10.1007/s00122-018-3122-6
Li H, Zhou Y, Xin W, Wei Y, Zhang J, Guo L (2019) Wheat breeding in northern China: achievements and technical advances. Crop J 7(6):718–729. https://doi.org/10.1016/j.cj.2019.09.003
Li J, Yang J, Li Y, Ma L (2020) Current strategies and advances in wheat biology. Crop J 8(6):879–891. https://doi.org/10.1016/j.cj.2020.03.004
Li H, Li C, Liu X, Yang Z (2022) A golgi-located transmembrane nine protein gene TMN11 functions in manganese/cadmium homeostasis and regulates growth and seed development in rice. Int J Mol Sci 23(24):15883
Liu H, Li H, Hao C, Wang K, Wang Y, Qin L, An D, Li T, Zhang X (2020) TaDA1, a conserved negative regulator of kernel size, has an additive effect with TaGW2 in common wheat (Triticum aestivum L.). Plant Biotechnol J 18(5):1330–1342. https://doi.org/10.1111/pbi.13298
Liu D, Zhao D, Zeng J, Shawai RS, Tong J, Li M, Li F, Zhou S, Hu W, Xia X, Tian Y, Zhu Q, Wang C, Wang D, He Z, Liu J, Zhang Y (2022) Identification of genetic loci for grain yield-related traits in the wheat population Zhongmai 578/Jimai 22. J Integr Agric 22(7):1985–1999. https://doi.org/10.1016/j.jia.2022.12.002
Ma F, Xu Y, Wang R, Tong Y, Zhang A, Liu D, An D (2023) Identification of major QTLs for yield-related traits with improved genetic map in wheat. Front Plant Sci 14. https://doi.org/10.3389/fpls.2023.1138696
Maeda S, Dubouzet JG, Kondou Y, Jikumaru Y, Seo S, Oda K, Matsui M, Hirochika H, Mori M (2019) The rice CYP78A gene BSR2 confers resistance to Rhizoctonia solani and affects seed size and growth in Arabidopsis and rice. Sci Rep 9(1):587. https://doi.org/10.1038/s41598-018-37365-1
Niu M, Wang H, Yin W, Meng W, Xiao Y, Liu D, Zhang X, Dong N, Liu J, Yang Y, Zhang F, Chu C, Tong H (2022) Rice DWARF AND LOW-TILLERING and the homeodomain protein OSH15 interact to regulate internode elongation via orchestrating brassinosteroid signaling and metabolism. Plant Cell 34(10):3754–3772. https://doi.org/10.1093/plcell/koac196
Pagès H, Aboyoun P, Gentleman R, DebRoy S (2019) Biostrings: efficient manipulation of biological strings. R package version 2520
Pang Y, Liu C, Wang D, St. Amand P, Bernardo A, Li W, He F, Li L, Wang L, Yuan X, Dong L, Su Y, Zhang H, Zhao M, Liang Y, Jia H, Shen X, Lu Y, Jiang H, Wu Y, Li A, Wang H, Kong L, Bai G, Liu S (2020) High-resolution genome-wide association study identifies genomic regions and candidate genes for important agronomic traits in wheat. Mol Plant 13(9):1311–1327. https://doi.org/10.1016/j.molp.2020.07.008
Qin R, Zeng D, Yang C, Akhter D, Alamin M, Jin X, Shi C (2018) LTBSG1, a New Allele of BRD2, regulates panicle and grain development in rice by brassinosteroid biosynthetic pathway. Genes 9(6):292
Qu Y, Wu P, Hu J, Chen Y, Shi Z, Qiu D, Li Y, Zhang H, Zhou Y, Yang L, Liu H, Zhu T, Liu Z, Zhang Y, Li H (2020) Molecular detection of the powdery mildew resistance genes in winter wheats DH51302 and Shimai 26. J Integr Agric 19(4):931–940. https://doi.org/10.1016/S2095-3119(19)62644-4
Quarrie SA, Steed A, Calestani C, Semikhodskii A, Lebreton C, Chinoy C, Steele N, Pljevljakusić D, Waterman E, Weyen J, Schondelmaier J, Habash DZ, Farmer P, Saker L, Clarkson DT, Abugalieva A, Yessimbekova M, Turuspekov Y, Abugalieva S, Tuberosa R, Sanguineti MC, Hollington PA, Aragués R, Royo A, Dodig D (2005) A high-density genetic map of hexaploid wheat (Triticum aestivum L.) from the cross Chinese Spring × SQ1 and its use to compare QTLs for grain yield across a range of environments. Theor Appl Genet 110(5):865–880. https://doi.org/10.1007/s00122-004-1902-7
Rasheed A, Xia X (2019) From markers to genome-based breeding in wheat. Theor Appl Genet 132(3):767–784. https://doi.org/10.1007/s00122-019-03286-4
Ren D, Rao Y, Wu L, Xu Q, Li Z, Yu H, Zhang Y, Leng Y, Hu J, Zhu L, Gao Z, Dong G, Zhang G, Guo L, Zeng D, Qian Q (2016) The pleiotropic ABNORMAL FLOWER AND DWARF1 affects plant height, floral development and grain yield in rice. J Integr Plant Biol 58(6):529–539. https://doi.org/10.1111/jipb.12441
Saini DK, Srivastava P, Pal N, Gupta PK (2022) Meta-QTLs, ortho-meta-QTLs and candidate genes for grain yield and associated traits in wheat (Triticum aestivum L.). Theor Appl Genet 135(3):1049–1081. https://doi.org/10.1007/s00122-021-04018-3
Sani Shawai R, Liu D, Li L, Chen T, Li M, Cao S, Xia X, Liu J, He Z, Zhang Y (2023) QTL mapping for pre-harvest sprouting in a recombinant inbred line population of elite wheat varieties Zhongmai 578 and Jimai 22. Crop J 11(3):863–869. https://doi.org/10.1016/j.cj.2022.12.001
Shiferaw B, Smale M, Braun H-J, Duveiller E, Reynolds M, Muricho G (2013) Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur 5(3):291–317. https://doi.org/10.1007/s12571-013-0263-y
Singh K, Saini DK, Saripalli G, Batra R, Gautam T, Singh R, Pal S, Kumar M, Jan I, Singh S, Kumar A, Sharma H, Chaudhary J, Kumar K, Kumar S, Singh VK, Singh VP, Kumar D, Sharma S, Kumar S, Kumar R, Sharma S, Gaurav SS, Sharma PK, Balyan HS, Gupta PK (2022) WheatQTLdb V2.0: a supplement to the database for wheat QTL. Mol Breeding 42(10):56. https://doi.org/10.1007/s11032-022-01329-1
Song L, Wang R, Yang X, Zhang A, Liu D (2023) Molecular markers and their applications in marker-assisted selection (MAS) in bread wheat (Triticum aestivum L.). Agriculture 13(3):642
Spindel J, Wright M, Chen C, Cobb J, Gage J, Harrington S, Lorieux M, Ahmadi N, McCouch S (2013) Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet 126(11):2699–2716. https://doi.org/10.1007/s00122-013-2166-x
Stewart CNJ, Via LE (1993) A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR amplifications. Biotech 14:748–750
Sun C, Dong Z, Zhao L, Ren Y, Zhang N, Chen F (2020) The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol J 18(6):1354–1360. https://doi.org/10.1111/pbi.13361
Tshikunde NM, Mashilo J, Shimelis H, Odindo A (2019) Agronomic and physiological traits, and associated quantitative trait loci (QTL) affecting yield response in wheat (Triticum aestivum L.): a review. Front Plant Sci 10. https://doi.org/10.3389/fpls.2019.01428
Wang Z, Dhakal S, Cerit M, Wang S, Rauf Y, Yu S, Maulana F, Huang W, Anderson JD, Ma X-F, Rudd JC, Ibrahim AMH, Xue Q, Hays DB, Bernardo A, St. Amand P, Bai G, Baker J, Baker S, Liu S (2022) QTL mapping of yield components and kernel traits in wheat cultivars TAM 112 and Duster. Front Plant Sci 13. https://doi.org/10.3389/fpls.2022.1057701
Wickham H (2016) ggplot2: Elegant graphics for data analysis. Springer-Verlag, New York
Xie Q, Sparkes DL (2021) Dissecting the trade-off of grain number and size in wheat. Planta 254(1):3. https://doi.org/10.1007/s00425-021-03658-5
Xiong H, Zhou C, Fu M, Guo H, Xie Y, Zhao L, Gu J, Zhao S, Ding Y, Li Y, Zhang J, Wang K, Li X, Liu L (2022) Cloning and functional characterization of Rht8, a “Green Revolution” replacement gene in wheat. Mol Plant 15(3):373–376. https://doi.org/10.1016/j.molp.2022.01.014
Xu Y, Li P, Yang Z, Xu C (2017) Genetic mapping of quantitative trait loci in crops. Crop J 5(2):175–184. https://doi.org/10.1016/j.cj.2016.06.003
Xu H, Sun H, Dong J, Ma C, Li J, Li Z, Wang Y, Ji J, Hu X, Wu M, Zhao C, Qin R, Wu J, Ni F, Cui F, Wu Y (2022) The brassinosteroid biosynthesis gene TaD11-2A controls grain size and its elite haplotype improves wheat grain yields. Theor Appl Genet 135(8):2907–2923. https://doi.org/10.1007/s00122-022-04158-0
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J (2006) The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci U S A 103(51):19581–19586. https://doi.org/10.1073/pnas.0607142103
Yang Z, Wang Z, Hu Z, Xin M, Yao Y, Peng H, You M, Su Z, Guo W (2020) Comparative analysis of the genomic sequences between commerical wheat varieties Jimai 22 and Liangxing 99. Acta Agron Sin 46(12):1870–1883. (in Chinese with English abstract)
Yao FQ, Li XH, Wang H, Song YN, Li ZQ, Li XG, Gao X-Q, Zhang XS, Bie XM (2021) Down-expression of TaPIN1s Increases the Tiller Number and Grain Yield in Wheat. BMC Plant Biol 21(1):443. https://doi.org/10.1186/s12870-021-03217-w
Zhang X, Jia H, Li T, Wu J, Nagarajan R, Lei L, Powers C, Kan C-C, Hua W, Liu Z, Chen C, Carver BF, Yan L (2022) TaCol-B5 modifies spike architecture and enhances grain yield in wheat. Science 376(6589):180–183. https://doi.org/10.1126/science.abm0717
Zhang J, Debernardi JM, Burguener GF, Choulet F, Paux E, O’Connor L, Enk J, Dubcovsky J (2023a) A second-generation capture panel for cost-effective sequencing of genome regulatory regions in wheat and relatives. Plant Genome 16(1):e20296. https://doi.org/10.1002/tpg2.20296
Zhang J, Li C, Zhang W, Zhang X, Mo Y, Tranquilli GE, Vanzetti LS, Dubcovsky J (2023b) Wheat plant height locus RHT25 encodes a PLATZ transcription factor that interacts with DELLA (RHT1). Proc Natl Acad Sci U S A 120(19):e2300203120. https://doi.org/10.1073/pnas.2300203120
Zhao B, Wu TT, Ma SS, Jiang DJ, Bie XM, Sui N, Zhang XS, Wang F (2020) TaD27-B gene controls the tiller number in hexaploid wheat. Plant Biotechnol J 18(2):513–525. https://doi.org/10.1111/pbi.13220
Zhou YH, Zhang YW, Wen YJ, Wang SB, Zhang YM (2022) QTL.gCIMapping: QTL genome-wide composite interval mapping. R package version 3.4. https://CRAN.R-project.org/package=QTL.gCIMapping
Funding
This work was supported by Shandong Province Agricultural Fine Seeds Project (grant number 2022LZGC001, 2021LZGC013), the National Natural Science Foundation of China (grant number 32072052 and 32201863) and the Taishan Scholars Program.
Author information
Authors and Affiliations
Contributions
SL conceived the research. YP, LW, LL and SL wrote the manuscript; YP, LW, LL, XW, DW, MZ, CM, HZ, QY, YL, YL, XK, HZ, XS, YZ and SL performed experiments and field trait evaluation; YP and SL analyzed the data; all authors contributed in writing and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11032_2024_1496_MOESM1_ESM.xlsx
Supplementary Material 1: Table S1. The traits performances of the two parents and the RIL population under each environment. Table S2. Mapped QTL for the nine evaluated traits. Table S3. The KASP primers for SNPs linked with the 12 QTL
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pang, Y., Wang, L., Li, L. et al. Genotype selection identified elite lines through quantitative trait loci mapping of agronomically important traits in wheat. Mol Breeding 44, 56 (2024). https://doi.org/10.1007/s11032-024-01496-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-024-01496-3