
Abstract
Apoptosis is one of the fundamental phenomena behind successful radiation and chemotherapy treatments. Non-invasive imaging of apoptosis can offer an early diagnosis of disease and the true efficiency of an ongoing treatment procedure. The present study describes an attempt to develop 99mTc-labeled 2-methyl-2-pentylmalonic acid ([99mTc] 8) as a new SPECT based apoptosis imaging agent. An optimized chemical and radiosynthesis procedure provided desired product [99mTc] 8 with high radiochemical yield (84%, n = 3) and radiochemical purity (>99%) as determined by radio HPLC. Biodistribution data indicated that the radiotracer has a rapid clearance from blood and other background tissues. High testes accumulation confirmed the ability of the radiotracer to detect testicular apoptosis in mice.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Apoptosis is an organized fundamental energy dependent cell death process that is triggered by internal or external signals [1]. The possible triggers include anti-hormonal therapy [2], immune reaction [3], radiation exposure, chemotherapy [4] and an abandonment of the growth factor [5]. The process involves a contraction of cytoplasm [6], protein cleavage [7], membrane blebbing, cutting of the intracellular components and formation of apoptotic bodies [8]. Normally, apoptosis plays key role in homeostasis, regulation of the immune system, embryogenesis, hormone dependent atrophy and normal cell turnover [9]. However, an abnormality in the process of cell death can be a factor in many medical conditions, including neurodegenerative disease, cancer development, viral or toxin induced hepatitis, bone marrow or organ transplant rejection, HIV, diabetes, or cardiovascular disease [10]. Many anti-cancer and therapeutic drugs are designed on the basis of the apoptotic cell death mechanism and their efficiency can be estimated by monitoring the ongoing apoptosis rate in the targeted organ [11]. Hence, molecular imaging of apoptosis using single positron emission computed tomography (SPECT) or positron emission tomography (PET), can offer an early detection of the cancer, monitoring the staging of the cancer, effectiveness of ongoing treatment procedures and the development of novel drugs to initiate or inhibit apoptosis [12]. The exposure of phosphatidylserine (PS) to an external cellular environment is one of the hallmarks of apoptosis [13, 14] which can be detected through radio-labeled annexin-V. Annexin-V (molecular weight = 36 kDa) is a Ca2+ dependent phospholipid binding protein that has high affinity for phosphatidylserine [15]. Annexin-V labeled with 123/124I, 18F, and 99mTc has been extensively studied for SPECT or PET based imaging of apoptosis [16]. 99mTc-labeled annexin-V has been used for the SPECT based detection of diverse medical disorders, such as acute cardiac rejection, myocardial infarction [17], organ transplant rejection studies [18], monitoring therapeutic effects of many anti-cancer drugs [19,20,21,22], chemotherapy effects, and the detection of cardiomyocyte death [23]. However, this radiolabeled protein has displayed some serious limitations, including a low clearance rate owing to the large structure, high kidney and liver uptakes and binding to apoptotic as well as necrotic cells [24]. Hence, 99mTc-labeled annexin-V is not specific and true imaging agent for apoptosis detection [25].
Recently, [18F] ML-10 (2-(5-[18F]fluoropentyl)-2-methyl malonic acid) was synthesized and used for PET based imaging of apoptosis [26, 27]. [18F] ML-10 displayed many advantages as compared with 99mTc-labeled Annexin-V, including the selective binding to apoptotic cells versus necrotic cells, the detection of early apoptosis, intracellular accumulation causing a high signal to background ratio, excellent pharmacokinetics and excretion profiles. Moreover, seven clinical trials are in progress to show its potential as a specific apoptosis imaging agent [28]. Herein, we are introducing 99mTc-labeled 2-methyl-2-pentylmalonic acid ([99mTc] 8), which is a structural analog of [18F] ML-10, for SPECT based apoptosis imaging. The major advantage of [99mTc] 8 is that it can be synthesized using a low cost procedure without the need of a cyclotron facility. Moreover, 99mTc is a well-established radionuclide with a favorable half life (t1/2 = 6 h) and suitable gamma energy (E = 141 keV) [29]. The apoptosis imaging will be possible on easily available SPECT modality [30, 31]. In this report, we present a complete synthesis procedure of 99mTc-labeled 2-methyl-2-pentylmalonic acid ([99mTc] 8) and biodistribution study to test its efficiency as a specific apoptosis imaging agent.
Experimental
Materials and instruments
All chemicals were purchased from Sigma-Aldrich Korea. Chemicals were analytically pure and used without further purification. All column chromatography purifications were performed using high purity silica gel (40–60 μm). 13C and 1H NMR spectra were acquired on JEOL NMR spectrometer (500 MHz) using dimethyl sulfoxide (DMSO-d6) and chloroform-d (CDCl3) as a solvent. The chemical shifts (δ) were reported in the units of parts per million (ppm) by using tetramethylsilane (0.0 ppm) as an internal standard. The abbreviations used for splitting patterns were as follows: singlet (s), doublet (d), doublet of doubles (dd), and multiplet (m). For quality control, HPLC analysis of products were carried out using Agilent Technologies 1290 HPLC system equipped with Eclipse XDB-C18 column (5 μm, 4.6 × 250 mm). For the purification of products, Agilent Technologies 1260 preparative HPLC system was used. Preparative HPLC system was equipped with Eclipse XDB-C18 column (7 μm, 21.2 × 150 mm). Agilent ESI-TOF was used for mass spectroscopy. 99Mo/99mTc generator (280 mCi) was supplied by Samyoung Unitech Co., Korea.
Synthesis of 2-methyl-2-pentylmalonic acid
Synthesis of 5-hydroxypentyl-4-methylbenzenesulfonate (1)
Compound 1,5-pentanediol (1.0 g, 9.5 mmol), N,N-Diisopropylethylamine (1.8 g, 13.9 mmol) and methylbenzenesulfonyl chloride (1.8 g, 9.5 mmol) were sequentially dissolved in chloroform at 20 °C. The reaction was carried out at room temperature for 4 h and then quenched by adding 1.0 M aqueous acetic acid. The crude product was extracted with 50 mL of chloroform, the extracts were dried over magnesium sulfate (MgSO4) and excess amount of solvent was removed under reduced pressure. The crude product was purified using silica gel column (hexane/ethyl acetate = 7:3) to give compound 1 as a colorless oil (1.7 g, 70%). 1H NMR (500 MHz, CDCl3) δ 7.73 (d, 2H), 7.38 (d, 2H), 4.06 (t, 2H), 3.57 (t, 2H), 2.45 (s, 3H), 1.72–1.61 (m, 2H), 1.54–1.46 (m, 2H), 1.42–1.35 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 144.8, 133.05, 129.9, 127.9, 70.62, 62.48, 31.85, 28.65, 23.70, 21.75; HRMS ([M+H]+) calculated for C12H19O4S+: 259.0968; found 259.0969.
Synthesis of 5-azidopentan-1-ol (2)
Compound 1 (1.5 g, 5.8 mmol) and sodium azide (1.13 g, 17.4 mmol) were dissolved in 10 mL of dry dimethylformamide (DMF). The reaction mixture was stirred for 2 h at 100 °C. After cooling down to room temperature, the crude product was diluted with ethyl acetate and then washed with distilled water. The combined organic phase, dried over MgSO4 and solvent was removed under reduced pressure. The crude product was purified using silica gel column (hexane/ethyl acetate = 5:5) to give pure product 2 as a light yellow oil (0.64 g, 85%). 1H NMR (500 MHz, CDCl3) δ 3.68 (t, 2H), 3.22 (t, 2H), 1.69–1.64 (m, 2H), 1.57–1.50 (m, 2H), 1.42–1.36 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 62.955, 50.622, 31.858, 28.653, 22.058; HRMS ([M+H]+) calculated for C5H12N3O+:130.0937; found 130.0939.
Synthesis of 5-azidopentyl 4-methylbenzenesulfonate (3)
Pyridine (0.73 g, 9.37 mmol) and 4-methylbenzenesulfonyl chloride (1.3 g, 6.9 mmol) were added to a stirred solution of compound 2 (0.60 g, 4.65 mmol) in chloroform (15 mL). The reaction was carried out at room temperature for 5 h. After the reaction was completed, the crude product was diluted with dichloromethane (DCM) and then washed with deionized water. The combined organic phase, was dried over MgSO4 and solvent was removed under reduced pressure. The crude product was purified using silica gel column (hexane/ethyl acetate = 8:2) to give product 3 (0.97 g, 75%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, 2H), 7.38 (d, 2H), 4.07 (t, 2H), 3.25 (t, 2H), 2.45 (s, 3H), 1.67–1.62 (m, 2H), 1.59–1.50 (m, 2H), 1.40–1.34 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 144.9, 133.05, 129.7, 127.9, 70.24, 51.20, 28.59, 28.46, 22.76, 21.73; HRMS ([M+H]+) calculated for C12H18 N3O3S+:284.1024; found 284.1027.
Synthesis of diethyl 2-methyl-2-(5-azidopentyl) malonate (4)
To the solution of compound 3 (0.83 g, 2.9 mmol) and diethyl methylmalonate (0.5 g, 2.9 mmol) in dry dimethylformamide (10 mL), reagent sodium hydride (0.23 g, 5.8 mmol, 60% in mineral oil) was added under nitrogen gas atmosphere at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, warmed to room temperature and then subjected to stirring at 50 °C for additional 5 h. The reaction was quenched by the addition of 1 M HCl and the crude product was extracted with diethyl ether. The combined organic layer was dried over magnesium sulfate and solvent removed under reduced pressure The crude product was purified using a silica gel column (ethyl acetate/hexane = 1:9) to give product 4 (0.5 g, 60%). 1H NMR (500 MHz, CDCl3) δ 4.15 (q, 4H), 3.23 (t, 2H), 1.83–1.76 (m, 2H), 1.62–1.54 (m, 2H), 1.36 (s, 3H, Me) 1.39–1.29 (m, 2H), 1.25–1.16 (m, 2H), 1.19 (t, 6H); 13C NMR (125 MHz, CDCl3) δ 172.4, 61.2, 53.6, 51.3, 35.3, 28.6, 27.0, 23.9. 19.9, 14.1; HRMS ([M+H]+) calculated for C13H24 N3O4 +:286.1601; found 286.1602.
Synthesis of diethyl 2-(5-aminopentyl)-2-methylmalonate (5)
Compound 4 (0.5 g, 1.7 mmol), triphenylphosphine (0.7 g, 2.55 mmol) and water (0.31 g, 17 mmol) were dissolved in 10 mL of tetrahydrofuran (THF). The reaction mixture was stirred at 50 °C for 36 h. After the reaction was completed, the crude product was diluted with chloroform and washed with distilled water. The combined organic phase was dried over magnesium sulfate and purified using silica gel column (Ethyl acetate/Methanol/triethylamine = 40/4/1) to give pure product 5 (0.4 g, 88%). 1H NMR (500 MHz, CDCl3) δ 4.16 (q, 4H), 2.67 (t, 2H), 1.82–1.75 (m, 2H), 1.63–1.55 (m, 2H), 1.36 (s, 3H, Me) 1.38–1.29 (m, 2H), 1.24–1.16 (m, 2H), 1.18 (t, 6H); 13C NMR (125 MHz, CDCl3) δ 172.5, 61.1, 53.6, 41.9, 35.4, 33.0, 27.1, 24.1, 19.8, 14.1; HRMS ([M+H]+) calculated for C13H26NO4 +: 260.1817; found 260.1818.
Synthesis of diethyl 2-(5-(bis(pyridin-2-ylmethyl)amino)pentyl)-2-methylmalonate (6)
To a solution of compound 5 (0.25 g, 0.964 mmol) in 10 mL of acetonitrile, 2-bromomethyl pyridine hydrobromide (1.24 g, 4.9 mmol) and triethylamine (0.974 g, 9.6 mmol) were added gradually. The reaction mixture was heated to reflux for 15 h. After the completion of reaction, acetonitrile was removed under reduced pressure and crude product was purified by using a silica gel column (Ethyl acetate/triethylamine = 40/1) to give pure product 6 (0.27 g, 65%). 1H NMR (500 MHz, CDCl3) δ 8.45 (d, 1H), 7.66 (t, 1H), 7.47 (d, 1H),7.16 (t, 1H), 4.14 (q, 4H), 3.77 (s, 2H), 2.47 (t, 2H), 1.82–1.75 (m, 2H), 1.53–1.43 (m, 2H), 1.31 (s, 3H, Me) 1.38–1.29 (m, 2H), 1.24–1.16 (m, 2H), 1.18 (t, 6H); 13C NMR (125 MHz, CDCl3) δ 172.5, 159.9, 148.8, 136.5, 122.9, 122.0, 61.1, 60.4, 54.3, 53.6, 35.4, 27.6, 27.2, 24.1, 19.8, 14.1; HRMS ([M+H]+) calculated for C25H36N3O4 +:442.2661; found 442.2662.
Synthesis of 2-(5-(bis(pyridin-2-ylmethyl)amino)pentyl)-2-methylmalonic acid (7)
Sodium hydroxide (2 M, 5 mL) was added to a solution of diethyl 2-(5-(bis(pyridin-2-ylmethyl)amino)pentyl)-2-methylmalonate 6 (0.25 g, 0.56 mmol) in ethyl alcohol (5 mL). The reaction mixture was heated to 75 °C under constant stirring. After 2 h, the reaction was stopped and pH of the reaction mixture was adjusted to 4 by adding 2 M HCl. The compound was extracted with chloroform and purified by a preparative HPLC (flow rate: 10 mL/min, HPLC solvents: 0.1% formic acid in acetonitrile (solvent B) and 0.1% formic acid in water (solvent A), eluents gradient: 0–2 min: 100%A/0%B; 2–13 min: a linear gradient to 50%A/50%B from 100%A/0%B; 13–25 min: a linear gradient to 5%A/95%B from 50%A/50%B; 25–30 min: a linear gradient to 0%A/100%B from 5%A/95%B, R t = 18.51 min) to give pure compound 7 (0.175 g, 70%). 1H NMR (500 MHz, CDCl3) δ 8.5 (d, 1H), 7.7 (t, 1H), 7.5 (d, 1H),7.19 (t, 1H), 3.9 (s, 2H), 2.6 (t, 2H), 1.85–1.65 (m, 2H), 1.65–1.45 (m, 2H), 1.4 (s, 3H, Me), 1.35–1.15 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 172.5, 160.0, 148.8, 136.6, 122.9, 122.0, 60.4, 54.4, 53.6, 35.5, 27.6, 26.9, 24.2, 19.8; HRMS ([M+H]+) calculated for C21H28N3O4 +:386.1035; found 386.1036.
Synthesis of compound (8)
The precursor [Re(CO)3(H2O)3]+ was synthesized according to published method [31] by using commercially available bromopentacarbonylrhenium. Compound 7 (0.05 g, 0.13 mmol) and equimolar amount of [Re(CO)3 (H2O)3]+ in 1 mL of saline was transferred to a 5 mL glass vial. The reaction mixture was purged with nitrogen gas and vial was sealed. The reaction mixture was stirred at 95 °C for 1 h. After cooling down to room temperature, the crude product was purified by preparative HPLC (flow rate: 10 mL/min, HPLC solvents: 0.05 M TEAP (tetraethyl ammonium phosphate) buffer (solvent B) and methanol (solvent A), eluents gradient: 0–5 min: 0%A/100%B; 5–8 min: a linear gradient to 25%A/75%B from 0%A/100%B; 8–11 min: a linear gradient to 35%A/65%B from 25%A/75%B; 11–30 min: a linear gradient to 0%A/100%B from 35%A/65%B, R t = 19.53 min) to give pure compound Re(CO)3-[2-(5-(bis(pyridin-2-ylmethyl) amino)pentyl)-2-methylmalonicacid] (8) (0.07 g, 81%). 1H NMR (500 MHz, CDCl3) δ 8.81 (d, 1H), 7.71 (dd, 1H), 7.47 (d, 1H),7.20 (dd, 1H), 4.08 (s, 2H), 2.49 (t, 2H), 1.90–1.82 (m, 2H), 1.63–1.55 (m, 2H), 1.40 (s, 3H, Me), 1.31–1.20 (m, 2H); HRMS ([M+H]+) calculated:657.0422; found 657.0421.
Radiosynthesis of compound [99mTc] 8
The intermediate precursor [99mTc(CO)3(H2O)3]+ was synthesized according to the published procedure [35]. The pH of the [99mTc(CO)3(H2O)3] + was adjusted to 7.0 using 1 M HCl. To a solution of compound 7 (1.0 mg, 100 μL) in saline, a freshly prepared [99mTc (CO)3(H2O)3]+ (6.04 m Ci, 500 μL) was added. The reaction mixture was sealed in glass vial and heated to 95 °C for 30 min. After cooling down to room temperature, the crude product was purified by preparative HPLC to give the desired product [99mTc] 8. The preparative HPLC solvent gradient was same as that of the product 8 and the retention time of radiolabeled compound was 20.41 min. Radiochemical yield and radiochemical purity of radiotracer was determined by radio HPLC system. The data were reported as an average of three independent experimental results.
Serum stability of radiolabeled compound [99mTc] 8
To the 500 μL of human serum, 50 μL of [99mTc] 8 (0.022 mCi) was added and the mixture was incubated at 37 °C for 12 h. An aliquot 100 μL was drawn at different time points (0.5, 2, 6, and 12 h) and added to 100 μL of acetonitrile to precipitate the serum protein. The mixture was centrifuged at 4000 rpm, at 5 °C for 5 min and serum protein was removed. The radiochemical stability of labeled product was determined by analyzing supernatant using radio HPLC. All animal experimental procedures were approved by the Institutional Animal Ethical Committee and performed according to the guidelines prescribed by the committee.
Measurement of partition coefficient (log P) value
A pure solution of [99mTc] 8 in saline (20 μCi, 100 μL) was added to the mixture of deionized water (3.9 mL) and n-octanol (4 mL). The mixture was transferred into 50 mL separating funnel and was shaken for 10 min. The organic and aqueous phases were allowed to separate. Equal volumes of aqueous and organic layers were collected in separate measuring vials and the radioactivity of each vial was measured using a gamma counter. The partition coefficient (P = radioactivity in octanol/radioactivity in water) was reported as log P plus standard deviation based on the results of three measurements.
Formulation of [99mTc] 8
The crude product ([99mTc] 8) was purified by using preparative HPLC. A fraction containing the desired radiolabeled product was diluted with deionized water (35 mL). The diluted product was loaded with a preconditioned SepPak C18 cartridge. The product trapped in the cartridge was flushed with 0.5 mL of ethanol and the product in ethanol was diluted with 4.5 mL of saline for in vivo experiment.
Biodistribution study
Total 30 ICR male mice (6-week old) were purchased from Orientbio Co., Ltd (Jeonbuk, Korea Republic). ICR mice were randomly divided into five groups (five animals per group). Each mice was injected with an aqueous solution of [99mTc] 8 (100 μL, 1 μCi) through the tail vein. At given time point (10, 30, 60, 120, and 360 min) five mice were sacrificed under anesthesia, the blood and the organs of interest (brain, lungs, heart, liver, kidneys, spleen, testes and intestines) were collected. The collected blood and organs were weighed and the accumulated radioactivity was measured by using a gamma counter. The final biodistribution data were reported in terms of the percentage injected dose per gram of organ or blood (%ID/g). All animal experimental procedures were approved by the Institutional Animal Ethical Committee and performed according to the guidelines prescribed by the committee.
Results and discussion
Results of synthesis and radiosynthesis
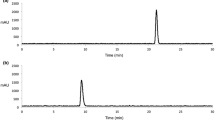
The desired radiotracer, 99mTc-labeled 2-methyl-2-pentylmalonic acid ([99mTc] 8) was synthesized via a multi-step procedure as described in Schemes 1 and 2. The precursor 2-(5-(bis(pyridin-2-ylmethyl) amino)pentyl)-2-methyl malonic acid (7) was designed with bis(pyridin-2-ylmethyl) amine (BPA) as a tridentate ligand. BPA is lipophilic ligand and in vivo biodistribution of radiotracer largely depends upon structure and the lipophilicity of the chelating ligand [31, 32]. Moreover, the coordination chemistry of BPA is thoroughly established, it can form a very stable complex with 99mTc (CO)3 [31] against trans-chelation by other ligands in vivo [33].Compound 8 was synthesized and characterized for the confirmation of the desired radiolabeled product [99mTc] 8. [99mTc(CO)3(H2O)3]+ precursor was synthesized with high radiochemical yield (93–95%) as determine by radio HPLC and used without further purification. Ligand 7 was heated with precursor [99mTc(CO)3(H2O)3]+ for 30 min to get desired product [99mTc] 8. Purification of crude product [99mTc] 8 was carried out by using preparative radio HPLC. The retention time of product [99mTc] 8 was 20.41 min as compared with standard compound 8 (R t = 19.53 min). The radiochemical yield of compound [99mTc] 8 was 84% (n = 3) and the radiochemical purity was more than 99%, as determined by radio HPLC (Fig. 1).

Synthesis of ligand 7 (a) 4-methylbenzenesulfonyl chloride, N,N-Diisopropyl ethyl amine, chloroform, room temperature, 4 h; (b) DMF (dimethylformamide), sodium azide, 100 °C, 2 h; (c) 4-methylbenzenesulfonyl chloride, pyridine, chloroform, room temperature, 5 h; (d) diethyl methylmalonate, sodium hydride, dimethylformamide, 50 °C, 5 h; (e) triphenylphosphine, H2O, tetrahydrofuran, 50 °C, 36 h; (f) 2-bromomethyl pyridine, triethylamine, reflux, 15 h; (g) 2 M NaOH, 75 °C, 2 h

Synthesis and proposed structure of 99mTc-labeled 2-methyl-2-pentylmalonic acid [99mTc] 8 and standard compound 8 (h) [99mTc/Re (CO)3(H2O)3]+, 95 °C, 0.5 or 1 h

Radio-HPLC chromatogram (a) crude product [99mTc] 8, (b) purified product [99mTc] 8, (c) purified product 8
Results of serum stability of [99mTc] 8
Radiotracer showed excellent stability in human serum under physiological conditions. The radiochemical purity of [99mTc] 8 was >99% after 12 h incubation (Fig. 2). The results indicated that the radiotracer was suitable for in vivo biodistribution study.

Radio-HPLC chromatogram of product [99mTc] 8 for in vitro stability at (a) 0.5 h (b) 2 h (c) 6 h and (d) 12 h
Results of partition coefficient (log P)
The partition coefficient (log Po/w) of [99mTc] 8 is −1.78 ± 0.4, suggesting that the complex has low lipophilicity [41]. It can be seen that the 99mTc-chelateion and charge have a significant effect on the log Po/w value of the labeled compound [99mTc] 8. The low partition coefficient is also associated with lower retention of radiolabeled complex in the blood and other background tissues [39].
Results of biodistribution study of [99mTc] 8
The biodistribution data of [99mTc] 8 in terms of percentage injected dose per gram (% ID/g) are summarized in Table 1. After an intravenous injection, the [99mTc] 8 was distributed throughout the body and did not show specific uptake in any internal organ. The compound showed rapid clearance from blood pool towards the kidneys. High uptake in the kidneys indicates excretion of the radiotracer via urine [40]. The organs including heart, spleen, lungs, and liver showed an uptake during initial time point that decreased quickly with the passage of time. The fast blood clearance and low accumulation of radioactivity in normal tissues is related to the structure and hydrophilic nature of radiotracer [42]. These results also confirmed in vivo stability of complex towards re-oxidation [43].The low brain uptake values (0.04 ± 0.21%ID/g after 10 min and 0.02 ± 0.15%ID/g after 360 min) suggested that the radiotracer could not cross the blood brain barrier because of low lipophilicity [38]. Previously reported radioiodinated analog of ML-10 showed high lipophilic character. The uptake in the blood and other non specific tissues was quite high. These radiotracers could not be used for SPECT based apoptosis imaging because of the high background signal [34, 38]. However, [99mTc] 8 showed a better biodistribution profile as compared with radioiodinated analogs. The high accumulation of radiotracer in the kidneys may restrict its use in the kidneys, however, low hepatobiliary excretion would be helpful to identify intestinal tumors [36].The PET imaging and biodistribution study using [18F] ML-10 on healthy male mice showed a high uptake of radiotracer in the testes as compared with normal tissue such as leg muscle. This is because the cellular apoptosis is a constant feature in the adult male mice testes [38]. Even a fluorescent dye labeled ML-10 analog showed high uptake in testes and visualization of testicular apoptosis was demonstrated [37]. Recently, testes uptake for radioiodinated analog of ML-10 was calculated to determine its efficiency to detect testicular apoptosis [38]. Keeping in the view, the uptake values of [99mTc] 8 in testes were determined and compared with uptake values in leg muscle. High testes uptake (0.71 ± 0.25%ID/g) was observed at initial time point and the value was 2.5-fold higher than that of normal muscle (Fig. 3). The maximum testicular uptake (0.85 ± 0.14%ID/g) was observed at 120 min post injection and the ratio between testicular to muscular uptakes was also quite high (3.7). These results confirmed the ability of radiotracer ([99mTc] 8) to detect cellular apoptosis in vivo [38].

Testes-to- muscle ratio of [99mTc] 8 uptake versus time
Conclusions
In this study, the radiosynthesis and initial biological evaluation of 99mTc-labeled 2-methyl-2-pentylmalonic acid [99mTc] 8 was reported. Radiotracer was synthesized with high radiochemical yield and radiochemical purity. Biodistribution study in healthy male mice showed better results as compared with radioiodinated analogs. The radiotracer demonstrated fast blood clearance and high uptakes in the testes as compared with normal muscle. The results warrant further study.
References
Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73(4):1907–1916
Berke G (1995) The CTL’s kiss of death. Cell 81(1):9–12
Collins MK, Perkins GR, Rodriguez Tarduchy G, Nieto MA, López Rivas A (1994) Growth factors as survival factors: regulation of apoptosis. BioEssays 16(2):133–138
Chan A, Reiter R, Wiese S, Fertig G, Gold R (1998) Plasma membrane phospholipid asymmetry precedes DNA fragmentation in different apoptotic cell models. Histochem Cell Biol 110(6):553–558
Egger L, Madden DT, Rheme C, Rao RV, Bredesen DE (2007) Endoplasmic reticulum stress-induced cell death mediated by the proteasome. Cell Death Differ 14(6):1172–1180
Martin S, Reutelingsperger CP, McGahon AJ, Rader JA, Schie RC, LaFace DM, Green DR (1995) Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bc1-2 and Abl. J Exp Med 182(5):1545–1556
Martin SJ, Green DR (1995) Protease activation during apoptosis: death by a thousand cuts? Cell 82(3):349–352
Verheij M (2008) Clinical biomarkers and imaging for radiotherapy-induced cell death. Cancer Metast Rev 27(3):471
Green DR, Kroemer G (2005) Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest 115(10):2610–2617
Kartachova M, Haas RL, Olmos RA, Hoebers FJ, van Zandwijk N, Verheij M (2004) In vivo imaging of apoptosis by 99 m Tc-annexin V scintigraphy: visual analysis in relation to treatment response. Radiother Oncol 72(3):333–339
Blankenberg FG (2008) In vivo imaging of apoptosis. Cancer Biol Ther 7(10):1525–1532
Nguyen QD, Aboagye EO (2010) Imaging the life and death of tumors in living subjects: preclinical PET imaging of proliferation and apoptosis. Integr Biol 2(10):483–495
Zwaal RF, Comfurius P, Bevers EM (2005) Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci 62(9):971–988
Zhivotovsky B, Gahm A, Ankarcrona M, Nicotera P, Orrenius S (1995) Multiple proteases are involved in thymocyte apoptosis. Exp Cell Res 221(2):404–412
Rongen GA, Oyen WJ, Ramakers BP, Riksen NP, Boerman OC, Steinmetz N, Smits P (2005) Annexin A5 scintigraphy of forearm as a novel in vivo model of skeletal muscle preconditioning in humans. Circulation 111(2):173–178
Belhocine TZ, Tait JF, Vanderheyden JL, Li C, Blankenberg FG (2004) Nuclear medicine in the era of genomics and proteomics: lessons from annexin V. J Proteome Res 3(3):345–349
Blankenberg FG, Kalinyak J, Liu L, Koike M, Cheng D, Goris ML, Green A, Vanderheyden JL, Tong DC, Yenari MA (2006) 99 mTc-HYNIC-annexin V SPECT imaging of acute stroke and its response to neuroprotective therapy with anti-Fas ligand antibody. Eur J Nucl Med Mol Imaging 33(5):566–574
Lorberboym M, Feldbrin Z, Hendel D, Blankenberg FG, Schachter P (2009) The use of 99 mTc-recombinant human annexin V imaging for differential diagnosis of aseptic loosening and low-grade infection in hip and knee prostheses. J Nucl Med 50(4):534–537
Rouzet F, Hernandez MD, Hervatin F, Sarda-Mantel L, Lefort A, Duval X, Louedec L, Fantin B, Le Guludec D, Michel JB (2008) Technetium 99m–labeled annexin V scintigraphy of platelet activation in vegetations of experimental endocarditis. Circulation 117(6):781–789
Kietselaer BL, Narula J, Hofstra L (2007) The annexin code: revealing endocarditis. Eur Heart J 28(8):948
Tahara N, Imaizumi T, Virmani R, Narula J (2009) Clinical feasibility of molecular imaging of plaque inflammation in atherosclerosis. J Nucl Med 50(3):331–334
Narula J, Acio ER, Narula N, Samuels LE, Fyfe B, Wood D, Fitzpatrick JM, Raghunath PN, Tomaszewski JE, Kelly C, Steinmetz N (2001) Annexin-V imaging for noninvasive detection of cardiac allograft rejection. Nat Med 7(12):1347–1352
Peker C, Sarda-Mantel L, Loiseau P, Rouzet F, Nazneen L, Martet G, Vrigneaud JM, Meulemans A, Saumon G, Michel JB, Le Guludec D (2004) Imaging apoptosis with 99mTc-Annexin-V in experimental subacute myocarditis. J Nucl Med 45(6):1081–1086
Callahan MK, Williamson P, Schlegel RA (2000) Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ 7(7):645
Elvas F, Vangestel C, Rapic S, Verhaeghe J, Gray B, Pak K, Stroobants S, Staelens S (2015) Characterization of [99mTc] duramycin as a SPECT imaging agent for early assessment of tumor apoptosis. Mol Imaging Biol 17(6):838–847
Reshef A, Shirvan A, Akselrod-Ballin A, Wall A, Ziv I (2010) Small-molecule biomarkers for clinical PET imaging of apoptosis. J Nucl Med 51(6):837–840
Reshef A, Shirvan A, Waterhouse RN, Grimberg H, Levin G, Cohen A, Ulysse LG, Friedman G, Antoni G, Ziv I (2008) Molecular imaging of neurovascular cell death in experimental cerebral stroke by PET. J Nucl Med 49(9):1520–1528
Höglund J, Shirvan A, Antoni G, Gustavsson SÅ, Långström B, Ringheim A, Sörensen J, Ben-Ami M, Ziv I (2011) 18F-ML-10, a PET tracer for apoptosis: first human study. J Nucl Med 52(5):720–725
Cohen A, Shirvan A, Levin G, Grimberg H, Reshef A, Ziv I (2009) From the Gla domain to a novel small-molecule detector of apoptosis. Cell Res 19(5):625–637
Sobrio F, Médoc M, Martial L, Delamare J, Barré L (2013) Automated radiosynthesis of [18F] ML-10, a PET radiotracer dedicated to apoptosis imaging, on a TRACERLab FX-FN module. Mol Imaging Biol 15(1):12–18
Lazarova N, James S, Babich J, Zubieta J (2004) A convenient synthesis, chemical characterization and reactivity of [Re (CO)3(H2O)3]Br: the crystal and molecular structure of [Re (CO)3 (CH3 CN)2Br]. Inorg Chem Commun 7(9):1023–1026
Mallia MB, Mittal S, Sarma HD, Banerjee S (2016) Modulation of in vivo distribution through chelator: synthesis and evaluation of a 2-nitroimidazole–dipicolylamine–99m Tc (CO)3 complex for detecting tumor hypoxia. Bioorg Med Chem Lett 26(1):46–50
Liu G, Dou S, He J, Vanderheyden JL, Rusckowski M, Hnatowich DJ (2004) Preparation and properties of 99mTc (CO)3+-labeled N, N-bis (2-pyridylmethyl)-4-aminobutyric acid. J Bioconjugate Chem 15(6):1441–1446
Bauwens M, De Saint-Hubert M, Cleynhens J, Brams L, Devos E, Mottaghy FM, Verbruggen A (2012) Radioiodinated phenylalkyl malonic acid derivatives as pH-sensitive SPECT tracers. PLoS ONE 7(6):38428
Park SH, Seifert S, Pietzsch HJ (2006) Novel and efficient preparation of precursor [188Re (OH2)3(CO)3]+for the labeling of biomolecules. Bioconjugate Chem 17(1):223–225
Mirshojaei SF, Gandomkar M, Najafi R, Ebrahimi SS, Babaei MH, Shafiei A, Talebi MH (2011) Radiolabeling, quality control and biodistribution of 99mTc-cefotaxime as an infection imaging agent. J Radioanal Nucl Chem 287(1):21–25
Kadirvel M, Fairclough M, Cawthorne C, Rowling EJ, Babur M, McMahon A, Birkket P, Smigova A, Freeman S, Williams KJ, Brown G (2014) Detection of apoptosis by PET/CT with the diethyl ester of [18F] ML-10 and fluorescence imaging with a dansyl analogue. Bioorg Med Chem 22(1):341–349
Jeon J, Shim HE, Mushtaq S, Kang JA, Nam YR, Yoon S, Kim HR, Choi DS, Jang BS, Park SH (2016) Radiosynthesis and in vivo evaluation of [125I] 2-(4-iodophenethyl)-2-methylmalonic acid as a potential radiotracer for detection of apoptosis. J Radioanal Nucl Chem 308(1):23–29
McAfee JG, Gagne G, Subramanian G, Schneider RF (1991) The localization of indium-111-leukocytes, gallium-67-polyclonal IgG and other radioactive agents in acute focal inflammatory lesions. J Nucl Med 32(11):2126–2131
Ibrahim IT (2009) Preparation of 99mTc-metronidazole as a model for tumor imaging. J Radioanal Nucl Chem 281(3):669–674
Wang J, Tian Y, Duan X, Yang J, Mao H, Tan C, Wu W (2012) Synthesis, radiolabeling and biodistribution studies of [99mTc (CO)3 (MN-TZ-BPA)]+ in tumor-bearing mice. J Radioanal Nucl Chem 292(1):177–181
Banerjee SR, Pullambhatla M, Foss CA, Falk A, Byun Y, Nimmagadda S, Mease RC, Pomper MG (2013) Effect of chelators on the pharmacokinetics of 99mTc-labeled imaging agents for the prostate-specific membrane antigen (PSMA). J Med Chem 56(15):6108–6114
Zenati K, Saied NM, Asmi A, Saidi M (2017) Synthesis and biological evaluation of 99mTc labeled aryl piperazine derivatives as cerebral radiotracers. J Radioanal Nucl Chem 312(1):67–74
Acknowledgements
This work was supported by The National Research Foundation of Korea grant funded by the Korea government (Grant Nos. 2012M2B2B1055245 and 2012M2A2A6011335) and Korea Atomic Energy Research Institute.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mushtaq, S., Jeon, J., Kang, J.A. et al. Radiosynthesis and preliminary biological evaluation of 99mTc-labeled 2-methyl-2-pentylmalonic acid as an apoptosis imaging agent. J Radioanal Nucl Chem 313, 207–215 (2017). https://doi.org/10.1007/s10967-017-5275-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5275-1