
Abstract
The lowest energy structures and electronic properties of ErSi n (n = 3–10) and their anions were probed using the ABCluster global search technique combined with the PBE, TPSSh and B3LYP schemes. The lowest energy energies of neutral ErSi n (n = 3–10) can be regarded as substituting a Si atom of the lowest energy structure of Sin+1 with a Er atom. The additional electron effects on the geometries are very strong, resulting the lowest energy structures of ErSi − n with n > 6 are different from their neutral counterparts. Starting from n = 7, the potential energy surfaces of ErSi − n are very flat, resulting isomeric arrangements occur and functional dependence of the predicted most stable structures exist. The AEAs, VDEs and simulated PES of ErSi n (n = 3–10) are reported. Introducing Er to Si cluster can significantly improve photochemical reactivity of the cluster. The 4f electron of Er atom in ErSi4, ErSi − n (n = 4, 7–10) prefers to take part in bonding. The total magnetic moments of ErSi n and their anions are mainly provided by the 4f electrons of Er atom. The dissociation energies of Er from ErSi n and their anions were evaluated to inspect relative stability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Silicon is not only a poor magnetic material due to absence of unpaired electrons, but also a poor photonic material because of its very short non-radiative lifetime and indirect band gap. However, doping lanthanide (Ln) metal atoms into Si clusters can improve their magnetic and optical properties because Ln atom not only has unpaired 4f-electrons, but also exhibits a number of intense and relatively narrow luminescence bands in the visible light and near infrared. Erbium as a dopant has been used in such optical fields as fiber amplifier, lasers, photomedicine and laser surgery, and upconversion luminescent material. Erbium doped Si clusters not only can make a silicon-based optical source: an electrically pumped optical amplifier, an injection laser or a LED (light emitting diode) [1], but also can have potential applications in the field of magnetism and spintronics. Knowledge of the ground and low-lying electronic states of neutral and anionic Er-doped silicon clusters is very important for understanding the properties and applications of these materials. With this motivation, we have done a detailed study of structure, thermochemistry, electron affinity and magnetic moment of erbium-doped silicon clusters ErSi n (n = 3–10) and their Anions using density functional theory.
Many theoretical calculations and simulations were accomplished for Ln atom doped Si clusters including LuSi n (n = 1–12) [2, 3], YbSi n (n ≤ 13) [4,5,6,7], HoSi n (n ≤ 20) [8,9,10,11], TbSi n (n ≤ 13) [12], GdSi n (n ≤ 13) [13, 14], EuSi n (n ≤ 13) [15, 16], SmSi n (n ≤ 10) [17, 18], PrSi n (n ≤ 21) [19, 20], LaSi n (n ≤ 21) [21, 22], YSi n (n ≤ 20) [23], and ScSi n (n ≤ 20) [24, 25] since the photoelectron spectroscopy (PES) of LnSi − n (Ln = Lu, Yb, Ho, Tb, Gd, Eu, Sm, Pr, Sc, and Y) was recorded to probe their electronic structures and properties [26,27,28,29]. The results of theoretical simulation and experimental observation revealed that (i) in light of the patterns of the PES, the LnSi n can be divided into three types: A, B, and AB [27]. And in light of the theoretical calculations, the 4f-electrons of Ln atom in A type involve scarcely in bonding, while in B type, the 4f-electrons participate in bonding [7, 11, 14, 18, 19]. (ii) The most stable structures of small size LnSi n cluster can be regarded as substituting a Si atom of the most stable structure of Sin+1 with a Ln atom. That is, substitutional structure [2, 3, 7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. For the negatively charged ions of A type, their ground-state structures are also substitutional structure. While for negatively charged ions of B type, their ground-state structures are different from their neutral counterparts when n ≥ 7 [11, 14, 19]. Dissimilar transition metal atom encapsulated in Si clusters, the magnetic moment of which is quenched [26, 27], the magnetic moment of Ln atom encapsulated in Si clusters is not quenched and is approximately identical to that of the free Ln atom [9, 11, 14, 16, 30].
Recently, our research group [3, 7, 11, 16, 18, 19] evaluated the most stable structures of SmSi n , EuSi n , GdSi n , HoSi n , YbSi n , and LuSi n (n ≤ 10) and their anions, as well as their adiabatic electron affinities (AEAs) by using different density functional theory, and saw that the AEAs predicted by the PBE, TPSSh, and B3LYP are agreement with experimental ones. In this study, we focused on Er-doped Si clusters to search the lowest-energy structures of ErSi n (n ≤ 10) and their anions, and to predict their properties such as relative stability, AEA, vertical detachment energy (VDE), simulated PES, HOMO–LUMO gap, charge transfer, and magnetic moment with the goal of understanding which type (A, B, or AB) ErSi n clusters belong to and how their properties are different from those of other Ln atom-doped silicon clusters and bare silicon clusters. Our theoretical calculations and simulations will give specific instructions for the exploration of medium-size clusters and intense motivation for experimental exploration of these significant ErSi n clusters and their negatively charged ions.
Methods
The computations were implemented using the PBE [31], TPSSh [32], and B3LYP [33, 34] methods. The basis sets employed for geometry optimization are LARGE basis sets which consists of cc-pVTZ basis set [35] for Si atoms and relativistic small-core ECP28MWB segmented valence basis sets [36] for Er atom. Analyses of frequency were carried out using the three methods to warrant that the geometries reported in this study are local minima on the potential energy surface. Because the diffuse functions are important for the negatively charged ions, the LARGE basis sets are augmented, and marked as aug-LARGE (which consists of aug-cc-pVTZ basis set [35] for Si atoms and relativistic small-core ECP28MWB segmented valence basis sets [36] augmented by diffuse functions 2pdfg with exponents 0.028 and 0.015 (p), 0.032 (d), and 0.05 (f,g) [37] for Er atom). Eventually, the calculations of single-point energy, METHOD/aug-LARGE//METHOD/LARGE (METHOD = PBE, TPSSh, and B3LYP), were carried out and used for calculations of properties such as AEA, VDE, and dissociation energies (DEs). The geometry optimizations with LARGE basis sets are justifiable in respect that the structural parameters optimized with the LARGE basis sets are identical to those optimized with aug-LARGE basis sets [18]. On the other hand, the AEAs predicted by the METHOD/aug-LARGE//METHOD/LARGE (METHOD = PBE, TPSSh, and B3LYP) are agreement with experimental data. For instance, the average absolute deviations of the PBE, TPSSh, and B3LYP from experimental data are 0.07 (for 19 AEAs of LuSi6–9, YbSi4–8,10, and EuSi3–11), 0.06 (for 27 AEAs of LuSi6–9, YbSi4–8,10, EuSi3–11, and SmSi3–10), and 0.10 eV (for 27 AEAs of LuSi6–9, YbSi4–8,10, EuSi3–11, and SmSi3–10) [3, 7, 16, 18], respectively. All of the calculations were performed by means of GAUSSIAN 09 software package [38].
To search for the lowest-energy structure for the ErSi n (n = 3–10) and their negatively charged ions, an ABCluster global search strategy [39] combined with the Gaussian 09 program is employed to choose the initial geometries. The ABCluster uses the “artificial bee colony” (ABC) algorithm to perform the global optimization. Firstly, 100 initial isomers for n ≤ 7 and 300 isomers for n ≥ 8 generated by ABCluster are optimized one by one by means of the PBE scheme combined with SMALL basis sets (which consists of 6-31G basis set for Si atoms and ECP57MWB basis set [40] for Er atom) and with spin multiplicities of doublet state. Secondly, the structures with the energy differences within 0.8 eV from the lowest-energy structure from the first step are selected and reoptimized by means of the PBE functional combined with LARGE basis sets. Thirdly, the structures of PBE/LARGE with the energy value within 0.8 eV from the lowest energy structure are optimized by means of the TPSSh and B3LYP method with LARGE basis sets. In addition to the initial isomers of the ABCluster, the ‘substitutional structure’, which are generated from replacing each Si atom of the lowest-energy structure of Sin+1 with a Er atom, is also considered owing to the fact that (1) the ground states structure for small LnSi n (Ln = Lu, Yb, Ho, Gd, Eu, Sm, Pr, La) clusters belong to substitutional structure [2, 3, 7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. (2) The possibility of missing the global minimum of the potential energy surface is always exist. For small size clusters, it is can be solved by an extensive search with a global optimization scheme. However, with the size of the cluster increases, finding the true global minima becomes increasingly a challenge because of the much increased number of low-lying isomers. It is to say that it is impossible to make an “ergodic” sampling on the potential surface of large clusters by computer simulation. (3) For LnSi n clusters with n ≤ 7, the 100 initial isomers generated by the ABCluster approach include all of the substitutional structures. However starting from n = 8, even though 500 initial isomers generated by the ABCluster approach don’t include all of the substitutional structures. In short, the selection of the initial isomer to take into account these two types is necessary to ensure the lowest-energy structure can be picked into initial isomers as much as possible. Besides, the spin multiplicity of triplet and quintuplet is taken into account for neutral ErSi n , and quartet and sextuplet is considered for the negatively charged ions. The results show that the spin multiplicity of the neutral ErSi n (n ≤ 10) is spin triplet ground state with the exception of ErSi and ErSi4. The most stable structure of ErSi is spin quintuplet ground state. The triplet and quintuplet state compete with each other for the ground state of neutral ErSi4. For anion ErSi − n (n ≤ 10), the spin multiplicity is quartet state excluded ErSi− which is sextuplet state. Despite a lot of isomers of ErSi n (n = 3–10) and their negatively charged ions are calculated, only several picked geometries are presented.
Results and Discussion
Structures
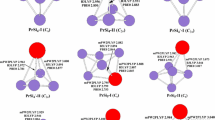
The geometries of ErSi n (n = 3–10) and their anions optimized by means of the three methods are shown in Fig. 1. The datum of spin (S) and operator S2 is listed in Table S1 of Supporting Information. The lowest-energy structure of ErSi3 and its anion is predicted to be rhombus with 3B1 and 4A2 ground state, respectively. For ErSi4, at the TPSSh level, the S2 operator analyses reveal that the spin contamination exists for the ErSi 4 -I isomer of 3A″ electronic state because the expectation value S2 of 2.49 is greater than 10%. The S2 can be expanded for pure states with higher multiplicities. In quintuplet state, the most stable structure C2v-symmetry ErSi 4 -II of 5B2 electronic state is obtained, which is more stable in energy than that of ErSi 4 -I by 0.06 eV at the TPSSh level. While at the B3LYP and PBE levels, the ErSi 4 -I structure is more stable in energy than that of ErSi 4 -II by 0.12 and 0.06 eV, respectively. It is to say that triplet and quintuplet state compete with each other for the ground state of neutral ErSi4. For negatively charged ion ErSi4−, the distorted trigonal bipyramid (ErSi − 4 -I) is calculated to be the lowest-energy structure with 4A″ ground state, which is more stable in energy than the ErSi − 4 -II of 6B2 electronic state by 0.25, 0.31, and 0.28 eV at the METHOD = TPSSh, B3LYP, and PBE levels (METHOD/aug-LARGE//METHOD/LARGE), respectively. The lowest-energy structure of ErSi5 and its anion is evaluated to be C s -symmetry face-capped trigonal bipyramid with 3A′ and 4A′ ground state, respectively. Two isomers are reported for ErSi6. The lowest-energy structure is predicted to be C2v-symmetry pentagonal bipyramid (ErSi 6 -I) of 3A2 ground state, which is more stable in energy than that of ErSi 6 -II, C s -symmetry pentagonal bipyramid of 3A″ electronic state, by 0.84, 0.57, and 0.76 eV at the TPSSh, B3LYP, and PBE levels, respectively. On the other hand, the S2 operator analysis shows that the spin contamination occurs for the ErSi 6 -II isomer of triplet electronic state at the TPSSh, B3LYP and PBE levels of the theory. Calculations of quintuplet state are performed. The results show that spin contamination disappear, but the energies of quintuplet and triplet state differ little from each other at the TPSSh, B3LYP, and PBE levels. For negatively charged ion ErSi6−, at the TPSSh, and PBE levels, the ErSi − 6 -I of 4A2 electronic state is more stable in energy than that of the ErSi − 6 -II of 4A″ electronic state by 0.17 and 0.11 eV, respectively. However at the B3LYP level, both are nearly degenerately, the ErSi − 6 -I is only less stable in energy than that of the ErSi − 6 -II by 0.05 eV. Five geometries for ErSi7 are presented. The most stable structure is evaluated to be enantiomers (ErSi 7 -I and ErSi 7 -II). It is to say that the ground state structure of ErSi7 possesses optical activity. They can be obtained via replacing a Si atom of the most stable distorted bicapped octahedron of Si8 [41, 42] with a Er atom. The second lowest-energy ErSi 7 -III isomer is higher in energy than the enantiomers by 0.12, 0.23, and 0.14 eV at the TPSSh, B3LYP and PBE levels, respectively. The ErSi 7 -IV isomer can be viewed as attaching a Si atom to the face of the lowest-energy structure of ErSi6. Energetically, it and ErSi 7 -V of 3A′ electronic state is much higher than that of the enantiomers at the three levels of theory. The S2 operator analyses for ErSi 7 -III and ErSi 7 -IV of triplet state at the three levels, and ErSi 7 -V at the TPSSh level show that the spin contamination occurs. Calculations of quintuplet state indicate that the energies of the quintuplet and triplet state, again, differ little from each other. For negatively charged ion ErSi7−, the energies of the ErSi − 7 -III and the enantiomers (ErSi − 7 -I and ErSi − 7 -II) differ little from each other. The ErSi − 7 -III structure is only more stable in energy than the enantiomers by 0.08, 0.11, and 0.04 eV at the TPSSh, B3LYP and PBE levels, respectively. It is to say that the potential energy surface of the ErSi7− is very flat, and that many isomeric arrangements are possible, analogous to LuSi7− [3]. The isomers ErSi − 7 -IV and ErSi − 7 -V of 4A′ electronic state are much higher in energy than the structure ErSi − 7 -III at the three levels of theory. For ErSi8, two isomers are reported. The lowest-energy structure is predicted to be bicapped pentagonal bipyramid (ErSi 8 -I) with 3B2 ground state. The co-apex di-trigonalbipyramind (ErSi 8 -II) of 3A electronic state is higher in energy than that of ErSi 8 -I by 0.53, 0.55 and 0.37 eV at the TPSSh, B3LYP, and PBE levels, respectively. At the same time, the spin contamination exists for the ErSi 8 -II of 3A electronic state. In quintuplet state, the energy differs little from that of triplet state at the three levels. For anion ErSi8−, at the TPSSh and B3LYP hybrid density functional levels, the bicapped pentagonal bipyramind (ErSi − 8 -I) of 4B2 electronic state is less stable in energy than the co-apex di-trigonalbipyramid (ErSi − 8 -II) of 4A electronic state by 0.19 and 0.20 eV, respectively. While at the PBE pure density functional level of theory, the ErSi − 8 -I is more stable than the ErSi − 8 -II by 0.11 eV. Three isomers for ErSi9 are presented. The C s symmetry ErSi 9 -I of 3A′ ground state is predicted to be the most stable structure, which belongs to substitutional structure and is similar to the ground state structure of LuSi9 [3]. The ErSi 9 -II isomer is also substitutional structure. Energetically, it is less stable than the ErSi 9 -I by 0.09, 0.20, and 0.12 eV at the TPSSh, B3LYP and PBE levels, respectively. On the other hand, the spin contamination exists for ErSi 9 -II. Although the spin contamination disappears in quintuplet state, the energy of quintuplet state is above that of triplet state. The C2v symmetry bicapped antitetragonal prism (ErSi 9 -III) of 3B2 electronic state is 0.23, 0.18, and 0.15 eV above the ErSi 9 -I in energy at the TPSSh, B3LYP and PBE levels, respectively. For negatively charged ion ErSi9−, the energies of tetra-capped trigonal prism (ErSi − 9 -II) and bicapped antitetragonal prism (ErSi − 9 -III) differ little from each other. At the TPSSh and PBE levels, the ErSi − 9 -II is more stable than the ErSi − 9 -III by 0.06 and 0.09 eV, respectively, while less stable by 0.07 eV at the B3LYP level. The energy of ErSi − 9 -I isomer is much higher than the structures of ErSi − 9 -II and ErSi − 9 -III. For example, the ErSi − 9 -I is higher in energy than the ErSi − 9 -II by 0.50, 0.50, and 0.43 eV at the TPSSh, B3LYP, and PBE levels, respectively. For ErSi10, two isomers are reported. The distorted tricapped tetragonal antiprim (ErSi 10 -I) is predicted to be the lowest-energy structure with 3A″ ground state. It can be viewed as substituting a Si atom of the most stable distorted tricapped tetragonal antiprim of Si11 [42] with an Er atom. The co-apex di-face-capped-trigonal-bipyramid (ErSi 10 -II) of 3A″ electronic state is not only higher in energy but also spin contamination. For example, it is 0.74, 0.36, and 1.03 eV above the ErSi 10 -I at the TPSSh, B3LYP, and PBE levels, respectively. And the energies of quintuplet state differ little from those of triplet state at the three levels of theory. For anion ErSi10−, at the TPSSh and B3LYP hybrid density functional levels, the ErSi − 10 -I of 4A″ electronic state is more stable in energy than the ErSi − 10 -II of 4A″ electronic state by 0.33 and 0.36 eV, respectively While less stable by 0.11 eV at the PBE pure density functional level of theory.


Isomers of ErSi n (n = 3–10) clusters and their anions (Er atoms are shown in red in the online version). The Er–Si bond distances are in Å
It can be drawn from the discussion that the substitutional structures are predicted to be the lowest energy structures for neutral ErSi n (n = 3–10), analogous to other Ln (Ln = Sm, Eu, Gd, Ho, Yb, Lu) atom-doped small silicon cluster. When the neutral ErSi n obtained an electron, the charge effects of this additional electron on the lowest energy structures are very strong. As a result, the lowest energy structures of ErSi − n are different from those of neutral counterparts starting from n = 7. On the other hand, starting from n = 7, the potential energy surfaces of ErSi − n are very flat, resulting isomeric arrangements occur and functional dependence of the predicted most stable structures exist.
Relative Stability
The energies of Er atom dissociated from ErSi n and their anions are calculated to examine the relative stabilities. The dissociation energies (DEs) of the lowest energy structure for ErSi n (n = 3–10) and their anions calculated at the METHOD/aug-LARGE//METHOD/LARGE (METHOD = B3LYP, TPSSh, and PBE) levels are shown in Figs. 2 and 3, respectively. So as to make easy comparisons, the DEs of YbSi n , LuSi n and their anions calculated at the TPSSh level are plotted in Fig. S1 of Supporting Information. The higher the DEs, the more stable the cluster. From Figs. 2, 3 and S1 we can see that for the three methods, the variation trends of DE curves are nearly identical. The order of DEs are B3LYP < PBE < TPSSh for the neutral, and B3LYP < PBE ≈ TPSSh for the anion. Similarly to LnSi4 and LnSi7 (Ln = Lu, Yb) and their anions, ErSi4 and ErSi7 and their anions are less stable, while ErSi5,ErSi8, ErSi5− and ErSi9−are more stable analogous to YbSi5, YbSi8, YbSi5− and YbSi9−. The DEs of neutral ErSi n and their anions are larger than those of YbSi n and their anions, and much smaller than those of LuSi n and their anions, respectively. The reason as described in Ref. [3] is that an electron of Lu atom occupies 5d orbital, of which feature iseasy to deforme and polarize, leading to increasing of components of covalent bond and the largest DEs of LuSi n . Although Er and Yb atoms have no 5d electrons, the 4f-electrons of Er atom in ErSi n are apt to remove to 5d orbitals and participate in bonding (see valence configurations below). The 4f-electrons of Yb atom in YbSi n participate hardly in bonding [7]. Therefore, the DEs of YbSi n and their anions are less than the DEs of ErSi n and their anions, respectively.

Dissociation energy with ZPVE correction for the reaction ErSi n → Er + Si n versus the number of atoms n for ErSi n clusters

Dissociation energy with ZPVE correction for the reaction ErSi − n → Er + Si − n versus the number of atoms n for ErSi − n clusters
Electronic Property
Electron affinity is not only one of the important electronic property but also a key spectral data and vitally important for use in the chemical cycle to determine bond dissociation energies. Two different types of energy separations, AEA and VDE, are predicted. The AEA and VDE is defined as the following formulas:
The AEA and VDE calculated at the METHOD/aug-LARGE//METHOD/LARGE (METHOD = B3LYP, TPSSh, and PBE) levels are listed in Table 1. From Table 1 we can see that the AEAs and VDEs predicted by the three schemes are very close to each other. There are no experimental data for comparison. So as to make easy comparisons, the AEAs of ErSi n , YbSi n , and LuSi n (n = 3–10) calculated at the TPSSh level are plotted in Fig. S2, as well as experimental AEAs of YbSi n . From Fig. S2 we can see that (1) the AEAs of LuSi n are larger than those of ErSi n and YbSi n . The AEAs of ErSi3, ErSi5 and ErSi6 are very close to those of YbSi3, YbSi5 and YbSi6. The AEAs of ErSi4,7–10 are averagely larger than those of YbSi4,7–10 by 0.24 eV. The reason is that the Lu atom includes an unpaired d-electron which interacts strongly with the extra electron. The 4f electrons of Yb atom in anion YbSi − n [7] and Er atom in anion ErSi −3,5,6 (see valence configurations below) are nearly unchanged and participate hardly in bonding, resulting AEAs of ErSi3,5,6 are nearly identical to those to YbSi3,5,6. While the part of 4f electrons of Er atom in anion ErSi −4,7–10 removed to 5d orbital (see valence configurations below) and then the 5d electrons participate in bonding. So The AEAs of ErSi4,7–10 are larger than those of YbSi4,7–10. Our calculations may give strong motivation for experimental investigations of Er-doped Si clusters and their anions.
PES as a powerful tool can probe the structure of clusters because it is susceptible to variations of the structure. Only TPSSh simulated PES is done and exhibited in Fig. 4. In the PES simulation the relative energies of the orbitals (∆E n ) is firstly calculated by means of the formulation:

Simulated photoelectron spectra for the most stable structure of ErSi − n clusters at the TPSSh level of theory
Then, the first peak related with the HOMO is placed in the VDE position, and other peaks related with deeper orbitals are moved to higher binding energies according to the data of -∆E n . Ultimately, a Gaussian FWHM (full width at half maximum) of 0.20 eV is adopted for fit all of these peaks. As can be seen from in Fig. 4, the simulated ErSi3− PES shows five peaks located in 1.66, 2.43, 3.31, 4.33, and 4.71 eV in the range of ≤ 5.0 eV, respectively. There are four major peaks for ErSi4− located in 2.31, 2.58, 2.83 and 3.67 eV, respectively. Six major peaks located in 1.92, 2.86, 3.16, 3.84, 4.16, and 4.34 eV are observed in the simulated PES of ErSi5−. There are seven major peaks for ErSi6− centered at 1.85, 2.43, 3.34, 3.85, 4.31, 4.60 and 4.95 eV, and six major peaks for ErSi7− centered at 2.38, 2.78, 3.13, 3.89, 4.52, and 4.96 eV, respectively. The simulated PES of ErSi8− exhibits five peaks centered at 2.96, 3.49, 4.00, 4.30 and 4.69 eV, respectively. Three peaks for ErSi9− are situated at 2.69, 3.38 and 3.93 eV, respectively. The simulated PES of ErSi10− in the range of ≤ 5.0 eV shows five peaks situated at 3.25, 3.67, 4.36, 4.61, and 4.84 eV, respectively. We think that these simulations will give strong motivation for experimental investigations of Er-doped Si clusters and their anions.
HOMO–LUMO gaps not only as a vital physical property can mirror the electronic properties but also as a key chemical property can mirror the chemical reactivity, especially for photochemical reaction. The curves of HOMO–LUMO gaps versus cluster size for the lowest energy geometries of ErSi n (n = 3–10) evaluated by using the three schemes are shown in Fig. 5. The HOMO–LUMO gaps of YbSi n , LuSi n , and Si n predicted at the TPSSh level are shown in Fig. S3 of Supporting Information in order to make easy comparisons. From Fig. 5, we can see that the curves of HOMO–LUMO gap evaluated by the TPSSh, B3LYP, and PBE are by and large in parallel, and the orders are B3LYP > TPSSh > PBE. The reason is that the energy of HOMO and LUMO evaluated in Kohn–Sham (KS) molecular orbital approximations go through by and large the alike quantity upshift, while Hartree–Fock (HF) scheme shift the LUMO up a much higher energy levels than the HOMO up [43]. On the other hand, the pure density functional PBE method doesn’t contain HF components. And the HF components of hybrid B3LYP scheme are larger than those of hybrid TPSSh method. Consequently, the orders are B3LYP > TPSSh > PBE. From Fig. S3 of Supporting Information, we can see that the curve of ErSi n parallels by and large with that of YbSi n , but not that of LuSi n . The HOMO–LUMO gaps of ErSi n are larger than those of YbSi n , but smaller than that of Si n with the exception of Si3. The HOMO–LUMO gap of Si3 is very close to that of ErSi3 because the ground state structure of Si3 is triplet state, not singlet state, leading to smaller the HOMO–LUMO gap. The smaller the HOMO–LUMO gap, the more easily the ErSi n inclines to set off photochemical reaction. Consequently, the photochemical activity of Er-doped Si n (n = 4–10) clusters is stronger than that of pure Si clusters, but weaker than that of YbSi n . This property can be used to manufacture novel functional materials such as optical materials, semiconductive materials, and environmental photocatalysis materials.

HOMO-LUMO gaps (eV) of ErSi n (n = 3–10) calculated at the three levels
Moreover, the natural population analysis (NPA) charges, valence configurations, and magnetic moments for the lowest energy structure are evaluated to further understand the interaction between Er and Si clusters. The values predicted with the three schemes differ little from each other. Consequently, only TPSSh data are listed in Tables S2 and S3 of Supporting Information. Similar to other Ln in small LnSi n (n = 3–10) clusters [3, 7, 11, 14, 16, 18, 19], the Er atom acts as an electron donor. The valence configurations of Er in ErSi n (n = 3, 5–10) are 6 s0.22−0.544f11.79−11.965d0.32−0.886p0.07−0.20. This indicates that the 4f electrons of Er in ErSi n (n = 3, 5–10) are hardly participate in bonding. For ErSi4, the 4f11.53−11.58 valence configuration shows that a 4f electron prefers to take part in bonding. In the case of negatively charged ions ErSi − n with n = 4, 7–10, their 4f11.18−11.49 valence configurations indicate the 4f electrons are also participate in bonding. It is to say that the ErSi n clusters belong to AB type. The 4f electrons of Er atom provide the total magnetic moments for ErSi n clusters and their anions as can be seen from Table S3.
To further understand ErSi n clusters belong to AB type, the PES of ErSi − n , YbSi − n , and LuSi − n (n = 4–7) are simulated at the TPSSh level and shown in Fig. S4 of Supporting Information. According to the classification of Grubisic et al. [27], spectra of A type are distinguished by a low electron binding energy feature. The representative is spectra of EuSi − n (Eu: half-filled 4f-electrons) and YbSi − n (Yb: full-filled 4f-electrons). Spectra of B type are lack of comparably low electron binding energy peaks. The representative is spectra of GdSi − n and LuSi − n (both have a 5d-electron). Some of spectra of AB resemble those of A, and others resemble those of B. In this study, we only choose YbSi n /YbSi − n and LuSi n /LuSi − n as representative for comparison. As can be seen from Fig. S4, the spectra of ErSi5− and ErSi6− resemble that of YbSi5− and YbSi6−, respectively. Spectra of ErSi4− resemble that of LuSi4−. While spectra of ErSi7− is spectral superposition of YbSi7− and LuSi7−.
Conclusions
The lowest energy structures and electronic properties of ErSi n (n = 3–10) clusters and their negatively charged ions were systematically studied using the ABCluster global search technique combined with the PBE, TPSSh and B3LYP schemes. The results revealed that the ground state is triplet electronic state for ErSi n (n = 3–10) excluded ErSi4, for which the triplet and quintuplet state compete with each other. For anion ErSi − n (n = 3–10), the spin multiplicity is quartet state. the lowest energy energies of neutral ErSi n (n = 3–10) are substitutional structures, similar to other Ln metal atoms-doped small silicon cluster such as LnSi n (Ln = Sm, Eu, Gd, Ho, Yb, Lu). When the neutral ErSi n gained an electron, the charge effects of this additional electron on the lowest energy structures are very strong. As a result, the lowest energy structures of ErSi − n are different from those of neutral counterparts starting from n = 7. On the other hand, starting from n = 7, the potential energy surfaces of ErSi − n are very flat, resulting isomeric arrangements occur and functional dependence of the predicted most stable structures exist. The DEs of neutral ErSi n and their anions are larger than those of YbSi n and their anions, and much smaller than those of LuSi n and their anions, respectively. The AEAs, VDEs and simulated PES of ErSi n (n = 3–10) are reported. Evaluation of HOMO–LUMO gap revealed that Er-doped Si cluster can significantly improve photochemical reactivity of the cluster. And the improved effects are weaker than those of the introducing Yb atom to Si cluster. The NPA analyses revealed that the 4f electron of Er atom in ErSi4, ErSi4−, ErSi7−, ErSi8−, ErSi9−, and ErSi10− prefers to take part in bonding. That is, ErSi n clusters belong to AB type. The total magnetic moments of ErSi n and their anions are mainly provided by the 4f electrons of Er atom.
References
A. J. Kenyon (2005). Semicond. Sci. Technol. 20, R65–R84.
T. T. Cao, L. X. Zhao, X. J. Feng, Y. M. Lei, and Y. H. Luo (2009). J. Mol. Struct. THEOCHEM 895, 148–155.
S. He and J. C. Yang (2017). J. Clust. Sci. 28, 2309–2322.
R. N. Zhao, Z. Y. Ren, P. Guo, J. T. Bai, C. H. Zhang, and J. G. Han (2006). J. Phys. Chem. A 110, 4071–4079.
R. N. Zhao, J. G. Han, J. T. Bai, F. Y. Liu, and L. S. Sheng (2010). Chem. Phys. 372, 89–95.
R. N. Zhao, J. G. Han, J. T. Bai, and L. S. Sheng (2010). Chem. Phys. 378, 82–87.
X. H. Xie, D. S. Hao, and J. C. Yang (2015). Chem. Phys. 461, 11–19.
T. G. Liu, W. Q. Zhang, and Y. L. Li (2014). Front. Phys. 9, 210–218.
L. Y. Hou, J. C. Yang, and Y. M. Liu (2016). J. Mol. Model. 22, 193.
R. N. Zhao and J. G. Han (2014). RSC Adv. 4, 64410–64418.
L. Y. Hou, J. C. Yang, and Y. M. Liu (2017). J. Mol. Model. 23, 117.
Y. Z. Bai, G. F. Zhao, X. F. Shen, J. M. Sun, and Y. X. Wang (2011). Acta Phys. Chim. Sin. 27, 39–46.
T. G. Liu, G. F. Zhao, and Y. X. Wang (2011). Phys. Lett. A 375, 1120–1127.
J. C. Yang, Y. T. Feng, X. H. Xie, H. W. Wu, and Y. M. Liu (2016). Theor. Chem. Acc. 135, 204.
G. F. Zhao, J. M. Sun, Y. Z. Gu, and Y. X. Wang (2009). J. Chem. Phys. 131, 114312.
J. C. Yang, J. Wang, and Y. R. Hao (2015). Theor. Chem. Acc. 134, 81.
C. G. Li, L. J. Pan, P. Shao, L. P. Ding, H. T. Feng, D. B. Luo, and B. Liu (2015). Theor. Chem. Acc. 134, 34.
X. H. Xie, D. S. Hao, Y. M. Liu, and J. C. Yang (2015). Comput. Theor. Chem. 1074, 1–8.
Y. T. Feng, J. C. Yang, and Y. M. Liu (2016). Theor. Chem. Acc. 135, 258.
Y. T. Feng and J. C. Yang (2017). J. Mol. Model. 23, 180.
Q. Peng and J. Shen (2008). J. Chem. Phys. 128, 084711.
T. T. Cao, X. J. Feng, L. X. Zhao, X. Liang, Y. M. Lei, and Y. H. Luo (2008). Eur. Phys. J. D 49, 343–351.
S. Jaiswal, V. P. Babar, and V. Kumar (2013). Phys. Rev. B 88, 085412.
J. Lu, J. C. Yang, Y. I. Kang, and H. M. Ning (2014). J. Mol. Model. 20, 2114.
N. Borshch and S. Kurganskii (2014). J. Appl. Phys. 116, 124302.
A. Grubisic, H. P. Wang, Y. J. Ko, and K. H. Bowen (2008). J. Chem. Phys. 129, 054302.
A. Grubisic, Y. J. Ko, H. P. Wang, and K. H. Bowen (2009). J. Am. Chem. Soc. 131, 10783–10790.
M. Ohara, K. Miyajima, A. Pramann, A. Nakajima, and K. Kaya (2002). J. Phys. Chem. A 106, 3702–3705.
K. Koyasu, J. Atobe, S. Furuse, and A. Nakajima (2008). J. Chem. Phys. 129, 214301.
V. Kumar, A. K. Singh, and Y. Kawazoe (2006). Phys. Rev. B 74, 125411.
J. P. Perdew, K. Burke, and M. Ernzerhof (1996). Phys. Rev. Lett. 77, 3865–3868.
V. N. Staroverov, G. E. Scuseria, J. Tao, and J. P. Perdew (2003). J. Chem. Phys. 119, 12129–12137.
A. D. Becke (1993). J. Chem. Phys. 98, 5648–5652.
C. Lee, W. Yang, and R. G. Parr (1988). Phys. Rev. B 37, 785–789.
D. E. Woon and T. H. Dunning Jr. (1993). J. Chem. Phys. 98, 1358–1371.
X. Y. Cao and M. Dolg (2002). J. Mol. Struct. THEOCHEM 581, 139–147.
A. A. Buchachenko, G. Chałasiński, and M. M. Szczęśniak (2007). Struct. Chem. 18, 769–772.
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox Gaussian 09, Revision C.01 (Gaussian Inc., Wallingford CT, 2010).
J. Zhang and M. Dolg (2015). Phys. Chem. Chem. Phys. 17, 24173–24181.
M. Dolg, H. Stoll, A. Savin, and H. Preuss (1989). Theor. Chim. Acta. 75, 173–194.
J. C. Yang, W. G. Xu, and W. S. Xiao (2005). J. Mol. Struct. THEOCHEM 719, 89–102.
Xl Zhu and X. C. Zeng (2003). J. Chem. Phys. 118, 3557–3570.
E. J. Baerends, O. V. Gritsenkoab, and R. V. Meer (2013). Phys. Chem. Chem. Phys. 15, 16408–16425.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21263010), by Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (Grant No. NMGIRT-A1603), and by the Inner Mongolia Natural Science Foundation (Grant No. 2015MS0216).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Yang, J. & Cheng, L. Probing Structure, Thermochemistry, Electron Affinity and Magnetic Moment of Erbium-Doped Silicon Clusters ErSi n (n = 3–10) and Their Anions with Density Functional Theory. J Clust Sci 29, 301–311 (2018). https://doi.org/10.1007/s10876-018-1336-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-018-1336-z