
Abstract
The total energies, growth patterns, equilibrium geometries, relative stabilities, hardnesses, intramolecular charge transfer, and magnetic moments of HoSi n (n = 12–20) clusters have been reexamined theoretically using two different density functional schemes in combination with relativistic small-core Stuttgart effective core potentials (ECP28MWB) for the Ho atoms. The results show that when n = 12–15, the most stable structures are predicted to be exohedral frameworks with a quartet ground state, but when n = 16–20, they are predicted to be endohedral frameworks with a sextuplet ground state. These trend in stability across the clusters (gauged from their dissociation energies) was found to be approximately the same regardless of the DFT scheme used in the calculations, with HoSi13, HoSi16, HoSi18, and HoSi20 calculated to be more stable than the other clusters. The results obtained for cluster hardness indicated that doping the Ho atom into Si13 and Si16 leads to the most stable HoSi n clusters, while doping Ho into the other Si n clusters increases the photochemical sensitivity of the cluster. Analyses of intracluster charge transfer and magnetic moments revealed that charge always shifts from the Ho atom to the Si n cluster during the creation of exohedral HoSi n (n = 12–15) structures. However, the direction of charge transfer is reversed during the creation of endohedral HoSi n (n = 16–20) structures, which implies that Ho acts as an electron acceptor when it is encapsulated in the Si n cage. Furthermore, when the most stable exohedral HoSi n (n = 12–15) structures are generated, the 4f electrons of Ho are virtually unchanged and barely participate in intracluster bonding. However, in the most stable endohedral HoSi n (n = 16–20) frameworks, a 4f electron does participate in bonding. It does this by transferring to the 5d orbital, which hybridizes with the 6s and 6p orbitals and then interacts with Si valence sp orbitals. Meanwhile, the total magnetic moments of the HoSi n (n = 16–20) clusters are considerably higher than those of HoSi n (n = 12–15). Interestingly, the endohedral HoSi16 and HoSi20 clusters can be viewed as the most suitable building blocks for novel high-density magnetic storage nanomaterials and for novel optical and optoelectronic photosensitive nanomaterials, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the past few years, extensive theoretical and experimental research has been performed on silicon clusters due to their distinctive chemical structures and bonding, as well as their wide range of applications in the microelectronics industry [1–14]. The ground-state structures of small (Si2–7) clusters have been determined using theoretical [1–3] and experimental [10–14] methods. Especially for large molecular clusters, it was found that the ground-state geometry can depend upon the method and basis set used to calculate it. Many calculations [4–8] have indicated that the most stable structures for medium-sized silicon clusters with less than 27 Si atoms can be categorized into two types: one is TTP (a tricapped trigonal prism structure); the other is SS (a structure containing a sixfold-puckered hexagonal ring and six-atom tetragonal bipyramid).
In recent years, rare earth metal (REM)-doped silicon clusters have attracted increasing attention because they can be used to miniaturize electronic equipment and they can be employed as self-assembled materials with many unusual optical, electronic, and magnetic properties [15–20]. In particular, doping lanthanide (Ln) atoms into a silicon cluster is regarded as a promising way to modify the magnetic properties of silicon. Since the 4f electrons in some REMs such as Sm and Eu barely participate in the bonding in their corresponding REMSi n clusters, these REM atoms can retain their atomic magnetic moments when they are incorporated into REMSi n clusters. On the other hand, in other REM atoms such as Pr, the 4f electrons do participate in the bonding in the REMSi n cluster; here, a 4f electron is transferred to the 5d orbital and becomes involved in bonding. Nevertheless, the total magnetic moment of the REM atom in REMSi n differs little from that of the isolated REM atom. However, when late-REM atoms are included in silicon clusters, such as the HoSi16–20 clusters studied in this paper, the total magnetic moment increases when a 4f electron is transferred to the 5d orbital. REM atoms differ from transition metal (TM) atoms in this regard; upon doping TM atoms into semiconductor clusters such as Si n and Ge n , the magnetic moment can be quenched [21, 22]. In addition to the magnetic moment, the implantation of REM atoms into silicon clusters can result in clusters with excellent optical properties; for example, doping an erbium atom into a silicon microcrystal yields a silicon-based optical source [23].
Although studies on REM-doped silicon clusters are still relatively rare, interest in their potential applications has stimulated a fair amount of research interest in these clusters over the past few years. Nakajima and co-workers [15, 24] were the first to investigate TbSi n −, HoSi n −, and LuSi n − (6 ≤ n ≤ 20) clusters experimentally using PES (photoelectron spectra) and a chemical probe. Their results suggested that when a Ho atom is encapsulated in a Si cage, the cage is incomplete when the number of Si atoms is less than 16. Bowen and co-workers [16] then explored the properties of HoSi n −, PrSi n −, GdSi n −, SmSi n −, EuSi n −, and YbSi n − (3 ≤ n ≤ 13) using PES. These REM-doped silicon clusters were categorized into two or three groups based on their appearance. In light of our experience [25–27], we believe that dividing the clusters into two groups is more reasonable than dividing them into three groups. Group A contains EuSi n −, YbSi n −, and SmSi n −, in which the 4f electrons of the REM atom barely participate in bonding. Group B contains HoSi n −, GdSi n −, and PrSi n −, in which the 4f electrons of the REM atom do participate in bonding. Spurred on by these experimental results, some theoretical studies have been carried out on REM-doped Si n species [28–37].
Recently, Zhao et al. [38] investigated the geometries and properties of HoSi n (n = 10–20) clusters theoretically using X3LYP functionals in combination with large-core potentials (ECP56MHF) for Ho atoms and 6-31G basis sets for Si atoms. The use of large-core (4f-in-core) basis sets for Ho atoms is unreasonable because the main difference between the Ln elements is that they all contain different numbers of electrons in their 4f shells. At the same time, the 4f electrons are valence electrons that can participate in bonding. Thus, 4f-in-core basis sets cannot accurately reflect the interaction between the Ln atom and the surrounding atoms, especially in a system with strongly interactions. Even if the 4f electrons are not involved in bonding, the electron configuration and the shapes of the 4f orbitals will be affected, so the electronic state will also be influenced. However, the ground-state structure is ultimately determined by the electronic state, and sometimes the energy differences between various electronic states are significant, so it is necessary to use small-core (4f-in-valence) basis sets to calculate structures and properties. We therefore reexamined the total energies, structures, and electron properties such as the growth pattern, structure, hardness, population analysis, and magnetic moment of each HoSi n (n = 12–20) cluster using two carefully selected density functionals in combination with relativistic small-core potential (ECP) basis sets for the Ho atom and cc-pVDZ for the Si atom in order to probe their unusual size-dependent electronic properties as well as the critical size of a silicon cluster encapsulating a Ho atom. This information should help to guide the development of new cluster-assembled materials. Two different functionals were utilized in this work to check whether the results of the theoretical determination of the most stable structures were dependent on the functional used in the calculations, as this phenomenon has been shown to occur for Si n species [7, 8].
Computational details
The calculations were performed using the Gaussian 09 software at the DFT level with the PBE0 [39] and B3LYP [40, 41] functionals in combination with the cc-pVDZ basis set [42] for Si atoms and relativistic small-core potentials (ECP28MWB) [43] (named SEG/ECP) for Ho atoms [44]. All stationary-point isomers of the HoSi n (n = 12–20) clusters were identified by calculating their vibrational frequencies with the two schemes. The optimized structures were obtained as local minima and ZPVE (zero-point vibrational energy) corrections were performed.
To search for the ground-state structures, a large number of isomers were studied in order to ensure that we did not overlook the lowest-energy isomers. At small cluster sizes, this approach is feasible. However, as the cluster size increases it becomes more difficult. There are two main reasons for this: one is the increase in the number of low-lying isomers as cluster size increases; the other is that large clusters cannot be efficiently and accurately optimized. In spite of this, the rules derived from calculations of smaller REM-doped Si n clusters can be applied to calculate medium and large REMSi n clusters. Cao et al. [33] performed a global search using a genetic algorithm (GA) to study LuSi n (n = 1–12) clusters, and concluded that the most stable structures were those obtained by replacing a Si atom in the lowest-energy structure of the pure Si n+1 cluster with a Lu atom (a “substitutional structure”). Wang et al. [29] studied LaSi6, CeSi6, YbSi6, and LuSi6 with the Saunders “Kick” global stochastic method and found that the most stable structures were the substitutional structures. We reported that the most stable structures of EuSi n , SmSi n , and YbSi n (n ≤ 11) are also substitutional structures [25–27]. Liu et al. [37] noted that the ground-state structures of HoSi n (n = 1–12, except for n = 7 and 10) were substitutional structures. In fact, the isomer reported by Liu et al. [37] (7A in Fig. 1) is less stable than the substitutional structure (7B in Fig. 1) by 0.15 and 0.17 eV when calculated at the B3LYP and PBE0 levels, respectively. For HoSi10, when the isomer presented by Liu et al. [37] as the initial geometry (10A in Fig. 1) is optimized at the B3LYP level of theory, structure 10B (shown in Fig. 1) is ultimately obtained. At the PBE0 level, structure 10B in the 4A″ state is more stable than 10A by 0.26 eV. Structure 10B, similar to the most stable structure of EuSi10 [25], can be viewed as that obtained by replacing a Si atom in the ground-state structure of Si11 [5] with a Ho atom. All of these results show that the most stable exohedral structures of neutral RESi n clusters can be viewed as the ground-state structure of Si n+1 with a Si atom swapped for an REM atom. Accordingly, only two families of initial geometries were taken into account in the optimization process in the work reported in the present paper: exohedral isomers (i.e., prolate structures that can be constructed by substituting a Si atom in the ground-state structure of Si n+1 with a Ho atom); and near-spherical geometries (obtained by a constrained search for structures similar to a fullerene cage). In order to find the most stable structures of HoSi n clusters as accurately as we could, previously reported ground-state structures of pure Si n clusters [4, 6–9], especially those of TTP and SS type, were considered when constructing prolate HoSi n (n = 12–20) structures. Furthermore, the spin multiplicities of the quartet and sextuplet states were considered, noting that the electronic state is a quartet state if the 4f electrons of the Ho atom barely participate in bonding, while the electronic state is a sextuplet state if a 4f electron does participate in bonding. Although a large number of isomers were obtained, only a few selected isomers are presented in this paper.

Geometries of HoSi7 and HoSi10. Isomers 7A and 10A are taken from [37]. 7B can be regarded as being derived from the ground-state structure of Si8 [3] but with a Si atom replaced with a Ho atom.10B can be viewed as being derived from the ground-state structure of Si11 [5] but with a Si atom replaced with a Ho atom
Results and discussion
Lowest-energy structures and isomers
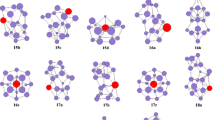
The isomers of the HoSi n (n = 12–20) clusters that were optimized with the PBE0 and B3LYP methods are shown in Fig. 2. The total and relative energies of the low-lying isomers are listed in Table 1.

The stable geometries of the isomers of the HoSi n (n = 12–20) clusters, as obtained at the B3LYP level of theory. The optimized geometries obtained at the PBE0 level of theory are similar to those yielded by B3LYP
There are three competing isomers for the ground-state structure of Si13. At the CCSD(T)/6-31G(d)//MP2/6-31G(d) level, it is predicted to be a distorted TTP with an additional rhombus capping the edge of the prism [6]. At the quantum Monte Carlo level, it is predicted to be a C 3v -symmetry capped trigonal antiprism [4], and at the MP2/aug-cc-pVTZ//B3LYP/6-31+G(d) level, it is predicted to have C 2v symmetry [9]. The most stable structure, 12A, of HoSi12 can be viewed as a capped Si trigonal antiprism [4] with one of the Si atoms replaced with a Ho atom. Liu et al. [37] reported that the quartet-state isomer 12B was the lowest-energy structure. Zhao et al. [38] noted that the semiclosed quartet-state isomer 12C was the most stable structure. Energetically, the isomers 12B (with quartet and sextuplet states) and 12C (with a sextuplet state) are less stable than the quartet-state isomer 12A.
For the isomer 12C (quartet state), our result shows that spin contamination occurs due to the expectation value (4.75) of the total spin (S 2). This may be expanded in terms of pure states with higher multiplicities. On the other hand, the valence configuration (6s 0.444f 10.045d 2.456p 0.78, as obtained at the B3LYP level) of the quartet state has five orbitals occupied by a single electron because of the transfer of one 4f electron. Energetically, both the quartet and sextuplet states are nearly identical, as can be seen from Table 1. These results indicate that the isomer 12C is essentially in the sextuplet state. That is, the result of performing 4f-in-valence calculations is different from that produced by 4f-in-core calculations. A similar situation is observed for the quartet-state isomers 13B, 13C, 14C, 15D, 16A, 17A, 18A, 19A, and 20A; we do not delve any more deeply into this observation here.
The ground-state structure of Si14 is a face-capped distorted TTP with an additional rhombus capping an edge of the prism [6]. For HoSi13, isomers 13A and 13B are constructed by replacing a Si in the ground-state Si14 [6] with a Ho atom. 13A (quartet) is the global minimum. 13B is the most stable when there is a sextuplet electronic state. Zhao et al. [38] reported that isomer 13C was the most stable structure, but it is much higher in energy than the quartet-state 13A.
The most stable structure of Si15 is a TTP with a tricapped trigonal antiprism [6]. Isomers 14A and 14B of HoSi14 are constructed by replacing a Si in the most stable structure of Si15 [6] with a Ho atom. 14A (quartet) was found to be the global minimum, and 14B to be the most stable when there is a sextuplet electronic state. Isomer 14C is taken from [38]. Isomer 14C is considered to be higher in energy than the quartet-state isomer 14A.
There are three competing isomers for the ground-state structure of Si16; these contain a TTP motif, an SS motif, and two fused pentagonal prisms, respectively [7]. For HoSi15, isomers 15A, 15B, and 15C are constructed by substituting a Si in the most stable Si16 structure with a Ho atom. The structure of 15D, with an incompletely encapsulated He atom, is taken from [39]. The sextuplet state of this isomer is less stable than the most stable (quartet-state) structure, 15A, by about 0.83 and 1.03 eV when calculated at the B3LYP and PBE0 levels, respectively.
For HoSi16, two isomers are reported. The prolate 16B is constructed by substituting a Si in the ground-state structure of Si17 containing an SS motif [7] with a Ho atom. The endohedral isomer 16A is taken from [39]. Its sextuplet state is more stable in energy than the quartet and sextuplet states of 16B by 0.02 eV (B3LYP) or 0.32 eV (PBE0) and by 0.32 eV (B3LYP) or 0.30 eV (PBE0), respectively.
Isomers containing SS and TTP motifs compete to be the most stable structure of Si18 [7]. Isomers 17B and 17C of HoSi17 are constructed by replacing an Si atom in the most stable Si18 structure with a Ho atom. The endohedral 17A is almost the most stable of the structures, as reported previously [38]. Its sextuplet state is the ground-state structure at the B3LYP level, while it is less stable at the PBE0 level than the sextuplet-state isomer 17C by 0.23 eV.
For HoSi18, two isomers are identified. One is the prolate 18B, which is constructed by replacing a Si in the most stable structure of Si19 (containing an SS motif) [7] with a Ho atom. Another is the endohedral 18A, which is analogous to the most stable structure reported previously [38]. Its sextuplet state is predicted to be the ground state.
For HoSi19, there are two isomers. The prolate 19B is obtained by replacing a Si in the ground-state structure of Si20 (containing an SS motif) [7] with a Ho atom. The cakelike 19A is taken from [39]. Energetically speaking, its sextuplet state was calculated to be the most stable structure.
Two isomers are also reported for HoSi20. The prolate 20B is generated by replacing a Si in the most stable structure of Si21 (containing an SS motif) [8] with a Ho atom. The quartet and sextuplet states of isomer 20B are much higher in energy than the sextuplet-state isomer 20A with an encapsulated Ho when calculated at the B3LYP and PBE0 levels.
From the discussion above, we can conclude that, starting from n = 16, the sextuplet-state Si n clusters with an encapsulated Ho atom are predicted to be the most stable structures (aside from when the isomers of HoSi17 are calculated using the PBE0 method, when the most stable structure is found to be an exohedron in the sextuplet state). This small-core results obviously differ from the predictions obtained when using large-core basis sets for Ho atoms [38]. Although the geometries obtained are similar, their electronic states are different. The large-core results suggest that, when n < 16, exohedral quartet-state clusters are the ground states for HoSi n . The quartet state for an endohedral and semiclosed geometry generally presents spin contamination. It is therefore crucial to inspect the spin contamination for clusters that include REM (and TM) atoms more closely.
Relative stabilities
To examine the relative stabilities of the most stable isomers of the HoSi n (n = 12–20) clusters, the binding energy per atom (BEPA) (HoSi n → Ho + nSi), where BEPA(HoSi n ) = [n E(Si) + E(Ho) – E(HoSi n )]/(n + 1), was calculated at the B3LYP and the PBE0 levels for each isomer. A plot of BEPA against cluster size (Fig. 3) reveals that, when calculations are performed at the PBE0 level, the most stable isomers of HoSi13, HoSi16, HoSi18, and HoSi20 are slightly more stable than suggested by the smoothly increasing trend. In addition, the most stable isomer of HoSi15 is also more stable when calculations are performed at the B3LYP level.

Binding energy per atom (BEPA) for the most stable isomer of each HoSi n (n = 12–20) cluster, as calculated using the PBE0 and the B3LYP methods
Apart from the BEPA, the dissociation energies (DEs) of the structures also illustrate their relative stabilities. DE1 [defined as the energy required for the disproportionation reaction 2HoSi n → HoSi n+1 + HoSi n−1, DE1(HoSi n ) = E(HoSi n+1) + E(HoSi n−1) – 2E(HoSi n )] as a function of cluster size is shown in Fig. 4. From Fig. 4, we can see that HoSi15, HoSi17, and HoSi19 are less stable than the other clusters because they are local minima at the PBE0 level, while HoSi14, HoSi17, and HoSi19 are less stable than the other clusters when calculated at the B3LYP level.

DE1 (eV) of HoSi n (n = 13–19) versus the number of atoms n
Other measures of the cluster stability include DE2(HoSi n ) = E(Si n ) + E(Ho) − E(HoSi n ), DE3(HoSi n ) = E(HoSi n−1) + E(Si) − E(HoSi n ), and DE4(Si n ) = E(Si n−1) + E(Si) − E(Si n ). The values of these parameters calculated at the B3LYP and the PBE0 levels of theory for the clusters of interest are sketched in Figs. 5 and 6, respectively. Analyses of the DE2 curve in Fig. 5 show that, at the B3LYP level, HoSi13, HoSi16, HoSi18, and HoSi20 are more stable than the clusters because their DE2 values are local maxima. HoSi20 is the most stable of these clusters because it has the largest DE2 value. On the other hand, analyses of the DE3 and DE4 curves reveal that HoSi13, HoSi15, HoSi16, HoSi18, and HoSi20 are more stable than the other clusters because their DE3 values are larger than their DE4 values. In other words, it is more energetically favorable to connect a Si atom to HoSi n−1 to form HoSi n rather than to add it to Si n−1 to form Si n . At the PBE0 level, the results of the analyses of DE2 are the same as those of the analyses of DE3 and DE4, and HoSi13, HoSi16, HoSi18, and HoSi20 are more stable than the other clusters.

DE2, DE3, and DE4 (eV) of the HoSi n (n = 12–20) clusters versus the number of Si atoms n, as calculated at the B3LYP level

DE2, DE3, and DE4 (eV) of the HoSi n (n = 12–20) clusters versus the number of Si atoms n, as calculated at the PBE0 level
From the discussion above, we can see that the relative stabilities of the clusters showed the same general trend regardless of the particular dissociation energy parameter used to judge the stability and the particular functional used to calculate the dissociation energy. HoSi13, HoSi16, HoSi18, and HoSi20 were found to be more stable than the other clusters. It should be noted that HoSi16, HoSi18, and HoSi20 could be used as building blocks for nanomaterials because of their cage-like structures—especially HoSi20, which is not only the most stable of these clusters but also features a Ho atom that is fully encapsulated in a Si20 fullerene-like framework.
Hardness
The hardness reflects the ability of a species to participate in a chemical reaction. The hardness—defined as the difference between the HOMO and the LUMO energies—of the most stable structure of each HoSi n (n = 12–20) cluster was evaluated at the PBE0 and the B3LYP levels, and the hardness values for all of the clusters of interest are sketched in Fig. 7. To facilitate comparison, the hardnesses of Si n clusters are also shown in Fig. 7. From Fig. 7, we can see that the hardness curves obtained at the two levels of theory are very similar. Also, the hardness of each HoSi n (n = 12–20) cluster is smaller than that of its corresponding Si n species except for HoSi13 and HoSi16; in other words, doping Ho into Si13 and Si16 results in clusters with good chemical stability. Accordingly, due to its particularly high chemical stability and relative stability, HoSi16 may be viewed as a cluster that is especially appropriate for use as a building block for high-density magnetic storage nanomaterials (its magnetic moment is 5 μB; see the “Charge transfer and magnetic moment” section). Doping a Ho atom into Si n (n = 12–20), with the exception of the n = 13 and 16 clusters, increases the photochemical sensitivity of the cluster. Therefore, given its high relative stability and photochemical sensitivity, the cluster HoSi20, in which Ho is completely encapsulated by Si atoms, can be viewed as a particularly suitable building block for novel optical and optoelectronic photosensitive nanomaterials.

The HOMO–LUMO gaps of the most stable structures of the clusters HoSi n and Si n (n = 12–20), as calculated with the PBE0 and the B3LYP methods
Note that, when using small-core basis sets, HoSi16 does not have the largest predicted HOMO–LUMO gap among all of the clusters, in contrast to the result obtained when using large-core basis sets for the Ho atom [38].
Charge transfer and magnetic moment
Natural population analyses (NPA) of the ground-state structures were conducted using the PBE0 and B3LYP methods to further understand the interaction between the Ho atom and the Si n cluster. The charge on and the NPA valence configuration of the Ho atom in each cluster are listed in Table 2.
These data indicate that the charge on and NPA valence configuration of Ho calculated using the B3LYP method are usually the same as those obtained with the PBE0 method. The valence configurations of 6s 0.15–0.464f 10.96–10.985d 0.47–0.876p 0.12–0.25 for the most stable exohedral structures of the clusters HoSi n (n = 12–15) reveal that the 4f electrons are almost unchanged upon the creation of the HoSi n cluster and barely participate in bonding. However, for the most stable endohedral structures of the clusters HoSi n (n = 16–20), the valence configurations of 6s 0.27–0.424f 9.99–10.015d 3.95–5.036p 1.41–1.74 show that the 4f shell loses an electron during the creation of the HoSi n cluster; this electron is transferred to the 5d orbital and participates in bonding. In addition to the charge transfer from the 4f to the 5d orbital, charge transfer occurs from the 6s to the 5d and the 6p orbitals, leading to spd hybridization.
Table 2 also shows that charge is always transferred from the Ho atom to the Si n cluster for exohedral HoSi n (n = 12–15) clusters, but the charge is transferred in the opposite direction for endohedral HoSi n (n = 16–20) clusters, which reveals that Ho acts as an electron acceptor in Si n clusters in which Ho is fully encapsulated. In fact, the charge on the REM (or TM) atom in REMSi n clusters in which the REM atom is at the center of the Si n cage is negative, and the smallest number of silicon atoms for REMSi n clusters with a negative charge on the REM atom is seen as the threshold value of n for the REM atom to be inserted into the silicon cage. The change transferred (2.68–4.23e) during the generation of the endohedral HoSi n (n = 16–20) clusters reveals that the bonding between the Ho and the Si n cage is ionic, because the charge is transferred from the valence sp orbitals on silicon to the spd hybridized orbitals on Ho. Again, it should be noted that the small-core results for charge transfer are different from those predicted using large-core basis sets for the Ho atom: that the charge is always transferred from the Si atoms to the Ho atom in HoSi n (n = 12–20).
As far as species containing REM atoms are concerned, magnetism is one of their most interesting properties. The magnetic moments of the 6s, 4f, 5d, and 6p states for Ho, the total magnetic moment of Ho, and the total magnetic moment of the most stable structure of each HoSi n (n = 12–20) cluster are listed in Table 3. From the data shown, we can conclude that, for all of the clusters, the magnetic moments calculated using the B3LYP method are almost identical to those yielded by the PBE0 method. For each of the exohedral HoSi n (n = 12–15) clusters, the total magnetic moment is 3 μB, which comes from the 4f state (3.01–3.02 μB) of the Ho atom. For each of the endohedral HoSi n (n = 16–20) clusters, the magnetic moment is 5 μB, which derives mainly from the 4f state (3.98–3.99 μB) of Ho, followed by the 5d state (0.02–0.37 μB) and the 3s and 3p states (0.62–0.99 μB) of the Si atoms. There is no contribution from the 6s and 6p states of the Ho atom. In other words, for the endohedral structures of the HoSi n (n = 16–20) clusters, there is electron transfer from 4f to 5d. As a result, not only do the 4f electrons participate in bonding, but the total magnetic moment of each HoSi n (n = 16–20) cluster also increases.
Conclusions
The total energies, equilibrium geometries, relative stabilities, hardnesses, intramolecular charge transfer, and magnetic moments of the clusters HoSi n (n = 12–20) have been reexamined theoretically using the B3LYP and PBE0 functionals in combination with the cc-pVDZ basis set for the Si atoms and relativistic small-core Stuttgart effective core potentials (ECP28MWB) for the Ho atom. The results were obtained:
-
When n = 12–15, the most stable structures of the HoSi n clusters are predicted to have exohedral geometries and a quartet ground state, and can be constructed by replacing a Si atom in the ground-state structure of Si n+1 with a Ho atom. However, the most stable structures were found to be endohedral frameworks with a sextuplet ground state when n = 16–20.
-
Relative cluster stabilities were evaluated based on different dissociation energy parameters, which were calculated with the B3LYP and PBE0 functionals. Regardless of the particular dissociation parameter considered, and the functional used to calculate it, the trend in relative stability across the clusters was the same. HoSi13, HoSi16, HoSi18, and HoSi20 were calculated to be more stable than the other clusters.
-
In light of the results for cluster hardness, the most stable HoSi n clusters are achieved when a Ho atom is doped into Si13 and Si16 clusters. Doping Ho into the other Si n clusters increases their photochemical sensitivity.
-
Analyses of the intracluster charge transfer revealed that there is charge transfer from the Ho atom to the Si n cluster when HoSi n (n = 12–15) clusters with exohedral structures are created. However, charge transfer occurs in the opposite direction when HoSi n (n = 16–20) clusters with endohedral structures are created. This shows that Ho acts as an electron acceptor when the Ho atom is encapsulated in the Si n cage.
-
Magnetic moment analysis of the clusters showed that the 4f electrons remain almost unchanged during the creation of the most stable exohedral structures of the HoSi n (n = 12–15) clusters, barely participating in the intracluster bonding. However, a 4f electron does participate in the bonding within the most stable endohedral frameworks of the HoSi n (n = 16–20) clusters; in these structures, a 4f electron is transferred to the 5d orbital, which hybridizes with the 6s and 6p orbitals and then interacts with the Si valence sp orbitals. Also, the total magnetic moments of the HoSi n (n = 16–20) clusters are considerably higher than those of the HoSi n (n = 12–15) clusters.
The results imply that, due to its especially high chemical stability and relative stability, the endohedral HoSi16 cluster is particularly well suited for use as a building block in novel high-density magnetic storage nanomaterials. On the other hand, due to its prominent relative stability and photochemical sensitivity, the HoSi20 cluster (in which Ho is completely encapsulated) appears to be a highly suitable building block for novel optical and optoelectronic photosensitive nanomaterials.
References
Raghavachari K (1986) J Chem Phys 84:5672–5686
Li BX, Cao PL, Zhan SC (2003) Phys Lett A 316:252–260
Vasiliev I, Öğüt S, Chelikowsky JR (1997) Phys Rev Lett 78:4805–4808
Grossman JC, Mitáš L (1995) Phys Rev Lett 74:1323–1326
Zhu X, Zeng XC (2003) J Chem Phys 118:3558–3570
Zhu X, Zeng XC, Lei YA, Pan B (2004) J Chem Phys 120:8985–8995
Yoo S, Zeng XC (2005) J Chem Phys 123:164303-1–164303-6
Yoo S, Zeng XC (2006) J Chem Phys 124:054304-1–054304-6
Nigam S, Majumder C, Kulshreshtha SK (2006) J Chem Phys 125:074303-1–074303-11
Li S, Zee RJV, Weltner W Jr (1994) J Chem Phys 100:7079–7086
Xu CS, Taylor TR, Burton GR, Neumark DM (1998) J Chem Phys 108:1395–1406
Ohara M, Koyasu K, Nakajima A, Kaya K (2003) Chem Phys Lett 371:490–497
Honea EC, Ogura A, Peale DR, Félix C, Murray CA, Raghavachari K, Sprenger WO, Jarrold MF, Brown WL (1999) J Chem Phys 110:12161–12171
Li S, Zee RJV, Weltner W Jr, Raghavachari K (1995) Chem Phys Lett 243:275–280
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) J Chem Phys 129:214301-1–214301-7
Grubisic A, Ko YJ, Wang H, Bowen KH (2009) J Am Chem Soc 131:10783–10790
Li J, Wang G, Yao C, Mu Y, Wan J, Han M (2009) J Chem Phys 130:164514-1–164514-9
Zhao G, Sun J, Gu Y, Wang Y (2009) J Chem Phys 131:114312-1–114312-7
Peng Q, Shen J (2008) J Chem Phys 128:084711-1–084711-11
Liu T, Zhao G, Wang Y (2011) Phys Lett A 375:1120–1127
Dhaka K, Bandyopadhyay D (2015) RSC Adv 5:83004–83012
Li Y, Tam NM, Claes P, Woodham AP, Lyon JT, Ngan VT, Nguyen MT, Lievens P, Fielicke A, Janssens E (2014) J Chem Phys A 118:8198–8203
Kenyon AJ (2005) Semicond Sci Technol 20:R65–R84
Ohara M, Miyajima K, Pramann A, Nakajima A, Kaya K (2002) J Chem Phys A 106:3702–3705
Yang JC, Wang J, Hao YR (2015) Theor Chem Accounts 134:81-1–81-11
Xie XH, Hao DS, Liu YM, Yang JC (2015) Comput Theor Chem 1074:1–8
Xie XH, Hao DS, Yang JC (2015) Chem Phys 461:11–19
Li CG, Pan LJ, Shao P, Ding LP, Feng HT, Luo DB, Liu B (2015) Theor Chem Accounts 134:34-1–34-11
Wang HQ, Li HF (2014) RSC Adv 4:29782–29793
Zhao RN, Han JG, Bai JT, Sheng LS (2010) Chem Phys 378:82–87
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) Chem Phys 372:89–95
Zhao RN, Ren ZY, Guo P, Bai JT, Zhang CH, Han JG (2006) J Phys Chem A 110(11):4071–4079
Cao TT, Zhao LX, Feng XJ, Lei YM, Luo YH (2009) J Mol Struct 895:148–155
Cao TT, Feng XJ, Zhao LX, Liang X, Lei YM, Luo YH (2008) Eur Phys J D 49:343–351
Kumar V, Singh AK, Kawazoe Y (2006) Phys Rev B 74:125411-1–125411-5
Wang J, Liu Y, Li YC (2010) Phys Chem Chem Phys 12:11428–11431
Liu TG, Zhang WQ, Li YL (2014) Front Phys 9:210–218
Zhao RN, Han JG (2014) RSC Adv 4:64410–64418
Adamo C, Barone V (1999) J Chem Phys 110:6158–6170
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Woon DE, Dunning TH Jr (1993) J Chem Phys 98:1358–1371
Cao X, Dolg M (2002) J Mol Struct THEOCHEM 581:139–147
Frisch MJ, Trucks GW, Schlegel HB et al (2010) Gaussian 09, revision C.01. Gaussian Inc., Wallingford
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 21263010), by the Program for Innovative Research Team in Universities of the Inner Mongolia Autonomous Region (grant no. NMGIRT-A1603), and by the Inner Mongolia Natural Science Foundation (grant no. 2015MS0216).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hou, L., Yang, J. & Liu, Y. Reexamination of structures, stabilities, and electronic properties of holmium-doped silicon clusters HoSi n (n = 12–20). J Mol Model 22, 193 (2016). https://doi.org/10.1007/s00894-016-3058-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-016-3058-1