
Abstract
The equilibrium geometries and properties such as adiabatic electron affinities (AEAs), simulated photoelectron spectra (PES), dissociation energies, relative stabilities, HOMO–LUMO gaps, charges transfer, and magnetic moments of PrSi n (n = 3–9) and their anions have been made a detailed study by means of the ABCluster global search technique combined with density functional methods. The structure optimization is carried out with three exchange correlation functionals (B3LYP, PBE0, and mPW2PLYP). The ground state structures predicted by mPW2PLYP are thought to be trustworthy. The experimental PES of PrSi4 − is reassigned in light of the theoretical results, and the experimental AEAs of 2.0 ± 0.1 eV are obtained. The mPW2PLYP AEAs of PrSi n are in excellent agreement with the experimental values. The average absolute deviations from experiment are only 0.05 eV, and the maximum deviations are 0.10 eV. The accordance between the experimental PES and the theoretical simulations indicates that the ground state structures of PrSi − n (n = 4–9) are trustworthy. Doping Pr atom to Si n (n = 3–9) clusters raises the photochemical sensitivity. A large proportion of the total magnetic moments for all of these species are contributed by Pr atom.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Introducing rare earth (RE) metal atoms into semiconductor clusters in the past decade, especially silicon, has been a subject of greater interest in respect that doping RE atom into silicon clusters can alter significantly their structures, properties, and stabilities [1–20].
There have been some previous studies on introducing RE atoms into silicon clusters. Bowen et al. [1, 2] presented the PES of RESi − n (RE = Eu, Sm, Yb, Pr, Gd, and Ho 3 ≤ n ≤ 17) and found that they can be divided into two categories based on their appearance. The spectra of EuSi n , YbSi n , and SmSi n belong to group “A”, and the spectra of PrSi n , GdSi n , and HoSi n fall into group “B”. In the previous investigation [18–20], we found that the 4f electron of Eu, Yb, and Sm atoms in Si n surrounding hardly participates in bonding. In this work, we can find that the 4f electron of Pr atoms participates in bonding. More specifically, a 4f electron of Pr atom removed to 5d orbital, and then the 5d electron participates in bonding. That is actually similar to Gd atom which itself contains a 5d electron; that is, the 4f or 5d electron of group “B” atom in the clusters prefers to take part in bonding. While for the group “A”, the 4f electron hardly participates in bonding. On the aspect of the experiment, Nakajima et al. [3, 4] have firstly explored the TbSi − n , LuSi − n , and HoSi − n (6 ≤ n ≤ 20) clusters by using PES. Urged by these experimental observations, some theoretical simulations have been achieved for RESi n clusters. The equilibrium geometries and properties such as relative stabilities, magnetic moments, charge transfers, HOMO–LUMO gaps, and adiabatic electron affinities (AEAs) of neutral SmSi n and YbSi n (n ≤ 13) and their charged ions were calculated by using various density functional theory (DFT) methods [8–12]. The growth behavior of the ground state structures for LuSin, HoSi n , LaSi n , and GdSi n (n ≤ 21) was also investigated by means of DFT schemes [13–17]. Recently, we evaluated the ground state structures and electron affinities of SmSi n , EuSi n , and YbSi n (3 < n<11) and their anions by means of several DFT techniques including B3LYP, wB97X, PBE0, PBE, and B2PLYP and found that the theoretical AEAs calculated by these methods agree with the experimental values [18–20].
In this study, the ground state structures and properties including AEAs, relative stabilities, dissociation energies (DEs), simulated PES spectra, HOMO–LUMO gaps, charges transfers, and magnetic moments of neutral PrSi n (n = 3–9) and their anions are explored with the aim of understanding how their properties are different from that spectra belong to “A”. The simulated PES spectra and calculated AEAs are compared with experimental ones in order to not only verify the reliability of the predicted results but also aid the reassignment of experimental PES. This work will also provide specific guidance for further investigation of medium-size clusters.
2 Theoretical methods
The calculations are carried out at the level of the DFT with the B3LYP [21, 22], PBE0 [23], and mPW2PLYP [24] functional. The basis sets used in the geometry optimization process are cc-pVTZ [25] for Si atoms and the segmented (SEG) Gaussian valence basis sets and relativistic small-core potentials (ECP28MWB) [26] (denoted as SEG/ECP) for Pr atoms. At the B3LYP and PBE0 levels, calculations of harmonic frequency for neutral PrSi n (n = 3–9) and their anions were done to assure that the optimized isomers are local minima. Then, the SEG basis sets of Pr were augmented by diffuse functions 2pdfg with exponents 0.028 and 0.015 (p), 0.032 (d), and 0.05 (f, g) [27] (denoted as aug-SEG/ECP), which aug-cc-pVTZ basis sets of Si [26] were used in the single-point energies calculations. Finally, the energies at 0 K are gained by adding the zero-point vibration energy (ZPVE) (the mPW2PLYP ZPVE employed that of the PBE0). The GAUSSIAN 09 codes [28] are used to perform all of the calculations.
The initial geometries are obtained by using the ABCluster global search method [29] combined with the GAUSSIAN 09 codes. The first step is achieved at the B3LYP level with relativistic large-core effective core potentials (ECP53MWB) [30, 31] for Pr atoms and 6-31G basis set for Si atoms. More than 100 initial geometries of each PrSi n clusters are generated for n ≤ 7, and more than 300 configurations are generated for n ≥ 8. The second step, the top ten lowest energy structures from the first step, and those with their energy differences within 0.8 eV from the lowest energy structure, are selected and optimized again by means of the B3LYP with the SEG/ECP basis set for Pr and the cc-pVTZ basis sets for Si atoms. Finally, the structures from the second step with their energy differences within 0.8 eV are optimized by means of the remaining two methods. The “substitutional structure”, which can be regarded as substituting a Si atom of the ground state structure of Sin+1 with a Pr atom, is also taken into account in respect that the ground state structures of YbSi n , SmSi n , and EuSi n are substitutional structure [18–20]. The global search, after all, is a mathematical scheme, and it is almost impossible to take an “ergodic” sampling on the potential energy surfaces for large clusters, especially for heteroatom clusters. Our experience is that, all of the “substitutional structures” are included when 100 configurations are generated from the ABCluster global search technique. However, starting from n = 8, only part of the “substitutional structures” are included when 300 (even up to 500) configurations are generated from the ABCluster global search method.
Furthermore, the spin multiplicities of quartet and sextuplet states were taken into account for neutral PrSi n (n ≤ 5). And triplet and quintuplet state were considered for their anions. The results show that the quartet state is predicted to be the ground state structure for the neutral with the exception of PrSi which is sextuplet state. The ground state structure for anions PrSi − n with n = 1–3 is calculated to be the quintuplet. The reason can be attributed to the ground state structure of Si n with n = 1–3 is triplet electronic state. Starting from n = 4, the ground state structure is triplet electronic state. Although many isomers are obtained, the ground state structures are mainly presented.
3 Results and discussion
3.1 The ground state structures of PrSi n and their anions
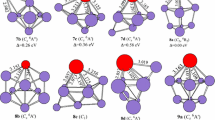
The isomers optimized at the B3LYP, PBE0, and mPW2PLYP levels are shown in Fig. 1 for PrSi n (n = 3–9) species and their anions. For PrSi3, the ground state structure (shown in Fig. 1) is calculated to be an approximate planar rhombus with quartet electronic state, which is more stable than that of sextuplet by 0.77, 0.61, and 0.41 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. For anion, the approximate planar rhombus PrSi − 3 -I (see Fig. 1) of triplet electronic state is more stable than that of PrSi − 3 -II isomer (see Fig. 1) by 0.39 and 0.01 eV at the B3LYP and PBE0 level, respectively. It is noted that for PrSi − 3 -I, the spin contamination occurs at the B3LYP and PBE0 levels due to the expectation value [2.69 (B3LYP) and 2.71 (PBE0)] of the total spin (S2) as can be seen from Table 1. At the mPW2PLYP level, the trigonal pyramid PrSi − 3 -II of quintuplet electronic state is evaluated to be the ground state structure. It is more stable than that of PrSi − 3 -I by 0.15 eV in energy.


Geometries of PrSi n (n = 3–9) and their anions in which red color (online) is Pr atom. The Pr–Si bond lengths are shown in Å
For neutral PrSi4, two isomers which compete with each other for the ground state structure are reported. The C s -symmetry trigonal bipyramind PrSi 4 -I of 4A″ electronic state, and the C 2v -symmetry PrSi 4 -II of 4A1 electronic state are shown in Fig. 1. The energy differences between PrSi 4 -I and PrSi 4 -II are only 0.07, 0.01, and −0.02 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. For anion PrSi4 −, the trigonal bipyramind PrSi − 4 -I with triplet electronic state (approximately C 3 v -symmetry) is calculated to be the ground state structure. The C 2v -symmetry PrSi − 4 -II of 3A1 electronic state is less stable in energy than that of PrSi − 4 -I by 0.56, 0.54, and 0.80 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. Furthermore, the PrSi − 4 -II is spin contamination at the B3LYP and mPW2PLYP levels. Their quintuplet isomers are less stable in energy than the ground state structure PrSi − 4 -I. For example, the C 2v -symmetry isomer of 5A1 electronic state, analogous to PrSi − 4 -II (not shown in Fig. 1), is less stable than the ground state PrSi − 4 -I structure by 0.48, 0.60, and 0.74 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively.
Two face-capped trigonal bipyramind with C s -symmetry for neutral PrSi5 is presented. At the mPW2PLYP level, the PrSi 5 -I of 4A′ electronic state is predicted to be the ground state structure, which is more stable than the PrSi 5 -II of 4A″ by 0.17 eV. While at the B3LYP and PBE0 levels, the PrSi 5 -II isomer, analogous to the ground state structure of YbSi5, SmSi5, and EuSi5 [18–20], is more stable in energy than that of PrSi 5 -I by 0.33 and 0.23 eV, respectively. For anion, the isomer PrSi − 5 -I of 3A′ electronic state is predicted to be the ground state structure, which is more stable than the PrSi − 5 -II by 0.10, 0.30, and 0.70 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. In addition, the PrSi − 5 -II is spin contamination at the B3LYP, PBE0, and mPW2PLYP levels.
Xu et al. [8] reported that the ground state structure of PrSi6 and its anion is C 2v - and C 5v -symmetry pentagonal bipyramid, respectively. Our result is the same as their outcome.
For PrSi7, four isomers are presented. The C 1-symmetry PrSi 7 -I isomer which can be regarded as being derived from the distorted bicapped octahedron of Si8 [32] by substituting a Si with a Pr atom is similar to the ground state structure of EuSi 7 [20], SmSi 7 [19], and YbSi 7 [18]. The approximate C s -symmetry PrSi 7 -II isomer is analogous to the most stable structure of GdSi7 [17]. The isomers PrSi 7 -III and PrSi 7 -IV possess 4A″ and 4A electronic state. At the mPW2PLYP and PBE0 levels, the PrSi 7 -II structure is more stable in energy than those of PrSi 7 -I, PrSi 7 -III, and PrSi 7 -IV by 0.05, 0.07, and 0.22 eV, and 0.05, 0.07, and 0.13 eV, respectively. At the B3LYP level, the PrSi 7 -I structure is more stable in energy than the isomers of PrSi 7 -II, PrSi 7 -III, and PrSi 7 -IV by 0.05, 0.01, and 0.27 eV, respectively. The energies of PrSi 7 -I, PrSi 7 -II and PrSi 7 -III isomers are almost equal. Their energy differences fall in 0.07 eV. These indicate that the potential energy surface of PrSi 7 is flat and that accurate prediction of structures requires advanced quantum mechanical investigations. For anion PrSi − 7 , four isomers are also reported. At the mPW2PLYP level, the energies of PrSi − 7 -II structure of 3A′, PrSi − 7 -III of 3A′, and PrSi − 7 -IV of 3A2 are nearly equal. The energy differences among them are within 0.02 eV. The PrSi − 7 -I isomer is less stable than that of PrSi − 7 -III by 0.07 eV in energy. At the B3LYP level, the PrSi − 7 -IV structure is more stable in energy than the isomers of PrSi − 7 -I, PrSi − 7 -II, and PrSi − 7 -III by 0.10, 0.05, and 0.08 eV, respectively. At the PBE0 level, the PrSi − 7 -II structure is more stable in energy than those of PrSi − 7 -I, PrSi − 7 -III, and PrSi − 7 -IV by 0.18, 0.09, and 0.04 eV in energy, respectively. Although the energies of PrSi − 7 -II, PrSi − 7 -III, and PrSi − 7 -IV isomers are almost degenerated, the PrSi − 7 -III structures are assigned to the ground state structure based on the following fact: Compared to experimental PES, the simulated PES of PrSi − 7 -III is more consistent than that of PrSi − 7 -II and PrSi − 7 -IV (see Sect. 3.3).
For PrSi8, three isomers are presented. The C 1-symmetry PrSi 8 -I geometry is predicted to be the ground state structure at the B3LYP, PBE0, and mPW2PLYP levels. This result differs from those of YbSi8, SmSi8, and EuSi8, of which ground state structure is the C 2v -symmetry bicapped pentagonal bipyramid [18–20]. The PrSi 8 -II of 4A″ electronic state and co-apex trigonal bipyramind PrSi 8 -III is less stable in energy than that of PrSi 8 -I by 0.22, 0.25, and 0.03 eV, and 0.22, 0.25, and 0.26 eV, respectively. For anion, three isomers are also presented. The C 2-symmetry co-apex trigonal bipyramind PrSi − 8 -III of 3A state is predicted to be the ground state structure. Energetically, it is more stable than the PrSi − 8 -I and PrSi − 8 -II by 0.34, 0.33, and 0.25 eV, and 0.17, 0.22, and 0.04 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. The ground state structure of PrSi − 8 differs from that of YbSi − 8 , SmSi − 8 , and EuSi − 8 , which is substitutional structure with C 2 v -symmetry [18–20].
For PrSi9, two geometries are presented. The C 2 v -symmetry bicapped antitetragonal prism of 4A1 state, PrSi 9 -I, is predicted to be the ground state structure at the mPW2PLYP level. It is more stable than the PrSi 9 -II isomer of 4A″ electronic state by 0.26 eV in energy. At the B3LYP and PBE0 levels, the PrSi 9 -II isomer, analogous to the ground state structure of GdSi9, YbSi9, SmSi9, and EuSi9 [17–20], is calculated to be the most stable structure. It is more stable than that of PrSi 9 -I by 0.37 and 0.25 eV, respectively. For anion PrSi − 9 , the C 2 v -symmetry bicapped antitetragonal prism of 3B 2 electronic state is predicted to be the ground state at the B3LYP, PBE0, and mPW2PLYP levels. It is, again, different from that of YbSi − 9 , SmSi − 9 , and EuSi − 9 , of which ground state structure is substitutional structure with C 3 v -symmetry [18–20].
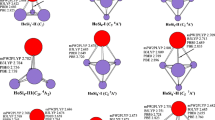
From discussion above, we can conclude that (1) the functional dependence on the evaluated the ground state structure is seen for PrSi −3 , PrSi5, PrSi 7 , PrSi − 7 , and PrSi 9 . The mPW2PLYP scheme can be trustworthy based on the following fact: (1) The CCSD(T) method was adopted for geometry optimization of PrSi n (n = 1–2) and its anion in order to check reliability of methods (The geometries are shown in Fig. 2 and the total energies are listed in Table 2). From Table 1, we can see that the ground state structures predicted by the mPW2PLYP scheme are the same as those evaluated by the CCSD(T) method, while the ground state structures of PrSi and PrSi2 predicted by the B3LYP and PBE0 methods differ from those of CCSD(T). (2) The electron affinities predicted by the mPW2PLYP are excellent in agreement with those of experimental data (see Sect. 3.2). (3) The simulated PES of the ground state structure predicted by the mPW2PLYP scheme is in accord with the experimental PES (see Sect. 3.3). It is to say that the methods including perturbative correlation part are very important as the species including f-electron (or d-electron) participating in bonding are treated [It is noted that the 4f electrons of Pr atom participate in bonding (see Sect. 3.6)]. (2) The extra electron effects on the ground state structure is intense. The ground state structures for PrSi3, PrSi6, and PrSi 8 differ from those of their anions. For PrSi4 and PrSi 7 , the ground state structures are undetermined because their potential energy surfaces are flat. The ground state structures of PrSi5 − and PrSi − 9 are unchanged compared to its neutrals. (3) Starting from n = 7, the ground state structures of PrSi n and their anions differ from those of YbSi n , SmSi n , and EuSi n .

Ground state structures for PrSi, PrSi2, and their anions
3.2 AEAs
The AEAs of PrSi n (n = 3–9) are calculated and listed in Table 3. From Table 3, we can see that the theoretical AEAs of PrSi4 deviated from experimental values (1.6 ± 0.1 eV) [1] by 0.40, 0.43, and 0.49 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. In fact, the PES of PrSi4 − recorded with 266 nm photons has a very small hump in low binding energy region before the first major peak, and Grubisic et al. assigned it as onset [1]. Based on the our calculated results, we reassigned the first major peak as being due of the transition from the ground state of the anion to the ground states of the neutral and obtained the experimental value of 2.0 ± 0.1 eV. The very small hump is probably because of the existence of low-lying isomers in the experiment. In this way, the average absolute deviations from experiment for PrSi n (n = 4–9) are by 0.15, 0.16, and 0.05 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. The largest deviations are 0.59, 0.67, and 0.10 eV, respectively. That is, the mPW2PLYP theoretical AEAs are in excellent agreement with the experimental data taken from Ref. [1].
In order to probe the effect of spin–orbit coupling (SOC), the effect of SOC is calculated via single-point calculations using the mPW2PLYP geometries and segmented all-electron relativistic Sapporo-DKH3-TZP basis sets with all-diffuse functions (Sapporo-DKH3-TZP-all) for Pr and Si atoms [33, 34], and using Hartree–Fock method via the Douglas–Kroll–Hess Hamiltonian (both with and without spin–orbit corrections). The AEAs with SOC corrections for PrSi n (n = 4–9) are listed in Table 4. From Table 4, we can see that the average absolute deviations from experiment are by 0.15, 0.16, and 0.06 eV at the B3LYP, PBE0, and mPW2PLYP levels, respectively. That is, the AEAs with SOC correction differ little from the results without SOC.
3.3 Simulated PES spectra
The anion PES spectra are simulated at the mPW2PLYP level on the basis of theoretically generalized Koopman theorem [35]. These simulated PES spectra and experimental ones taken from Ref. [1] are shown in Fig. 3. First step for the simulation is calculated the relative energies of the orbitals (ΔE n ) using the formula: ΔE n = ΔE HOMO-n − E HOMO. Secondly, the first peak associated with the HOMO is placed at the VDE (vertical detachment energy) plot, and the peaks of the deeper orbitals are moved to higher binding energy. Thirdly, these peaks are suited with a unit-area Gaussian function of 0.20 eV FWHM (full widths at half maximum). To quantitatively compare theoretical intensities with experimental ones are not possible in respect that the nonadiabatic interactions and anharmonic resonances are not included in calculations. The locations and the amounts of distinct peaks of simulated PES for PrSi − 4 -I, PrSi − 5 -I, PrSi − 7 -III and PrSi − 8 -III in the range of ≤4.5 eV general accord with experimental ones as can be seen from Fig. 3. And the positions of the first two peaks of simulated PES of PrSi − 6 and PrSi − 9 are in accord with experimental ones. The agreement of locations and the amounts of distinct peaks between simulated and experimental PES reveals that the ground state structures of PrSi − n (n = 4–9) reported in this paper are trustworthy.

Experimental photoelectron spectra (PES) (taken from Ref. [1], copyright 2009 American Society) and simulated PES at the mPW2PLYP level for the anions PrSi − n
3.4 DEs
The DEs of PrSi n and their anions (defined as the energy required in the reactions PrSi n → Pr + Si n for neutral and PrSi − n → Pr + Si − n for anion) are calculated at the mPW2PLYP level and drawn in Figs. 4 and 5, respectively. The DEs of SmSi n and EuSi n (n = 3–9) and their anions [19, 20] are also, respectively, shown in Figs. 4 and 5 in order to facilitate comparison. The higher values of the DEs show that the cluster bonding of a Pr atom is stable. As can be seen from Figs. 4 and 5, the DEs of EuSi n and SmSi n (n = 3–9) are smaller than that of PrSi n . Although Pr atom has no 5d electrons, the 4f electron transfers to 5d orbital when Pr atom interacts with silicon clusters and then participates in bonding. The profiles of d-orbital are facilely deformed and tend to ionic polarization. The ionic bonding weakens, and the covalent bond strengthens due to the result of ionic polarization, and therefore causes a relatively large DE of PrSi n . The same variation trends of DE curves exist on PrSi n , EuSi n , and SmSi n . The DEs of PrSi4 and PrSi 7 are local minima, and the DEs of PrSi5 and PrSi8 are local maxima. This result accords with that of ASi n (A = Li, Na) and is interpreted by the parallelism between the EA and the DE of Si n because the binding of an Pr to the Si n species results in electronic charge transfer from the Pr atom to Si n , similar to the conditions of binding of an alkali atom to the Si n species [36]. The DEs of EuSi − n and SmSi − n (n = 3–9) are smaller than the DEs of PrSi − n . When n = 4–7, the DEs of PrSi − n are different little from each other, but when n = 8 and 9, the DEs of PrSi − n are larger than those of the others. The DEs of PrSi − n are larger than those of corresponding neutral for n = 7−9, smaller for n = 3 and 5, and almost equal for n = 4 and 6. The explanation will be seen in Sect. 3.6.


3.5 HOMO–LUMO gaps
HOMO–LUMO gaps can be served as an important criterion to reflect the chemical reactivity of molecules in a sense, especially for RE-doped silicon clusters which have fine photochemical sensitivity. The HOMO–LUMO gaps for the most stable structures of PrSi n (n = 3−9) predicted by the mPW2PLYP method are tabulated in Fig. 6, along with the HOMO–LUMO gaps of EuSi n , SmSi n , and Si n [19, 20] for comparison. From Fig. 6, we can conclude that similar to EuSi n and SmSi n , doping Pr atom to silicon species raises the photochemical sensitivity due to the fact that the HOMO–LUMO gap of PrSi n (n = 3–9) is smaller than that of Si n with the same n. But the effect of raising photochemical sensitivity is not as good as the doping Eu or Sm to silicon species. The photochemical sensitivity of PrSi6 is better than that of its neighboring clusters.

3.6 Charge transfer and magnetic moment
NPA (natural population analysis) is conducted with the mPW2PLYP method in order to further understand the interaction between the Pr atom and the Si n species. The charges of Pr and NPA valence configurations are listed in Table 5. The magnetic moments of 6s, 4f, 5d, and 6p state for Pr, total magnetic moments of Pr, and total magnetic moments of the ground state of PrSi n (n = 3–9) and their anions are listed in Table 6. As can be seen from Table 5, the 4f shell of Pr in the cluster (except for PrSi3) is obviously changed. The charge transfer occurs largely not only from 6s to 5d but also 4f to 5d orbitals, resulting in hybridization between the 6s and 5d orbitals. That is, the 4f electrons migrated to 5d orbit and then participated in bonding. The theoretical charges of the Pr in PrSi n (n = 3–9) species (except for PrSi8) show that Pr atom acts as an electron donor and the characteristics of bonding between Pr and silicon clusters possess not only ionic bonds, but also covalent bonds in nature. Similar to anion EuSi − n and SmSi − n [19, 20], the majority of the additional electron’s charge in PrSi − n (n = 3–9) is found to be localized on the Si n species. And average charges of 0.47 a.u. go back to Pr atom from Si n compared to the neutral, which leads to decreasing of the ionic bond components and increasing of the covalent bond components. If the increased data are larger than the decreased data, the DEs of Pr from the PrSi − n will be larger than those of their neutral (for example, PrSi − 7 , PrSi8 −, and PrSi9 −). For PrSi3 − and PrSi5 −, the conditions are the opposite. And for PrSi4 − and PrSi6 −, the increased and decreased value differs little from each other. From Table 6, we can see that the total magnetic moments of PrSi3, PrSi5, and PrSi − n (n = 4–9) are contributed by Pr atom. And for the remaining species, in addition to a large proportion of magnetic moments that contributed by Pr atom, a small portion of magnetic moments are contributed by the silicon clusters.
4 Conclusions
We have investigated the equilibrium geometries and properties such as AEAs, simulated photoelectron spectra (PES), dissociation energies (DEs), relative stabilities, HOMO–LUMO gaps, charges transfer, and magnetic moments of PrSi n (n = 3–9) and their anions using the ABCluster global search technique combined with density functional methods. Prudently chosen DFT methods employed with aug-SEG/ECP basis set for Pr atoms are competent for the reliable prediction of the structures and properties of the PrSi n species. The mPW2PLYP results show that (1) starting from n = 7, the ground state structures of neutral PrSi n (n = 3–9) and their anions do not belong to “substitutional structure”. When binding an electron to the ground state structure of the neutral, the extra electron effect on the ground state structure is intense. The ground state structures for PrSi3 −, PrSi6 −, and PrSi8 − are different from their neutral ones. (2) The experimental PES of PrSi4 − has been reassigned based on the theoretical results. Assigning experimental value of 2.0 ± 0.1 eV to the AEA is more justifiable than to 1.6 ± 0.1 eV. The mPW2PLYP AEAs of PrSi n are in excellent agreement with the experimental data. The average absolute deviations from experiment are only 0.05 eV, and the maximum deviations are 0.10 eV. (3) The accordance between the experimental PES and the theoretical simulations indicates that the ground state structures of PrSi − n (n = 4–9) reported in this paper are trustworthy. (4) The DEs of Pr atom from PrSi n species and their anions are larger than those of Eu and Sm. (5) HOMO–LUMO gaps reveal that doping Pr atom to Si n (n = 3–9) species raises the photochemical sensitivity. But the effect of raising photochemical sensitivity is not as good as the effect of the doping Eu or Sm to silicon species. (6) Calculations of magnetic moments show that Pr atom contributes a large proportion of the total magnetic moments.
References
Grubisic A, Ko YJ, Wang HP, Bowen KH (2009) Photoelectron spectroscopy of lanthanide–silicon cluster anions LnSi − n (3 ≤ n ≤ 13; Ln = Ho, Gd, Pr, Sm, Eu, Yb): prospect for magnetic silicon-based clusters. J Am Chem Soc 131:10783–10790
Grubisic A, Wang HP, Ko YJ, Bowen KH (2008) Photoelectron spectroscopy of europium-silicon clusters anions, EuSi − n (3 ≤ n ≤ 17). J Chem Phys 129:054302-1–054302-5
Ohara M, Miyajima K, Pramann A, Nakajima A, Kaya K (2002) Geometric and electronic structures of terbium–silicon mixed clusters (TbSi n ; 6 ≤ n ≤ 16). J Phys Chem A 106:3702–3705
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) Anion photoelectron spectroscopy of transition metal- and lanthanide metal-silicon clusters: MSi − n (n = 6–20). J Chem Phys 129:214301-3–214301-7
Kenyon AJ (2005) Erbium in silicon. Semicond Sci Technol 20:R65–R84
Zhao RN, Han JG (2014) Geometrical stabilities and electronic properties of Sin (n = 12–20) clusters with rare earth holmium impurity: a density functional investigation. RSC Adv 4:64410–64418
Zhao GF, Sun JM, Gu YZ, Wang YX (2009) Density-functional study of structural, electronic, and magnetic properties of the EuSi n (n = 1–13) clusters. J Chem Phys 131:114312-1–114312-7
Xu W, Ji WX, Xiao Y, Wang SG (2015) Stable structures of LnSi6 − and LnSi6 (Ln = Pr, Eu, Gd, Tb, Yb), C2v or C5v? Explanation of photoelectron spectra. Comput Theor Chem 1070:1–8
Li CG, Pan LJ, Shao P, Ding LP, Feng HT, Luo DB, Liu B (2015) Structures, stabilities, and electronic properties of the neutral and anionic Si n Smλ (n = 1–9, λ = 0, −1) clusters: comparison with pure silicon clusters. Theor Chem Acc 134:34-1-11
Zhao RN, Ren ZY, Guo P, Bai JT, Zhang CH, Han JG (2006) Geometries and electronic properties of the neutral and charged rare earth Yb-doped Si n (n = 1–6) clusters: a relativistic density functional investigation. J Phys Chem A 110:4071–4079
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) A relativistic density functional study of Si n (n = 7–13) clusters with rare earth ytterbium impurity. Chem Phys 372:89–95
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) The medium-sized charged YbSi ± n (n = 7–13) clusters: a relativistic computational investigation. Chem Phys 378:82–87
Cao TT, Zhao LX, Feng XJ, Lei YM, Luo YH (2009) Structural and electronic properties of LuSi n (n = 1–12) clusters: a density functional theory investigation. J Mol Struct TheoChem 895:148–155
Liu TG, Zhang WQ, Li YL (2014) First-principles study on the structure, electronic and magnetic properties of HoSi n (n = 1–12, 20) clusters. Front Phys 9:210–218
Cao TT, Feng XJ, Zhao LX, Liang X, Lei YM, Luo YH (2008) Structure and magnetic properties of La-doped Sin (n = 1–12, 24) clusters: a density functional theory investigation. Eur Phys J D 49:343–351
Peng Q, Shen J (2008) Growth behavior of La@Si n (n = 1–21) metal-encapsulated clusters. J Chem Phys 128:084711-1-11
Liu TG, Zhao GF, Wang YX (2011) Structural, electronic and magnetic properties of GdSi n (n = 1–17) clusters: a density functional study. Phys Lett A 375:1120–1127
Xie XH, Hao DS, Yang JC (2015) Ytterbium doped silicon clusters YbSi n (n = 4–10) and their anions: structures, thermochemistry, and electron affinities. Chem Phys 461:11–19
Xie XH, Hao DS, Liu YM, Yang JC (2015) Samarium doped silicon clusters SmSi n (n = 3–10) and their anions: structures, thermochemistry, electron affinities, and magnetic moments. Comput Theor Chem 1074:1–8
Yang JC, Wang J, Hao YR (2015) Europium-doped silicon clusters EuSi n (n = 3–11) and their anions: structures, thermochemistry, electron affinities, and magnetic moments. Theor Chem Acc 134:81-1-11
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6170
Schwabe T, Grimme S (2006) Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effects. Phys Chem Chem Phys 8:4398–4401
Woon DE, Dunning TH Jr (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98:1358–1371
Cao X, Dolg M (2002) Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J Mol Struct Theochem 581:139–147
Buchachenko AA, Chalasiński G, Szcześniak MM (2007) Diffuse basis functions for small-core relativistic pseudopotential basis sets and static dipole polarizabilities of selected lanthanides La, Sm, Eu, Tm and Yb. Struct Chem 18:769–772
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V et al (2010) Gaussian 09 revision C.01. Gaussian Inc., Wallingford
Zhang J, Dolg M (2015) ABCluster: the artificial bee colony algorithm for cluster global optimization. Phys Chem Chem Phys 17:24173–24181
Dolg M, Stoll H, Savin A, Preuss H (1989) Energy-adjusted pseudopotentials for the rare earth elements. Theor Chim Acta 75:173–194
Dolg M, Stoll H, Preuss H (1993) A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor Chim Acta 85:441–450
Yang JC, Xu WG, Xiao WS (2005) The small silicon clusters Si n (n = 2–10) and their anions: structures, themochemistry, and electron affinities. J Mol Struct Theochem 719:89–102
Sekiya M, Noro T, Koga T, Shimazaki T (2012) Relativistic segmented contraction basis sets with core-valence correlation effects for atoms 57La through 71Lu: Sapporo-DK-nZP sets (n = D, T, Q). Theor Chem Acc 131:124-7-18
Noro T, Sekiya M, Koga T (2012) Segmented contracted basis sets for atoms H through Xe: Sapporo-(DK)-nZP sets (n = D, T, Q). Theor Chem Acc 131:112-4-18
Tozer DJ, Handy NC (1998) Improving virtual Kohn-Sham orbitals and eigenvalues: application to excitation energies and static polarizabilities. J Chem Phys 109:10180–10189
Sporea C, Rabilloud F, Cosson X, Allouche AR, Frécon M (2006) Theoretical study of mixed silicon-lithium clusters SinLip(+) (n = 1–6, p = 1–2). J Phys Chem A 110:6032–6038
Acknowledgements
We thank Prof. Kit H. Bowen for providing the clear experimental PES of PrSi n (n = 4–9). This work was supported by the National Natural Science Foundation of China (Grant No. 21263010), by Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (Gran No. NMGIRT-A1603), and by the Inner Mongolia Natural Science Foundation (Grant No. 2015MS0216).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Feng, Y., Yang, J. & Liu, Y. Study on the structures and properties of praseodymium-doped silicon clusters PrSi n (n = 3–9) and their anions with density functional schemes. Theor Chem Acc 135, 258 (2016). https://doi.org/10.1007/s00214-016-2017-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-2017-3