
Abstract
The geometries, electronic structures and properties including simulated photoelectron spectra (PES), adiabatic electron affinities (AEAs), and relative stability of LuSi n (n = 3–10) and their anions were investigated adopting the ABCluster global search technique combined with density functional methods. The results revealed that the most stable structures of neutral belong to “substitutional structure”, but not for their anions. The additional electron effects on the most stable structure are intense. The TPSSh AEAs of LuSi n (n = 6–9) agree excellently with the experimental data. The mean absolute error and the largest error are only 0.03 eV and 0.05 eV, respectively. The agreement between the experimental and theoretical PES indicates that the most stable structures of LuSi − n (n = 6–10) are trustworthy. The DEs and charge transfer are calculated to explain the relative stabilities. HOMO–LUMO gaps reveal that introducing Lu atom to Si n (n = 3–10) raises the photochemical sensitivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Silicon-based clusters, especially rare earth metal (REM) atom doped silicon clusters, have attracted wide interest in the past decade in respect that they can be used as building blocks of cluster-assembled nanotubes with novel photonic, magnetic, and electronic properties controlled by altering composition, size, and structure [1,2,3,4,5,6,7,8,9,10]. For example, silicon is a poor photonic material because of its very short non-radiative lifetime and indirect band gap, but erbium doped silicon microcrystal has been used as silicon-based optical source [11].
The photoelectron spectroscopy (PES) of REMSi − n (REM = Lu, Yb, Sm, Eu, Ho, Pr, Gd, and Tb 3 ≤ n ≤17) was recorded to probe their electronic structures and electron affinities [7,8,9,10]. In light of their appearance, the PES was divided into two types [7]. Urged by the experimental observations, some theoretical simulations for introducing REM atoms into silicon clusters have been achieved. For example, the growth behavior of the most stable structures of LuSin (n = 1–12), HoSi n (n = 1–12), and GdSi n (n = 1–17) had been studied at the level of density functional theory (DFT) and found that their ground state structures can be regarded as a substitution of REM (REM = Lu, Ho, and Gd) atom for a Si atom in the ground state structures of Sin+1 species [12,13,14]. The equilibrium geometries and properties including magnetic moments, relative stabilities, HOMO–LUMO gaps, charge transfers, and adiabatic electron affinities (AEAs) of neutral SmSi n and YbSi n (n ≤ 13) and their anions was evaluated at the levels of DFT [1, 15,16,17,18]. The most stable structure of Sm@Si20, Tm@Si20, Gd@Si20 −, and Eu@Si20 was evaluated to be fullerene-like silicon geometry and retain significant magnetic moments [19, 20]. Recently, we have predicted the ground state structures and AEAs of neutral YbSi n , SmSi n , and EuSi n (3 < n <11) and their anions at the levels of DFT with B3LYP, wB97X, PBE0, PBE, and B2PLYP functional and found that the calculated AEAs by these methods agree with the experimental values [21,22,23]. We prudently chose four DFT scheme in this work to predict the most stable structure and properties including simulated PES, AEAs, and relative stability of LuSin (n = 3–10) and their anions with the target of comprehending how their properties are diverse from that of not only bare Si clusters but also the others REMSi n species, and with the aim of verifying the reliability of theoretical evaluated results via comparing the simulated PES and theoretical AEAs with experimental ones and to help the reapportionment of experimental PES with a featureless very rounded and long tail.
Theoretical Methods
The computations are performed at the level of the DFT with the PBE [24], TPSSh [25, 26], B3LYP [27, 28], and wB97XD [29] functional. The basis sets used for geometry optimizations are the cc-pVTZ [30] for Si atoms and relativistic small-core potentials (ECP28MWB) [31] combined with segmented (SEG) Gaussian valence basis sets for Lu atoms (named as SEG/ECP). The harmonic frequency calculations are done at the four different levels of theory to guarantee that the optimized geometries are local minimal points. The optimized geometries in each method are used for all single-point calculations. Then, the SEG basis sets of Lu atoms are augmented by diffuse functions 2pdfg with exponents 0.028 and 0.015 (p), 0.032 (d), and 0.05 (f, g) [32] (named as aug-SEG/ECP) because the diffuse functions are important for the anions. Finally, the aug-SEG/ECP of Lu and aug-cc-pVTZ basis sets of Si [30] are used in the single-point energies calculations. The optimizations with cc-pVTZ and SEG/ECP basis sets are reasonable because the structural parameters optimized with them are equal to those optimized with aug-cc-pVTZ and aug-SEG/ECP basis sets [22]. The GAUSSIAN 09 codes [33] are used to carry out all of the calculations.
To choose the four methods is for the following reasons: the popular PBE method is the representative of pure density functionals; both TPSSh and B3LYP are the typical representative of the many hybrid density functionals because the exchange–correlation functionals of them are completely different; the wB97XD method as a hybrid density functional used here is to consider the effect of long-range corrections and dispersion corrections. The results show that the TPSSh method is reliable.
The ABCluster global search method [34] combined with the GAUSSIAN 09 codes is adopted to obtain the initial geometries. The first step is done using the PBE with relativistic large-core effective core potentials (ECP60MWB) basis set [35] for Lu atoms and 6-31G basis set for Si atoms. For n ≤ 7, more than 100 initial isomers of each LuSi n are generated, and for n ≥ 8 more than 300 isomers are generated. Then, the top-eight lowest energy geometries from the first step, and those with their energy differences within 0.8 eV from the lowest energy geometry are selected and optimized again by using the PBE with the SEG/ECP basis set for Lu and the cc-pVTZ basis sets for Si atoms. Finally, the geometries from the second step with their energy differences within 0.8 eV from the lowest energy geometry are optimized by using the remaining three schemes. Apart from considering the isomers of ABCluster scheme, the “substitutional structure” (which can be regarded as substitution of Lu atom for a Si atom in the most stable structure of Sin+1 cluster) is also considered. The reason is that the most stable structure of SmSi n , EuSi n , and YbSi n is substitutional structure [21,22,23], and the global search method is impossible to make an “ergodic” sampling on the potential energy surface of large clusters by computer simulation (especially for heteroatom clusters). Our experience is that the 100 configurations generated from the ABCluster method include all of the “substitutional” isomers when n ≤ 7, from n = 8, even though the 500 configurations generated from ABCluster method do not include all of the “substitutional” structures (For example, the ground state structures of LuSi8 and LuSi9 − belong to “substitutional”, but cannot be found by ABCluster method). Furthermore, the spin multiplicities of doublet and quartet are considered for neutral LuSi n (n = 1–3) in respect that the ground states of Si, Si2, and Si3 are triplet. While singlet and triplet are considered for anions LuSi − n with n = 1–3. The result reveals that the doublet state is evaluated to be the ground state for the neutral excluded LuSi which is quartet state, and that the singlet state is predicted to be the ground state for the anions excluded LuSi− and LuSi2 − which are triplet state. Although many isomers of LuSi n (n = 3–10) and their anions are obtained, only the most stable structures are presented (some of low-lying isomers are shown in Supporting Information).
Results and Discussion
Neutral and Anionic Geometries
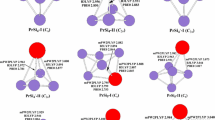
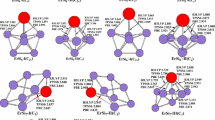
The geometries optimized at the PBE, TPSSh, B3LYP and wB97XD levels are shown in Fig. 1 for LuSi n (n = 3–10) clusters and their anions. For LuSi, the ground state structure is predicted to be \( ^{4} \sum \) electronic state, which is more stable in energy than that of doublet by 0.30, 0.31, 0.40, and 0.57 eV at the PBE, TPSSh, B3LYP, and wB97XD levels of theory, respectively. For LuSi−, the \( ^{3} \prod \) state is more stable in energy than that of \( ^{1} \sum \) by 0.24, 0.29, 0.34, and 0.46 eV, at the PBE, TPSSh, B3LYP, and wB97XD levels of theory, respectively. The most stable structure of LuSi2 is calculated to be C 2v symmetry with 2B2 electronic ground state. At the B3LYP and wB97XD levels, the 1A1 electronic state is more stable in energy than the 3B2 by 0.09 and 0.21 eV for anion LuSi2 −, respectively. While at the PBE and TPSSh levels, the \( ^{1} {\text{A}}_{1} \) electronic state is less stable in energy that the \( ^{3} {\text{B}}_{2} \) by 0.06 and 0.09 eV, respectively. In this case, we adopted the singlet-point energy calculations at the CCSD(T) level in combination with cc-pVTZ-DK basis sets for Si atoms [30] and DKH2 basis sets [36] for Lu atoms, and employed the spin-free, one-electron Douglas-Kroll-Hess Hamiltonian [37,38,39,40]. The results show that the C 2v -symmetry LuSi2 − of triplet is more stable in energy than that of singlet by 0.20, 0.21, 0.27, and 0.29 eV with the PBE, TPSSh, B3LYP, and wB97XD geometry, respectively.


The geometries of neutral LuSi n (n = 3–10) and their anions in which the Lu atom is in black color. The Lu–Si bond lengths are shown in Å
The most stable structure of LuSi3 and its anion is predicted to be C 2v symmetry with \( ^{2} {\text{A}}_{1} \) and \( ^{1} {\text{A}}_{1} \) ground state, respectively. For LuSi4 and its anion, The most stable structures which can be viewed as a substitution of Lu for a Si atom in the ground state trigonal bipyramind of Si5 [41] are calculated to be C s -symmetry with \( ^{2} {\text{A}}^{\prime } \) and C 3v -symetry with \( ^{1} {\text{A}}_{1} \) ground state, respectively. For LuSi5, two geometries which compete with each other for the most stable structure are presented. Both are C s symmetry with \( ^{2} {\text{A}}^{\prime } \) electronic state. At the wB97XD level, the LuSi5 −I is more stable in energy than that of LuSi5-II by 0.10 eV, but less stable by 0.01, 0.11, and 0.15 eV at the B3LYP, PBE, and TPSSh levels, respectively. Similarly to the case of LuSi2 −, the CCSD(T) is adopted. Using the CCSD(T) method with the PBE, TPSSh, B3LYP, and wB97XD geometry, the LuSi5-I isomer is only more stable than the LuSi5-II by 0.05, 0.05, 0.05, and 0.01 eV, respectively. For anion LuSi5 −, two isomers of \( ^{1} {\text{A}}^{\prime } \) electronic state are also presented. At the B3LYP and wB97XD levels, the LuSi5 −-I isomer is more stable than the LuSi5 −-II by 0.05 and 0.35 eV, but less stable by 0.14 and 0.16 eV at the PBE and TPSSh levels, respectively. At the CCSD(T) level, the LuSi5 −-I isomer is more stable in energy than the LuSi5 −-II by 0.17, 0.19, 0.16, and 0.13 eV with the PBE, TPSSh, B3LYP, wB97XD geometry, respectively. The most stable structures of LuSi6 and its anion can be reviewed as a substitution of Lu atom for a Si atom in the ground state pentagonal bipyramind of Si7 [41], but the substitutional position is different. They are C 2v symmetry with \( ^{2} {\text{A}}_{1} \) ground state and C 5v symmetry with \( ^{1} {\text{A}}_{1} \) ground state, respectively.
Cao et al. [12] reported that two isomers compete with each other for the most stable structure of LuSi7. Our results are same as their conclusions. The structure LuSi7-I is more stable in energy than that of LuSi7-II by 0.02, 0.00, and 0.02 eV at the PBE, TPSSh, and CCSD(T) (with the PBE geometry) levels of theory, respectively. At the B3LYP and wB97XD levels, the isomer LuSi7-II is more stable in energy than that of LuSi7-I by 0.05 eV. For anion LuSi7 −, at the B3LYP, wB97XD, and CCSD(T) (with the PBE geometry) levels of theory, The structure LuSi7 −-I is more stable in energy than that of LuSi7 −-II by 0.14, 0.19, and 0.02 eV, respectively. At the PBE and TPSSh levels of theory, both are nearly degenerate (the energy difference between isomer LuSi7 −-I and LuSi7 −-II is only 0.04 and 0.00 eV, respectively). Based on the agreement between the experimental and simulated PES, the LuSi7 −-I is assigned to the most stable structure (see “Relative stability” section). The most stable structure of LuSi8, analogous to YbSi8, SmSi8, and EuSi8 [21,22,23], is C 2v -symmetry bicapped pentagonal bipyramid with \( ^{2} {\text{A}}_{1} \) ground state. For anion LuSi8 −, the C2-symmetry co-apex bi-trigonalbipyramind of \( ^{1} {\text{A}} \) ground state is predicted to be the most stable structure, which differs from those of YbSi8 −, SmSi8 −, and EuSi8 − [21,22,23]. The most stable structure of LuSi9 and its anions, similar to YbSi9, SmSi9, and EuSi9 and their anions [21,22,23], is C s -symmetry substitutional geometry with \( ^{2} {\text{A}}^{\prime } \) ground state and C 3v -symmetry tetra-capped trigonal prism, respectively. For LuSi10, two geometries compete with each other for the most stable. Cao et al. [12] reported that the isomer LuSi10-II is the ground state structrue. At the PBE and TPSSh levels, it is slightly more stable in energy than that of LuSi10-I structure by 0.02 and 0.06 eV, but less stable by 0.07 and 0.11 eV at the wB97XD and B3LYP levels, respectively. For anion LuSi10 −, the most stable structure is predicted to be the co-apex di-face-capped-trigonalbipyramind with \( ^{1} {\text{A}}^{\prime } \) ground state, which differs from those of YbSi8 −, SmSi8 −, and EuSi8 − [21,22,23]. The change of geometry is obvious compared with its neutral.
From the discussion above, we can see that, (1) the substitutional structures are calculated to be the ground state structure for neutral LuSi n (n ≤ 10), which reappear the previous conclusion presented by Cao et al. [12] The potential energy surfaces of LuSi5, LuSi7 and LuSi10 are very flat. As a result, isomeric arrangement is possible and functional dependence of the predicted lowest-energy isomer occurs. (2) The charge effects on the most stable structures are intense. Starting from n = 6, the most stable structures of LuSi − n (n = 3–10) differ from those of their neutrals. And the most stable structures of LuSi8 − and LuSi10 − do not belong to substitutional structure. The reason is that the Lu atom includes an unpaired d-electron which is easily polarized. Polarization results in the d-orbital deformation. The change of charge affects the degree of polarization and deformation, especially for 5d-orbitals. So the charge effect on the geometries is very strong.
Simulated PES and AEAs
The PES is generally sensitive to change of geometry. The PES is simulated at the TPSSh level of theory based on the Koopman theorem [42]. In the PES simulation, the orbital relative energies (ΔE n = EHOMO-n − EHOMO) are calculated. The first peaks regarding the HOMO are located at the VDE plot, and the others are moved to higher binding energy. The peaks are suited with a unit-area Gaussian function of 0.40 eV FWHM (full widths at half maximum). These simulated and experimental ones [9] are shown in Fig. 2. From Fig. 2 we can see that the locations and the amounts of different peaks of simulated PES for LuSi6 −, LuSi8 − and LuSi10 − in the range of ≤6.0 eV accord with those of experimental PES. The positions of the first two peaks of LuSi7 −-I accord with experimental ones. The experimental PES of LuSi9 − shows a featureless long and very rounded tail. As a result, the positions of the first two peaks of its experimental PES are unsharp and seem to be inconsistent with those of simulated PES. In fact, if the peaks are suited with 0.50 eV FWHM, the simulated PES of LuSi9 − accord with experimental one (see Fig. 3). The agreement of positions and the amounts of different peaks between simulated and experimental PES reveals that the most stable structures of LuSi − n (n = 6–10) are trustworthy.

Simulated photoelectron spectra for the LuSi − n (n = 3–10) clusters with the TPSSh method. Experimental spectra of LuSi − n (n = 6–10) are taken from Ref. [9]

Simulated photoelectron spectra for the LuSi9 − at the TPSSh level of theory with a unit-area Gaussian function of 0.50 eV full widths at half maximum
The theoretical and experimental AEAs of LuSi n (n ≤ 10) are listed in Table 1. The AEAs MAE (mean absolute error) for LuSi n (n = 6–10) is 0.26, 0.17, 0.20, and 0.21 eV at the PBE, TPSSh, B3LYP, and wB97XD levels of theory, respectively. The largest error is that of LuSi10, which is off by 0.51–0.88 eV. If LuSi10 is removed, the mean absolute error is 0.11, 0.03, 0.12, and 0.13 eV, and the largest error is 0.19, 0.05, 0.16 and 0.22 eV at the PBE, TPSSh, B3LYP, and wB97XD levels of theory, respectively. That is, the TPSSh AEA is in excellent concord with the experimental values. In this case, we dare to evaluate the AEA of LuSi10 is 2.99 ± 0.03 eV, and no experimental data are reliable. Our theoretical calculations will provide valuable reference for further experimental researches of LuSi10.
Relative Stability
Comparisons of DEs of various clusters examine relative stability of clusters. The larger the DEs, the more stable the cluster. The DEs of LuSi n and their anions (DEs = E(Lu) + E(Si n ) − E(LuSi n ) for the neutral and DEs = E(Lu) + E(Si − n ) − E(LuSi − n ) for the anion) are calculated at the TPSSh level of theory, and along with DEs of YbSi n , EuSi n and SmSi n (n = 3–10) and their anions [21,22,23] drawn in Figs. 4 and 5, respectively, to facilitate comparison. From Figs. 4 and 5, we can see that the DEs of LuSi n (n = 3–10) are larger than those of YbSi n , EuSi n and SmSi n . The reason is that Lu atom has 5d electrons, profiles of which are facilely deformed and polarized, resulting in increasing of components of covalent bond and causing a larger DEs of LuSi n . The same variation trends of DE curves exist on LuSi n , YbSi n , EuSi n , and SmSi n . When n = 4 and 7, the DEs are local minima, but local maxima when n = 5 and 8. The DEs of LuSi − n (n = 3–10) are also larger than those of YbSi − n , EuSi − n and SmSi − n . While the variation trends of LuSi − n are different from those of YbSi − n , EuSi − n and SmSi − n . When n = 4, 6 and 9, the DEs of LuSi − n are local maxima. For YbSi − n , EuSi − n and SmSi − n , they are not local maxima when n = 4 and 6. The reason is that the most stable structures of LuSi4 − and LuSi6 − are η3-(Si4)Lu− and η5-(Si6)Lu− geometry, but η2-(Si4)REM− and η4-(Si6)REM− for REMSi4 − and REMSi6 −(REM = Yb, Eu, and Sm) [21,22,23], respectively. When n = 5 and 7, the DEs of LuSi − n are local minima. The DEs of LuSi − n are larger than those of corresponding neutral for n = 7–10, smaller for n = 3 and 5, and almost equal for n = 4 and 6. To explain this phenomenon the NPA (natural population analysis) charges and valence configurations of Lu are calculated at the TPSSh level of theory and listed in Table 2. The Lu atom in LuSi n and their anions acts as an electron donor and the feature of bonding between Lu and silicon clusters possesses not only ionic bonds, but also covalent bonds in nature. When the LuSi n obtained an extra electron, the majority of the extra electron’s charge of LuSi − n is located in the Si clusters. And 0.32 a.u. mean charges of LuSi − n (n = 3–10) excluded LuSi9 − go back to Lu atom from Si cluster, which results in the ionic bond component decrease and the covalent bond component increase. If the increased covalent bond is larger than the decreased ionic bond, then the DEs of Lu from the LuSi − n are larger than those of their neutral (for instance, LuSi7 −, LuSi8 −, and LuSi10 −). The conditions are the opposite for LuSi3 − and LuSi5 −. And for LuSi4 − and LuSi6 −, the reduced and increased value differ litter from each other. The charge of Lu in LuSi9 − is larger than that in LuSi9. As a result, the DEs of Lu from LuSi9 − is larger than that of Lu from LuSi9.


HOMO–LUMO gaps are not only an important physical property, but also an important index in a sense to reflect the chemical reactivity of compounds, especially for photochemical sensitivity. The HOMO–LUMO gaps for the most stable structures of LuSi n (n = 3–10) calculated by the TPSSh method is shown in Fig. 6, and along with the HOMO–LUMO gaps of YbSi n [21], EuSi n , SmSi n and Si n [22] for comparison. From Fig. 6, we can conclude that, similar to YbSi n , EuSi n and SmSi n , introducing Lu atom to Si n species raises the photochemical sensitivity due to the fact that the HOMO–LUMO gap of LuSi n (n = 3–10) is smaller than that of Si n with the same n. And the effect of raising photochemical sensitivity for LuSi7 is the most obvious. For REMSi n (REM = Yb, Eu, and Sm), the REMSi6 is the most obvious.

Conclusions
The equilibrium geometries, electronic structures and properties including simulated PES, AEAs, and relative stabilities of LuSi n (n = 3–10) and their anions have been inspected adopting the ABCluster global search technique combined with density functional methods. Prudently chosen DFT schemes adopted with aug-SEG/ECP basis set for Lu atoms are competent of reliably prediction the structures and properties for the LuSi n species. The results revealed that the most stable structures of neutral LuSi n (n = 3–10) belong to “substitutional structure”, but not for their anions. When adding an electron to the most stable structure of the neutral, the charge effects on the most stable structure is intense. Starting from n = 6, the most stable structures of LuSi − n (n = 3–10) differ from those of their neutrals. The TPSSh AEAs of LuSi n (n = 6–9) are in excellent agreement with the available experimental values. The mean absolute error and the largest error is only 0.03 and 0.05 eV, respectively. For LuSi10, the theoretical AEA of 2.99 ± 0.03 eV may be able to challenge the experimental value of 3.70 ± 0.0043 eV reported previously. The agreement between the experimental and theoretical PES indicates that the most stable structures of LuSi − n (n = 6–10) presented in this paper are trustworthy. The DEs of Lu atom from LuSi n (n = 3–10) and their anions are larger than those of Yb, Eu and Sm. The variation trends of LuSi n are the same as those of YbSi n , EuSi n and SmSi n , but the variation trends of LuSi − n are different from those of YbSi − n , EuSi − n and SmSi − n . The analyses of HOMO–LUMO gaps reveal that introducing Lu atom to Si n (n = 3–10) species raises the photochemical sensitivity, especially for LuSi7. The TPSSh charge transfer is calculated to explain the relative stabilities.
References
C. G. Li, L. J. Pan, P. Shao, L. P. Ding, H. T. Feng, D. B. Luo, and B. Liu (2015). Theor. Chem. Acc. 134, 34-1–34-11.
R. N. Zhao and J. G. Han (2014). RSC Adv. 4, 64410–64418.
G. F. Zhao, J. M. Sun, Y. Z. Gu, and Y. X. Wang (2009). J. Chem. Phys. 131, 114312-1–114312-7.
Q. Peng and J. Shen (2008). J. Chem. Phys. 128, 084711-1–084711-11.
L. Y. Hou, J. C. Yang, and Y. M. Liu (2016). J. Mol. Model. 22, 193-1–193-10.
M. Ohara, K. Miyajima, A. Pramann, A. Nakajima, and K. Kaya (2002). J. Phys. Chem. A 106, 3702–3705.
A. Grubisic, Y. J. Ko, H. P. Wang, and K. H. Bowen (2009). J. Am. Chem. Soc. 131, 10783–10790.
A. Grubisic, H. P. Wang, Y. J. Ko, and K. H. Bowen (2008). J. Chem. Phys. 129, 054302-1–054302-5.
K. Koyasu, J. Atobe, S. Furuse, and A. Nakajima (2008). J. Chem. Phys. 129, 214301-1–214301-7.
K. Koyasu, J. Atobe, M. Akutsu, M. Mitsui, and A. Nakajima (2007). J. Phys. Chem. A 111, 42–49.
A. J. Kenyon (2005). Semicond. Sci. Technol. 20, R65–R84.
T. T. Cao, L. X. Zhao, X. J. Feng, Y. M. Lei, and Y. H. Luo (2009). J. Mol. Struct. THEOCHEM 895, 148–155.
T. G. Liu, W. Q. Zhang, and Y. L. Li (2014). Front. Phys. 9, 210–218.
T. G. Liu, G. F. Zhao, and Y. X. Wang (2011). Phys. Lett. A 375, 1120–1127.
R. N. Zhao, Z. Y. Ren, P. Guo, J. T. Bai, C. H. Zhang, and J. G. Han (2006). J. Phys. Chem. A 110, 4071–4079.
R. N. Zhao, J. G. Han, J. T. Bai, F. Y. Liu, and L. S. Sheng (2010). Chem. Phys. 372, 89–95.
R. N. Zhao, J. G. Han, J. T. Bai, and L. S. Sheng (2010). Chem. Phys. Lett. 378, 82–87.
W. Xu, W. X. Ji, Y. Xiao, and S. G. Wang (2015). Comput. Theor. Chem. 1070, 1–8.
V. Kumar, A. K. Singh, and Y. Kawazoe (2006). Phys. Rev. B 74, 125411-1–125411-5.
J. Wang, Y. Liu, and Y. C. Li (2010). Phys. Chem. Chem. Phys. 12, 11428–11431.
X. H. Xie, D. S. Hao, and J. C. Yang (2015). Phys. Chem. 461, 11–19.
X. H. Xie, D. S. Hao, Y. M. Liu, and J. C. Yang (2015). Comput. Theor. Chem. 1074, 1–8.
J. C. Yang, J. Wang, and Y. R. Hao (2015). Theor. Chem. Acc. 134, 81.
J. P. Perdew, K. Burke, and M. Ernzerhof (1996). Phys. Rev. Lett. 77, 3865–3868.
J. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria (2003). Phys. Rev. Lett. 91, 146401-1–146401-4.
V. N. Staroverov, G. E. Scuseria, J. Tao, and J. P. Perdew (2003). J. Chem. Phys. 119, 12129–12137.
A. D. Becke (1993). J. Chem. Phys. 98, 5648–5652.
C. Lee, W. Yang, and R. G. Parr (1988). Phys. Rev. B 37, 785–789.
J. D. Chai and M. H. Gordon (2008). Phys. Chem. Chem. Phys. 10, 6615–6620.
D. E. Woon and T. H. Dunning Jr. (1993). J. Chem. Phys. 98, 1358–1371.
X. Y. Cao and M. Dolg (2002). J. Mol. Struct. THEOCHEM. 581, 139–147.
A. A. Buchachenko, G. Chalasin´ski, and M. M. Szezes´niak (2007). Struct. Chem. 18, 769–772.
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, (2010).
J. Zhang and M. Dolg (2015). Phys. Chem. Chem. Phys. 17, 24173–24181.
M. Dolg, H. Stoll, A. Savin, and H. Preuss (1989). Theor. Chim. Acta. 75, 173–194.
M. Dolg (2011). J. Chem. Theory Comput. 7, 3131–3142.
M. Douglas and N. M. Kroll (1974). Ann. Phys. (NY) 82, 89–155.
B. A. Hess (1985). Phys. Rev. A 32, 756–763.
B. A. Hess (1986). Phys. Rev. A 33, 3742–3748.
G. Jansen and B. A. Phys (1989). Rev. A 39, 6016–6017.
J. C. Yang, W. G. Xu, and W. S. Xiao (2005). J. Mol. Struct. THEOCHEM 719, 89–102.
D. J. Tozer and N. C. Handy (1998). J. Chem. Phys. 109, 10180–10189.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 21263010), by Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (Gran No. NMGIRT-A1603), by the Inner Mongolia Natural Science Foundation (Grant No. 2015MS0216), and by the Science and Research Foundation of Higher Education of Inner Mongolia (Grant No. NJZY16419).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, S., Yang, J. Study on Structure and Property of Lutetium Introduced Silicon Clusters LuSi n (n = 3–10) and Their Anions with Density Functional Theory. J Clust Sci 28, 2309–2322 (2017). https://doi.org/10.1007/s10876-017-1225-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-017-1225-x