
Abstract
This review explores the pivotal role of the nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome in the pathogenesis of diabetes and its complications, highlighting the therapeutic potential of various oral hypoglycemic drugs targeting this pathway. NLRP3 inflammasome activation, triggered by metabolic stressors like hyperglycemia, hyperlipidemia, and free fatty acids (FFAs), leads to the release of pro-inflammatory cytokines interleukin-1β and interleukin-18, driving insulin resistance, pancreatic β-cell dysfunction, and systemic inflammation. These processes contribute to diabetic complications such as nephropathy, neuropathy, retinopathy, and cardiovascular diseases (CVD). Here we discuss the various transcriptional, epigenetic, and gut microbiome mediated regulation of NLRP3 activation in diabetes. Different classes of oral hypoglycemic drugs modulate NLRP3 inflammasome activity through various mechanisms: sulfonylureas inhibit NLRP3 activation and reduce inflammatory cytokine levels; sodium–glucose co-transporter 2 inhibitors (SGLT2i) suppress inflammasome activity by reducing oxidative stress and modulating intracellular signaling pathways; dipeptidyl peptidase-4 inhibitors mitigate inflammasome activation, protecting against renal and vascular complications; glucagon-like peptide-1 receptor agonists attenuate NLRP3 activity, reducing inflammation and improving metabolic outcomes; alpha-glucosidase inhibitors and thiazolidinediones exhibit anti-inflammatory properties by directly inhibiting NLRP3 activation. Agents that specifically target NLRP3 and inhibit their activation have been identified recently such as MCC950, Anakinra, CY-09, and many more. Targeting the NLRP3 inflammasome, thus, presents a promising strategy for managing diabetes and its complications, with oral hypoglycemic drugs offering dual benefits of glycemic control and inflammation reduction. Further research into the specific mechanisms and long-term effects of these drugs on NLRP3 inflammasome activity is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a common metabolic disease marked by persistent high blood sugar levels, resulting from deficiencies in insulin production, insulin effectiveness, or a combination of both. This condition significantly contributes to morbidity and mortality worldwide, primarily due to its association with various complications such as cardiovascular disease, neuropathy, nephropathy, and retinopathy (Galicia-Garcia, et al. 2020).
The underlying mechanisms of diabetes involve intricate interactions between metabolic and inflammatory pathways. Among these, the NLRP3 (nucleotide-binding oligomerization domain (NOD)-like receptor protein 3) inflammasome has emerged as a critical player in the inflammatory processes linked to diabetes. The NLRP3 inflammasome, a multiprotein complex within the innate immune system, detects cellular stress and damage-associated molecular patterns. Upon activation, it facilitates the cleavage of pro-caspase-1 into active caspase-1, leading to the maturation and secretion of pro-inflammatory cytokines IL-1β and IL-18. These cytokines are pivotal in promoting inflammatory responses that exacerbate insulin resistance and β-cell dysfunction, key features of both type 1 and type 2 diabetes (T2DM). Thus, targeting the NLRP3 inflammasome represents a promising therapeutic strategy for mitigating inflammation-driven diabetic complications (Gora et al. 2021).
Current treatment strategies for diabetes focus on lifestyle modifications, oral medications, and insulin therapy. Lifestyle changes include diet, exercise, and weight management. Insulin therapy, crucial for managing type 1 and advanced type 2 diabetes, includes different formulations to meet both basal and mealtime requirements. Continuous glucose monitoring and the use of insulin pumps enhance blood sugar management. Oral hypoglycemic agents, integral to current treatment strategies for diabetes management, encompass several drug classes such as sulfonylureas, biguanides, alpha-glucosidase inhibitors, thiazolidinediones, dipeptidyl peptidase-4 inhibitors (DPP-4i), glucagon-like peptide-1 receptor agonists (GLP-1RA), and sodium–glucose co-transporter-2 inhibitors (SGLT2i). These drugs primarily aim to regulate blood glucose levels by several mechanisms, such as inducing insulin secretion, improving insulin sensitivity, and reducing glucose absorption or reabsorption. Recent studies have indicated that these drugs may also exert anti-inflammatory effects, potentially influencing the NLRP3 inflammasome pathway. Metformin, a widely used biguanide, inhibits NLRP3 inflammasome activation through AMP-activated protein kinase (AMPK)-dependent and independent pathways, thereby reducing inflammatory cytokine production and improving insulin sensitivity (Xian et al. 2021). GLP-1RA, DPP-4i, and SGLT2i are the major players that exhibit anti-inflammatory actions via activation of NLRP3 inflammasome through AMPK/mTOR pathway modulation against reactive oxygen species (ROS)-mediated interleukin-1β secretion (IL-1β) (Ma et al. 2021; Arab et al. 2021; Chen et al. 2020a). Thiazolidinediones, known as PPARγ agonists and alpha-glucosidase inhibitors are also under investigation for their ability to suppress NLRP3 inflammasome activity but requires much further investigation to strengthen the evidence for their action (Chen et al. 2020b; Li et al. 2019).
This review attempts to present a comprehensive analysis of how various classes of oral hypoglycemic drugs interact with the NLRP3 inflammasome pathway. By synthesizing current research findings, this review will highlight the potential therapeutic implications of these interactions, offering new insights into the holistic management of diabetes. Understanding these mechanisms could lead to more effective strategies for preventing and treating inflammation-related complications in diabetic patients.
Mechanisms of NLRP3 inflammasome activation in diabetes
Pathophysiology of NLRP3
The inflammasome is a multiprotein complex crucial to the innate immune system, responsible for detecting pathogenic microorganisms and cellular stress, thereby activating inflammatory responses. The NLRP3 inflammasome is one of the extensively researched inflammasomes and can be activated by various stimuli, including microbial infections, metabolic disturbances, and environmental irritants. These stimuli include pathogen-associated molecular patterns (PAMPs) like bacterial lipopolysaccharide (LPS) and viral RNA, damage-associated molecular patterns (DAMPs) such as ATP, uric acid crystals, and amyloid-beta, as well as environmental and metabolic stimuli including silica, asbestos, cholesterol crystals, and free fatty acids (FFAs) (Zheng et al. 2020). In diabetes, several DAMPs activate the NLRP3 inflammasome, a critical mediator of inflammation implicated in disease progression. High glucose levels, a hallmark of diabetes, serve as a prominent DAMP triggering NLRP3 activation in pancreatic β-cells, endothelial cells, and immune cells. Advanced glycation end products (AGEs), formed under hyperglycemic conditions, engage receptor-mediated pathways such as RAGE to activate NLRP3 (Fang et al. 2022). Concurrently, elevated FFAs in obesity-associated diabetes induce NLRP3 inflammasome in adipose tissue and pancreatic islets (Xu et al. 2022). Islet amyloid polypeptide (IAPP), accumulating in T2DM, contributes to NLRP3 activation in pancreatic β-cells (Mori et al. 2021). Lipid derivatives like ceramides and free cholesterol, associated with lipotoxicity in various tissues, also stimulate NLRP3 inflammasome (Ouyang et al. 2023; Xue, et al. 2022). Furthermore, mitochondrial dysfunction and increased ROS production in diabetes exacerbate NLRP3 activation (Liu et al. 2020). These DAMPs collectively highlight the intricate role of NLRP3 inflammasome in diabetes pathogenesis, underscoring its potential as a therapeutic target to mitigate inflammation and improve disease outcomes.
The activation of the NLRP3 inflammasome involves a two-step process: priming and activation. Priming is initiated by recognizing pathogen- or damage-associated molecular patterns (PAMPs or DAMPs) through receptors such as Toll-like receptors (TLRs) or cytokine receptors (Shen et al. 2022; Habas et al. 2022). This recognition triggers signaling pathways, including MAPK (mitogen-activated protein kinase) and NF-κB, leading to the upregulation of NLRP3 and pro-IL-1β, preparing the cell for activation (Cai et al. 2021; Escartín-Gutiérrez et al. 2023).
Activation occurs in response to a second signal, often involving potassium efflux from the cell. This drop in potassium triggers the assembly of the NLRP3 inflammasome complex, where NLRP3 oligomerizes and recruits the adaptor protein ASC and pro-caspase-1 (Wang et al. 2020a). This complex formation leads to the activation of caspase-1, which then cleaves pro-IL-1β and pro-IL-18 into their active forms, IL-1β and IL-18 (Lee et al. 2012). These cytokines are released to mediate inflammatory responses. IL-1β induces fever, inflammation, and tissue destruction, while IL-18 enhances the inflammatory response by inducing IFN-γ production (Lu 2023). Understanding the molecular mechanisms underlying inflammasome activation provides valuable insights into potential therapeutic targets for chronic inflammatory diseases such as diabetes (Fig. 1).

The process of assembly and activation of the NLRP3 inflammasome involves multiple stages. Before activation, priming of NLRP3 is necessary. This priming phase involves the binding of PAMPs or DAMPs to TLR/CD36 receptors, which triggers the promoters of NLRP3 and IL-1β transcription. In addition, during priming, NLRP3 is de-ubiquitinated. The ubiquitination and phosphorylation of ASC (apoptosis-associated speck-like protein containing a CARD) are also crucial for inflammasome assembly. Furthermore, disruption of lysosomes and subsequent cathepsin release further promotes NLRP3 inflammasome activation. Once assembled and activated, the inflammasome initiates pro-caspase-1 maturation and active form, which then activates pro-IL-1β and pro-IL-18 into IL-1β and IL-18. It is observed that active form of NLRP3 conjugates with ASC through PYD–PYD interactions, while pro-caspase-1 conjugates with ASC via CARD–CARD (caspase recruitment domain) interaction
Epigenetic regulation of NLRP3 inflammasome activation in diabetes
Recent progress in molecular biology has emphasized the significance of epigenetic mechanisms in the development of diabetes. Epigenetics involves inheritable changes in gene expression without changes to the DNA sequence itself. These alterations, which include DNA methylation, histone modifications, and the regulation by non-coding RNAs, affect the expression of genes linked to glucose metabolism, insulin sensitivity, and inflammation (Rosen et al. 2018).
Specifically, epigenetic regulation of the NLRP3 inflammasome, a critical component of innate immunity implicated in diabetes complications, has garnered attention. Understanding how epigenetic changes modulate NLRP3 expression and activation offers novel insights into potential therapeutic strategies for managing diabetic inflammation and its associated sequelae (Fig. 2). This review explores current research on epigenetic mechanisms governing NLRP3 inflammasome activation in diabetes, underscoring their significance in disease pathophysiology and therapeutic development (Raneros et al. 2021).

The diagram depicts how different modes of epigenetic regulations such hypermethylation, histone modification through HAT, HMT, and HDAC, miRNA and lncRNA influence the activation of NLRP3 inflammasome in diabetes pathology. Specific enzymes and RNA molecules have been mentioned that either promote (in black text) or (in red text) inhibit inflammasome activation. DNMT DNA methyltransferase, HMT histone methyltransferase, HAT histone methyltransferase, HDAC histone deacetyltransferase, miR microRNA, lncRNA long non-coding RNA
DNA methylation
DNA methylation refers to the incorporation of a methyl group to cytosine residues primarily in CpG dinucleotides, resulting in gene silencing. In the context of diabetes and NLRP3 inflammasome regulation, atypical DNA methylation of key inflammatory genes has been reported in several studies. Hypomethylation of CpG sites within the NLRP3 gene promoter has been observed in diabetic conditions, leading to enhanced NLRP3 expression and inflammasome activation. Chen et al. aimed to identify genetic markers for diabetic retinopathy (DR) through gene expression and DNA methylation analysis. Differential gene expression and methylation profiles revealed enrichment in immune and inflammatory responses, highlighting TGFB1, CCL2, and TNFSF2 as pivotal genes associated with NLRP3 inflammation. Promoter hypomethylation of these genes correlated with increased mRNA levels and HbA1c in T2DM patients, suggesting their role as potential therapeutic targets for DR detection and treatment (Chen et al. 2020). Zhou et al. aimed to investigate methylation levels in NLRP3, AIM2, and ASC gene promoters and their association with type 2 diabetes mellitus (T2DM) and vascular complications in a Southern Han Chinese population. Lower methylation in AIM2 promoters increased T2DM risk, while reduced methylation in NLRP3 and AIM2 correlated with higher risks of microvascular and macrovascular complications in T2DM patients. Methylation levels in ASC CpG sites mediated age-related vascular complications in T2DM (Zhou, et al. 2018). In a study investigating glucocorticoid (GC) resistance in idiopathic nephrotic syndrome (INS), NLRP3 gene promoter methylation levels were compared between GC-resistant and GC-sensitive patients with minimal change disease (MCD) or focal segmental glomerulosclerosis (FSGS). Methylation was significantly lower in GC-resistant patients, distinguishing them from GC-sensitive ones. This finding suggests NLRP3 methylation as a potential diagnostic marker for GC resistance in INS (Lucafò et al. May 2021).
Meng et al. conducted a study that concluded that N6-methyladenosine (m6A) methylation via Methyltransferase-Like 14 (METLL14) regulates lncRNAs and is crucial in diabetic cardiomyopathy (DCM). METTL14-mediated m6A modification suppresses pyroptosis by decreasing lncRNA TINCR and NLRP3 levels. This study highlights the role of m6A methylation in regulating pyroptosis and DCM progression through the METTL14-TINCR-NLRP3 axis (Meng et al. 2022). Liu et al. found that total flavones of Abelmoschus manihot (TFA) alleviated podocyte injury in diabetic kidney disease by modulating m6A modification through METTL3. TFA reduced NLRP3 inflammasome activation, and regulated PTEN/PI3K/Akt signaling, decreasing levels of gasdermin D, IL-1β, IL-18, and enhancing nephrin, ZO-1, and podocalyxin (Liu et al. 2021). Studies have demonstrated that hypermethylation of the NLRP3 promoter region correlates with decreased gene expression and reduced inflammasome activation in certain cell types. In diabetes-associated periodontitis (PD), upregulated METTL3 expression under high glucose conditions increased NLRP3 m6A methylation, enhancing its stability via IGF2BP3 binding. METTL3 knockdown attenuated pyroptosis, suggesting its role in PD pathogenesis (Zhou, et al. 2024). In diabetic retinopathy (DR), high glucose reduces miR-20a expression while increasing IL-1β, IL-18, caspase-1, and NLRP3, promoting pyroptosis in RPE cells. DNMT1-induced miR-20a methylation enhances TXNIP expression, exacerbating pyroptosis. Inhibiting DNMT1 or TXNIP alleviates pyroptosis and inflammation, suggesting potential therapeutic targets for DR (Xi et al. 2023). In the study conducted by Xi et al., high glucose was found to inhibit the PI3K/Akt pathway, upregulating DNMT1/DNMT3a and TXNIP, which decreased autophagy, increased apoptosis, and potentially activated the NLRP3 inflammasome, exacerbating inflammation in DPN (Zhang et al. 2021). In diabetic nephropathy (DN), WTAP regulates NLRP3 inflammasome activation through m6A methylation of NLRP3 mRNA, promoting cell pyroptosis and inflammation. Enhanced Wilm’s tumor-1-associated protein (WTAP) expression in HG-treated HK-2 cells and DN patients correlates with increased NLRP3 inflammasome components and pro-inflammatory cytokines. WTAP modulation influences these processes, implicating it as a potential therapeutic target for DN management (Lan et al. 2022).
Histone modification
Histone acetyltransferases (HATs) add acetyl groups to histone lysine residues, loosening chromatin structure and facilitating gene expression. Conversely, histone deacetylases (HDACs) remove these groups, leading to transcriptional repression. In diabetes, dysregulated histone acetylation affects NLRP3 inflammasome activity. HDAC inhibition has shown to mitigate NLRP3 activation and IL-1β secretion in macrophages under high glucose. Conversely, heightened histone H3 acetylation at the NLRP3 promoter exacerbates inflammasome activation in DN models, underscoring the role of histone modifications in modulating NLRP3-mediated inflammation.
Microtubule Affinity Regulating Kinase 4 (MARK4) regulates NLRP3 inflammasome activation, triggering IL-1β and IL-18 release. E74-like ETS Transcription Factor 3 (ELF3) modulates MARK4 in endothelial cells, influencing NLRP3 activation by high glucose. High glucose elevates ELF3, enhancing NLRP3 inflammasome activity via reduced SET8 and H4K20me1 levels. These findings suggest histone modifications and ELF3 influence diabetic endothelial inflammation through MARK4-NLRP3 pathways (Wang et al. 2020b). NLRP3, in macrophages, undergoes acetylation regulated by SIRT2, an NAD+-dependent deacetylase. This acetylation switch influences aging-associated inflammation and insulin resistance. Studies using aging models and a co-culture system suggest targeting SIRT2 and NLRP3 deacetylation could reverse these conditions, indicating the potential reversibility of aging-related chronic inflammation and insulin resistance (He et al. Mar. 2020). Magupalli et al. found that NLRP3 inflammasome activation in diabetes involves HDAC6 at the microtubule-organizing center (MTOC), promoting inflammatory response and pyroptosis. HDAC6 facilitates NLRP3 transport, essential for its activation and subsequent cytokine maturation (Magupalli, et al. 2020). CMap analysis of renal biopsy data from DN patients identified CAY10603, an HDAC6 inhibitor, as a promising drug candidate. In diabetic mice and cell models, CAY10603 reversed tubular injury and reduced NLRP3 inflammasome activation in tubular cells and macrophages, suggesting therapeutic potential for DN treatment (Hou 2022). Delayed wound healing in diabetes involves sustained NLRP3 inflammasome activation and IL-1β expression. Study by Karnam et al. suggests that HDAC6 overexpression hindered healing, while HDAC6 inhibition with Tubastatin A (TSA) promoted wound repair by reducing inflammation and enhanced collagen deposition and angiogenesis (Karnam et al. 2020). Ginsenoside Rg1, a key component of ginseng, shows promise in treating type 1 diabetes mellitus (T1DM) by modulating NLRP3 inflammasome activity and oxidative stress. In the study using STZ-induced diabetic mice, Rg1 reduced blood glucose, inflammation markers, and liver enzyme levels, while promoting insulin secretion and activating the Nrf2/ARE pathway to enhance antioxidant defenses. Rg1 also influenced histone H3K9 methylation in liver and pancreas (Gao et al. 2020). Sun et al. found that NLRP3 inflammasome activation mediated inflammatory response via caspase-1 activation in spontaneously hypertensive rats (SHR). Histone acetylation increased NFκB activation, promoting vascular smooth muscle cell (VSMC) phenotype switching and proliferation. Inhibition of histone acetylation prevented these effects (Sun et al. 2017). Set7, a histone methyltransferase drives histone methylation on NF-kB promotes inflammation and vascular dysfunction in T2DM patients during a clinical trial. Increased Set7 expression correlates with NLRP3 inflammasome activation, oxidative stress, and impaired dilation (Paneni et al. 2015; Singla and Narasimhulu 2023). In another research study, EzH2 (Enhancer of Zeste Homolog 2) regulated gene expression through H3K27me3 histone methylation, while Egr1 (Early Growth Response Protein 1) controlled NLRP3 inflammasome expression. Lysophosphatidic acid (LPA) decreased EzH2 and H3K27me3 levels, leading to increased Egr1 expression and NLRP3 activation, which exacerbated podocyte damage and diabetic nephropathy (Kim et al. 2023). Study conducted by Dai et al. revealed that inhibiting S-adenosylhomocysteine (SAH) hydrolase (SAHH) with adenosine dialdehyde (ADA) worsens diabetic nephropathy by increasing plasma SAH and enhancing podocyte injury through NLRP3 inflammasome activation. SAHH inhibition also elevates TXNIP via reduced EZH2 activity and EGR1 activation, contributing to inflammation and oxidative stress (Dai et al. 2021).
Non-coding RNA
Non-coding RNAs (ncRNAs), such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), have emerged as key epigenetic regulators of gene expression. miRNAs are short ncRNAs that bind with complementary sequences in target mRNAs, and results in the degradation or translational repression of mRNA.
In diabetes, specific miRNAs have been identified as regulators of NLRP3 inflammasome components. In diabetic kidney disease (DKD), downregulation of kidney-enriched miR-10a/b contributes to NLRP3 inflammasome activation. Reduced miR-10a/b levels enhance NLRP3 inflammasome assembly and pro-inflammatory cytokine release in diabetic kidneys. Restoring miR-10a/b expression mitigates renal inflammation and albuminuria in DKD models, highlighting a potential therapeutic approach to inhibit NLRP3-mediated kidney inflammation (Ding et al. 2021). In high-glucose and high-fat (HGHF)-induced diabetic mice, miR-223-3p expression is reduced while NLRP3 expression is elevated in cardiac microvascular endothelial cells (MCMECs). Overexpression of miR-223-3p decreases NLRP3 levels, Bax, and caspase-3 proteins, and suppresses apoptosis. This suggests miR-223-3p regulates NLRP3 to mitigate endothelial cell injury in diabetic hearts (Deng et al. 2020). In diabetic rat models, adenovirus-mediated overexpression of miR-24 suppresses the NLRP3-related inflammatory pathway, mitigating vascular remodeling. This intervention reduces neointimal hyperplasia, promotes reendothelialization, and alters expression of inflammatory markers and vascular structural proteins, indicating miR-24’s potential in alleviating diabetic vascular complications (Fan et al. 2020). miR-20b-3p was investigated in DR progression using rat models induced by streptozotocin. Overexpression of miR-20b-3p and silencing thioredoxin-interacting protein (TXNIP) improved visual function, reduced inflammation, microvascular damage, permeability, apoptosis, and angiogenesis in retinal tissues. miR-20b-3p targeted TXNIP, suggesting a therapeutic avenue for DR treatment (Wang et al. 2020). Mesenchymal stem cell (MSC)-derived small extracellular vesicles (sEVs) mitigate DR. In diabetic rats, intravitreal injection of MSC-sEVs suppressed NLRP3 inflammasome activation, reduced microglial activation and inflammatory cytokine levels in retinal tissues, improving histological morphology and blood–retinal barrier function. Mechanistically, miR-22-3p within MSC-sEVs was crucial for inhibiting NLRP3 inflammasome activation, highlighting MSC-sEVs as a potential DR therapy (Chen et al. 2024). DN involves renal tubular epithelial cell pyroptosis. Human umbilical cord MSCs (UC-MSCs) reduce inflammation and kidney damage in DN by upregulating miR-342-3p, which inhibits NLRP3/Caspase-1-mediated pyroptosis (Zheng et al. 2023). MicroRNA-21 (miR-21) induced by Angiotensin II (AngII) promotes NLRP3 inflammasome activation in liver fibrosis. Angiotensin-(1–7) (Ang-(1–7)) inhibits miR-21, NLRP3 inflammasome, and fibrosis, suggesting therapeutic potential in liver disease (Ning et al. 2016).
lncRNAs, on the other hand, exert their regulatory effects through diverse mechanisms, including chromatin remodeling, transcriptional regulation, and post-transcriptional processing. In DN, several lncRNAs have been implicated in modulating NLRP3 inflammasome activation and downstream inflammatory responses. Silencing lncRNA AC040162.3 mitigates HCV-induced T2DM by modulating the miR-223-3p/NLRP3 axis. Elevated lncRNA AC040162.3 and NLRP3, alongside reduced miR-223-3p, were observed in HCV-T2DM. Targeting lncRNA AC040162.3 enhanced miR-223-3p expression, reducing apoptosis and pyroptosis while improving cell viability in MIN6 cells (Niu et al. 2023). In DN research, LncRNA SNHG16 and Toll-like receptor-4 (TLR4) have been scrutinized for their roles. Liu et al. examined SNHG16’s influence on diabetic renal injury (DRI) through TLR4 and its mechanisms. Silencing SNHG16 in HG-induced HRMCs reduced cytokine secretion, ROS, MDA, fibrosis, and enhanced SOD/GSH levels. SNHG16 was found to interact with EIF4A3 to stabilize TLR4 mRNA. Notably, TLR4 overexpression countered the effects of SNHG16 knockdown, underscoring the potential of targeting the SNHG16/EIF4A3/TLR4 axis in treating DRI (Liu et al. 2023). Results from another showed SNHG16 upregulation in diabetic conditions and its interaction with miR-212-3p to modulate NF-κB signaling, highlighting its potential as a biomarker for T2DM (Huang et al. 2023). In DR, elevated levels of the lncRNA HOTAIR are associated with increased NLRP3 inflammasome activation and oxidative stress induced by high glucose. Knockdown of lncRNA HOX transcript antisense intergenic RNA (HOTAIR) mitigated NLRP3 inflammasome components, IL-1β maturation, and pyroptosis in human retinal endothelial cells, indicating its regulatory role via Nrf2 inhibition (You et al. 2023). Another study explored how the lncRNA HOTAIR influenced high glucose-induced pyroptosis in cardiomyocytes, a hallmark of DCM. HOTAIR levels were reduced under high glucose conditions, correlating with increased pyroptosis and inflammatory markers. HOTAIR overexpression mitigated these effects by upregulating SIRT3 via FUS, suggesting a potential therapeutic target for DCM (Xiong and Zhou May 2023). Li et al. aimed to assess the diagnostic value of MALAT1 and NLRP3 expression in T2DM patients with lower extremity atherosclerosis disease (LEAD). MALAT1 and NLRP3 levels were significantly elevated in T2DM with LEAD compared to T2DM alone, with MALAT1 correlating positively with NLRP3. MALAT1 shows promise as a biomarker for T2DM with LEAD (Li et al. 2024a). In DN, sterile inflammation involving pyroptosis is pivotal. lncRNA NEAT1 upregulation correlates with increased pyroptosis. NEAT1 regulates pyroptosis via miR-34c, affecting NLRP3, caspase-1, and IL-1β expressions. This suggests NEAT1’s role in DN pathogenesis through inflammasome modulation (Zhan et al. Jul. 2020).
Role of NLRP3 inflammasome in diabetic complications
Cardiovascular diseases (CVD) represent the leading cause of morbidity and mortality in individuals with diabetes. Activation of the NLRP3 inflammasome in response to metabolic stressors promotes endothelial dysfunction, vascular inflammation, and atherosclerosis (Fig. 3). Inflammasome-mediated release of IL-1β and IL-18 exacerbates vascular inflammation, contributing to plaque instability and thrombotic events characteristic of coronary artery disease (CAD) and peripheral arterial disease in diabetic patients. Cigarette smoke was found to trigger sterile inflammation via the NLRP3 inflammasome, contributing to vascular complications such as atherosclerosis. The study conducted by Mehta et al., examined NLRP3 activation in smokers with CAD compared to controls. Elevated inflammasome markers and oxidative stress in smokers suggested a significant role in disease progression, highlighting smoking’s impact on vascular health (Mehta et al. 2020). Pereira et al. investigated how mitochondrial DNA (mDNA) contributes to diabetes-related endothelial dysfunction and vascular inflammation via NLRP3 inflammasome activation. They found that diabetic conditions increased ROS generation and activated caspase-1 and IL-1β in C57BL/6 mice, which was mitigated in Nlrp3–/–mice. Endothelial dysfunction was exacerbated by diabetic mDNA, activating NLRP3 through mitochondrial ROS and calcium influx. Patients with T1DM showed elevated circulating mDNA and inflammasome activation. These findings underscore the role of NLRP3 inflammasome in diabetes-related vascular complications (Pereira 2020). Inflammation-induced damage to endothelial cells is critical in diabetes-related atherosclerosis (AS). NLRP3 activation in endothelial cells triggers inflammasome formation, leading to caspase-1 activation and IL-1β secretion, promoting immune responses in AS. In vitro, NLRP3-depleted human aortic endothelial cells resisted apoptotic cell death and reduced ROS production when exposed to oxidized LDL. In diabetic mice, endothelial-specific NLRP3 depletion attenuated AS severity, suggesting a role for endothelial NLRP3 in diabetes-associated AS development (Huang et al. 2020).

The diagram illustrates how the major pathophysiology of diabetes in the form of insulin resistance, hyperlipidemia, and hyperglycemia results in the release of DAMPs in the circulation which results in the activation of inflammasome activation and contribute to the development of several diabetic complications such as chronic kidney disease, atherosclerosis, fatty liver, and retinopathies
Diabetic neuropathy, a common complication of diabetes, results from nerve damage attributed to chronic hyperglycemia and inflammation. Activation of the NLRP3 inflammasome in peripheral nerves and dorsal root ganglia contributes to neuroinflammation and neuronal dysfunction. IL-1β released upon inflammasome activation induces neuroinflammatory responses and sensitizes nociceptive neurons, exacerbating pain perception and sensory deficits associated with DN. The study aimed to investigate the role of the TXNIP/NLRP3 inflammasome pathway in sciatic nerves of T2DM rats. Rats were induced into a T2DM model and treated with resveratrol, observing improvements in blood glucose levels, body weight, sciatic nerve function, and histological changes in nerve structure. Lower levels of TXNIP, NLRP3, caspase-1, and IL-1β proteins were detected in resveratrol-treated rats compared to untreated diabetic rats, suggesting potential therapeutic benefits of targeting this pathway in diabetic peripheral neuropathy (DPN) (Han et al. 2022). The study conducted by Han et al. investigated the role of the NLRP3 pathway in diabetic neuropathic pain (DNP) and its regulation by TET2 in dorsal root ganglion (DRG). Using a streptozotocin-induced DNP mouse model, researchers found NLRP3 pathway activation in DRG, alleviated by the NLRP3 inhibitor MCC950. They observed increased TET2 expression and DNA demethylation in DRG of diabetic mice. RNA sequencing revealed elevated Txnip expression, which mediates NLRP3 activation. Knocking down TET2 reduced Txnip mRNA and inhibited NLRP3 inflammasome expression/activation, thereby alleviating pain sensitivity in DNP. These findings suggest TET2-mediated epigenetic regulation of the TXNIP/NLRP3 pathway in DNP pathogenesis (Chen et al. 2022). Diabetic foot ulcers (DFUs) are prevalent and severe complications of diabetes, posing significant risks to patients. Neutrophil extracellular traps (NETs), implicated in delayed wound healing in diabetes, are regulated by the NLRP3/caspase-1/GSDMD pathway. Inhibition of GSDMD, such as with disulfiram, accelerates DFU healing by mitigating NET formation, offering a promising therapeutic strategy (Yang et al. 2023).
Diabetic nephropathy, characterized by progressive kidney damage and decline in renal function, is a major cause of end-stage renal disease (ESRD) worldwide. The NLRP3 inflammasome activation in renal cells, including podocytes, tubular epithelial cells, and mesangial cells, promotes renal inflammation, fibrosis, and glomerular dysfunction. IL-1β and IL-18 released by activated inflammasomes contribute to the pathogenesis of DN by promoting pro-inflammatory cytokine production, leukocyte infiltration, and extracellular matrix deposition within the kidney (Williams et al. 2022). In DN, CD36 promotes NLRP3 inflammasome activation through mitochondrial ROS (mtROS) in renal tubular cells. High glucose triggers NLRP3 activation, IL-1β secretion, and cell apoptosis, processes attenuated by CD36 knockdown or MitoTempo treatment. CD36 inhibition in diabetic mice reduces nephropathy-related inflammation and apoptosis by enhancing AMPK activity and mitochondrial fatty acid oxidation (FAO) (Hou et al. 2021). In DN, tubular injury and epithelial-to-mesenchymal transition (EMT) are pivotal in renal deterioration. In HK-2 cells, high glucose (HG) and TGF-β1 induce EMT via NLRP3 activation and ROS production, mechanisms inhibited by NLRP3 silencing or antioxidant treatment. These findings propose NLRP3 as a therapeutic target in DN to mitigate EMT and renal fibrosis (Song et al. 2018).
Diabetic retinopathy, a sight-threatening complication of diabetes, is characterized by vascular dysfunction and retinal inflammation. Activation of the NLRP3 inflammasome in retinal cells and infiltrating immune cells exacerbates retinal inflammation and vascular damage. IL-1β and IL-18 released by the inflammasome contribute to the breakdown of the blood–retinal barrier, neovascularization, and retinal cell death observed in DR (Zheng et al. 2023). It was observed that retinal endothelial cells (RECs) are vulnerable to diabetes-induced vascular damage. The P2X7/NLRP3 pathway amplifies inflammation via ATP feedback loop, leading to pyroptosis and apoptosis in DR. The study evaluated 3TC, a nucleoside reverse transcriptase inhibitor, for its anti-inflammatory effects in vitro (HG and LPS-treated RECs) and in vivo (streptozotocin-induced diabetic mice). Results indicated 3TC reduced inflammasome-related proteins, apoptosis, and pyroptosis, suggesting potential therapeutic benefits in diabetic retinal injury (Kong et al. 2022). Another study aimed to assess vitamin D levels in vitreous and serum, and NLRP3 inflammasome pathway expression in proliferative diabetic retinopathy (PDR). Elevated NLRP3 and VEGF levels were found in PDR vitreous, contrasting with decreased vitamin D levels. Vitamin D showed a negative correlation with NLRP3 expression, suggesting potential therapeutic implications for PDR management (Lu et al. 2021).
In reproductive health, gestational diabetes and associated inflammatory responses contribute to pregnancy complications, including macrosomia and preeclampsia, involving inflammasome activation and cytokine dysregulation (Gayatri et al. 2024). Exposure to fructose (Fru) during pregnancy induced insulin resistance (IR) in mice, persisting postpartum and affecting offspring. Fru increased gestational weight gain, blood glucose, insulin levels, and inflammatory markers like IL-6 and TNF-α. Mechanistically, Fru activated the NF-κB–NLRP3 pathway, suggesting its role in promoting IR and inflammation in gestational diabetes mellitus (GDM) (Liu, et al. 2022). Study by Wu et al. investigated how the NLRP3 inflammasome, regulated by hydrogen sulfide (H2S) synthetases, contributes to GDM pathogenesis via placental inflammation. Clinical placenta samples from GDM and healthy pregnancies showed increased NLRP3 and cleaved caspase-1, inversely correlated with reduced CBS and CSE. H2S treatment suppressed NLRP3 activation, suggesting a potential therapeutic avenue (Wu et al. 2022).
Role of gut microbiota in regulation of inflammasome
Emerging research underscores the significant influence of the gut microbiota on NLRP3 inflammasome activity. For example, short-chain fatty acids (SCFAs) like butyrate, produced by gut bacteria, can inhibit NLRP3 activation. Butyrate achieves this by inhibiting HDACs, which subsequently downregulate inflammatory responses. This anti-inflammatory effect is particularly relevant in diabetes management, where chronic inflammation exacerbates disease progression (Roshanravan et al. 2020). Sodium butyrate (NaB), a gut microbiota metabolite, inhibits cardiac fibroblast differentiation into myofibroblasts by deactivating the NLRP3/Caspase-1 pyroptosis pathway. This reduction in NLRP3 inflammasome activity lowers inflammatory responses and curbs pathological remodeling, highlighting butyrate’s potential in managing inflammation-related cardiac conditions (Dong et al. 2024).
In the context of diabetes, particularly T2DM, gut dysbiosis—an imbalance in the gut microbial community—has been linked to increased NLRP3 inflammasome activity. This dysbiosis often features a reduction in beneficial SCFA-producing bacteria such as those from the Lachnospiraceae family, coupled with an increase in pro-inflammatory bacteria. The resultant decrease in SCFA production, including butyrate, leads to reduced inhibition of the NLRP3 inflammasome, thereby heightening inflammation and insulin resistance (Zhao et al. 2024).
Studies involving NLRP3-deficient mice further illuminate this relationship. These mice exhibit less severe diabetes symptoms and reduced intestinal lesions compared to their wild-type counterparts, highlighting the inflammasome’s role in maintaining both intestinal and systemic homeostasis. The interplay between the gut microbiota and the NLRP3 inflammasome is multifaceted, involving various pathways mediated by microbial metabolites and host immune responses.
In addition, gut microbiota composition directly impacts glucose metabolism and insulin sensitivity. Certain gut bacteria can influence the production of lipopolysaccharides (LPS), which activate the NLRP3 inflammasome and contribute to insulin resistance (Li et al. Jul. 2020). Therefore, maintaining a balanced gut microbiome through diet and probiotics, or fecal microbiota transplantation (FMT) emerges as a potential strategy to modulate NLRP3 activity and mitigate diabetes-related inflammation. Gut microbiota dysbiosis increases the expression of the NLRP3 inflammasome, contributing to age-related atrial fibrillation (AF). In a FMT rat model, aged rats’ microbiota increased AF susceptibility and atrial fibrosis in young hosts. Inhibiting NLRP3 with MCC950 reduced AF susceptibility, highlighting the microbiota-NLRP3 inflammasome link (Zhang et al. 2022). The NLRP3 inflammasome triggers inflammation and insulin resistance in obesity. NLRP3 deficiency in mice protects against high-fat diet-induced left ventricle remodeling, reduces systemic inflammation, and modifies gut microbiota and metabolic profiles, including lower ceramide and altered fatty acid levels. These changes mitigate liver steatosis and benefit lipid metabolism (Sokolova et al. 2020).
Various specific gut bacteria and their metabolites have been identified as influential in this process. Akkermansia muciniphila for instance, produce SCFAs like butyrate, which possess anti-inflammatory properties that modulate NLRP3 inflammasome activity. Butyrate specifically inhibits NLRP3 activation by enhancing mitochondrial function and reducing reactive oxygen species (ROS) production (Siwen et al. 2021). Deleting the NLRP3 gene delays T1D progression. Lactobacillus plantarum NC8 and its metabolite acetate alleviate T1D by inhibiting NLRP3, reducing Th1/Th17 cells, and decreasing pancreatic macrophages in T1D mice (Zhang et al. 2023). NLRP3 inflammasome activation in macrophages reduces inflammation. In mice, E. faecalis alleviates colitis and prevents colitis-associated colorectal cancer, highlighting its potential as a therapeutic strategy (Chung et al. 2019).
In diabetic patients, the gut microbiota composition is often altered, leading to an imbalance favoring pro-inflammatory bacteria. Increased levels of Escherichia coli have been associated with heightened inflammation through LPS, which triggers the NLRP3 inflammasome by binding to toll-like receptors (TLRs) on immune cells. This promotes the activation of NLRP3 and the release of inflammatory cytokines (Wang et al. 2023). Pregnancy-induced hypertension (PIH) correlates with gut microbiota dysbiosis and elevated NLRP3 levels. PIH patients exhibited differences in specific bacteria such as increased Prevotellaceae, and decreased Bacteroides, alongside increased pro-inflammatory cytokines compared to healthy controls (Wu et al. 2023). Sanziguben polysaccharides (SZP) improved DN in db/db mice by reducing albuminuria and insulin resistance. SZP regulated gut microbiota, decreasing Proteobacteria, Klebsiella, and Escherichia–Shigella, and inhibited the TLR4/NF-κB/NLRP3 pathway, thereby alleviating DN-associated inflammation (Wang et al. 2023). Recent studies have explored the link between gut microbiota and IgA nephropathy (IgAN). Both human patients and a mouse model of IgAN showed gut microbiota dysbiosis, particularly reduced Bifidobacterium levels correlated with disease severity. Probiotic treatments, especially those containing Bifidobacterium, improved dysbiosis and mitigated IgAN symptoms via the NLRP3/ASC/Caspase-1 pathway inhibition (Tan et al. 2022). Feng in his study explored how antibiotics affect the intestinal barrier in mice. It found that antibiotics disrupt gut microbiota, reduce short-chain fatty acids, and impair tight junction proteins like ZO-1. This disruption increases intestinal permeability and activates cellular mechanisms such as NLRP3 inflammasome and autophagy pathways, impacting gastrointestinal health (Feng et al. 2019,; Lathakumari et al. 2024). This study examined how antibiotics affect NLRP3 inflammasome activation in microbiome-depleted mice. Xiaoyaosan treatment improved depressive and anxious behaviors by restoring gut flora diversity, reducing LPS-producing bacteria like Bacteroidaceae, and suppressing NLRP3, ASC, and CASPASE-1 expression in the colon, thereby alleviating anxiety and depression (Hao 2021).
In conclusion, the composition of the gut microbiome and the metabolic products of specific bacterial species significantly impact NLRP3 inflammasome activity in diabetes. This intricate interplay underscores the potential of targeting the gut microbiota for therapeutic strategies to modulate inflammation and improve metabolic health in diabetic patients. Continued research into these microbial interactions and their mechanisms promises to provide novel insights into treatments for diabetes and related inflammatory conditions.
Classes of oral hypoglycemic drugs and their impact on NLRP3 inflammasome
Biguanides (e.g., metformin)
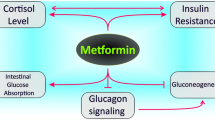
Biguanides, especially metformin, lower glucose levels through multiple mechanisms. Metformin primarily reduces hepatic glucose production by inhibiting gluconeogenesis via AMP-activated protein kinase (AMPK) activation, which decreases gluconeogenic enzyme expression. It also enhances insulin sensitivity in muscle cells, increasing glucose uptake and utilization through AMPK activation and improved insulin receptor signaling. In addition, metformin decreases intestinal glucose absorption, further reducing blood glucose levels. Beyond glucose control, metformin improves lipid profiles by lowering triglycerides and LDL cholesterol. Importantly, it does not cause hypoglycemia since it does not stimulate insulin secretion. These multifaceted effects make metformin a cornerstone in managing T2DM and offer cardiovascular health benefits (Foretz et al. 2023).
Metformin shows cardioprotective effects against ischemia/reperfusion (I/R) injury by activating the AMPK pathway, which suppresses NLRP3 inflammasome activation. This reduces myocardial infarct size, apoptosis, fibrosis, and inflammation. In neonatal rat ventricle myocytes (NRVMs), metformin increased cell viability and decreased inflammation. The AMPK inhibitor Compound C negated these benefits, highlighting AMPK’s role in metformin’s action (Zhang, et al. 2020) Tang et al. investigated metformin’s effect on diabetes-accelerated atherosclerosis and its targeting of NLRP3 inflammasome. Using apoE−/−male mice and THP-1-differentiated macrophages, results showed that diabetes increased plasma lipids, pro-inflammatory interleukin-1β, macrophage infiltration, and atherosclerosis, linked to NLRP3 inflammasome activation and thioredoxin dysregulation. Metformin alleviated these effects via AMPK activation and thioredoxin regulation, reducing NLRP3 inflammasome activation and atherosclerosis progression (Tang et al. 2019). Metformin reduces the risk of ARDS in severe COVID-19 by inhibiting NLRP3 inflammasome activation and interleukin-1β production in macrophages. This action mitigates ARDS induced by lipopolysaccharides and SARS-CoV-2. Metformin achieves this by blocking ATP-dependent mtDNA synthesis, independently of AMPK or NF-κB pathways (Xian et al. 2021). NLRP3 inflammasome-mediated pyroptosis is crucial in preeclampsia, linked with TLR4/NF-κB signaling and trophoblastic glycometabolism. Metformin, an NF-κB inhibitor, may help treat preeclampsia by suppressing pyroptosis and correcting glycometabolic reprogramming and oxidative stress via the TLR4/NF-κB/PFKFB3 pathway. These findings suggest metformin’s therapeutic potential in preeclampsia (Zhang et al. 2021; Jia et al. 2024). Alzokaky et al. investigates Metformin’s role in preventing doxorubicin (DOX)-induced cardiotoxicity in male albino mice. DOX increased markers of cardiac damage and inflammation, including the NLRP3 inflammasome. Metformin significantly reduced these effects, lowering the expression of the NLRP3 inflammasome and other inflammatory biomarkers. This suggests metformin protects heart function by inhibiting the HMGB1/NF-κB inflammatory pathway, which reduces inflammasome activity (Alzokaky et al. 2023). Recent studies show that NLRP3 activation leads to ovarian granulosa cells (OGCs) pyroptosis in polycystic ovary syndrome (PCOS) mice. While metformin reduces insulin resistance in PCOS women, its impact on OGC pyroptosis was unclear. This study found that metformin reduces LPS-induced pyroptosis markers and inflammation in OGCs through the miR-670-3p/NOX2/ROS pathway, suggesting a protective role against PCOS (Zhou et al. 2023). Metformin accelerates wound healing by inhibiting NLRP3 inflammasome activation through the AMPK/mTOR pathway in rat skin defect models, enhancing angiogenesis and M2 macrophage polarization (Qing et al. 2019). Metformin and resveratrol both inhibit NLRP3 inflammasome activation in adipose tissue under high glucose conditions. They protect mitochondrial integrity by regulating Drp1 phosphorylation via AMPK, and suppress ER stress markers like IRE1α and eIF2α. This reduces inflammation and apoptosis, preserving adipose function in diabetic mice and 3T3-L1 adipocytes (Li et al. 2016).
Despite its benefits, metformin can cause gastrointestinal issues such as diarrhea and nausea. A significant concern is the risk of lactic acidosis, particularly in individuals with renal impairment, which can limit its use in certain populations. These side effects necessitate careful patient monitoring and may restrict the drug’s broader application in targeting the NLRP3 pathway (Tarry-Adkins et al. 2021).
Metformin exerts potent anti-inflammatory effects by targeting the NLRP3 inflammasome across diverse conditions such as cardiac injury, diabetic atherosclerosis, ARDS, preeclampsia, doxorubicin-induced cardiotoxicity, PCOS, and wound healing. Through AMPK activation and other pathways, metformin suppresses NLRP3 inflammasome activation, mitigating inflammation and associated tissue damage. These findings highlight metformin’s therapeutic potential in modulating inflammatory responses crucial to multiple pathological processes (Table 1).
Sulfonylurea
Sulfonylureas are oral hypoglycemic agents primarily used to treat T2DM. They function by stimulating insulin secretion in pancreatic islet cells. This is achieved through the inhibition of ATP-sensitive potassium channels present on the cell membrane. Binding of sulfonylureas to the SUR1 subunit of the K+-ATP channels causes these channels to close, leading to cell membrane depolarization. This opens up voltage-gated calcium channels and influx of calcium ions. The increased intracellular calcium concentration promotes the exocytosis of insulin-containing granules. Sulfonylureas are classified into first and second generations, with second-generation drugs (e.g., glipizide, glyburide) being more potent and having fewer side effects. Common side effects include hypoglycemia and weight gain. They are contraindicated in patients with type 1 diabetes or diabetic ketoacidosis (Thulé and Umpierrez 2014).
Sulfonylureas, known for their insulin-secretion role in T2DM, are explored as NLRP3 inflammasome inhibitors to protect pancreatic β-cells. Study conducted by Hill et al. identified that nine newly synthesized sulfonylureas effectively inhibit NLRP3 activation in murine macrophages, with six compounds demonstrating potent nanomolar inhibition. These findings suggest a promising dual therapeutic approach for managing diabetes and its inflammatory complications (Hill et al. 2017). New N-sulfonylurea derivatives, including compound 4b, were developed by Narros-Fernández et al. through modifying MCC950’s hexahydroindacene structure. Compound 4b exhibited potent inhibition of IL-1β release and reduced caspase-1 activation and ASC oligomerization. In silico predictions indicated compound 4b’s safety, and in vitro studies confirmed its non-toxic effects. In vivo, compound 4b demonstrated significant anti-inflammatory effects and alleviated mechanical hyperalgesia in a mouse model of gout (Narros-Fernández et al. 2022). In another study, AMS-17, a newly developed tertiary sulfonylurea compound, effectively inhibited NLRP3 inflammasome expression and downstream inflammatory cytokines in microglia. AMS-17 also suppressed LPS-induced microglial activation and phagocytosis in vitro, as well as microglial activation in mouse brain in vivo. These findings suggest AMS-17 holds promise as a therapeutic candidate for neuroinflammatory diseases (Zhang et al. 2022a). The study by Dwivedi and Jena aimed to investigate the impact of glibenclamide (GLB), dimethyl fumarate (DMF), and their combination on NLRP3 inflammasome and Nrf2/ARE signaling in diabetic NAFLD. GLB inhibits NLRP3 inflammasome, while DMF activates Nrf2/ARE pathway, both ameliorating diabetes-induced NAFLD markers in rats. This suggests potential therapeutic strategies for fatty liver diseases involving inflammatory and oxidative stress pathways (Dwivedi and Jena 2023). The drug glibenclamide acts as a potent inhibitor of the NLRP3 inflammasome, a key mediator in neurotoxicity induced by the solvent metabolite 2,5-hexanedione (HD). Glibenclamide reduces NLRP3 inflammasome activation, leading to decreased caspase-1 activity and interleukin-1β maturation. This inhibition correlates with preservation of neurofilament protein L (NF-L), prevention of demyelination and axon degeneration, and modulation of microglial polarization from M1 to M2 phenotype (Hou et al. 2020). Further, glibenclamide (GLB) exhibits neuroprotective effects beyond its role in reducing brain edema. It inhibits the NLRP3 inflammasome activation by blocking the SUR1-TRPM4 channel, independent of its edema-preventing function. This mechanism mitigates neuroinflammation, improves cognitive function, and reduces neuropathological damage in a rat model of cardiac arrest-induced brain injury, as well as in vitro conditions mimicking neuroinflammatory processes (He et al. 2022). In a study, gliclazide, effectively targeted inflammation in atherosclerotic plaque. The nanoghost (NG) formulation inhibited key inflammatory markers like TNFα and suppressed NLRP3, MyD88, NOS, IL-1β, IL-18, and caspases 1/3/8/9 in LPS-primed monocytes. This action suggests a significant role in modulating inflammasome activation and related cell signaling pathways involved in atherosclerosis progression (Karami et al. 2023).
Sulfonylureas, with side effects like hypoglycemia and weight gain, may worsen inflammation linked to NLRP3 inflammasome activation. Their long-term efficacy is compromised due to potential β-cell dysfunction and cardiovascular risks, making them less suitable as reliable therapeutic targets for NLRP3 inflammasome-related conditions (Scheen 2021).
Sulfonylurea drugs, including glibenclamide and gliclazide, have shown potent inhibition of the NLRP3 inflammasome across various studies. These drugs effectively suppress inflammasome activation, reduce inflammatory cytokines, and modulate cellular signaling pathways involved in inflammatory diseases like diabetes, neuroinflammation, NAFLD, and atherosclerosis. This highlights their potential as dual-action therapeutics for managing both metabolic disorders and associated inflammatory complications.
Sodium–glucose co-transporter 2 (SGLT2) inhibitors
SGLT2 inhibitors, employed in the treatment of T2DM, inhibit the co-transporterSGLT2 in the proximal renal tubules, thereby decreasing glucose reabsorption and enhancing glucose excretion in urine. This mechanism lowers blood glucose levels independently of insulin and promotes weight loss. Important pharmacological details include reduced cardiovascular events, improved renal outcomes, and blood pressure reduction. SGLT2 inhibitors like dapagliflozin, empagliflozin, and canagliflozin are key therapies in managing diabetes and associated cardiovascular and renal complications (Gronda et al. 2022).
SGLT2 inhibitors decrease cardiovascular events in T2DM, possibly through mechanisms involving the NLRP3 inflammasome and IL-1β release (Hu et al. 2023). In a study comparing empagliflozin with sulfonylurea, empagliflozin reduced IL-1β secretion more significantly, associated with increased serum β-hydroxybutyrate and decreased insulin levels, indicating potential cardioprotective effects via NLRP3 inflammasome inhibition (Kim et al. 2020). In study conducted by Li et al., IL-17A was found to induce oxidative stress and enhance inflammatory signaling in human aortic smooth muscle cells (SMC) through TRAF3IP2-mediated activation of the NLRP3/caspase-1 pathway, leading to secretion of IL-1β and IL-18. Empagliflozin (EMPA), an SGLT2 inhibitor, attenuated these effects by reducing oxidative stress, NLRP3 activation, and cytokine secretion, thereby inhibiting SMC proliferation and migration. These findings underscore EMPA’s potential therapeutic role in treating vascular proliferative diseases by targeting inflammatory and oxidative pathways in SMC (Sukhanov et al. 2021). EMPA improved metabolic abnormalities and reduced renal injury by suppressing NLRP3 inflammasome activity and enhancing the HO-1–adiponectin axis, highlighting its potential for renal protection beyond diabetes management (Ye et al. 2022). Dapagliflozin, an SGLT2 inhibitor, was studied using an ischemia/reperfusion-induced fibrosis model, revealing it reduces tricarboxylic acid cycle metabolite accumulation, inflammation, and activates immunomodulatory itaconate. Notably, dapagliflozin blocks NLRP3 inflammasome activation, highlighting its potential in treating progressive chronic kidney disease through metabolic and inflammatory modulation (Ke et al. 2022). Dapagliflozin also inhibits NLRP3 inflammasome activation via modulation of NHE1/NCX signaling pathways, reducing intracellular calcium and sodium levels, which in turn promotes autophagy and decreases inflammation in cardiomyocytes (Yu et al. 2022). Dapagliflozin (DPZ) also shows potential as a treatment for ulcerative colitis (UC) by mitigating colonic damage induced by acetic acid in rats, comparable to 5-ASA. DPZ improved colon tissue integrity, survival rates, and reduced disease severity by modulating NLRP3 inflammasome activity through NFκB/AMPK pathways, inhibiting IL-1β and IL-18 release. Further clinical studies are needed to validate its efficacy in UC management (El-Rous et al. 2021). Dapagliflozin (Dapa), a SGLT-2 inhibitor, shows potential as an antidepressant by targeting the NLRP3 inflammasome pathway and ET-1/ETBR signaling. In a rat model of depression, Dapa administration reduced NLRP3 inflammasome activation and restored ETBR expression, enhancing neuroplasticity and preserving blood–brain barrier integrity, which collectively contributed to improved behavioral outcomes (Muhammad et al. 2021). Canagliflozin (CAN), a hypoglycemic drug, was found to transcriptionally inhibit NLRP3 inflammasome-related proteins by blocking NF-κB signaling. CAN promoted autophagy through upregulation of Bif-1, crucial for autophagosome formation. These findings suggest a non-hypoglycemic mechanism by which CAN may mitigate NLRP3 inflammasome activation and inflammation, offering potential therapeutic insights for pneumonia patients with diabetes (Niu 2022).
However, SGLT2 inhibitors are associated with an increased risk of urinary tract infections and genital infections due to their mechanism of action. In addition, there is a risk of diabetic ketoacidosis, especially in patients with type 1 diabetes. These side effects and risks require careful patient selection and monitoring, limiting the widespread use of SGLT2 inhibitors for targeting the NLRP3 pathway (Saisho 2020).
SGLT2 inhibitors like empagliflozin and dapagliflozin demonstrate multifaceted effects on the NLRP3 inflammasome across various clinical contexts. They attenuate IL-1β and IL-18 release, reduce oxidative stress, modulate inflammatory pathways, and enhance metabolic and renal protective mechanisms. These findings highlight their potential therapeutic benefits in cardiovascular diseases (CVD), chronic kidney disease, ulcerative colitis, and even depression, suggesting broader applications beyond diabetes management. Further research is crucial to validate their efficacy and safety in diverse patient populations.
Dipeptidyl peptidase-4 (DPP-4) inhibitors
Dipeptidyl peptidase-4 (DPP-4) inhibitors, like sitagliptin and saxagliptin, treat T2DM by blocking DPP-4 enzyme activity, which degrades incretin hormones like GLP-1 and GIP. This inhibition prolongs the action of GLP-1 and GIP, enhancing glucose-dependent insulin secretion and suppressing glucagon release, thereby lowering blood glucose levels after meals. They are well-absorbed orally, metabolized hepatically, and generally have favorable safety profiles with low risks of hypoglycemia. Beyond glycemic control, they may preserve beta-cell function and have potential cardiovascular and renal benefits, making them valuable in comprehensive diabetes management strategies (Saini et al. 2023).
Soluble DPP4 (sDPP4) released from visceral adipose tissue increases in obesity, contributing to vascular aging by inducing endothelial cell senescence and dysfunction via the PAR2–COX-2–TP axis, leading to NLRP3 inflammasome activation. In obese patients, elevated sDPP4 correlates with impaired microvascular function. Blocking sDPP4 activity reverses endothelial dysfunction, suggesting sDPP4 as a potential therapeutic target for mitigating vascular aging (Valencia et al. 2022). Recent findings show saxagliptin (Sax) and sitagliptin (Sit), DPP-4 inhibitors used in diabetes treatment, promote murine BC 4T1 metastasis via a ROS–NRF2–HO-1–NF-κB–NLRP3 axis in immune-competent BALB/c mice. These drugs activate ROS-dependent NF-κB signaling, elevate metastasis-related genes, and alter immune cell infiltration and cytokine production within BC microenvironments, suggesting implications for managing DPP-4 inhibitor-induced BC metastasis through targeted intervention in tumor immune-suppressive microenvironments (Li et al. 2021). Saxagliptin (SAX) and vildagliptin (VIL), DPP4 inhibitors, effectively mitigate DXR-induced kidney damage by reducing inflammatory markers, NLRP3 inflammasome activation, and tubulointerstitial injury. This highlights their potential therapeutic role in protecting against DXR-induced renal toxicity (Mostafa et al. 2021). Another study also revealed that Saxa, attenuated diabetic DN in type 2 (BTBR) and type 1 (Akita) diabetic mice by targeting the NLRP3 inflammasome pathway. It reduced the levels of apoptosis-associated speck-like protein 1, NLRP3, TNFα, and Caspase-1 in kidney and adipose tissues, indicating suppression of inflammasome activation and associated inflammation pathways (Birnbaum et al. 2016). Gemigliptin restored autophagy markers such as ULK1 and the LC3II/LC3I ratio in human NASH patients, a NASH mouse model, and HepG2 cells under NASH-mimicking conditions. Interestingly, gemigliptin treatment in NASH mice also showed restoration of AMPK activity, crucial for autophagy induction. Furthermore, gemigliptin reduced liver lipid accumulation, inflammation, and fibrosis by promoting ULK1-dependent autophagy and alleviating inflammasome activation. These findings suggest gemigliptin’s potential therapeutic role in NASH through AMPK-independent pathways involving autophagy modulation and inflammasome regulation (Song et al. 2023). Saxagliptin showed protective effects against ethanol-induced gastric injury in rats without causing hypoglycemia. It enhanced gastric mucosal autophagy through AMPK/mTOR pathway modulation, reduced apoptotic markers, and suppressed NLRP3 inflammasome activation and oxidative stress. These findings highlight saxagliptin’s potential as a therapeutic agent for ethanol-induced gastropathy (Arab et al. 2021).
Teneligliptin, a DPP-4 inhibitor, demonstrated therapeutic potential against diabetes-related cognitive impairment (CI) in mice. It improved behavioral deficits in memory and learning tests, reduced lipid abnormalities, suppressed inflammation, oxidative stress, and inhibited NLRP3 inflammasome and ER stress pathways in the hippocampus of diabetic mice, suggesting a beneficial role in managing DM-associated CI (Wang and Zhang 2023). High-fat (HF) diet-fed mice showed compromised intestinal barrier function and dysbiosis, allowing increased liver permeability to lipopolysaccharides (LPS). This disrupted gut–liver axis exacerbated pro-inflammatory responses (Tlr4 and Nlrp3 upregulation), contributing to non-alcoholic fatty liver disease (NAFLD) susceptibility.
Treatment with a PPAR-alpha agonist and DPP-4 inhibitor (linagliptin) restored gut microbiota balance, improved intestinal barrier integrity, and reduced liver steatosis through anti-inflammatory actions, suggesting promising therapeutic avenues for NAFLD management (Silva-Veiga et al. 2022). DPP-4 inhibitors, such as sitagliptin and NVPDPP728, demonstrated anti-atherosclerotic effects by reducing NLRP3, TLR4, and IL-1β expression in human macrophages exposed to oxidized LDL. This effect was associated with increased GLP-1R expression and involved inhibition of PKC activity. These findings highlight a potential mechanism for DPP-4 inhibitors in modulating inflammation and immune responses in atherosclerosis (Dai et al. 2014). Vildagliptin, a DPP-4 inhibitor used clinically for diabetes, protects against FFA-induced endothelial dysfunction by inhibiting LDH release, ROS generation, and NAPHD oxidase NOX-4 expression in endothelial cells. It preserves mitochondrial function, suppresses NLRP3 inflammasome proteins (NLRP3, ASC, p20, HMGB-1), and activates AMPK, thereby reducing IL-1β and IL-18 cytokines. This dual action suggests vildagliptin improves both glucose control and vascular health (Qi et al. 2019). Lina, a DPP4 inhibitor, and GLP-1 receptor activation by exenatide (EX) were compared for their effects on infarct size (IS), inflammasome activation, and cardiac remodeling in WT and db/db diabetic mice post-myocardial infarction. Both treatments reduced IS and improved ejection fraction similarly. Lina showed stronger attenuation of inflammasome activation, collagen expression, and apoptosis in db/db mice, attributed to p38-mediated upregulation of miR-146b suppressing TLR4 expression, distinct from GLP-1 receptor activation alone (Birnbaum et al. 2019). Anagliptin, a DPP4 inhibitor used to treat T2DM, was studied for its effects on high glucose-induced endothelial dysfunction. It preserved cell viability, reduced LDH release, and mitigated mitochondrial ROS and NOX-4 expression in HUVECs. Anagliptin also inhibited TXNIP and NLRP3 inflammasome activation, including IL-1β and IL-18 maturation, via SIRT1-dependent mechanisms (Jiang et al. 2019).
DPP-4 inhibitors help modulate the NLRP3 inflammasome and exhibit anti-inflammatory actions. Generally well-tolerated, DPP-4 inhibitors have been linked to an increased risk of pancreatitis and severe joint pain. Although these side effects are relatively rare, they pose significant concerns for the continuous use of DPP-4 inhibitors, particularly when considering their long-term impact on patient health and safety (Rai et al. 2019).
Dipeptidyl peptidase-4 (DPP-4) inhibitors demonstrate significant therapeutic potential across various clinical pathologies by modulating the NLRP3 inflammasome pathway. These inhibitors, including sitagliptin, saxagliptin, and vildagliptin, have shown to suppress inflammasome activation, reduce inflammatory cytokines like IL-1β and IL-18, and improve conditions such as DN, atherosclerosis, non-alcoholic fatty liver disease, and endothelial dysfunction. The underlying mechanisms involve the inhibition of key inflammasome components (NLRP3, ASC, and caspase-1) and pathways (AMPK and PKC), highlighting their multifaceted role in managing inflammation-driven complications in metabolic and cardiovascular diseases.
GLP-1 agonists
GLP-1 agonists mimic glucagon-like peptide-1, enhancing insulin secretion and inhibiting glucagon release in a glucose-dependent manner. They slow gastric emptying, reducing postprandial glucose spikes, and promote satiety, aiding in weight loss. These effects collectively improve glycemic control in diabetes. Pharmacologically, GLP-1 agonists activate GLP-1 receptors on pancreatic beta cells, leading to increased cyclic AMP (cAMP) and insulin secretion. They are administered subcutaneously and have varying half-lives, influencing dosing frequency. Common agents include exenatide, liraglutide, and semaglutide. Side effects can include gastrointestinal issues like nausea and an increased risk of pancreatitis (Drucker 2018).
Zhang et al. found that glucagon-like peptide-1 receptor agonist (GLP-1RA) alleviates diabetic neuropathic pain by inhibiting brain microglia activation and suppressing the NLRP3 inflammasome. Positron emission tomography/computed tomography and RNA sequencing revealed reduced microglia activation and NLRP3 expression in DNP rats treated with GLP-1RA, indicating its potential therapeutic effect (Zhang et al. 2022b). Dulaglutide, a GLP-1 receptor agonist, protected against high glucose-induced endothelial dysfunction in T2DM by inhibiting NLRP3 inflammasome activation. It reduced reactive oxygen species, protein carbonyl, NOX-4, and TXNIP expression, and prevented IL-1β and IL-18 maturation in endothelial cells. SIRT1 mediated these protective effects (Luo et al. 2019). A GLP-1 receptor agonist effectively reduced weight, suppressed eosinophilic airway inflammation, and decreased airway hyper-responsiveness in obese asthma mice. This was achieved by inhibiting NLRP3 inflammasome activity and IL-1β expression. The results suggest GLP-1 receptor agonists as a potential novel treatment for asthma in obese individuals (Hur et al. 2021). Xia et al. demonstrated that liraglutide, a GLP-1 analog, reduced intimal hyperplasia after coronary stent implantation in diabetic pigs by regulating glycemic variability, NLRP3 inflammasome activity, and IL-10 levels. The treatment decreased inflammation and improved artery healing, suggesting that liraglutide’s cardioprotective effects were mediated through GLP-1 receptor-dependent pathways (Xia 2020). Another study investigated the effects of GLP-1 receptor agonists liraglutide and semaglutide on podocyte survival under high glucose conditions using mouse kidney podocyte MPC5 cells. It demonstrated that both agonists increased podocyte survival by suppressing the NLRP3 inflammasome pathway and reducing pyroptosis-related gene and protein expressions, along with inflammatory factors IL-1β and IL-18. The findings suggest that liraglutide and semaglutide may offer protective benefits for podocytes in pyroptosis and diabetic kidney disease (Li et al. 2024b). Liraglutide, known for its neuroprotective effects crossing the blood–brain barrier, was studied for its mechanisms in ischemic stroke. In a rat model of middle cerebral artery occlusion (MCAO), liraglutide reduced brain edema, infarct size, neurological deficits, and neuronal apoptosis while enhancing neuron survival. These effects were mediated through GLP-1 receptor activation, promoting M2 polarization, activating Nrf2, and inhibiting NLRP3 activation in microglial cells (Tu et al. 2023). GLP-1 agonists like liraglutide show promise in treating non-alcoholic steatohepatitis (NASH), a critical stage in non-alcoholic fatty liver disease (NAFLD). They inhibit NLRP3 inflammasome activation, reducing pyroptosis and improving liver function in NASH by enhancing mitophagy and mitigating mitochondrial dysfunction and oxidative stress (Yu et al. 2019). Semaglutide, a long-acting GLP-1 receptor agonist, attenuates pressure overload-induced cardiac hypertrophy in rats. It enhances cardiac mitophagy, reducing left ventricular hypertrophy (LVH) and suppressing NLRP3 inflammasome activation. However, inhibition of mitophagy with hydroxychloroquine reverses semaglutide’s beneficial effects on hypertrophy and inflammasome activation (He et al. 2024).
GLP-1 receptor agonists exhibit anti-inflammatory effects by modulating the AMPK/mTOR pathway, impacting the NLRP3 inflammasome. While effective, these drugs can cause gastrointestinal side effects, including nausea and vomiting. There are also concerns about their association with pancreatitis and a potential risk of thyroid cancer. These side effects can limit the long-term use of GLP-1 RAs in patients, despite their promising role in targeting inflammation associated with diabetes (Wan et al. 2024).
In summary, GLP-1 receptor agonists exert diverse therapeutic effects by targeting the NLRP3 inflammasome across multiple clinical pathologies. From alleviating diabetic neuropathic pain and endothelial dysfunction to reducing airway inflammation in asthma and cardiac hypertrophy in pressure overload, these agents demonstrate potential in improving outcomes through their anti-inflammatory and tissue-protective actions.
Alpha-glucosidase inhibitors
Alpha-glucosidase inhibitors like acarbose and miglitol reduce postprandial blood glucose levels by inhibiting alpha-glucosidase enzymes in the small intestine. These enzymes normally break down complex carbohydrates into simple sugars for absorption. By slowing this process, alpha-glucosidase inhibitors delay glucose absorption, thereby reducing the rise in blood sugar after meals. They are effective in T2DM management and are typically used in combination with other antidiabetic medications. Side effects include gastrointestinal discomfort due to undigested carbohydrates reaching the colon. These drugs do not cause hypoglycemia and are contraindicated in patients with inflammatory bowel disease or intestinal disorders (Derosa and Maffioli 2012).
Acarbose effectively inhibited NLRP3 inflammasome activation induced by high glucose in rat aortic endothelial cells (RAECs). This inhibition was associated with decreased Nox4-dependent superoxide production, a known regulator of NLRP3 activation. Acarbose treatment also restored levels of junction proteins ZO-1 and VE-Cadherin, reducing vascular hyperpermeability. In diabetic rats, acarbose administration improved vascular integrity and endothelial function, as evidenced by reduced vascular leakage and enhanced vasodilatory responses. These findings suggest that acarbose protects against endothelial barrier dysfunction by directly targeting NLRP3 inflammasome activation and oxidative stress, highlighting its potential therapeutic role in preventing cardiovascular complications in diabetes (Li et al. 2019).
Recent studies suggest that alpha-glucosidase inhibitors might also have anti-inflammatory effects that could influence the NLRP3 inflammasome pathway. However, their efficacy in directly modulating NLRP3 activity is not well-established. The main limitations of alpha-glucosidase inhibitors include gastrointestinal side effects like flatulence, diarrhea, and abdominal discomfort, which can significantly impact patient adherence to therapy. These side effects, coupled with the relatively modest impact on HbA1c levels, may limit their utility as a primary strategy for targeting the NLRP3 inflammasome in diabetes management (Khalili and Safavipour 2020).
Thiazolidinediones (TZDs)
Thiazolidinediones (TZDs) such as pioglitazone and rosiglitazone act by activating peroxisome proliferator-activated receptor gamma (PPARγ), primarily in adipose tissue and other organs. This activation enhances insulin sensitivity by promoting adipocyte differentiation, reducing circulating free fatty acids, and improving glucose uptake in skeletal muscle and liver. Beyond glycemic control, TZDs exert anti-inflammatory effects by inhibiting nuclear factor-kappa B (NF-κB) and reducing pro-inflammatory cytokine production.
Lobeglitazone, a thiazolidinedione, exhibits anti-inflammatory properties, reducing LPS-induced NLRP3 activation, cytokine production, hepatocyte inflammation, and hepatic stellate cell HSC activation. It also suppresses liver fibrosis by inhibiting TGF-β secretion and CTGF expression (Seo et al. 2023) Pioglitazone inhibits the mitochondrial pyruvate carrier (MPC). This inhibition enhances NLRP3 inflammasome activation and IL-1β secretion in macrophages by skewing ATP-linked mitochondrial oxygen consumption toward cytosolic lactate production. This metabolic shift exacerbates NLRP3 activation in response to MSU crystals, potentially increasing susceptibility to auto-inflammatory diseases like gout in diabetic patients (Chen et al. 2023).
Lobeglitazone demonstrates anti-inflammatory effects by inhibiting NLRP3 activation and cytokine production, mitigating liver inflammation, and suppressing fibrosis markers. In contrast, pioglitazone’s inhibition of MPC enhances NLRP3 activation via metabolic shifts, potentially exacerbating inflammation and auto-inflammatory conditions like gout in diabetic individuals. The studies exploring action of TZDs on inflammasome activation are limited and require further investigation.
Despite their efficacy in treating T2DM, TZDs are associated with adverse effects including weight gain, fluid retention, heat failure, and increased fracture risk, requiring careful consideration in patient management to balance benefits and risks, making their use less favorable despite their potential benefits in reducing NLRP3 inflammasome activity (Hauner 2002; Yasmin and Jayaprakash 2017).
NLRP3 inhibitors: recent advances in drug development
Various inhibitors, ranging from small molecules to biologics, have shown promise in preclinical studies by attenuating NLRP3-mediated inflammation and associated pathologies. Understanding the mechanisms and potential of NLRP3 inhibitors holds promise for developing novel treatments against a spectrum of inflammatory diseases (Table 2).
A randomized controlled trial (RCT) done on NAFLD patients in 2021 found increased NLRP3 inflammasome activation compared to healthy controls. Anthocyanins significantly reduced this activation, highlighting their potential as a safe and effective anti-inflammatory treatment for NAFLD (Zhu et al. 2022). A first-in-human study on the oral NLRP3 inhibitor DFV890 in healthy participants found it generally well-tolerated with no serious adverse events. DFV890 showed effective pharmacokinetics and pharmacodynamics, supporting its further development (Gatlik et al. 2024). ZYIL1, an NLRP3 inhibitor, was well-tolerated in healthy subjects, showing rapid absorption and dose-proportional exposure. Both single and multiple doses led to significant IL-1β and IL-18 inhibition, supporting its potential for treating inflammatory disorders (Parmar et al. 2023). A clinical trial in 2020 analyzed colchicine’s impact on NLRP3 inflammasome activation in COVID-19 patients. Colchicine reduced serum Casp1p20 and IL-18, decreasing hospitalization duration and oxygen needs, demonstrating its safety and efficacy (Amaral et al. 2023). A study on ST-segment elevation myocardial infarction (STEMI) patients undergoing PCI showed that adding colchicine reduced NLRP3 levels compared to placebo, though the reduction was not statistically significant, suggesting potential anti-inflammatory benefits and safety (Karim et al. 2024). A phase 1B study on heart failure patients with reduced ejection fraction (HFrEF) found that the NLRP3 inhibitor dapansutrile (OLT1177) was safe and well-tolerated, with some improvements in left ventricular ejection fraction and exercise time.(Wohlford et al. 2020) In a phase 1b clinical trial, the NLRP3 inhibitor selnoflast was well-tolerated in ulcerative colitis patients, achieving effective IL-1β inhibition. However, PD biomarkers showed no significant therapeutic effects (Klughammer et al. 2023). In a RCT of Behçet’s disease, zinc gluconate (30 mg/day) significantly reduced NLRP3 inflammasome expression and IL-1β levels, improving genital ulcers and inflammation, suggesting its potential as an effective adjuvant therapy (Faghfouri et al. 2022). In elderly postmenopausal osteoporosis patients, Drynaria fortunei improved bone mineral density and lipid profiles, significantly reduced NLRP3 inflammasome activity, and downregulated Notch1, showing potential as a therapeutic option (Lu et al. 2022).
There are several other potential candidates that are still under preclinical screening that are being investigated extensively for their ability to target NLRP3 inflammasome activation. The NLRP3 inhibitor MCC950 reduced atherosclerotic lesions, inflammation, and oxidative stress, and improved vascular function and cognitive function in diabetic mice, suggesting therapeutic potential (Sharma et al. 2020; Ward et al. 2019). But in a later study conducted by Hong et al., inhibiting NLRP3 with MCC950 in diabetic mice unexpectedly worsened renal inflammation, oxidative stress, and pathology, leading to increased mesangial expansion and glomerulosclerosis, indicating a lack of renoprotective effects in this context (Hong et al. 2019). Nlrp3-deficient mice show protection against nephropathy, and IL-1R antagonism via Anakinra reverses the disease, indicating therapeutic potential (Shahzad et al. 2015). CY-09 inhibited NLRP3, reducing inflammation, apoptosis, and fibrosis in diabetic mice and in vitro, suggesting potential as a therapeutic for diabetic kidney disease (Yang and Zhao 2023). Parthenolide (PN), an NLRP3 inflammasome inhibitor, reduced inflammation markers and improved insulin resistance, suggesting it as a potential therapeutic for obesity-induced insulin resistance (Chinta et al. 2022). Isoliquiritigenin (ILG), a chalcone from Glycyrrhiza uralensis, inhibits NLRP3 inflammasome independent of TLR4. It surpasses parthenolide in potency. ILG inhibits AIM2 (absent in melanoma 2) inflammasome and ASC oligomerization, reducing IAPP-induced IL-1β production more effectively than glyburide. ILG mitigates HFD-induced obesity, hypercholesterolemia, insulin resistance, liver steatosis, and adipose tissue inflammation, indicating potential for treating NLRP3 inflammasome-associated diseases (Honda et al. 2014).
Memantine, known for its Alzheimer’s treatment, shows promise in diabetic retinopathy (DRET) by inhibiting glutamate-induced NLRP3 inflammasome activation via TXNIP. It improves retinal structure, reduces oxidative stress markers, and lowers inflammatory cytokines, suggesting therapeutic potential pending clinical trials (El-Sayed et al. 2023). BAY 11–7082, a selective NLRP3 inflammasome inhibitor, was tested in high-fat, high-fructose diet-fed mice. It attenuated diabetes-associated metabolic abnormalities and organ injury by inhibiting NLRP3 activation, caspase-1, and interleukin production, suggesting therapeutic promise against diabetic complications (Chiazza et al. 2015). In chronic metabolic diseases, NATx0, a nitroalkene-Trolox™ derivative, inhibited NF-kB and NLRP3 inflammasome activation, reducing inflammation in vitro and improving glucose tolerance in diet-induced obese mice, highlighting its potential as a therapeutic candidate (Dapueto et al. 2021). Rosuvastatin, known for its antioxidant and anti-inflammatory properties, attenuated LPS-induced cardiac injury in mice and H9C2 cells. It improved cardiac function, reduced apoptosis and oxidative stress markers, and inhibited the NLRP3/TLR4 pathway (Ren et al. 2021).
Oridonin, known for anti-inflammatory properties, attenuated renal fibrosis and preserved kidney function in DN. It inhibited TXNIP/NLRP3 and NF-κB pathways while activating PPARγ, suggesting therapeutic promise (Huang et al. xxxx). Loganin shows promise in treating diabetic kidney disease (DKD) by reducing blood glucose, urea nitrogen, and creatinine levels in vivo. It inhibits pyroptosis-related proteins and cytokines, and in vitro studies confirm its efficacy in reducing pyroptosis in HK-2 cells via NLRP3 inflammasome inhibition and ROS reduction (Kong et al. 2023). Schisandrin B, a traditional Chinese medicine with anti-inflammatory and antioxidant properties, potentially reduces P2X7 receptor expression in diabetic rats. It ameliorates cardiovascular dysfunctions and chronic inflammation (Zhang et al. 2023). The study explored astragaloside IV (AS-IV) as a protector against DN by enhancing klotho expression. AS-IV mitigated podocyte pyroptosis via NLRP3 inhibition through the NF-κB pathway, suggesting therapeutic potential for DN (He et al. 2023). Breviscapine (Bre) mitigates podocyte injury in DN. In diabetic mice, Bre preserves renal function, enhances podocyte viability, and reduces apoptosis. It inhibits NF-κB/NLRP3 signaling, attenuating pyroptosis and improving renal outcomes in diabetes-induced podocyte injury (Sun et al. 2023).
Montelukast (Mon) was found to significantly reduce the renal expression of NLRP3 (NOD-like receptor family pyrin domain containing 3) in diabetic rats. This action suggests that Mon’s renoprotective effects in DN may be mediated, at least in part, by its ability to inhibit NLRP3 inflammasome activation (Awad et al. 2024). Piperine (PIP) demonstrated significant benefits in streptozotocin (STZ)-induced diabetic rats by reducing glycemia, oxidative stress markers, and lipid profiles. It improved aortic structure, function, and reduced inflammation through TXNIP-NLRP3 pathway modulation (Amin et al. 2023). L-ergothioneine, a mitochondrial antioxidant, mitigates placental inflammation in GDM by reducing mitochondrial superoxide production and NLRP3 inflammasome activation (McElwain et al. 2024). Quercetin, a natural flavonoid, mitigates pulmonary dysfunction induced by diabetes in rats by restoring glucose and insulin levels, improving arterial blood gases, reducing oxidative stress markers, inhibiting NLRP3-mediated pyroptosis, and alleviating pathological lung changes (El-Shaer et al. 2023). Other such agents include triptolide, taxifolin, suramin, and salidroside that have been extensively been studied for their action toward NLRP3 mediate inflammasome pathway (Iwasa, et al. 2023; Lv et al. 2023; Oda et al. 2023; Zhou et al. 2023).
The abovementioned therapeutic agents influence NLRP3 inflammasome pathway to ameliorate all major complication of diabetes. There are several other agents that are under research that have not been mentioned due to the extensive list and vast research data available.
Challenges and future implications
Developing inhibitors that specifically target the NLRP3 inflammasome without affecting other essential inflammatory pathways is a significant challenge. Off-target effects can lead to unintended consequences and undermine therapeutic efficacy. Efficient delivery of NLRP3 inhibitors to target tissues, such as pancreatic β-cells, renal glomeruli, and retinal cells, remains a challenge. Overcoming barriers like cell membrane permeability and systemic distribution is crucial for effective treatment outcomes. Diabetes is characterized by multifaceted pathophysiological mechanisms, including chronic inflammation, oxidative stress, and metabolic dysregulation. Targeting NLRP3 inflammasome alone may not suffice to address the complexity of diabetic complications comprehensively. Long-term safety profiles of NLRP3 inhibitors need thorough evaluation to mitigate potential adverse effects, including immune suppression, increased susceptibility to infections, and interference with normal immune responses. Moving from preclinical models to clinical trials involves navigating regulatory hurdles, optimizing dosage regimens, and demonstrating robust efficacy and safety profiles in diverse patient populations with different stages and types of diabetes. Advancements in biomarker research can facilitate the identification of patient subgroups most likely to benefit from NLRP3 inflammasome-targeted therapies. Personalized treatment approaches based on genetic, molecular, and clinical profiling hold promise for optimizing therapeutic outcomes.
The use of oral hypoglycemic agents (OHAs) to target the NLRP3 inflammasome is limited by several factors, primarily their side effects. Metformin can cause gastrointestinal disturbances and lactic acidosis, particularly in patients with renal impairment. Thiazolidinediones are associated with weight gain, fluid retention, heart failure, and an increased risk of bone fractures. GLP-1 receptor agonists may lead to gastrointestinal issues, pancreatitis, and potential thyroid cancer, while DPP-4 inhibitors carry risks of pancreatitis and severe joint pain. SGLT2 inhibitors are linked to urinary tract infections, genital infections, and diabetic ketoacidosis, especially in type 1 diabetes. In addition, alpha-glucosidase inhibitors often cause gastrointestinal discomfort, which can impact patient adherence. Beyond these side effects, a significant limitation is the indirect nature of these drugs’ effects on the NLRP3 inflammasome. Most OHAs primarily focus on glucose regulation, with their anti-inflammatory actions being secondary. This indirect targeting may require combination therapies to achieve significant results. Furthermore, variability in patient response and the need for individualized treatment plans add to the complexity of using OHAs as therapeutic agents for NLRP3 inflammasome modulation. Understanding these limitations is crucial for developing effective and safe strategies for managing inflammation-related complications in diabetes.
Combining NLRP3 inhibitors with existing diabetes treatments, such as insulin sensitizers, GLP-1 agonists, and SGLT-2 inhibitors, may offer synergistic effects and enhance therapeutic efficacy. This approach could address multiple pathological pathways simultaneously and improve patient outcomes. Development of biologic agents, including monoclonal antibodies and gene therapies targeting NLRP3 inflammasome components, represents a cutting-edge approach. These therapies can provide highly specific inhibition with potentially lower off-target effects compared to small molecule inhibitors.
Advancements in nanotechnology enable targeted delivery of NLRP3 inhibitors to specific cells and tissues affected in diabetes. Nanoformulations can enhance drug stability, improve bioavailability, and reduce systemic toxicity, thereby optimizing therapeutic outcomes. Collaborative efforts between basic researchers, clinicians, and pharmaceutical industries are essential for translating promising preclinical findings into clinically viable therapies. Continuous refinement of experimental models and clinical trial designs will accelerate the translation of NLRP3-targeted therapies from bench to bedside.
In conclusion, while targeting the NLRP3 inflammasome in diabetes presents substantial challenges related to specificity, delivery, safety, and clinical translation, ongoing research holds immense potential for innovative therapeutic approaches. Future applications focusing on precision medicine, combination therapies, biological agents, nanotechnology, and translational research are poised to revolutionize the management of diabetes and its associated complications. These efforts underscore the critical role of interdisciplinary collaboration and technological innovation in advancing diabetes therapeutics toward more effective and personalized treatments.
Conclusion
The activation of the NLRP3 inflammasome is crucial in the progression of diabetes and its complications. This inflammasome mediates inflammation, exacerbating insulin resistance and beta-cell dysfunction, and contributing to complications such as neuropathy, nephropathy, retinopathy, and CVD. Understanding the mechanisms behind NLRP3 inflammasome activation in diabetes is essential for developing effective therapeutic strategies.
Oral hypoglycemic drugs, beyond lowering blood glucose levels, also exhibit anti-inflammatory properties by targeting the NLRP3 inflammasome. Sulfonylureas inhibit NLRP3 activation and downstream inflammatory pathways, protecting pancreatic β-cells and offering dual benefits in diabetes management. SGLT2 inhibitors reduce NLRP3 inflammasome activation through mechanisms like oxidative stress modulation and autophagy, showing promise in treating DN and CVD. DPP4 inhibitors suppress NLRP3 inflammasome activity, reducing inflammation and oxidative stress, providing potential therapeutic benefits in DN and cardiovascular conditions. GLP-1 receptor agonists inhibit NLRP3 inflammasome activation, improving endothelial function, reducing inflammation, and offering protective benefits in diabetic complications such as neuropathy and CVD. Alpha-glucosidase inhibitors target NLRP3 inflammasome activation and oxidative stress, protecting against endothelial dysfunction and vascular complications in diabetes. Thiazolidinediones exhibit varying effects on NLRP3 inflammasome activation; some reduce inflammation and fibrosis, while others may exacerbate inflammasome activity under certain conditions, highlighting the need for further research.
The anti-inflammatory effects of these oral hypoglycemic drugs represent a promising avenue for mitigating the inflammatory burden in diabetes and its complications. By targeting the NLRP3 inflammasome, these therapies not only improve glycemic control but also address the underlying inflammatory mechanisms driving diabetic complications. Future research should continue to explore these mechanisms to develop more effective and comprehensive treatment strategies for diabetes. The dual role of these drugs in managing both hyperglycemia and inflammation underscores their potential in offering a multifaceted approach to diabetes treatment. As our understanding of the interplay between inflammation and diabetes deepens, these therapeutic strategies could significantly improve patient outcomes, emphasizing the importance of continued research and development in this field.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Alzokaky AA et al (2023) Metformin ameliorates doxorubicin-induced cardiotoxicity targeting HMGB1/TLR4/NLRP3 signaling pathway in mice. Life Sci 316:121390
Amaral NB et al (2023) Colchicine reduces the activation of NLRP3 inflammasome in COVID-19 patients. Inflamm Res off J Eur Histamine Res Soc 72(5):895–899
Amin FM, Shehatou GSG, Nader MA, Abdelaziz RR (2023) Piperine mitigates aortic vasculopathy in streptozotocin-diabetic rats via targeting TXNIP-NLRP3 signaling. Life Sci 314:121275
Arab HH, Ashour AM, Gad AM, Mahmoud AM, Kabel AM (2021) Activation of AMPK/mTOR-driven autophagy and inhibition of NLRP3 inflammasome by saxagliptin ameliorate ethanol-induced gastric mucosal damage. Life Sci 280:119743
Awad AM, Elshaer SL, Gangaraju R, Abdelaziz RR, Nader MA (2024) Ameliorative effect of montelukast against STZ induced diabetic nephropathy: targeting HMGB1, TLR4, NF-κB, NLRP3 inflammasome, and autophagy pathways. Inflammopharmacology 32(1):495–508
Birnbaum Y, Bajaj M, Qian J, Ye Y (2016) Dipeptidyl peptidase-4 inhibition by Saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res & Care 4(1):e000227
Birnbaum Y, Tran D, Bajaj M, Ye Y (2019) DPP-4 inhibition by linagliptin prevents cardiac dysfunction and inflammation by targeting the Nlrp3/ASC inflammasome. Basic Res Cardiol 114(5):35
Cai J et al (2021) NLRP3 inflammasome mediated pyroptosis is involved in cadmium exposure-induced neuroinflammation through the IL-1β/IkB-α-NF-κB-NLRP3 feedback loop in swine. Toxicology 453:152720
Chen H et al (2020) Identification of NLRP3 inflammation-related gene promoter hypomethylation in diabetic retinopathy. Invest Ophthalmol vis Sci 61(13):12
Chen H, Tran D, Yang H-C, Nylander S, Birnbaum Y, Ye Y (2020a) Dapagliflozin and ticagrelor have additive effects on the attenuation of the activation of the NLRP3 inflammasome and the progression of diabetic cardiomyopathy: an AMPK–mTOR interplay. Cardiovasc Drugs Ther 34(4):443–461
Chen Y et al (2020b) Discovery and optimization of 4-oxo-2-thioxo-thiazolidinones as NOD-like receptor (NLR) family, pyrin domain-containing protein 3 (NLRP3) inhibitors. Bioorg Med Chem Lett 30(7):127021
Chen W et al (2022) The upregulation of NLRP3 inflammasome in dorsal root ganglion by ten-eleven translocation methylcytosine dioxygenase 2 (TET2) contributed to diabetic neuropathic pain in mice. J Neuroinflammation 19(1):302
Chen L-C et al (2023) Inactivation of mitochondrial pyruvate carrier promotes NLRP3 inflammasome activation and gout development via metabolic reprogramming. Immunology 169(3):271–291
Chen Y et al (2024) MSC-derived small extracellular vesicles alleviate diabetic retinopathy by delivering miR-22-3p to inhibit NLRP3 inflammasome activation. Stem Cells 42(1):64–75
Chiazza F et al (2015) Targeting the NLRP3 inflammasome to Reduce Diet-induced Metabolic Abnormalities in Mice. Mol Med 21(1):1025–1037
Chinta PK, Tambe S, Umrani D, Pal AK, Nandave M (2022) Effect of parthenolide, an NLRP3 inflammasome inhibitor, on insulin resistance in high-fat diet-obese mice. Can J Physiol Pharmacol 100(3):272–281
Chung I-C et al (2019) Pretreatment with a heat-killed probiotic modulates the NLRP3 inflammasome and attenuates colitis-associated colorectal cancer in mice. Nutrients. https://doi.org/10.3390/nu11030516
Dai Y, Dai D, Wang X, Ding Z, Mehta JL (2014) DPP-4 inhibitors repress NLRP3 Inflammasome and Interleukin-1beta via GLP-1 receptor in macrophages through protein Kinase C pathway. Cardiovasc Drugs Ther 28(5):425–432
Dai X et al (2021) Epigenetic regulation of TXNIP-mediated oxidative stress and NLRP3 inflammasome activation contributes to SAHH inhibition-aggravated diabetic nephropathy. Redox Biol 45:102033
Dapueto R et al (2021) A novel nitroalkene vitamin E analogue inhibits the NLRP3 inflammasome and protects against inflammation and glucose intolerance triggered by obesity. Redox Biol 39:101833
Deng B, Hu Y, Sheng X, Zeng H, Huo Y (2020) miR-223-3p reduces high glucose and high fat-induced endothelial cell injury in diabetic mice by regulating NLRP3 expression. Exp Ther Med 20(2):1514–1520
Derosa G, Maffioli P (2012) α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci 8(5):899–906
Ding H et al (2021) MicroRNA-10 negatively regulates inflammation in diabetic kidney via targeting activation of the NLRP3 inflammasome. Mol Ther 29(7):2308–2320
Dong T, Huang D, Jin Z (2024) Mechanism of sodium butyrate, a metabolite of gut microbiota, regulating cardiac fibroblast transdifferentiation via the NLRP3/Caspase-1 pyroptosis pathway. J Cardiothorac Surg 19(1):208
Drucker DJ (2018) Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 27(4):740–756
Dwivedi DK, Jena GB (2023) Dimethyl fumarate-mediated Nrf2/ARE pathway activation and glibenclamide-mediated NLRP3 inflammasome cascade inhibition alleviate type II diabetes-associated fatty liver in rats by mitigating oxidative stress and inflammation. J Biochem Mol Toxicol 37(7):e23357
El-Rous MA, Saber S, Raafat EM, Ahmed AAE (2021) Dapagliflozin, an SGLT2 inhibitor, ameliorates acetic acid-induced colitis in rats by targeting NFκB/AMPK/NLRP3 axis. Inflammopharmacology 29(4):1169–1185
ElSayed MH et al (2023) Memantine mitigates ROS/TXNIP/NLRP3 signaling and protects against mouse diabetic retinopathy: histopathologic, ultrastructural and bioinformatic studies. Biomed Pharmacother 163:114772
El-Shaer NO, Hegazy AM, Muhammad MH (2023) Protective effect of quercetin on pulmonary dysfunction in streptozotocin-induced diabetic rats via inhibition of NLRP3 signaling pathway. Environ Sci Pollut Res 30(14):42390–42398
Escartín-Gutiérrez JR, Ponce-Figueroa M, Torres-Vega MÁ, Aguilar-Faisal L, Figueroa-Arredondo P (2023) Transcriptional activation of a pro-inflammatory response (NF-κB, AP-1, IL-1β) by the Vibrio cholerae cytotoxin (VCC) MONOMER THROUGH the MAPK signaling pathway in the thp-1 human macrophage cell line. Int J Mol Sci. https://doi.org/10.3390/ijms24087272
Faghfouri AH et al (2022) Regulation of NLRP3 inflammasome by zinc supplementation in Behçet’s disease patients: a double-blind, randomized placebo-controlled clinical trial. Int Immunopharmacol 109:108825
Fan Z, Yang J, Yang C, Zhang J, Cai W, Huang C (2020) MicroRNA-24 attenuates diabetic vascular remodeling by suppressing the NLRP3/caspase-1/IL-1β signaling pathway Corrigendum in /10.3892/ijmm.2022.5079. Int J Mol Med 45(5):1534–1542
Fang J et al (2022) Advanced glycation end products promote melanogenesis by activating NLRP3 inflammasome in human dermal fibroblasts. J Invest Dermatol 142(10):2591-2602.e8
Feng Y, Huang Y, Wang Y, Wang P, Song H, Wang F (2019) Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS ONE 14(6):e0218384
Foretz M, Guigas B, Viollet B (2023) Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinol 19(8):460–476
Galicia-Garcia U et al (2020) pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. https://doi.org/10.3390/ijms21176275
Gao Y et al (2020) Ginsenoside Rg1 protects mice against streptozotocin-induced type 1 diabetic by modulating the NLRP3 and Keap1/Nrf2/HO-1 pathways. Eur J Pharmacol 866:172801
Gatlik E et al (2024) First-in-human safety, tolerability, and pharmacokinetic results of DFV890, an oral low-molecular-weight NLRP3 inhibitor. Clin Transl Sci 17(5):e13789
Gayatri V, Krishna Prasad M, Mohandas S, Nagarajan S, Kumaran K, Ramkumar KM (2024) Crosstalk between inflammasomes, inflammation, and Nrf 2: implications for gestational diabetes mellitus pathogenesis and therapeutics. Eur J Pharmacol 963:176241
Gora IM, Ciechanowska A, Ladyzynski P (2021) NLRP3 inflammasome at the interface of inflammation, endothelial dysfunction, and type 2 diabetes. Cells. https://doi.org/10.3390/cells10020314
Gronda E et al (2022) Mechanisms of action of SGLT2 inhibitors and their beneficial effects on the cardiorenal axis. Can J Physiol Pharmacol 100(2):93–106
Habas A et al (2022) NPT1220-312, a TLR2/TLR9 small molecule antagonist, inhibits pro-inflammatory signaling, cytokine release, and NLRP3 inflammasome activation. Int J Inflam 2022(1):2337363
Han L, Xi G, Guo N, Guo J, Rong Q (2022) Expression and mechanism of TXNIP/NLRP3 inflammasome in sciatic nerve of type 2 diabetic rats. Dis Markers 2022(1):9696303
Hao W et al (2021) Xiaoyaosan improves antibiotic-induced depressive-like and anxiety-like behavior in mice through modulating the gut microbiota and regulating the NLRP3 inflammasome in the colon. Front Pharmacol. https://doi.org/10.3389/fphar.2021.619103
Hauner H (2002) The mode of action of thiazolidinediones. Diabetes Metab Res Rev 18(S2):S10–S15
He M et al (2020) An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab 31(3):580-591.e5
He Y et al (2022) Glibenclamide directly prevents neuroinflammation by targeting SUR1-TRPM4-Mediated NLRP3 inflammasome activation in microglia. Mol Neurobiol 59(10):6590–6607
He J et al (2023) Astragaloside IV attenuates high-glucose-induced impairment in diabetic nephropathy by increasing klotho expression via the NF-κB/NLRP3 Axis. J Diabetes Res 2023(1):7423661
He W, Wei J, Liu X, Zhang Z, Huang R, Jiang Z (2024) Semaglutide ameliorates pressure overload-induced cardiac hypertrophy by improving cardiac mitophagy to suppress the activation of NLRP3 inflammasome. Sci Rep 14(1):11824
Hill JR et al (2017) Sulfonylureas as concomitant insulin secretagogues and NLRP3 inflammasome inhibitors. ChemMedChem 12(17):1449–1457
Honda H et al (2014) Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J Leukoc Biol 96(6):1087–1100
Hong P et al (2019) NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflammation 16(1):121
Hou L et al (2020) Glibenclamide attenuates 2,5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol Lett 331:152–158
Hou Y, Wang Q, Han B, Chen Y, Qiao X, Wang L (2021) CD36 promotes NLRP3 inflammasome activation via the mtROS pathway in renal tubular epithelial cells of diabetic kidneys. Cell Death Dis 12(6):523
Hou Q et al (2022) Inhibition of HDAC6 With CAY10603 ameliorates diabetic kidney disease by suppressing NLRP3 inflammasome. Front Pharmacol. https://doi.org/10.3389/fphar.2022.938391
Hu J et al (2023) Dapagliflozin protects against dilated cardiomyopathy progression by targeting NLRP3 inflammasome activation. Naunyn Schmiedebergs Arch Pharmacol 396(7):1461–1470
Huang D, Gao W, Zhong X, Ge J (2020) NLRP3 activation in endothelia promotes development of diabetes-associated atherosclerosis. Aging 12(18):18181–18191
Huang L, Xiong S, Liu H, Zhang R, Wu Y, Hu X (2023) Silencing LncRNA SNHG16 suppresses the diabetic inflammatory response by targeting the miR-212-3p/NF-κB signaling pathway. Diabetol Metab Syndr 15(1):119
Huang G, Zhang Y, Zhang Y, Zhou X, Xu Y, Wei H, Chen X, Ma Y Oridonin attenuates diabetes‑induced renal fibrosis via the inhibition of TXNIP/NLRP3 and NF‑κB pathways by activating PPARγ in rats, Exp Clin Endocrinol Diabetes, vol. 0, no. AAM.
Hur J, Kang JY, Kim YK, Lee SY, Lee HY (2021) Glucagon-like peptide 1 receptor (GLP-1R) agonist relieved asthmatic airway inflammation via suppression of NLRP3 inflammasome activation in obese asthma mice model. Pulm Pharmacol Ther 67:102003
Iwasa M et al (2023) Taxifolin suppresses inflammatory responses of high-glucose-stimulated mouse microglia by attenuating the TXNIP–NLRP3 axis. Nutrients 15(12):2738
Jia R, Ma H, Hao H, Wang F, Yang H (2024) Metformin inhibits activation of NLRP3 inflammasome and inflammatory response in preeclamptic rats. Gene 919:148509
Jiang T, Jiang D, Zhang L, Ding M, Zhou H (2019) Anagliptin ameliorates high glucose- induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol Immunol 107:54–60
Karami Z, Mehrzad J, Akrami M, Hosseinkhani S (2023) Anti-inflammation-based treatment of atherosclerosis using Gliclazide-loaded biomimetic nanoghosts. Sci Rep 13(1):13880
Karim B et al (2024) Role of colchicine to reduce NLRP3 marker in STEMI patients undergo primary PCI: a randomised controlled clinical trial. Med J Malaysia 79(2):146–150
Karnam K et al (2020) HDAC6 inhibitor accelerates wound healing by inhibiting tubulin mediated IL-1β secretion in diabetic mice. Biochim Biophys Acta - Mol Basis Dis 1866(11):165903
Ke Q et al (2022) SGLT2 inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. FASEB J 36(1):e22078
Khalili N, Safavipour A (2020) Evaluation of the effects of acarbose on weight and metabolic, inflammatory, and cardiovascular markers in patients with obesity and overweight. Int J Prev Med 11(1):140
Kim SR et al (2020) SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun 11(1):2127
Kim D, Ban K-Y, Lee G-H, Jun H-S (2023) Lysophosphatidic acid induces podocyte pyroptosis in diabetic nephropathy by an Increase of Egr1 expression via downregulation of EzH2. Int J Mol Sci. https://doi.org/10.3390/ijms24129968
Klughammer B et al (2023) A randomized, double-blind phase 1b study evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of the NLRP3 inhibitor selnoflast in patients with moderate to severe active ulcerative colitis. Clin Transl Med 13(11):e1471
Kong H, Zhao H, Chen T, Song Y, Cui Y (2022) Targeted P2X7/NLRP3 signaling pathway against inflammation, apoptosis, and pyroptosis of retinal endothelial cells in diabetic retinopathy. Cell Death Dis 13(4):336
Kong X et al (2023) Loganin reduces diabetic kidney injury by inhibiting the activation of NLRP3 inflammasome-mediated pyroptosis. Chem Biol Interact 382:110640
Lan J, Xu B, Shi X, Pan Q, Tao Q (2022) WTAP-mediated N6-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett 27(1):51
Lathakumari RH, Vajravelu LK, Satheesan A, Ravi S, Thulukanam J (2024) Antibiotics and the gut microbiome: understanding the impact on human health. Med Microecol 20:100106
Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K (2012) Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62(1):194–204
Li A, Zhang S, Li J, Liu K, Huang F, Liu B (2016) Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol Cell Endocrinol 434:36–47
Li X-X, Ling S-K, Hu M-Y, Ma Y, Li Y, Huang P-L (2019) Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic Biol Med 145:175–186
Li W-Z, Stirling K, Yang J-J, Zhang L (2020) Gut microbiota and diabetes: from correlation to causality and mechanism. World J Diabetes 11(7):293–308
Li R et al (2021) Antidiabetic DPP-4 inhibitors reprogram tumor microenvironment that facilitates murine breast cancer metastasis through interaction with cancer cells via a ROS–NF-кB–NLRP3 Axis. Front Oncol. https://doi.org/10.3389/fonc.2021.728047
Li J, Wang C, Shao C, Xu J (2024a) Expression and diagnostic value of lncRNA MALAT1 and NLRP3 in lower limb atherosclerosis in diabetes. BMC Endocr Disord 24(1):28
Li X et al (2024b) GLP-1RAs inhibit the activation of the NLRP3 inflammasome signaling pathway to regulate mouse renal podocyte pyroptosis. Acta Diabetol 61(2):225–234
Liu Y, Bi X, Zhang Y, Wang Y, Ding W (2020) Mitochondrial dysfunction/NLRP3 inflammasome axis contributes to angiotensin II-induced skeletal muscle wasting via PPAR-γ. Lab Investig 100(5):712–726
Liu B-H et al (2021) Total flavones of abelmoschus manihot ameliorates podocyte pyroptosis and injury in high glucose conditions by targeting METTL3-dependent m(6)A modification-mediated NLRP3-inflammasome activation and PTEN/PI3K/Akt Signaling. Front Pharmacol 12:667644
Liu Y et al (2022) Fructose induces insulin resistance of gestational diabetes mellitus in mice via the NLRP3 inflammasome pathway. Front Nutr 9:839174
Liu Y et al (2023) LncRNA SNHG16 regulates RAS and NF-κB pathway-mediated NLRP3 inflammasome activation to aggravate diabetes nephropathy through stabilizing TLR4. Acta Diabetol 60(4):563–577
Lu L, Zou G, Chen L, Lu Q, Wu M, Li C (2021) Elevated NLRP3 inflammasome levels correlate with vitamin D in the vitreous of proliferative diabetic retinopathy. Front Med. https://doi.org/10.3389/fmed.2021.736316
Lu L, Wang Z, Zhang H, Liu T, Fang H (2022) Drynaria fortunei improves lipid profiles of elderly patients with postmenopausal osteoporosis via regulation of Notch1-NLRP3 inflammasome-mediated inflammation. Gynecol Endocrinol off J Int Soc Gynecol Endocrinol 38(2):176–180
Lu S et al (2023) Role of the inflammasome in insulin resistance and type 2 diabetes mellitus. Front Immunol. https://doi.org/10.3389/fimmu.2023.1052756
Lucafò M et al (2021) Hypomethylation of NLRP3 gene promoter discriminates glucocorticoid-resistant from glucocorticoid-sensitive idiopathic nephrotic syndrome patients. Clin Transl Sci 14(3):964–975
Luo X et al (2019) Dulaglutide inhibits high glucose- induced endothelial dysfunction and NLRP3 inflammasome activation. Arch Biochem Biophys 671:203–209
Lv C, Cheng T, Zhang B, Sun K, Lu K (2023) Triptolide protects against podocyte injury in diabetic nephropathy by activating the Nrf2/HO-1 pathway and inhibiting the NLRP3 inflammasome pathway. Ren Fail 45(1):2165103
Ma X et al (2021) GLP-1 receptor agonists (GLP-1RAs): cardiovascular actions and therapeutic potential. Int J Biol Sci 17(8):2050–2068
Magupalli VG et al (2020) HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369(6510):8995
McElwain CJ, Musumeci A, Manna S, McCarthy FP, McCarthy CM (2024) L-ergothioneine reduces mitochondrial-driven NLRP3 activation in gestational diabetes mellitus. J Reprod Immunol 161:104171
Mehta S, Vijayvergiya R, Dhawan V (2020) Activation of NLRP3 inflammasome assembly is associated with smoking status of patients with coronary artery disease. Int Immunopharmacol 87:106820
Meng L, Lin H, Huang X, Weng J, Peng F, Wu S (2022) METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis 13(1):38
Mori W, Kaneko N, Nakanishi A, Zako T, Masumoto J (2021) Insulin amyloid fibrils interact directly with the NLRP3, resulting in inflammasome activation and pyroptotic cell death. Int J Immunopathol Pharmacol 35:20587384211038356
Mostafa RE, Morsi AH, Asaad GF (2021) Anti-inflammatory effects of saxagliptin and vildagliptin against doxorubicin-induced nephrotoxicity in rats: attenuation of NLRP3 inflammasome up-regulation and tubulo-interstitial injury. Res Pharm Sci. https://doi.org/10.4103/1735-5362.323920
Muhammad RN, Ahmed LA, Abdul Salam RM, Ahmed KA, Attia AS (2021) Crosstalk among NLRP3 inflammasome, ETBR signaling, and miRNAs in stress-induced depression-like behavior: a modulatory role for SGLT2 inhibitors. Neurotherapeutics 18(4):2664–2681
Narros-Fernández P et al (2022) Synthesis and pharmacological evaluation of new N-sulfonylureas as NLRP3 inflammasome inhibitors: identification of a hit compound to treat gout. J Med Chem 65(8):6250–6260
Ning Z-W et al (2016) MicroRNA-21 mediates angiotensin II-induced liver fibrosis by activating NLRP3 inflammasome/IL-1β axis via targeting smad7 and spry1. Antioxid Redox Signal 27(1):1–20
Niu Y et al (2022) Canagliflozin ameliorates NLRP3 inflammasome-mediated inflammation through inhibiting NF-κB signaling and upregulating Bif-1. Front Pharmacol 13:820541
Niu B, Xia X, Ma L, Yao L, Zhang Y, Su H (2023) LncRNA AC040162.3 promotes HCV-induced T2DM deterioration through the miRNA-223–3p/NLRP3 molecular axis. Anal Cell Pathol 2023(1):5350999
Oda K, Miyamoto S, Kodera R, Wada J, Shikata K (2023) Suramin prevents the development of diabetic kidney disease by inhibiting NLRP3 inflammasome activation in KK-Ay mice. J Diabetes Investig 14(2):205–220
Ouyang H, Wang Y, Wu J, Ji Y (2023) Mechanisms of pulmonary microvascular endothelial cells barrier dysfunction induced by LPS: The roles of ceramides and the Txnip/NLRP3 inflammasome. Microvasc Res 147:104491
Paneni F et al (2015) Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet 8(1):150–158
Parmar DV et al (2023) Safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral NLRP3 inflammasome inhibitor ZYIL1: first-in-human phase 1 studies (single ascending dose and multiple ascending dose). Clin Pharmacol Drug Dev 12(2):202–211
Pereira CA et al (2020) Mitochondrial DNA promotes NLRP3 inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes. Front Physiol 10:1557
Qi Y, Du X, Yao X, Zhao Y (2019) Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif Cells Nanomed Biotechnol 47(1):1067–1074
Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J (2019) Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Transl Res 11(2):655–668
Rai P, Dwibedi N, Rowneki M, Helmer DA, Sambamoorthi U (2019) Dipeptidyl peptidase-4 inhibitors and joint pain: a retrospective cohort study of older veterans with type 2 diabetes mellitus. Am Heal Drug Benefits 12(5):223–231
Raneros AB, Bernet CR, Flórez AB, Suarez-Alvarez B (2021) An epigenetic insight into NLRP3 inflammasome activation in inflammation-related processes. Biomedicines. https://doi.org/10.3390/biomedicines9111614
Ren G, Zhou Q, Lu M, Wang H (2021) Rosuvastatin corrects oxidative stress and inflammation induced by LPS to attenuate cardiac injury by inhibiting the NLRP3/TLR4 pathway. Can J Physiol Pharmacol 99(9):964–973
Rosen ED et al (2018) Epigenetics and epigenomics: implications for diabetes and obesity. Diabetes 67(10):1923–1931
Roshanravan N et al (2020) Effects of oral butyrate and inulin supplementation on inflammation-induced pyroptosis pathway in type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Cytokine 131:155101
Saini K, Sharma S, Khan Y (2023) DPP-4 inhibitors for treating T2DM - hype or hope? An analysis based on the current literature. Front Mol Biosci. https://doi.org/10.3389/fmolb.2023.1130625
Saisho Y (2020) SGLT2 inhibitors: the star in the treatment of type 2 diabetes? Diseases 8(2):14
Scheen AJ (2021) Sulphonylureas in the management of type 2 diabetes: to be or not to be? Diabetes Epidemiol Manag 1:100002
Seo H-Y et al (2023) Lobeglitazone inhibits LPS-induced NLRP3 inflammasome activation and inflammation in the liver. PLoS ONE 18(8):e0290532
Shahzad K et al (2015) Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int 87(1):74–84
Sharma A et al (2020) Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 70(3):772–787
Shen J, Dai Z, Li Y, Zhu H, Zhao L (2022) TLR9 regulates NLRP3 inflammasome activation via the NF-kB signaling pathway in diabetic nephropathy. Diabetol Metab Syndr 14(1):26
Silva-Veiga FM et al (2022) Peroxisome proliferator-activated receptor-alpha activation and dipeptidyl peptidase-4 inhibition target dysbiosis to treat fatty liver in obese mice. World J Gastroenterol 28(17):1814–1829
Singla D, Narasimhulu C (2023) BMP-7 attenuates TLR4-NLRP3 inflammasome mediated pyroptosis in vascular smooth muscle cells in atherosclerotic plaques. Atherosclerosis 379:S15–S16
Siwen Q et al (2021) Akkermansia muciniphila alleviates dextran sulfate sodium (DSS)-induced acute colitis by NLRP3 activation. Microbiol Spectr 9(2):e00730-e821
Sokolova M et al (2020) NLRP3 inflammasome deficiency attenuates metabolic disturbances involving alterations in the gut microbial profile in mice exposed to high fat diet. Sci Rep 10(1):21006
Song S et al (2018) Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. J Mol Endocrinol 61(3):101–113
Song Y et al (2023) Gemigliptin, a DPP4 inhibitor, ameliorates nonalcoholic steatohepatitis through AMP-activated protein kinase-independent and ULK1-mediated autophagy. Mol Metab 78:101806
Sukhanov S et al (2021) The SGLT2 inhibitor Empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1β and IL-18 secretion. Cell Signal 77:109825
Sun H-J et al (2017) NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis 8(10):e3074–e3074
Sun L, Ding M, Chen F, Zhu D, Xie X (2023) Breviscapine alleviates podocyte injury by inhibiting NF-κB/NLRP3-mediated pyroptosis in diabetic nephropathy. PeerJ 11:e14826
Tan J et al (2022) Probiotics ameliorate IgA nephropathy by improving gut dysbiosis and blunting NLRP3 signaling. J Transl Med 20(1):382
Tang G et al (2019) Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE−/− mice. Biomed Pharmacother 119:109410
Tarry-Adkins JL, Grant ID, Ozanne SE, Reynolds RM, Aiken CE (2021) Efficacy and side effect profile of different formulations of metformin: a systematic review and meta-analysis. Diabetes Ther 12(7):1901–1914
Thulé PM, Umpierrez G (2014) Sulfonylureas: a new look at old therapy. Curr Diab Rep 14(4):473
Tu X, Chen P, Chen J, Ding Y, Chen Q, Shi S (2023) GLP-1R knockdown abrogates the protective effects of liraglutide on ischaemic stroke via inhibition of M2 polarisation and activation of NLRP3 inflammasome by reducing Nrf2 activation. Neuropharmacology 237:109603
Valencia I et al (2022) DPP4 promotes human endothelial cell senescence and dysfunction via the PAR2–COX-2–TP axis and NLRP3 inflammasome activation. Hypertension 79(7):1361–1373
Wan J, Ferrari C, Tadros M (2024) GLP-1RA essentials in gastroenterology: side effect management, precautions for endoscopy and applications for gastrointestinal disease treatment. Gastroenterol Insights 15(1):191–212
Wang W, Zhang J (2023) Teneligliptin alleviates diabetes-related cognitive impairment by inhibiting the endoplasmic reticulum (ER) stress and NLRP3 inflammasome in mice. Aging 16(9):8336–8347
Wang S, Du S, Lv Y, Wang W, Zhang F (2020) Elevated microRNA-20b-3p and reduced thioredoxin-interacting protein ameliorate diabetic retinopathy progression by suppressing the NLRP3 inflammasomes. IUBMB Life 72(7):1433–1448
Wang D et al (2020a) P2X7 receptor mediates NLRP3 inflammasome activation in depression and diabetes. Cell Biosci 10(1):28
Wang J et al (2020b) High glucose mediates NLRP3 inflammasome activation via upregulation of ELF3 expression. Cell Death Dis 11(5):383
Wang H et al (2023) Yersiniabactin-producing E. coli induces the pyroptosis of intestinal epithelial cells via the NLRP3 pathway and promotes gut inflammation. Int J Mol Sci. https://doi.org/10.3390/ijms241411451
Wang F et al (2023) Sanziguben polysaccharides improve diabetic nephropathy in mice by regulating gut microbiota to inhibit the TLR4/NF-κB/NLRP3 signalling pathway. Pharm Biol 61(1):427–436
Ward R et al (2019) NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res 142:237–250
Williams BM, Cliff CL, Lee K, Squires PE, Hills CE (2022) The Role of the NLRP3 inflammasome in mediating glomerular and tubular injury in diabetic nephropathy. Front Physiol 13:907504
Wohlford GF et al (2020) Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 Inhibitor dapansutrile in subjects With NYHA II-III systolic heart failure. J Cardiovasc Pharmacol 77(1):49–60
Wu W et al (2022) NLRP3 inflammasome activation in gestational diabetes mellitus placentas is associated with hydrogen sulfide synthetase deficiency. Exp Ther Med 23(1):94
Wu J, Zhang D, Zhao M, Zheng X (2023) Gut microbiota dysbiosis and increased NLRP3 levels in patients with pregnancy-induced hypertension. Curr Microbiol 80(5):168
Xi X, Wang M, Chen Q, Ma J, Zhang J, Li Y (2023) DNMT1 regulates miR-20a/TXNIP-mediated pyroptosis of retinal pigment epithelial cells through DNA methylation. Mol Cell Endocrinol 577:112012
Xia J et al (2020) A GLP-1 analog liraglutide reduces intimal hyperplasia after coronary stent implantation via regulation of glycemic variability and NLRP3 Inflammasome/IL-10 signaling in diabetic swine. Front Pharmacol 11:372
Xian H et al (2021) Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54(7):1463-1477.e11
Xiong J, Zhou Q (2023) The lncRNA HOTAIR attenuates pyroptosis of diabetic cardiomyocytes by recruiting FUS to regulate SIRT3 expression. Kaohsiung J Med Sci 39(5):458–467
Xu T, Sheng L, Guo X, Ding Z (2022) Free fatty acid increases the expression of NLRP3-caspase1 in adipose tissue macrophages in obese severe acute pancreatitis. Dig Dis Sci 67(6):2220–2231
Xue C et al (2022) The relationships between cholesterol crystals, NLRP3 inflammasome, and coronary atherosclerotic plaque vulnerability in acute coronary syndrome: an optical coherence tomography study. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2022.905363
Yang M, Zhao L (2023) The selective NLRP3-inflammasome inhibitor CY-09 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3- inflammasome activation. Curr Med Chem 30(28):3261–3270
Yang S et al (2023) Disulfiram accelerates diabetic foot ulcer healing by blocking NET formation via suppressing the NLRP3/Caspase-1/GSDMD pathway. Transl Res 254:115–127
Yasmin S, Jayaprakash V (2017) Thiazolidinediones and PPAR orchestra as antidiabetic agents: from past to present. Eur J Med Chem 126:879–893
Ye T et al (2022) Empagliflozin attenuates obesity-related kidney dysfunction and NLRP3 inflammasome activity through the HO-1–adiponectin axis. Front Endocrinol. https://doi.org/10.3389/fendo.2022.907984
You H, Li H, Gou W (2023) lncRNA HOTAIR promotes ROS generation and NLRP3 inflammasome activation by inhibiting Nrf2 in diabetic retinopathy. Medicine (baltimore) 102(37):e35155
Yu X et al (2019) Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur J Pharmacol 864:172715
Yu Y-W et al (2022) Sodium-glucose co-transporter-2 inhibitor of dapagliflozin attenuates myocardial ischemia/reperfusion injury by limiting NLRP3 inflammasome activation and modulating autophagy. Front Cardiovasc Med 8:768214
Zhan J-F, Huang H-W, Huang C, Hu L-L, Xu W-W (2020) Long non-coding RNA NEAT1 regulates pyroptosis in diabetic nephropathy via mediating the miR-34c/NLRP3 Axis. Kidney Blood Press Res 45(4):589–602
Zhang Y et al (2020) Gut microbiome-related effects of berberine and probiotics on type 2 diabetes (the PREMOTE study). Nat Commun 11(1):5015
Zhang J et al (2020) Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 12(23):24270–24287
Zhang Y et al (2021) Metformin corrects glucose metabolism reprogramming and NLRP3 inflammasome-induced pyroptosis via inhibiting the TLR4/NF-κB/PFKFB3 signaling in trophoblasts: implication for a potential therapy of preeclampsia. Oxid Med Cell Longev 2021(1):1806344
Zhang X et al (2021) TXNIP, a novel key factor to cause Schwann cell dysfunction in diabetic peripheral neuropathy, under the regulation of PI3K/Akt pathway inhibition-induced DNMT1 and DNMT3a overexpression. Cell Death Dis 12(7):642
Zhang Y et al (2022) Gut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc Res 118(3):785–797
Zhang C et al (2022a) Targeting NLRP3 signaling by a novel-designed sulfonylurea compound for inhibition of microglial inflammation. Bioorg Med Chem 58:116645
Zhang Q et al (2022b) Glucagon-like peptide-1 receptor agonist attenuates diabetic neuropathic pain via inhibition of NOD-like receptor protein 3 inflammasome in brain microglia. Diabetes Res Clin Pract 186:109806
Zhang Y et al (2023) Lactobacillus plantarum NC8 and its metabolite acetate alleviate type 1 diabetes via inhibiting NLRP3. Microb Pathog 182:106237
Zhang Z et al (2023) Schisandrin B alleviates diabetic cardiac autonomic neuropathy induced by p2x7 receptor in superior cervical ganglion via NLRP3. Dis Markers 2023(1):9956950
Zhao J et al (2024) New insights into the interplay between autophagy, gut microbiota and insulin resistance in metabolic syndrome. Biomed Pharmacother 176:116807
Zheng D, Liwinski T, Elinav E (2020) Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6(1):36
Zheng X, Wan J, Tan G (2023) The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in diabetic retinopathy. Front Immunol. https://doi.org/10.3389/fimmu.2023.1151185
Zheng S et al (2023) Human umbilical cord mesenchymal stem cells inhibit pyroptosis of renal tubular epithelial cells through miR-342-3p/caspase1 signaling pathway in diabetic nephropathy. Stem Cells Int 2023(1):5584894
Zhou Z et al (2018) Association analysis of NLRP3 inflammation-related gene promotor methylation as well as mediating effects on T2DM and vascular complications in a southern han chinese population. Front Endocrinol (lausanne). https://doi.org/10.3389/fendo.2018.00709
Zhou L-H et al (2023) Metformin inhibits ovarian granular cell pyroptosis through the miR-670–3p/NOX2/ROS pathway. Aging 15(10):4429–4443
Zhou J et al (2023) Salidroside protects pancreatic β-cells against pyroptosis by regulating the NLRP3/GSDMD pathway in diabetic conditions. Int Immunopharmacol 114:109543
Zhou X et al (2024) Inhibition of METTL3 alleviates NLRP3 inflammasome activation via increasing ubiquitination of NEK7. Adv Sci 11(26):e2308786
Zhu X, Lin X, Zhang P, Liu Y, Ling W, Guo H (2022) Upregulated NLRP3 inflammasome activation is attenuated by anthocyanins in patients with nonalcoholic fatty liver disease: a case-control and an intervention study. Clin Res Hepatol Gastroenterol 46(4):101843
Acknowledgements
The authors would like to thank Mr. Rahul H L Research Scholar, Department of Microbiology SRM Medical College Hospital and Research Centre for critically reading the manuscript and providing valuable comments.
Funding
The authors did not receive any financial support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows—study conception, design, draft manuscript preparation and Visualization: Abhishek Satheesan and Ria Murugesan. Writing review, editing, investigation, and methodology: Janardanan Kumar and Matcha Angelin. Supervision and project administration: Kakithakara Vajravelu Leela. Data collection, analysis and software: Venkata Chaithanya. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Satheesan, A., Kumar, J., Leela, K.V. et al. Review on the role of nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome pathway in diabetes: mechanistic insights and therapeutic implications. Inflammopharmacol 32, 2753–2779 (2024). https://doi.org/10.1007/s10787-024-01556-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-024-01556-2