
Abstract
Type 2 diabetes mellitus (T2DM) is the most common chronic metabolic condition. Several genetic and environmental factors are involved in developing T2DM. Aging, inflammation, and obesity are the main contributors to the initiation of T2DM. They cause chronic sterile meta-inflammation and insulin resistance, thereby making a person more susceptible to developing T2DM. Metformin, a natural cationic biguanide, is widely used as the first-line treatment of T2DM. The exact action mechanism behind the glucose-lowering effect of metformin is not clear, but, presumably, metformin utilizes a broad spectrum of molecular mechanisms to control blood glucose including decreasing intestinal glucose absorption, inhibition of the hepatic gluconeogenesis, decreasing insulin resistance, etc. Recent studies have shown that metformin exerts its effects through the inhibition of mitochondrial respiratory chain complex 1 and the AMP-activated protein kinase (AMPK) activation, but it has been identified in the other studies that AMPK is not the sole hub in metformin mode of action or there are other unknown mechanisms which are involved and yet to be explored. Therefore, here, we discuss the updated findings of the mechanism of action of metformin that contributes to the meta-inflammation and inflammaging action. It is proposed that figuring out the precise mechanism of action of metformin could improve its application in the fields of obesity, inflammation, aging, and inflammaging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic life-lasting condition that is characterized by high blood glucose and insufficient insulin to overcome the already existing insulin resistance. It causes many complications like neuropathy, nephropathy, and retinopathy, and reduces life expectancy. A sedentary lifestyle and genetic factors, besides some medications, infections, and endocrinopathies, make an individual more susceptible to developing T2DM. If exercise and change in lifestyle fail to control high blood glucose levels, the administration of some medications like metformin is inevitable [1,2,3].
Metformin is a natural biguanide compound derived from the French lilac (also known as Galega officinalis) and has been introduced as a medication for T2DM treatment in the 1950s and has been widely prescribed ever since. Metformin reduces both fasting and postprandial blood glucose, via inhibition of hepatic gluconeogenesis and alleviating the insulin resistance [4].
Recently, the pleiotropic effects of metformin have made it more attractive for researchers. Metformin has extensive effects on cell metabolism. It inhibits complex 1 in mitochondrial ETC and decreases ATP synthesis by mitochondria which in turn leads to an increase in AMP: ATP ratio and 5'adenosine monophosphate-activated protein kinase (AMPK) activation [5]. Recent studies have shown that AMPK acts as a cellular energy level sensor and is not activated just by ETC inhibition. Therefore, metformin can activate AMPK indirectly and independently of ETC. AMPK is known as the main target of metformin, which is activated by various pathways and has led to its perception as a glucose reducing drug in the first line of treatment of T2MD. AMPK and an increase in its activity cause a transformation from anabolic to catabolic state. Consequently, not only does activation of AMPK impede protein synthesis and lipogenesis, but it also increases autophagy and fatty acid β-oxidation [6] (Fig. 2). Therefore, understanding these mechanisms is important, because it can help us explore the other effects of this drug and utilize it for advanced purposes.
Glucose-lowering effect of metformin is the most important mechanism of action that reduces blood sugar by improving the sensitivity of insulin receptors and the translocation of GLut4 in the cell membrane. However, the hypoglycemic effects of metformin are exerted by inhibiting hepatic gluconeogenesis, inhibiting glucose uptake in the intestine, inhibiting the glucagon signaling pathway, and reducing cortisol [7]. Different findings are available on how metformin affects AMPK, in which LKB plays a major role in regulating its activity. Metformin inhibits gluconeogenesis and stops the de novo synthesis of glucose in the liver by regulating the glucagon hormone and cortisol, Glyceraldehyde-3-phosphate dehydrogenase inhibition, and modulating cellular redox [8]. However, this is not sufficient and the absorption of glucose from the intestine is reduced by metformin, which is known to be the most important effect today [9]. Among the functional mechanisms of metformin, some non-canonical signaling pathways have emerged that can define metformin with advanced effects such as anti-aging, anti-obesity, and anti-inflammation.
Obesity and aging are the two main contributing causes of T2DM. Chronic sterile inflammation is a common feature of aging and obesity. In aged individuals, SASP factors like IL-6, IL-1 β/α, and MCP-1, which are secreted from senescent cells, disrupt immune system homeostasis and lead to low-grade chronic inflammation. The same disruption occurs in obese individuals by secretion of cortisol and some other hormones as well as pro-inflammatory cytokines from adipose tissues [10, 11]. Furthermore, insulin sensitivity declines in obese and also aged individuals, making them more prone to developing diabetes [12].
In this review, an update of molecular mechanism of action of metformin based on glucose-lowering effect have been discussed. In the following, we have been focused on non-canonical signaling pathways and molecular mode of actions which underlie the advanced application of this anti-diabetic agent like as anti-obesity, anti-inflammation, and anti-aging.
Glucose-lowering effect of metformin
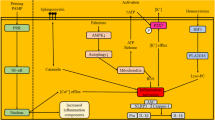
Metformin has an extensive effect on cells and, recently, we are witnessing a growing number of its applications, for example in cancer treatment, regenerative medicine, and recently, its anti-aging and anti-inflammatory effects have been further explored [13]. The glucose-lowering effect of metformin is a complicated process that has raised some controversies (Fig. 1). We will mention prevailing theories about the role of metformin in Gluconeogenesis, insulin sensitivity, and in the following paragraphs.

Glucose-lowering mechanisms of metformin. Metformin can control blood glucose level by reducing intestinal glucose absorption, blood cortisol level, insulin resistance, as well as the inhibition of glucagon signaling and gluconeogenesis
Improving insulin sensitivity
Insulin resistance is a complicated process in which insulin-dependent tissues, like adipose tissue and skeletal muscles, fail to respond properly to insulin and cannot read out the insulin message [10]. AMPK α2 is a primary determinant in insulin sensitivity of the whole body. AMPK α2 knock-out mice manifest a high blood glucose level after eating or oral glucose challenge [14]. Glut-4, a glucose transporter, is expressed in insulin-dependent tissues. It is so important in controlling the blood glucose level and causes glucose uptake in response to insulin. Glut-4 is present in the membrane of vesicles inside the cytoplasm of fat and muscle cells. Insulin stimulates translocation and fusion of Glut-4 carrier vesicles to the cell membrane [15]. Its trafficking and translocation get impaired in subjects suffering from insulin resistance. Reduction in number or ligand-binding efficiency of insulin receptor as well as decreased tyrosine kinase activity of the insulin receptor leads to insulin resistance [16]. PTP1B is another factor that influences insulin sensitivity. PTP1B is a negative modulator of insulin signaling and causes IRS-1 (Insulin Receptor Substrate 1) dephosphorylation and thereby blunts insulin signaling [17]. PTP1B also inhibits leptin signaling and causes obesity. Inhibition of PTP1B is a potential therapy for insulin resistance, obesity, and T2D [12]. In some studies, it has been identified that metformin could inhibit PTP1B [18]. The precise mechanism behind the pathophysiology of insulin resistance is not completely understood. However, in the next following paragraphs, we will introduce some of the causal factors in insulin resistance (Table 1).
Improving GLUT-4 translocation
Overeating high caloric foods increases free fatty acids (FFA), and on the other hand, it has been shown a high concentration of FFA induces insulin résistance [19, 20] (Table 1). Additionally, high fructose intake can also lead to developing insulin resistance [21]. Insulin sensitivity can be improved by medications like PTP1B inhibitors and also fibroblast growth factor 21 (FGF 21) [12, 16]. In diabetic persons, to deal with insulin resistance, the body increases insulin secretion and unfortunately long time exposure to a high level of insulin, by itself, exacerbates insulin resistance [16]. By decreasing the serum FFA level, metformin reduces insulin resistance [22]. It was shown that treatment with metformin is able to increase insulin receptor tyrosine phosphorylation activity in vitro [23, 24]. A study by Garabadu et al. reveled that metformin improves the hepatic PI3K/Akt/GLUT-4 signaling pathway in T2DM rats [25]. It was also shown that metformin increases Glut-4 translocation to the cell surface. Activated AMPK mediates this effect of contraction on GLUT-4 translocation and glucose transport in muscle. This increase in GLUT-4 is partly responsible for the increased insulin sensitivity [26]. Treatment with metformin activates P38 MAPK and causes a subsequent rise in glucose uptake through Glut-4 translocation. In insulin-resistant myotubes, insulin-stimulated activation of ERK and JNK is unchanged, but P38 MAPK activation is impaired [23]. In another study, the authors reported that metformin causes Glut-4 translocation to the cell surface via activating Cbl and subsequent CAP (Cbl-associated proteins) activation by AMPK [27]. Metformin improves insulin sensitivity and impaired Glut-4 trafficking, created by chronic hyperinsulinemia, in adipocytes through inhibiting Glut-4 endocytosis [28].
Reduction of intestinal glucose absorption
The PET-computed tomography (CT) scan has shown that metformin accumulates in the intestine in high concentrations, up to 30–300 times its concentration in the blood [29]. Intravenous administration of metformin is less effective in reducing the blood glucose level than the orally taken drug [30]. Horakova et al. shed light on the new aspect of glucose reduction by metformin. The authors emphasized that the role of AMPK in the acute glucose-lowering effect of metformin is negligible. They demonstrated that metformin hinders glucose transport through the intestinal lumen into the blood. They identified that metformin can make it easy for the body to decrease food-borne glucose elevation simply by inhibiting transepithelial glucose transport into blood [31]. Some researchers give special importance to the role of gut, gut–brain neural axis, and gut microbiome in the glucose-lowering effect of metformin [32,33,34,35,36]. However, this aspect of mechanism of action of metformin needs more studies.
Gluconeogenesis inhibition by mG3PD
In healthy individuals, gluconeogenesis is responsible for ~ 50% of EGP, endogenous glucose production, after overnight fasting, and it increases up to 90% of EGP after 40 h fasting [37, 38]. In a T2D person, relative glucagon increment and insulin resistance lead to a sustained glucose production in the liver. Therefore, in diabetes, we have unchecked and sustained gluconeogenesis [39]. Most scientists believe that metformin decreases the blood glucose level by inhibition of gluconeogenesis in the liver. G-6-Pase catalyzes the conversion of glucose-6-phosphate to glucose which finally is released from the liver. However, mice lacking liver-specific G-6-Pase have normal blood glucose even after long-term fasting [40]. While PEPCK is a crucial enzyme in gluconeogenesis, liver-specific PEPCK knock-out mice have a normal glucose level even after 24 h of fasting [41]. These observations are likely because of the compensatory increase of extra-hepatic gluconeogenesis [42].
FBPase, which catalyzes the conversion of fructose-1,6-bisphosphate to fructose-6-phosphate, is another important enzyme in the gluconeogenesis pathway. It is a promising target for the treatment of type 2 diabetes. MB07803 is a second-generation FBPase inhibitor that is in the phase Ib clinical trial. It is beneficial in controlling fasting blood glucose level, but in the postprandial state, it fails to decrease blood glucose as fast as metformin [39, 43]. By considering that FBPase inhibitors are more efficient in gluconeogenesis inhibition than metformin, inhibition of gluconeogenesis is not likely to solely be responsible for the acute glucose-lowering effect of metformin that can decrease blood glucose back to pre-eating level within less than 3 h [31].
Van Poelje et al. demonstrated that in diabetics, the increase in gluconeogenesis is twice as high as in normal individuals. Three months of treatment with metformin causes an about 36% decrease in gluconeogenesis and a 30% decrease in fasting plasma glucose concentration. Because in this study the statistical sample was small, further research using more subjects may yield different results [39]. There are several studies about the mechanism by which metformin inhibit gluconeogenesis. Cao et al. demonstrated metformin even at low concentrations through the activation of AMPK can suppress endogenous glucose production and gluconeogenic gene expression in hepatocytes. Interestingly, changing in cAMP level is dispensable from the metformin-mediated inhibition of gluconeogenesis [44].
Foretz et al. reported that in LKB−/− hepatocytes or primary hepatocytes lacking AMPK, metformin inhibits glucose production almost normally and so this inhibition is independent of AMPK or LKB1 [45]. In another study, authors determined that metformin exerts a glucose-lowering effect through inhibition of glycerophosphate dehydrogenase and change in redox status of cytoplasm [46]. What is universally accepted is that metformin inhibits gluconeogenesis, and by doing so, it results in blood glucose reduction. In addition to the AMPK signaling pathway, metformin inhibits gluconeogenesis through various pathways that yet to be identified.
Hormonal changes: cortisol and glucagon
It is demonstrated that in some of the diabetic subjects, there is an elevated level of cortisol. The overall flattening of the cortisol curve and elevated daily cortisol level makes it hard for a diabetic person to control blood glucose [47]. Glucocorticoids, like cortisol, exert their diabetogenic effects partially by increasing lipolysis (in adipocytes) and subsequent rise in plasma free fatty acid level and reduction of insulin responsiveness. Cortisol induces insulin resistance, impairs glucose uptake by skeletal muscles, and promotes hepatic gluconeogenesis [48]. It was shown that metformin administration decreases cortisol back to the normal level [49]. However, whether metformin has a direct effect on cortisol secretion is not clear. As mentioned in previous headings, metformin can potentially antagonize glucagon signaling by decreasing the cAMP level. Hormonal changes induced by metformin play a significant role in reducing blood glucose and importance of that needs further studies.
Metformin mode of action
Metformin is a positively charged natural biguanide compound. Despite the relatively long-term use of this drug, the precise mechanism of its action has not been fully elucidated and there are some disagreements among the scientists in this field [50]. Therefore, in the following paragraphs, we will introduce some of the common theories and some controversies about the metformin mode of action.
Complex 1 inhibition and AMPK activation
It is almost accepted that metformin exerts its effects by inhibiting mitochondrial electron transport chain, complex 1, and this inhibition occurs just at mM concentration. Nevertheless, the level of this drug in the blood and extracellular fluids does not exceed mM concentration. There are two explanations in the literature for the inhibitory effect of metformin on the mitochondrial complex 1. First, some scientists showed that, in the intact cells and in vivo studies, metformin triggers a signaling pathway which in turn causes inhibition of complex 1, although such a pathway has not yet been identified. On the other hand, thiol groups in several complex 1 subunits have been identified as the targets of post-translational modification, but how metformin affects the thiol groups has yet to be determined [51].
The second hypothesis, which is more popular, states that metformin accumulates in mitochondria and its concentration reaches to mM, and then it can inhibit complex 1 [5]. By inhibiting complex 1, metformin increases AMP:ATP ratio and activates AMPK (Fig. 2). However, there is not enough evidence to prove this.

AMPK Mode of Action. AMPK is activated by several enzymes such as TAK-1, LKB-1, and CaMKK2. AMPK has extensive effects inside the cells, some of which are depicted above
Metformin has an extensive role in cell metabolism and exerts most of its effects through AMPK (Fig. 2). AMPK increases glucose uptake by muscles via stimulating Glut-4 translocation to the cell membrane. AMPK also phosphorylates HMG-COA reductase and decreases cholesterol synthesis. AMPK lowers triglyceride and phospholipid synthesis as well as glycogen production in cells [52]. AMPK phosphorylates and inactivates acetyl-CoA carboxylase (ACC1/2), and inhibits the conversion of acetyl-CoA to malonyl-CoA and fatty acid synthesis. Malonyl-CoA is a potent inhibitor of CPT1. By removing the inhibition from CPT1, AMPK increases the acyl-CoA entrance into mitochondria via CPT1. Acyl-CoA in mitochondria undergoes beta-oxidation and produces NADH and FADH2 that enter the ETC. Besides, the produced acetyl-CoA can be utilized in the Krebs cycle [53]. Protein synthesis is an energy-consuming process and is more active in growing cells. AMPK, by phosphorylation of RNA polymerase I, reduces ribosomal RNA synthesis and subsequently decreases protein synthesis [54]. AMPK also inhibits protein synthesis via phosphorylation of eukaryotic translation elongation factor (eEF2). Also, by inhibition of mTORC1, it abrogates phosphorylation of Eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and p70 S6 kinase [55]. Phosphorylation of 4EBP1 and p70 S6 kinas is so important in protein synthesis. Furthermore, through the inactivation of mTORC1, AMPK increases autophagy. It also directly influences the autophagy by phosphorylating of Unc-51 like autophagy activating kinase 1 (ULK1) [56].
AMPK activation leads to CREB binding protein (CBP) phosphorylation in Ser436 via PKCι/λ and causes its dissociation from CREB co-activator complex and inhibition of gluconeogenic genes expression [57]. Furthermore, AMPK phosphorylates and activates peroxisome proliferator-activated receptor-gamma co-activator 1 α (PGC-1 α), which in turn causes mitochondrial biogenesis and reduces ROS production [58].
AMPK structure and subtypes
AMPK is a heterotrimeric enzyme with two regulatory subunits, β and γ, and a catalytic subunit, α. There are some isoforms of AMPK which are different in subcellular localization, function, and regulation. Therefore, it is important to know which AMPK is most affected by metformin and what effects it has on the cells. For example, AMPK α1 and AMPK α2, the two well-studied isoforms of AMPK, have some differences including the following:
-
(1)
AMPK α2 has a greater sensitivity to AMP level changes in comparison with AMPK α1 [59].
-
(2)
After activation, AMPK α2 is found in both cytoplasm and nucleus, but AMPK α1 is exclusive to the cytoplasm. Therefore, AMPK α2 can regulate gene expression and so it performs a more significant role [59].
-
(3)
LKB1 is a master upstream kinase of AMPK. It has been found that in the LKB1 lacking cardiac muscle cells under the ischemic condition, i.e., increased AMP:ATP ratio, AMPK α2 phosphorylation and activation do not occur, whereas, this deletion has no significant effect on AMPK α1 phosphorylation and activation. By considering these observations, it is presumable that the AMPK α1 activation mechanism is different from that of AMPK α2 [60].
-
(4)
AMPK α1 knock-out mice have almost the same amount of blood glucose; however, AMPK α2 lacking mice have impaired glucose homeostasis [61, 62]. Given the above explanations and what will be discussed below, it is assumed that metformin has a greater effect on AMPK α2 and there is not enough information about its effect on AMPK α1.
AMPK mechanism of activation
AMPK can be activated through various mechanisms which in turn activates various signaling pathways. Phosphorylation of Thr172 in the α subunit and binding of AMP to γ subunit are the two essential steps in AMPK activation. There are at least three kinases upstream of AMPK, namely STK11 also known as LKB1, CaMKK2, and TAK1, which can activate AMPK [63]. There are some theories about the activation of AMPK which are more valid about AMPK α2, but we have little information about AMPK α1 (Fig. 3).

AMPK Mechanism of Activation. A AMPK resides in the cytoplasm and its regulation is under the control of the LKB1 and phosphatases. LKB1 is permanently active and phosphorylate AMPK. Binding of AMP to the AMPK leads to a change in its conformation and prevents phosphatases activity. B LKB1 is inactive and is found in the nucleus. Stimuli such as reactive nitrogen species and metformin lead to activation of PKC-ζ, after activation and phosphorylation of SIRT1 at Ser47 committing SIRT1 to proteasome degradation, SIRT1 increases LKB1 deacetylation and activation. LKB1 is exported to the cytoplasm, and with the help of the co-activators such as MO25 and STRAD phosphorylates and activates the AMPK. C: AMPK is not free in the cytoplasm and it is resident on the surface of lysosomes and its activity depends on V-ATPase-Regulator-AXIN/LKB1 activation. When the cells are in the low energy state, AXIN puts LKB1 in the vicinity of the AMPK and activate it. On the other hand, LKB1/AXIN complex inhibits guanine nucleotide exchange factor (GEF) activity of the V-ATPase-Regulator, which leads to the breakdown of mTORC1 from the lysosomal surface and phosphorylation of AMPK by LKB1
The first mechanism suggests that LKB1 is active constantly in the cytoplasm and phosphorylates Thr172 in the α subunit, but phosphatases remove that phosphate group almost immediately. By attachment of AMP to γ subunit, AMPK undergoes a conformational change and the β subunit covers the phosphate group attached to Thr172 and prevents phosphatases’ access to that phosphate group [64] (Fig. 3). Hawley et al., using cell lines expressing mutant AMPK that was insensitive to AMP level, revealed that AMPK activation is dependent on AMP binding to γ subunit [65]. It has been demonstrated that metformin exerts its effects without increasing AMP:ATP ratio. However, this may be because of inefficient methods of measurement of AMP and ATP changes. Therefore, it is presumable that nM changes are enough for AMPK to be sensed. This mechanism is the most accepted notion about AMPK activation, but by considering it, we cannot explain energy independent activation of AMPK by CaMKK2 in hyperosmotic stress [66]. Moreover, in the LKB1 lacking cells, basal levels of phosphorylated and active AMPK are detectable and this suggests that AMPK can be phosphorylated from other pathways, as well [67].
The second mechanism holds that in endothelial cells, stimuli, such as nitrogen reactive species (RNS), blood pressure, and metformin, cause proto-oncogene c-Src activation which in turn activates PKCζ. The phosphorylated PKCζ translocate to the nucleus and phosphorylates LKB1 and causes its exportation to the cytoplasm and subsequent activation of AMPK [68]. Moreover, SIRT1 deacetylates LKB1 and enhances its nuclear exclusion and its association with STRAD and MO25, which are two accessory co-activators of LKB1 complex [69] (Fig. 3). This type of activation mechanism was observed in endothelial cells and has yet to be studied in other cell types.
The third mechanism regarding the AMPK activation states that the lysosomal surface protein complex, V-ATPase-Regulator-AXIN/LKB1, is essential for APMK activation. According to this mechanism, AMPK is considered as a lysosomal surface residential protein. Under low energy state, LKB1/AXIN complex associates with V-ATPase-Regulator and causes inhibition of GEF activity of the regulator. This leads to the dissociation of the mammalian target of mTORC1 from the lysosomal surface. On the other hand, this interaction enhances the phosphorylation of AMPK by LKB1 [70] (Fig. 3). This mechanism contradicts the previous studies, demonstrating that LKB1 is found in the nucleus [71] and AMPK has a uniform distribution in the cell cytoplasm before activation [72].
Gluconeogenesis inhibition: its relation with redox changes
Glycerol-3-phosphate and aspartate/malate shuttles are the two main mechanisms for the regeneration of NAD+ from NADH, which is necessary for glycolysis maintenance. In glycerol-3-phosphate shuttle, cGPD and mGPD play a major role. cGPD causes NAD+ production from NADH, and in parallel, mGPD converts FAD to FADH2. By doing so, high energy equivalents produced in the cytoplasm can be transferred into mitochondria and enter ETC, resulting in ATP production. In the malate/aspartate shuttle, aspartate is transported from mitochondria to cytosol in which it is converted to malate in two steps. NADH is used in this process. On the other hand, malate enters mitochondria where it is converted to aspartate in a two-step process which converts NAD+ to NADH. Glycerol-3-phosphate and lactate are the two reduced precursors for glucose production through gluconeogenesis. Acute and chronic treatments of hepatocytes with a low concentration of metformin efficiently cause a reduction in hepatic EGP. Metformin inhibits mG3PDH by about 50%. Cytosolic NADH/NAD+ ratio increases, whereas mitochondrial NADH/NAD+ ratio declines in metformin treatment. Inhibition of mGPD by metformin influences EGP using two mechanisms. First, this inhibition increases cytoplasmic NADH/NAD + ratio which in turn inhibits LDH and blocks the conversion of lactate to pyruvate and the subsequent glucose production using gluconeogenesis. On the other hand, the inhibition of mGPD impedes glucose production from glycerol. Hepatic mG3PDH Knockdown rats manifest impaired metformin-mediated increase in cytosolic redox state and gluconeogenesis inhibition [73, 74]. Recently, researchers challenged this model by showing that pharmaceutically inhibition of mG3PDH has an insignificant effect on gluconeogenesis [75]. Knocking down cGPD, an indispensable part of the glycerophosphate shuttle, has a minor effect on redox status in comparison with inhibition of mGPD and also is not able to reduce endogenous glucose production [74]. Furthermore, it was shown that a low concentration of metformin leads to inhibition of gluconeogenesis from both reduced and oxidized substrates by increasing these substrates utilization in glycolysis in a redox-independent manner [76].
cAMP reduction
Glucagon is a hormone secreted from alpha cells of islets of Langerhans in the pancreas and is responsible for keeping blood glucose in a normal range in fasting state. Insulin and glucagon are the chief parts of a feedback system for controlling the blood glucose level. Glucagon is a primary regulator of gluconeogenesis. In a fasting state, it stimulates gluconeogenesis and glycogenolysis and raises blood glucose. Cyclic AMP is the secondary messenger in glucagon signaling. Tengholm et al. revealed that acute reduction of gluconeogenesis by metformin is because of antagonizing glucagon signaling by reducing the level of cAMP and subsequent protein kinases A (PKA) activity [77]. cAMP activates PKA, and then, it phosphorylates AMPK in Ser485, an inhibitory phosphorylation, that abrogates AMPK effects. Metformin increases AMP:ATP ratio and thereby inhibits adenylyl cyclase which consequently causes a cAMP level reduction (Fig. 2). A mutated AMPK in S485 is not capable of being phosphorylated by PKA [78]. In contrast, AMPK phosphorylates and activates cAMP-specific 3’, 5’-cyclic phosphodiesterase 4 (PDE 4B), and causes a drop in cAMP level [79].
Functional and clinical dose of metformin
The maximum approved concentration of metformin for the treatment of type 2 diabetes is 2.5 g daily (35 mg/kg body weight). After oral administration, metformin is absorbed in the intestine by PMAT and OCT3. Then, it is transferred to the liver via the portal vein. Metformin concentration in the portal vein is reported to be about 40–70 µM. Hepatocytes uptake metformin by OCT1/3, and it is excreted from hepatocytes through multidrug and toxin extrusion 1 (MATE 1) transporter [80]. After hepatic uptake, the serum concentration of metformin reaches 10–40 µM [80]. Metformin is absorbed by renal epithelial cells via OCT2 and its excretion is performed by MATE1/2 [81]. In cultured primary hepatocytes, concentrations even were below 50 µM [82]. There are some disagreements about the rational and efficient concentration of metformin among researchers. In most studies, researchers have used a high concentration of metformin, 200–400 mg/kg in preclinical, and 250 µM to ≥ 2 mM in vitro studies, which are much more than metformin concentration in the blood after an approved dose is administered [80]. It is noteworthy that the concentration of metformin inside the hepatocytes reaches five times more than extracellular or serum concentration, but its exact molecular mechanism is unclear. For a good in vivo simulation, we must balance time and concentration. For example, 100 µM for 4 h or 80 µM for 24 h are more recommended [44, 83]. The minimum concentration of metformin for activating AMPK in skeletal muscle is much higher than its concentration in blood. However, metformin may accumulate in muscles through time [84]. For in vitro studies, we have to simulate clinical conditions and use appropriate doses to achieve reliable results. In the next section, we will discuss how metformin exerts its glucose-lowering effects.
Alleviation of obesity-induced meta-inflammation by metformin
Obesity is an increasingly important cause of disease worldwide [10]. Body adiposity is highly heritable; a recent study by Akbari et al. showed that GPR75 is corresponding to 1.8 kg/m2 lower BMI and 5.3 kg lower body weight, and knockdown of GPR75 in mice resulted in resistance to weight gain in a high-fat diet model which resulted in ameliorated glycemic control and insulin sensitivity [85]. Moreover, adipocytes act as endocrine cells and contribute to the secretion of inflammatory mediators [10] that suggest a potential association between GPR75 and the level of inflammatory cytokines. Since metformin act as glucose-lowering and improves insulin sensitivity might be able to modulate GPR75 expression as a therapeutic strategy for obesity and inflammation which needs further studies.
Mitochondrial dysfunction, ER (endoplasmic reticulum) stress, ROS overload, that are hallmarks of aging, as well as SASP are implicated in insulin resistance [10, 86]. By aging, cells undergo substantial changes and accumulation of inefficient organelles that impair cell response to stimuli. Metformin can reduce the secretion of SASP factors and age-related meta-inflammation [87].
Inflammaging is a sterile, chronic, low-grade inflammation that contributes to aging and is fueled by SASP factors [88,89,90]. Cell senescence was coined by professor Hayflick in 1961 and refers to irreversible cell cycle arrest after continuous passages in vitro and it is assumed that the same phenomenon occurs in the body. It is believed that cell senescence is a tumor-suppressor response. Senescent cells fail to undergo apoptosis thus accumulate in the body by age [88]. By secreting early SASP factors, senescent cells become detectable for the immune system; however, after a while, their secretory phenotype starts to change and shifts to the secretion of late SASP factors. In this stage, they elude the immune system and induce senescence to the neighboring cells and accelerate senescence propagation [88, 91].
Late SASP factors contain inflammatory factors like IL-6, IL-1 β/α, and MCP-1, and promote cancer incidence. They also cause insulin resistance and chronic inflammation [88, 91] (Table 1). Metformin, by inhibiting the NF-κβ pathway, decreases SASP factors production and mitigates inflammaging [87, 92].
SASP: an anti-aging target of metformin
Adipocytes act as endocrine cells and are able to secrete inflammatory mediators (like TNF-α), steroid hormones (such as estrogen and cortisol), leptin, and adiponectin. All of these, together, contribute to obesity-related insulin resistance [93, 94]. TNF-α receptor knock-out mice with diet-induced obesity are almost immune to developing insulin resistance [95]. Activation of AMPK by metformin and inhibition of NF-κβ can reduce the secretion of inflammatory cytokines such as TNF-α and reduces the inflammation, which will be discussed in the following sections [87, 92] (Fig. 4).

Meta-inflammation changes in adipose tissues. Monocytes can differentiate to the type-1 (M1) and type-2 (M2) macrophages in the exposure of different cytokines. As shown in the figure, in the lean adipose tissue monocyte differentiate mainly to M2 macrophages and through Th2 produces anti-inflammatory cytokines IL-10, TGF-β, PDL-1, and anginase 1. While in the obese state, they differentiate mostly to M1 macrophages and release, inflammatory factors such as TNF-α, IL-1α and β, IL-6, leukotriene B4 (LTB4), and nitric oxide (NO) which exacerbate the obesity-related inflammation
The first observation about the relationship between inflammation and glucose homeostasis dates back to 120 years ago when researchers observed that aspirin administration helps glucose control in diabetic patients [96]. Inflammatory mediators impair insulin responsiveness in different manners. JNK and IKKB phosphorylate insulin receptor and insulin response substrates (IRSs) in inhibitory sites and impede insulin signaling. Salicylates like aspirin, through inhibition of IKKB, improve insulin sensitivity. NF-κβ induces expression of SOCSs, suppressors of cytokine signaling, that are proteins that bind to insulin receptor and hinder IRS phosphorylation by it. TNF-α increases lipolysis in adipocytes, raises FFA, and reduces insulin sensitivity. An elevated level of TNF-α, IL-6, and MCP-1 is reported in diabetic subjects [97, 98] (Fig. 4; Table 1).
There are two subtypes of macrophages including pro-inflammatory, or M1 type macrophages, and anti-inflammatory, or M2 type macrophages. Each type can repolarize to the other one in response to extrinsic factors. For example, in tumor niches, a high level of lactate stimulates polarization to M2 macrophages type. By elimination of the lactate, they repolarized to M1 state [99]. M1 macrophages are dependent on glycolysis and Krebs cycle, while M2 macrophages rely on fatty acid oxidation. The other difference between M1 and M2 macrophages is their secreted factors. M2 cells produce anti-inflammatory cytokines like IL-10, whereas M1 cells release inflammatory factors such as TNF-α, IL-1α and β, IL-6, leukotriene B4 (LTB4), and nitric oxide (NO) [97] (Fig. 4). In lean adipose tissue, IL-4 level is high and leads to polarization of residential as well as newly attracted macrophages to M2 state. Adiponectin, secreted from adipocytes, helps the M2 state maintenance. In contrast, in obesity, adipose tissue undergoes substantial changes in cellular composition, and secreted mediators. Altogether, these changes are in favor of polarization to M1 macrophages [97, 100, 101]. MATE1 and OCTs are expressed strongly in macrophages and other inflammatory cells. Both OCT1 and OCT3 are expressed on monocytes, macrophages, dendritic cells, CD4+ T cells, and peripheral blood mononuclear cells [102]. By taking these into account, immune cells are a good target for metformin. Metformin inhibits NF-κβ signaling through the hindrance of IKKα1β activation via TAK1, independent of AMPK [87]. Furthermore, activation of AMPK by metformin inhibits NF-κβ signaling and alleviates inflammation [92]. Another study has shown that metformin could inhibit NLRP3 inflammasome through AMPK/mTOR signaling axis and consequently boosts M1–M2 type macrophages polarization and accelerates tissue and wound healing. This finding is important in diabetic patients with diabetic ulcers, but this application of metformin is yet to be studied precisely [103].
Conclusion
This review aims to concisely discuss the pharmacodynamics and pharmacokinetics of metformin. It was revealed that inhibition of hepatic gluconeogenesis by metformin through AMPK activation and inhibition of mG3PDH provides enough time for the body to manage the food-borne glucose increase. On the other hand, metformin can exert its glucose-lowering effect by increasing insulin sensitivity and inducing peripheral glucose disposal. Besides, it was demonstrated that metformin decreases glucose absorption from the intestine and it is enough for the body to deal with glucose increase after eating. Overall, it can be suggested that all of the mentioned mechanisms including gluconeogenesis inhibition, decreasing cortisol level, antagonizing glucagon signaling, and decreasing glucose absorption from the intestine are altogether involved in acute and chronic glucose-lowering effects of metformin. Besides the glucose-lowering effect of metformin, it reduces obesity, inflammaging, and age-related sterile meta-inflammation which by increasing insulin sensitivity is also beneficial for its glucose-lowering effect.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CaMKK2:
-
Calcium/calmodulin-dependent protein kinase kinase 2
- CPT1:
-
Carnitine palmitoyltransferase
- JNK:
-
C-Jun N-terminal kinase
- cGPD:
-
Cytosolic glycerol-3-phosphate dehydrogenase
- ETC:
-
Electron transport chain
- EGP:
-
Endogenous glucose production
- ERK:
-
Extracellular signal-regulated kinase
- GPR75:
-
G protein coupled receptor-75
- G-6-Pase:
-
Glucose -6-phosphatase
- GEF:
-
Guanine nucleotide exchange factor
- LKB:
-
Liver Kinase B
- mTORC1:
-
Mammalian target of rapamycin complex 1
- mGPD:
-
Mitochondrial glycerol-3-phosphate dehydrogenase
- MAPK:
-
Mitogen-activated protein kinase
- OCT3:
-
Organic cation transporter
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PMAT:
-
Plasma mono-amine transporter
- PKCζ:
-
Protein kinases Cζ
- PTP1B:
-
Protein-tyrosine phosphatase 1
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory proteins
- TAK1:
-
TGF-β activated kinase 1
- T2DM:
-
Type 2 diabetes mellitus
- STK11:
-
Serine/threonine kinase 11
References
Olokoba AB, Obateru OA, Olokoba LB. Type 2 diabetes mellitus: a review of current trends. Oman Med J. 2012;27(4):269–73.
DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019.
Shafiei-Irannejad V, Samadi N, Salehi R, Yousefi B, Zarghami N. New insights into antidiabetic drugs: possible applications in cancer treatment. Chem Biol Drug Des. 2017;90(6):1056–66.
Amirsaadat S, Jafari-Gharabaghlou D, Alijani S, Mousazadeh H, Dadashpour M, Zarghami N. Metformin and Silibinin co-loaded PLGA-PEG nanoparticles for effective combination therapy against human breast cancer cells. J Drug Deliv Sci Technol. 2021;61:102107.
Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol. 2019;10:294.
Kaneto H, Kimura T, Obata A, Shimoda M, Kaku K. Multifaceted mechanisms of action of metformin which have been unraveled one after another in the long history. Int J Mol Sci. 2021;22(5):2596.
LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev. 2021;42(1):77–96.
Agius L, Ford BE, Chachra SS. The metformin mechanism on gluconeogenesis and AMPK activation: the metabolite perspective. Int J Mo Sci. 2020;21(9):3240.
Kumar A, Bharti SK, Kumar A. Therapeutic molecules against type 2 diabetes: what we have and what are we expecting? Pharmacol Rep. 2017;69(5):959–70.
Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: review of the underlying molecular mechanisms. J Cell Physiol. 2019;234(6):8152–61.
Dehkordi AH, Abbaszadeh A, Mir S, Hasanvand A. Metformin and its anti-inflammatory and anti-oxidative effects; new concepts. J Renal Inj Prev. 2019;8(1):54–61.
Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov. 2002;1(9):696–709.
Harada N. Effects of metformin on blood glucose levels and bodyweight mediated through intestinal effects. J Diabetes Investig. 2020;11(6):1420.
Song T-J, Park C-H, In K-R, Kim J-B, Kim JH, Kim M, et al. Antidiabetic effects of betulinic acid mediated by the activation of the AMP-activated protein kinase pathway. PLoS ONE. 2021;16(4):e0249109.
Lauritzen HPMM. Insulin- and contraction-induced glucose transporter 4 traffic in muscle: insights from a novel imaging approach. Exerc Sport Sci Rev. 2013;41(2):77–86.
Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–223.
Yip SC, Saha S, Chernoff J. PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci. 2010;35(8):442–9.
Holland W, Morrison T, Chang Y, Wiernsperger N, Stith B. Metformin (Glucophage) inhibits tyrosine phosphatase activity to stimulate the insulin receptor tyrosine kinase. Biochem Pharmacol. 2004;67:2081–91.
Erdmann J, Kallabis B, Oppel U, Sypchenko O, Wagenpfeil S, Schusdziarra V. Development of hyperinsulinemia and insulin resistance during the early stage of weight gain. Am J Physiol Endocrinol Metab. 2008;294(3):E568–75.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277(52):50230–6.
Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab. 2005;2(1):5.
Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29(4 Pt 2):6s28-35.
Kumar N, Dey CS. Metformin enhances insulin signalling in insulin-dependent and-independent pathways in insulin resistant muscle cells. Br J Pharmacol. 2002;137(3):329–36.
Gunton JE, Delhanty PJ, Takahashi S, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab. 2003;88(3):1323–32.
Garabadu D, Krishnamurthy S. Metformin attenuates hepatic insulin resistance in type-2 diabetic rats through PI3K/Akt/GLUT-4 signalling independent to bicuculline-sensitive GABAA receptor stimulation. Pharm Biol. 2017;55(1):722–8.
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51(7):2074–81.
Lee JO, Lee SK, Kim JH, Kim N, You GY, Moon JW, et al. Metformin regulates glucose transporter 4 (GLUT4) translocation through AMP-activated protein kinase (AMPK)-mediated Cbl/CAP signaling in 3T3-L1 preadipocyte cells. J Biol Chem. 2012;287(53):44121–9.
He JH, Chen LX, Li H. Progress in the discovery of naturally occurring anti-diabetic drugs and in the identification of their molecular targets. Fitoterapia. 2019;1(134):270–89.
Wu T, Horowitz M, Rayner CK. New insights into the anti-diabetic actions of metformin: from the liver to the gut. Expert Rev Gastroenterol Hepatol. 2017;11(2):157–66.
Sum CF, Webster JM, Johnson AB, Catalano C, Cooper BG, Taylor R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet Med. 1992;9(1):61–5.
Horakova O, Kroupova P, Bardova K, Buresova J, Janovska P, Kopecky J, et al. Metformin acutely lowers blood glucose levels by inhibition of intestinal glucose transport. Sci Rep. 2019;9(1):6156.
Borg MJ, Bound M, Grivell J, Sun Z, Jones KL, Horowitz M, et al. Comparative effects of proximal and distal small intestinal administration of metformin on plasma glucose and glucagon-like peptide-1, and gastric emptying after oral glucose, in type 2 diabetes. Diabetes Obes Metab. 2019;21(3):640–7.
DeFronzo RA, Buse JB, Kim T, Burns C, Skare S, Baron A, et al. Once-daily delayed-release metformin lowers plasma glucose and enhances fasting and postprandial GLP-1 and PYY: results from two randomised trials. Diabetologia. 2016;59(8):1645–54.
Waise TMZ, Rasti M, Duca FA, Zhang SY, Bauer PV, Rhodes CJ, et al. Inhibition of upper small intestinal mTOR lowers plasma glucose levels by inhibiting glucose production. Nat Commun. 2019;10(1):714.
Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919–29.
Wang PY, Caspi L, Lam CK, Chari M, Li X, Light PE, et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452(7190):1012–6.
Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98(2):378–85.
Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254(5031):573–6.
van Poelje PD, Potter SC, Erion MD. Fructose-1, 6-bisphosphatase inhibitors for reducing excessive endogenous glucose production in type 2 diabetes. Handb Exp Pharmacol. 2011;203:279–301.
Mutel E, Gautier-Stein A, Abdul-Wahed A, Amigo-Correig M, Zitoun C, Stefanutti A, et al. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: induction of renal and intestinal gluconeogenesis by glucagon. Diabetes. 2011;60(12):3121–31.
She P, Burgess SC, Shiota M, Flakoll P, Donahue EP, Malloy CR, et al. Mechanisms by which liver-specific PEPCK knockout mice preserve euglycemia during starvation. Diabetes. 2003;52(7):1649–54.
Rines AK, Sharabi K, Tavares CD, Puigserver P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov. 2016;15(11):786–804.
van Poelje PD, Potter SC, Chandramouli VC, Landau BR, Dang Q, Erion MD. Inhibition of fructose 1,6-bisphosphatase reduces excessive endogenous glucose production and attenuates hyperglycemia in zucker diabetic fatty rats. Diabetes. 2006;55(6):1747.
Cao J, Meng S, Chang E, Beckwith-Fickas K, Xiong L, Cole RN, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J Biol Chem. 2014;289(30):20435–46.
Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–69.
Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–6.
Joseph JJ, Golden SH. Cortisol dysregulation: the bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann NY Acad Sci. 2017;1391(1):20–34.
Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am. 2014;43(1):75–102.
Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–9.
Grytsai O, Myrgorodska I, Rocchi S, Ronco C, Benhida R. Biguanides drugs: past success stories and promising future for drug discovery. Eur J Med Chem. 2021;224:113726.
Hou WL, Yin J, Alimujiang M, Yu XY, Ai LG, Yq B, et al. Inhibition of mitochondrial complex I improves glucose metabolism independently of AMPK activation. J Cell Mol Med. 2018;22(2):1316–28.
Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–85.
Maher M, Diesch J, Casquero R, Buschbeck M. Epigenetic-transcriptional regulation of fatty acid metabolism and its alterations in leukaemia. Front genet. 2018;9:405.
Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, et al. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA. 2009;106(42):17781.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41.
He L, Meng S, Germain-Lee EL, Radovick S, Wondisford FE. Potential biomarker of metformin action. J Endocrinol. 2014;221(3):363–9.
Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84.
Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–87.
Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur A-C, Dutta A, Prescott AR, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab. 2006;290(5):E780–8.
Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111(1):91–8.
Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279(2):1070–9.
Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48(7):e245.
Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282(45):32539–48.
Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11(6):554–65.
Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277(28):25226–32.
Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100(3):328–41.
Xie Z, Dong Y, Scholz R, Neumann D, Zou M-H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117(7):952–62.
Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–35.
Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20(3):526–40.
Dorfman J, Macara IG. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008;19(4):1614–26.
Balnis J, Korponay TC, Jaitovich A. AMP-activated protein kinase (AMPK) at the crossroads between CO2 retention and skeletal muscle dysfunction in chronic obstructive pulmonary disease (COPD). Int J Mol Sci. 2020;21(3):955.
Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–6.
Baur JA, Birnbaum MJ. Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab. 2014;20(2):197–9.
Saheki T, Iijima M, Li MX, Kobayashi K, Horiuchi M, Ushikai M, et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J Biol Chem. 2007;282(34):25041–52.
Alshawi A, Agius L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J Biol Chem. 2019;294(8):2839–53.
Tengholm A, Gylfe E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes Metab. 2017;19:42–53.
He L, Chang E, Peng J, An H, McMillin SM, Radovick S, et al. Activation of the cAMP-PKA pathway antagonizes metformin suppression of hepatic glucose production. J Biol Chem. 2016;291(20):10562–70.
Johanns M, Lai YC, Hsu MF, Jacobs R, Vertommen D, Van Sande J, et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat Commun. 2016;7:10856.
He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015;21(2):159–62.
Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820–7.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74.
Al-Oanzi ZH, Fountana S, Moonira T, Tudhope SJ, Petrie JL, Alshawi A, et al. Opposite effects of a glucokinase activator and metformin on glucose-regulated gene expression in hepatocytes. Diabetes Obes Metab. 2017;19(8):1078–87.
Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56(9):1898–906.
Akbari P, Gilani A, Sosina O, Kosmicki JA, Khrimian L, Fang Y-Y, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. 2021;373:6550.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12(3):489–98.
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5(1):99–118.
Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018;2018:1.
Salminen A. Feed-forward regulation between cellular senescence and immunosuppression promotes the aging process and age-related diseases. Ageing Res Rev. 2021;67:101280.
Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes. 2015;64(7):2289–98.
Xiang HC, Lin LX, Hu XF, Zhu H, Li HP, Zhang RY, et al. AMPK activation attenuates inflammatory pain through inhibiting NF-kappaB activation and IL-1beta expression. J Neuroinflammation. 2019;16(1):34.
Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–6.
Mohammadzadeh G, Zarghami N. Serum leptin level is reduced in non-obese subjects with type 2 diabetes. Int J Endocrinol Metab. 2013;11:3–10.
Hotamisligil GS. The role of TNFalpha and TNF receptors in obesity and insulin resistance. J Intern Med. 1999;245(6):621–5.
Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets. 2015;15(3):196–205.
McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41(1):36–48.
Ashoori M, Rahmati M, Ostadrahimi A, Fekri Aval S, Zarghami N. MicroRNAs and adipocytokines: promising biomarkers for pharmacological targets in diabetes mellitus and its complications. Biomed Pharmacother. 2017;93:1326–36.
Liao ZX, Fa YC, Kempson IM, Tseng SJ. Repolarization of M2 to M1 macrophages triggered by lactate oxidase released from methylcellulose hydrogel. Bioconjug Chem. 2019;30(10):2697–702.
Jing Y, Wu F, Li D, Yang L, Li Q, Li R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol Cell Endocrinol. 2018;461:256–64.
Hattori Y, Hattori K, Hayashi T. Pleiotropic benefits of metformin: macrophage targeting its anti-inflammatory mechanisms. Diabetes. 2015;64(6):1907–9.
Arimany-Nardi C, Minuesa G, Keller T, Erkizia I, Koepsell H, Martinez-Picado J, et al. Role of human organic cation transporter 1 (hOCT1) polymorphisms in lamivudine (3TC) uptake and drug-drug interactions. Front Pharmacol. 2016;7:175.
Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Transl Res. 2019;11(2):655–68.
Acknowledgements
This work was supported by the Department of Medical Biotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran (Grant number: 63438).
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation: MK and DJ; writing—review and editing: DJ; conceptualization and supervision: NZ.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khodadadi, M., Jafari-Gharabaghlou, D. & Zarghami, N. An update on mode of action of metformin in modulation of meta-inflammation and inflammaging. Pharmacol. Rep 74, 310–322 (2022). https://doi.org/10.1007/s43440-021-00334-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-021-00334-z