
Abstract
Glibenclamide (GLB) reduces brain edema and improves neurological outcome in animal experiments and preliminary clinical studies. Recent studies also suggested a strong anti-inflammatory effect of GLB, via inhibiting nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome activation. However, it remains unknown whether the anti-inflammatory effect of GLB is independent of its role in preventing brain edema, and how GLB inhibits the NLRP3 inflammasome is not fully understood. Sprague–Dawley male rats underwent 10-min asphyxial cardiac arrest and cardiopulmonary resuscitation or sham-operation. The Trpm4 siRNA and GLB were injected to block sulfonylurea receptor 1-transient receptor potential M4 (SUR1-TRPM4) channel in rats. Western blotting, quantitative real-time polymerase chain reaction, behavioral analysis, and histological examination were used to evaluate the role of GLB in preventing NLRP3-mediated neuroinflammation through inhibiting SUR1-TRPM4, and corresponding neuroprotective effect. To further explore the underlying mechanism, BV2 cells were subjected to lipopolysaccharides, or oxygen–glucose deprivation/reperfusion. Here, in rat model of cardiac arrest with brain edema combined with neuroinflammation, GLB significantly alleviated neurocognitive deficit and neuropathological damage, via the inhibition of microglial NLRP3 inflammasome activation by blocking SUR1-TRPM4. Of note, the above effects of GLB could be achieved by knockdown of Trpm4. In vitro under circumstance of eliminating distractions from brain edema, SUR1-TRPM4 and NLRP3 inflammasome were also activated in BV2 cells subjected to lipopolysaccharides, or oxygen–glucose deprivation/reperfusion, which could be blocked by GLB or 9-phenanthrol, a TRPM4 inhibitor. Importantly, activation of SUR1-TRPM4 in BV2 cells required the P2X7 receptor-mediated Ca2+ influx, which in turn magnified the K+ efflux via the Na+ influx-driven opening of K+ channels, leading to the NLRP3 inflammasome activation. These findings suggest that GLB has a direct anti-inflammatory neuroprotective effect independent of its role in preventing brain edema, through inhibition of SUR1-TRPM4 which amplifies K+ efflux and promotes NLRP3 inflammasome activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glibenclamide (GLB) is a sulfonylurea that has been used for decades to treat type 2 diabetes. Sulfonylurea receptor 1-transient receptor potential M4 (SUR1-TRPM4) heteromer is assembled by integrating SUR1 and TRPM4, in which sulfonylureas like GLB could bind to SUR1 and thereby prolong the closed states of TRPM4 channel [1], a pore-forming protein nonselectively conducting monovalent cations [2]. In recent years, it has been proved that GLB exerts neuroprotection on several neurologic disease models [3,4,5,6], potentially through inhibiting the SUR1-TRPM4 channel expressed in neurovascular units, and reducing consequential cell swelling, blood–brain barrier (BBB) disruption, and brain edema [3, 4]. Encouragingly, in several clinical studies, GLB has been shown to reduce brain edema and even improve neurological function [7,8,9]. More interestingly, even in some people who may not develop malignant brain edema [10, 11], sulfonylureas have shown neuroprotective effects, suggesting that they may have additional protective effects in addition to reducing brain edema. Indeed, animal studies have shown salutary roles of GLB in brain damages mainly characterized by neuroinflammation but not severe edema [12, 13], implying a neuroprotective mechanism of GLB apart from blocking the maladaptive influx of extracellular Na+ and water mediated by SUR1-TRPM4.
Inflammasomes, the key protein complexes of the host-defense system, play a critical role in the inherent inflammatory process in response to danger-associated molecular patterns (DAMPs) released during tissue injury [14], among which the canonical inflammasome nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) triggers the activation of the precursor of caspase-1 (pro-caspase-1), followed by the active caspase-1-mediated proteolytic cleavage of proinflammatory interleukin-1β (IL-1β) and consequential excessive inflammation [15]. Lamkanfi et al. [16] have corroborated that GLB functions upstream of NLRP3 to block NLRP3 inflammasome activation, suggesting a crucial role of GLB in restricting inflammation. However, it is unclear whether the anti-inflammatory effects of GLB contribute to the complementary neuroprotection in inflammatory brain damages as mentioned above. In view of our and other researchers’ findings that both SUR1-TRPM4 complex and NLRP3 inflammasome in microglia directly orchestrate the excessive neuroinflammation [12, 17], we assume that GLB might prevent the NLRP3 inflammasome activation in microglia and subsequent violent neuroinflammation through blocking SUR1-TRPM4, thus alleviating the neuroinflammatory injuries.
The mechanism underlying the potential link between SUR1-TRPM4 and NLRP3 inflammasome remains to be elucidated. After the priming phase initiated by ligating Toll-like receptor (TLR) with agonists such as lipopolysaccharides (LPS) [18], NLRP3 inflammasome is activated by DAMPs including adenosine triphosphate (ATP) which ligates the purinergic P2X7 receptor (P2X7R) [19, 20], a non-selective cation channel involved in the requisite decrease in intracellular K+ for NLRP3 activation [21, 22]. Previous studies have shown that opening P2X7R is not sufficient to reach the threshold of intracellular K+ to engage NLRP3, in need of the cooperation with other K+ channels in two-pore domain K+ channel (K2P) family and voltage-gated K+ channel (Kv) family [23,24,25]. However, the mechanism by which these channels synergistically launch the NLRP3 activation remains elusive. In consideration of the findings that GLB acts downstream of P2X7R [16], and the opening of above K+ channels is regulated by the depolarized membrane potential [26, 27], we hypothesize that SUR1-TRPM4 functions as an intermediate link between P2X7R and downstream K+ channels to gate the K+ channels via conducting Na+ inward current.
In the present study, we identified a neuroprotective role and mechanism of GLB in NLRP3-mediated neuroinflammation through blocking the SUR1-TRPM4 channel in microglia, independent of the previously known prevention of brain edema. Mechanistically, we revealed that the SUR1-TRPM4 complex served as a K+ efflux amplifier in activating the NLRP3 inflammasome in microglia, via mediating Na+ influx, contributing to the depolarized membrane potential, and thereby gating downstream K+ channels.
Materials and Methods
Animals
All animal experiments were performed in accordance with guidelines established by the Animal Care and Use Committee of Nanfang Hospital, Southern Medical University (Guangzhou, China). Male Sprague–Dawley rats (350–400 g) were purchased from the Experimental Animal Center of Southern Medical University. All animals in current study were housed in a specific pathogen-free facility under a strict 12-h light/dark cycle with free access to food and water.
Cell Line Culture and Treatments
Murine BV2 microglial cells and RAW264.7 macrophage cells were purchased from American Type Culture Collection (VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, NY, USA) containing 4.5 g/L glucose, 10% fetal bovine serum (FBS; Gibco), 100 IU/mL penicillin, and 100 mg/mL streptomycin, under a humidified atmosphere of 5% CO2 at 37 °C.
When BV2 or RAW264.7 cells reached 80 to 90% confluence, they were primed via oxygen–glucose deprivation/reperfusion (OGD/R) or 3-h LPS (1 μg/mL; Sigma-Aldrich, MO, USA) incubation. LPS was dissolved in sterile water, while the equal volume of sterile water was added as control. The 0.5-h incubation with NaN3 (1 mmol/L; Sigma-Aldrich), nigericin (10 μg/mL; MCE, NJ, USA), diazoxide (100 μmol/L; Sigma-Aldrich), or ATP (5 mmol/L; Roche, Basel, Switzerland) was conducted 6 h after the OGD, or LPS stimulation. Additionally, the following reagents were added into the medium 0.5 h preceding the intervention with ATP, NaN3, nigericin, or diazoxide: MCC950 (10 μmol/L; MCE) against NLRP3, PAP-1 (10 nmol/L; MCE) inhibiting Kv1.3, EGTA (4 mmol/L; Sigma-Aldrich) chelating extracellular Ca2+, Brilliant Blue G (BBG) (100 μmol/L; Sigma-Aldrich) against P2X7R, 9-phenanthrol (9-Ph) (50 μmol/L; Sigma-Aldrich) against TRPM4, quinine (100 μmol/L; MCE) inhibiting K2P family, and 0–200 μmol/L sulfonylureas (dissolved in 0.1–1% DMSO; Sigma-Aldrich) against SUR1 including GLB, gliclazide (GLZ), and glimepiride (GLM). Particularly, cells were pre‐incubated with transcription factor inhibitors such as Mithramycin A (2 μmol/L; MCE) blocking specificity protein 1 (Sp-1) and JSH23 (30 μmol/L; MCE) against nuclear factor kappa-B (NF-κB) for 1 h before challenged by LPS, or OGD.
Solutions Used in Ion-Replacement Experiments
The preparation of solutions was in accordance with the previous study [23]. Nominally K+-free solution contains (in mM) 150 NaCl, 2 CaCl2, 1 MgCl2, and 10 Hepes; Nominally K+-, Na+-, and Ca2+-free solution contains (in mM) 150 NMDG-HCl, 1 MgCl2, and 10 Hepes.
Asphyxial Cardiac Arrest Model in Rats and Treatments
Rats were randomly assigned to appropriate groups, and the 10-min asphyxial cardiac arrest/cardiopulmonary resuscitation (CA/CPR) model was performed as per our previous study [28]. In brief, the rats were anesthetized with isoflurane (4% for induction and 2% for maintenance; RWD, Shenzhen, China), followed by the orotracheal intubation with a 14G cannula (BD, Suzhou, China) and connection to a ventilator (RWD). Intravascular catheters (PE50; Smiths Medical, Ashford, UK) were inserted into the right femoral artery and vein for dynamic blood pressure monitoring and drug delivery, respectively. After 10-min stabilization, rats were chemically paralyzed by IV Vecuronium (2 mg/kg), and then the ventilator was disconnected for 10 min, typically leading to circulatory arrest in 5 min, which was characterized by the cessation of arterial pulse and a decrease of mean arterial pressure (MAP) to 20 mmHg below. At the end of 10-min asphyxia, cardiopulmonary resuscitation was initiated by effective ventilation with 100% oxygen and intravenous administration of epinephrine (0.01 mg/kg), simultaneously accompanied by the chest compressions (200 compressions/min). Additional doses of epinephrine (0.02 mg/kg) were given at 2-min intervals until the return of spontaneous circulation (ROSC), as defined by an increase of MAP to 60 mmHg above for at least 10 min. After spontaneous respiration recovery, rats were weaned from ventilator and extubated, but the rats that failed to ROSC within 5 min or were unable to be weaned from ventilator after 1-h observation were excluded from the continuing experiments.
GLB was dissolved in 0.05% dimethyl sulfoxide (DMSO) (2.5 μg/mL). After ROSC, GLB was administrated intraperitoneally with an initial dose of 10 μg/kg at 15 min and 4 maintenance doses of 1.2 μg per 6 h [28], whereas rats in the vehicle group received equivalent volume of 0.05% DMSO (Fig. S1A). Rats that underwent all procedures except asphyxial CA/CPR were used as sham control.
Intracerebroventricular Injection of siRNA solution
The Trpm4 in vivo-ready siRNA and the universal in vivo-negative control siRNA were purchased from Ambion (MA, USA). The sequences of Trpm4 siRNA were as follows: sense 5′-CGCUAGUAGCAGCAAAUCUtt-3′ and antisense 5′-AGAUUUGCUGCUACUAGCGtg-3′, which downregulated the expression of TRPM4 in vivo as per our previous study [29]. Forty-eight hours before induction of cardiac arrest, rats were anesthetized with pentobarbital, and 500 pmol of Trpm4 or negative control siRNA in a 10-μL Hamilton syringe (Hamilton Co, Reno, NV, USA) was injected into right lateral ventricle at a rate of 0.5 μL/min via a burr hole on the skull, according to the coordinates relative to bregma: 1.0 mm lateral, 1.5 mm posterior, and 3.5 mm below the horizontal plane of bregma.
Cell OGD/R Model
OGD/R on BV2 cells was performed as described previously [30]. The cells were cultured in the glucose-free DMEM (Gibco), and placed in a hypoxic chamber with 1% O2, 5% CO2, and 94% N2 at 37 °C for 3 h to mimic the hypoxic-ischemic injury, followed by the restoration with DMEM (containing 4.5 g/L glucose) at the normoxic condition to mimic the reperfusion. Control cells were maintained in DMEM (containing 4.5 g/L glucose) without oxygen deprivation.
Western Blotting
Western blotting was routinely performed as previously reported [31]. Rodent brain tissues, BV2 cells, and RAW264.7 cells were homogenized in RIPA lysis buffer (Beyotime, Shanghai, China) containing protease inhibitor cocktail. After denatured in loading buffer, the samples were subjected to SDS-PAGE and then transferred to PVDF membranes (Millipore, MA, USA). After blocked by 5% non-fat milk, the membranes were incubated overnight at 4 °C with the primary antibodies as below: mouse anti-β-actin (Proteintech, IL, USA), rabbit anti-GAPDH (Proteintech), rabbit anti-TRPM4 (Sigma-Aldrich), mouse anti-SUR1 (Abcam, Cambridge, UK), rabbit anti-NLRP3 (Novus, CO, USA), rabbit anti-caspase-1 p20 (Bioss, Beijing, China), rabbit anti-IL-1β p17 (Novus), rabbit anti-Kir6.1 (Abcam), and rabbit anti-Kir6.2 (Abcam), followed by the detection via the secondary antibodies (CST, MA, USA). The densities of protein blots were quantified by ImageJ software (NIH, MD, USA) and normalized to the level of β-actin or GAPDH.
Measurement of Gene Expression
The mRNA levels of SUR1, TRPM4, NLRP3, caspase-1, IL-1β, Kir6.1, Kir6.2, K2P family, Kv family, and GADPH were routinely measured by quantitative real-time polymerase chain reaction (qRT-PCR) [32]. Briefly, total RNA was isolated using Trizol Reagent (Thermo Fisher Scientific, MA, USA) and reverse transcribed to cDNA with the PrimeScript™ RT Master Mix Kit (Takara, Dalian, China) according to the manufacturer’s instructions. qRT-PCR was performed using the SYBR Green master mixes (Takara) and Roche LightCycler480 System. Relative changes of mRNA expression were normalized to the level of GADPH.
Neurologic Function Evaluation
The neurologic outcome of post-cardiac arrest rats was assessed at 24, 48, 72 h, and 7 days after ROSC, utilizing a scale of neurologic deficit scores (NDSs) in which 80 was considered normal, whereas 0 represented brain death [33]. The total NDS scale consisted of 7 components: general behavioral deficit, brain-stem function, motor assessment, sensory assessment, motor behavior, behavior, and seizures.
Behavioral Testing
The Morris water maze was carried out to evaluate short-term spatial learning and memory as described previously [17, 34]. The water maze apparatus is a circular tank filled with opaque water (divided into four quadrants, called Q1, Q2, Q3, Q4), and a hidden platform is submerged 2–3 cm for rats below the water surface and not visible to the rodents. Firstly, the rodents were trained to search for the platform on day 9–12 post-cardiac arrest at a frequency of four trials/day, orienting by referencing 3 external cues surrounding the tank. If the rodents did not find the platform in 60 s, they were manually placed on it for 15 s. Rodents’ movements were tracked by TSE VideoMot2 tracking system (Bad Homburg, Germany) to record the path and latency time taken to escape from 4 randomly assigned locations. After the training termed as acquisition trial, the probe trial was performed on the following day, when the rodents were allowed 60 s to explore the platform which has been removed. The percentage of total time that rodents spent in the target quadrant and the number of platform location crossings were recorded and analyzed.
Histological Examination
For detecting neuronal loss and the levels of certain markers, rats were deeply anesthetized and transcardially perfused with saline on day 14 post-surgery. The brains were fixed with 4% paraformaldehyde (PFA) overnight and embedded in paraffin, and the 4-μm-thick coronal slices located at 3.5 mm posterior to bregma were stained with cresyl violet (Beyotime) and then observed under an optical microscope (Olympus, Tokyo, Japan). Viable neurons in the hippocampal CA1 region had the characteristics of visible nucleus and intact cytoplasm with discernable Nissl staining, while those shrunken cell bodies surrounded by empty space were regarded as dead neurons.
Immunohistochemistry of hippocampus CA1 region from rats was conducted on day 14 post-surgery by incubation with antibodies against neuronal nuclei (NeuN; CST) for neurons, microtubule-associated protein 2 (MAP-2; Sigma-Aldrich) for dendrites, glial fibrillary acidic protein (GFAP; Abcam) for astrocytes, ionized calcium-binding adapter molecule-1 (Iba-1; Novus), and CD68 (Novus) for microglia. After the immunoperoxidase staining via secondary antibodies, the slides were counterstained with hematoxylin. In each brain section, 3 fields were randomly examined. The relative intensity of MAP-2 staining and the number of NeuN-, GFAP-, Iba-1-, or CD68-positive cells in hippocampus CA1 region were quantified using ImageJ software.
For immunofluorescence, rats were euthanized after 12 h post-cardiac arrest. Following fixation with 4% PFA, the brains were immersed in 15% and 30% sucrose at 4 °C for cryoprotection. With regard to the BV2 cells, they were fixed with 2% PFA for 10 min at room temperature. For immunofluorescent co-localization, the slides of brain sections and cells were incubated with the pairs of the following primary antibodies: rabbit anti-TRPM4, mouse anti-SUR1, and mouse anti-CD68. Afterwards, the slides were washed and detected with appropriate Alexa Fluor dye of secondary antibodies preceding counterstaining with DAPI in the dark. Finally, fluorescent signaling was observed under a confocal microscope (Olympus) and analyzed using ImageJ software.
Caspase-1 Activity Detection
BV2 cells were seeded in 96-well plates, of which the activated caspase-1 was evaluated utilizing a FAM-FLICA detection kit (Immunochemistry Technologies, MN, USA) as described previously with some adjustments [35]. The fluorescent probe FAM-YVAD-FMK (FLICA) was employed to irreversibly label in situ activated caspase-1 in living cells, and the green fluorescent signal directly reflected the caspase-1 activity at the time the reagent was added. Briefly, following the reconstitution with DMSO, FLICA was diluted with phosphate buffered saline (PBS), added to each sample and incubated for 1 h prior to the nuclear staining with Hoechst. FLICA excited from 490 to 495 nm and emitted from 515 to 525 nm. The images were captured with a fluorescence microscope and the fluorescence intensity of cleaved caspase-1 was quantified by the multiscan spectrum (BMG, Offenburg, Germany).
Measurement of Intracellular Ca2+ and Membrane Potential
The procedure was established as described previously [36, 37]. Experiments were typically conducted using BV-2 cells seeded into 96-well plates and allowed to adhere overnight. After treatment with LPS and indicated antagonists, the cultures were washed three times in buffer and loaded for 30 min in the dark at 37 °C, with 10 μmol/L of either cell-permeable Ca2+ fluorescent indicator Fluo-4AM (Beyotime) or membrane potential indicator Dioc5 (Abbkine, CA, USA). Cells were then washed again in buffer and all experiments were performed at room temperature in the dark. Using the multiscan spectrum, emission intensity was measured up to 15 min, and the decline in Dioc5 fluorescent intensity at 430 nm represents the depolarization of membrane potential. Stimulating drugs including ATP, diazoxide and nigericin were added 30 s after the first measurement.
Measurement of Intracellular K+ and Na+
This procedure was routinely performed as previously reported [38]. The BV-2 cells were seeded into 96-well plates to adhere overnight. After treatment with LPS and indicated antagonists, the cells were loaded with either potassium- or sodium-sensitive fluorescent dyes, PBFI-AM (5 μmol/L, Abcam), or SBFI-AM (5 μmol/L, Abcam), which were freshly prepared by combining with equal volume of 25% Pluronic F-127 (Solarbio, Beijing, China) for 40 min at 37℃. The change in 340 nm/380 nm ratio that represents the alteration of intracellular K+ or Na+ was analyzed with the multiscan spectrum. Stimulating drugs including ATP and diazoxide were added 30 s after the first measurement.
Measurement of Intracellular ATP
Cells were cultured in a 96-well plate and allowed to grow overnight. After treatment, the intracellular ATP level was measured using CellTiter-Glo luminescent cell viability assay kit (Promega, WI, USA) as previously reported [39]. The luminescence was measured to calculate the ATP concentration using the standard curve.
Co-immunoprecipitation
This assay was performed in line with our previous study [17]. BV2 and RAW264.7 cells were lysed in moderate lysis buffer (Genstar, Beijing, China) containing phenylmethylsulfonyl fluoride. After centrifuged at 14,000 g for 15 min, the supernatant was obtained and quantified, of which the portion containing 500 μg protein was immunoprecipitated with 1 μg rabbit anti-TRPM4 or mouse anti-SUR1 under rotation overnight at 4 °C. Following the rotary incubation with 30 μL protein A/G magnetic beads (Bimake, TX, USA) for another 20 min at room temperature and subsequent magnetic separation, the complexes were washed 5 times, and resuspended in 40 μL loading buffer prior to denatured at 100 °C. Finally, the protein complexes were subjected to Western blotting as described above, and the employed antibodies were as follows: rabbit anti-TRPM4, mouse anti-SUR1, rabbit anti-Kir6.1, and rabbit anti-Kir6.2. Whole cell lysates were used as an input control, and homophytic IgG was used as a negative control.
Statistical Analysis
All data were presented as means ± SD or medians and 25th to 75th percentiles (NDSs). Quantitative data were analyzed with one-way ANOVA followed by Tukey’s post hoc multiple comparison tests, or as indicated. NDSs were compared using Mann–Whitney U test. The data of escape latency in the water maze test were analyzed with repeated-measures ANOVA comprising treatment groups, time points, and groups × time interaction, followed by Tukey’s post hoc multiple comparison tests. SPSS 20.0 (IBM, NY, USA) and GraphPad Prism 8.0 (GraphPad, CA, USA) were used for statistical analyses. P < 0.05 was considered statistically significant.
Results
Blockage of SUR1-TRPM4 by GLB Prevents NLRP3 Inflammasome Activation in Microglia After Cardiac Arrest
Rats undergoing 10-min CA/CPR modeling (n = 58) or sham operation (n = 15) were randomly assigned to appropriate groups for different experimental purposes (Fig. S1B). Eleven rats failed to ROSC and were randomly distributed to the vehicle/control group (6/32) and the GLB/siRNA group (5/26), indicating that no significant difference in the success rates of CA/CPR was observed among groups. For resuscitated animals, 4 rats (2 in the vehicle/control group, 2 in the GLB/siRNA group) were unable to be weaned from ventilator, and there was no significant difference in the fail rate of removing ventilator among these groups. The time from asphyxia to cardiac arrest, the time required for ROSC, the epinephrine usage, and physiological variables including MAP, heart rate, and rectal temperature were all comparable among experimental groups (Table S1). There were also no statistical differences in laboratory results including PaCO2, PaO2, PH, and blood glucose level at baseline or after ROSC among these groups (Table S2).
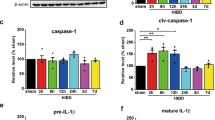
In the rat model of CA/CPR characterized by drastic neuroinflammation [17], we first addressed whether GLB exerted the anti-inflammatory effect via inhibiting the NLRP3 inflammasome activation mediated by the SUR1-TRPM4 channel in microglia. Sampling at 12 h post-cardiac arrest was determined according to our previous studies [17, 40], which showed that the SUR1 and TRPM4 protein levels significantly increased from 6 to 24 h after CA/CPR, accompanied by concurrent NLRP3 inflammasome activation. In this study, the levels of NLRP3, caspase-1 p20, and IL-1β p17 from hippocampus and cortex of rats were elevated at 12 h after CA/CPR, but reversed by pharmacological inhibition of SUR1 with GLB and gene knockdown of Trpm4 (Fig. 1A−D), implying that obstructing SUR1-TRPM4 might disturb the activation signal required for NLRP3 inflammasome. However, GLB could not suppress the upregulated SUR1 and TRPM4 at 12 h post-cardiac arrest (Fig. 1A, 1C), suggesting that GLB might inhibit the transport activity of this specific channel rather than reduce its expression as per our previous study [28]. As determined by the highly consistent immunofluorescent location in the hippocampus under confocal microscopy, SUR1 co-localized with TRPM4 suggesting on the expression of the SUR1-TRPM4 channel, and TRPM4 was also massively localized in cells expressing CD68 (Fig. 1E), collectively depicting the formation of the complex mainly in microglia. In sum, the above results elucidate that cardiac arrest fuels the activation of NLRP3 inflammasome during the acute phase, which can be reversed by GLB through blocking the SUR1-TRPM4 complex in microglia.

Blocking SUR1-TRPM4 with GLB reverses NLRP3 inflammasome activation in microglia after CA/CPR. A and C The protein levels of indicated molecules among the sham, vehicle, and GLB groups. B and D The protein levels of above molecules among the sham, control (treated with the negative control siRNA), and Trpm4 siRNA groups. E Confocal analysis of TRPM4 (green) and SUR1 (red) or CD68 (red) staining 12 h after ROSC. Scale bar = 50 μm. Statistical significances are determined with one-way ANOVA followed by Tukey’s post hoc test. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; #P < 0.05, ##P < 0.01, ### P < 0.001 versus vehicle or control. n = 5 per group
Inhibiting SUR1-TRPM4 Channel with GLB Improves Neurological Function After CA/CPR
We further explored whether blockage of the SUR1-TRPM4 channel with GLB improved neurologic outcomes in CA/CPR modeling rats. We used the NDSs to assess the neurologic function after CA/CPR and observed that rats in the vehicle group presented lower NDSs at 24, 48, and 72 h after ROSC than those in the GLB group, without prominent difference on day 7 (Fig. 2A), manifesting that the neurologic deficits caused by CA/CPR were alleviated by GLB.

Inhibiting SUR1-TRPM4 channel with GLB improves neurological function and neuropathological damage after CA/CPR. A NDSs of survived rats at 24, 48, and 72 h and on day 7 after ROSC. B–D The effect of GLB on post-cardiac arrest spatial memory deficits in Morris water maze test. E–H The effect of GLB on post-cardiac arrest neuropathological damage. Scale bar = 500 μm or 100 μm. Statistical significances are determined with one-way ANOVA followed by Tukey’s post hoc test or Mann–Whitney U test (NDSs), except the data of escape latency which are analyzed with repeated-measures ANOVA. Data are presented as mean ± SD, or medians and percentiles (NDSs). *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; #P < 0.05, ##P < 0.01, ### P < 0.001 versus vehicle. n = 5 or 9 per group
We also employed the Morris water maze test to gauge the effect of GLB on short-term spatial learning and memory ability after cardiac arrest. During the hidden-platform training on day 9–12 after surgery, rats in the sham, vehicle, and GLB groups all showed a gradual decline in the latency to find the hidden platform over time (F = 65.183, P = 0.000) (Fig. 2B). Besides, repeated-measure ANOVA revealed that escape latency differed significantly among the groups (F = 14.294, P = 0.000), with no significant interaction between groups and time points (F = 1.375, P = 0.241) (Fig. 2B). Tukey’s post hoc test showed that GLB substantially rescued the extended latency caused by CA/CPR on day 10–12 (Fig. 2B). In the probe trial of the Morris water maze test on day 13 after surgery, the frequency of crossing the platform area was decreased in the vehicle group compared with the sham group, whereas the propensity to reduction was largely reversed by GLB (Fig. 2C−D). Likewise, rats administrated with GLB spent markedly more time searching in the platform quadrant (Q3) in comparison with vehicle rats (Fig. 2C−D). These results collectively demonstrate that inhibiting SUR1-TRPM4 with GLB provides persistent neuroprotection and mitigates the spatial learning and memory deficiency derived from cardiac arrest.
GLB Ameliorates Neurological Injury After CA/CPR Through Targeting SUR1-TRPM4 Channel
To assess the profound effect of GLB on the histological damage caused by cardiac arrest, all the rats involved in the Morris water maze test were euthanized on day 14 after CA/CPR to conduct histological examination. The results of Nissl staining unraveled that CA/CPR elicited neuron loss in hippocampal CA1 region, a susceptible region to ischemia, whereas more viable neurons were reserved by GLB (Fig. 2E−F). Consistently, GLB-treated rats exhibited more NeuN + neurons in hippocampal CA1 region than vehicle-treated rats (Fig. 2G−H). We also set out to evaluate the injury to dendrite by immunostaining for MAP-2. As expected, GLB partially recovered the extensive loss of dendrites caused by CA/CPR in hippocampal CA1 region (Fig. 2G−H). Neuron loss is usually accompanied by glial activation to clean up the cell debris. Herein, we carried out the Iba-1, CD68, and GFAP immunohistochemical staining to verify the activation of microglia and astrocytes. As illustrated in Fig. 2G−H, dramatic increment of Iba-1-positive, GFAP-positive, and CD68-positive cells appeared in the post-cardiac arrest hippocampal CA1 region when compared to the sham controls. However, the activated glial cells were significantly reduced in the presence of GLB, as evidenced by the decreased immunoreactivities of Iba-1, CD68, and GFAP. Overall, the findings suggest that occluding SUR1-TRPM4 complex with GLB mitigates CA/CPR-induced neuropathological injury.
Signal Coupling of SUR1-TRPM4 Complex and NLRP3 Inflammasome Occurs in BV2 Cells Challenged with Various Injurious Stimuli
Due to the difficulty in completely eliminating the interference of brain edema in CA/CPR model [41], in vitro, we further explored the anti-inflammatory effect of GLB. To evaluate the connection between SUR1-TRPM4 complex and NLRP3 inflammasome, we first sought to examine the expression of key components of SUR1-TRPM4 complex and NLRP3 inflammasome in LPS- and OGD/R-primed BV2 cells, in which the latter mimicked in vivo CA/CPR model. As shown in the heatmaps (Fig. 3A), the mRNA levels of SUR1 (encoded by Abcc8), TRPM4, NLRP3, caspase-1, and IL-1β were remarkably elevated in post-modeling cells, but reversed by inhibiting Sp-1 and NF-κB with Mithramycin A and JSH23, suggesting the engagement of the two transcriptional factors in the priming stage of NLRP3 activation [42, 43]. Besides, no alteration was found in the levels of Kir6.1 and Kir6.2 (encoded by KcnJ8 and KcnJ11, respectively), which functioned as the components of KATP (SUR1/2-Kir6.x) channel, another specific target of sulfonylureas. Consistently, the massively increased protein levels of SUR1, TRPM4, and NLRP3 after modeling were determined by immunoblotting (Fig. 3B). Additionally, the fluorescence in Fig. 3C showed the obvious formation of SUR1-TRPM4 channel after modeling, as evidenced by the highly consistent localization of SUR1 and TRPM4.

Signal coupling of SUR1-TRPM4 complex and NLRP3 inflammasome in BV2 cells challenged with various injurious stimuli. A The heatmap reflecting the mRNA levels of indicated genes in LPS- or OGD-primed BV2 cells with or without transcriptional factor inhibitors. Mith A, Mithramycin A. B Western blotting bar graph of SUR1, TRPM4, and NLRP3 in LPS- or OGD-primed BV2 cells. C Confocal analysis of TRPM4 (green) and SUR1 (red) staining in LPS- or OGD-primed BV2 cells. Scale bar = 50 μm. D and E The immunofluorescence reflecting the caspase-1 activity in LPS- or OGD-primed BV2 cells challenged with ATP with or without MCC950. Scale bar = 50 μm. F and G The protein expression levels of caspase-1 p20 and supernatant IL-1β reflecting the NLRP3 activation in OGD-primed BV2 cells challenged with ATP with or without MCC950. All data in this figure are analyzed using one-way ANOVA followed by Tukey’s post hoc test. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ### P < 0.001 versus LPS- or OGD-primed BV2 cells challenged with ATP. n = 3 per group
Afterwards, the increment of caspase-1 activity was reflected by the higher fluorescent intensity and increased caspase-1 p20 in post-modeling BV2 cells treated with ATP (Fig. 3D−G). Likewise, the ATP-induced release of IL-1β into supernatant was also observed via Western blotting. Nevertheless, the increased caspase-1 p20 and supernatant IL-1β was eliminated through intervening in NLRP3 by MCC950 (Fig. 3F−G), suggesting that ATP mainly activates canonical NLRP3 inflammasome rather than non-NLRP3 inflammasomes. Taken together, these findings indicate the tight relation of SUR1-TRPM4 but not KATP channel with NLRP3 inflammasome activation in response to ATP in microglia challenged with excessive inflammation, or ischemia–reperfusion injury, further suggesting the critical role of SUR1-TRPM4 in NLRP3 activation in vivo.
Pharmacological Inhibition of the SUR1-TRPM4 Channel Reverses the Activation of NLRP3 Inflammasome in BV2 Cells
To further investigate the role of SUR1-TRPM4 channel in ATP-induced NLRP3 activation, we evaluated the expression of relevant molecules in ATP-treated BV2 cells challenged with LPS, when exposed to the concentration gradient of SUR1 or TRPM4 inhibitors. The results revealed that the NLRP3 inflammasome activation was almost abolished through dampening TRPM4 by 50 μmol/L of 9-Ph, as evaluated by the level of caspase-1 p20 and supernatant IL-1β returning to baseline (Fig. 4A). As indicated in Fig. 4B−D, the sulfonylureas including GLB, GLZ, and GLM also inhibited NLRP3 inflammasome activation in a concentration-dependent manner. Likewise, ATP-induced NLRP3 activation was diminished by 50 μmol/L of 9-Ph and 100 μmol/L of sulfonylureas in BV2 cells OGD/R model (Fig. 4E). In sum, closing the SUR1-TRPM4 channel with inhibitors such as GLB effectively prevents ATP-induced NLRP3 inflammasome activation in microglia challenged with excessive inflammation, or ischemia–reperfusion injury, in consistency with the results in CA/CPR model.

Pharmacological inhibition of SUR1-TRPM4 reverses the activation of NLRP3 inflammasome in BV2 cells. A–D Western blotting results manifesting the levels of NLRP3 inflammasome activation in LPS-primed BV2 cells challenged with ATP, when exposed to the concentration gradient of 9-Ph (A), GLB (B), GLM (C), and GLZ (D). E Western blotting results manifesting the levels of NLRP3 inflammasome activation in OGD-primed BV2 cells challenged with ATP, in the presence of indicated SUR1 or TRPM4 inhibitors. All data in this figure are analyzed using one-way ANOVA followed by Tukey’s post hoc test. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ### P < 0.001 versus OGD-primed BV2 cells challenged with ATP. n = 3 per group
Upstream P2X7R-Mediated Ca2+ Influx Modulates the Opening of SUR1-TRPM4 Required by NLRP3 Inflammasome Activation
We next set out to determine the electrophysiological mechanism by which the activated SUR1-TRPM4 channel delivered the activation signal to NLRP3 inflammasome (Fig. 5A). As assessed by the level of caspase-1 p20 subunit and released IL-1β, LPS-primed BV2 cells challenged with ATP showed apparent NLRP3 activation, whereas the activation was largely blunted due to the presence of BBG, a specific antagonist of P2X7R (Fig. 5B−C). Moreover, we observed a marked increase of NLRP3 inflammasome activation in LPS-treated BV2 cells with diazoxide opening SUR1-regulated channels, comparable to what was observed posterior to ATP incubation. The difference was that BBG could not eliminate caspase-1 cleavage and IL-1β release, but 9-Ph or GLB could, collectively demonstrating that SUR1-TRPM4 complex acts downstream of P2X7R. Thereafter, we found that EGTA depletion of extracellular Ca2+ was responsible for the inhibition of NLRP3 activation in the LPS-primed BV2 cells incubated with ATP, but the activation of NLRP3 inflammasome after diazoxide treatment showed no relation to extracellular Ca2+ concentration (Fig. 5D−E). We further detected the change in intracellular Ca2+ after various stimulations and interventions (Fig. 5F), and corroborated that the intracellular Ca2+ was significantly elevated given the stimulation with LPS and ATP, but was largely reversed when blocking the P2X7R by BBG or depleting extracellular Ca2+ by EGTA, illustrating that ATP-induced opening of P2X7R activates SUR1-TRPM4, through increasing intracellular Ca2+, the critical condition for TRPM4 channel opening [44].

The activation of SUR1-TRPM4 in BV2 cells involves upstream P2X7R-mediated Ca2+ influx, and participates in the NLRP3 activation mediated by the ATP decrease in BV2 cells. A The schematic of the hypothesis and experiment design in this chapter. B–E Western blotting results reflecting NLRP3 activation in LPS-primed BV2 cells challenged with ATP or diazoxide, in the presence of BBG, GLB, 9-Ph, or EGTA. F The change in intracellular Ca2+ of BV2 cells after various stimulations and interventions. G–J Western blotting results reflecting NLRP3 activation in LPS-primed BV2 cells challenged with ATP, nigericin, or NaN3, in the presence of GLB or 9-Ph. K The histogram to show the level of intracellular ATP in indicated groups. All data in this figure are analyzed using one-way ANOVA followed by Tukey’s post hoc test. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. n = 3 per group
Lack of SUR1-TRPM4 Channel Suppresses the NLRP3 Inflammasome Activation Mediated by the ATP Decrease in BV2 Cells
Nigericin, a K+ ionophore, elicits the NLRP3 inflammasome activation involving increased K+ efflux independent of P2X7R [45] and the decline of intracellular ATP, of which the latter is considered as a key trigger of NLRP3 inflammasome activation [46] and also another critical condition for TRPM4 channel opening [47]. We thus explored the role of SUR1-TRPM4 complex in the P2X7R-independent NLRP3 activation. The nigericin-treated BV2 cells showed increment of caspase-1 activation and mature IL-1β release, but the increase was abolished by 9-Ph or GLB (Fig. 5G−H). Likewise, GLB or 9-Ph abrogated the NLRP3 activation fueled by NaN3 (Fig. 5I−J), a metabolic toxin depleting intracellular ATP [48], and Fig. 5K verifies the efficacy of nigericin and NaN3 to reduce intracellular ATP. Overall, these results imply that SUR1-TRPM4 channel participates to the P2X7R-independent NLRP3 inflammasome activation following cytoplasmic ATP decrease in microglia, which may be another NLRP3 activation pathway involving SUR1-TRPM4 in vivo.
SUR1-TRPM4 Magnifies the K+ Efflux via the Na+ Influx-Driven Opening of Downstream K+ Channels in BV2 Cells
Previous studies have illuminated the engagement of multiple specific K+ channels belonging to K2P and Kv family [23,24,25] in the decline of intracellular K+ required for NLRP3 activation. Therefore, we further addressed the K+ efflux mechanism involving above channels in the NLRP3 inflammasome activation mediated by SUR1-TRPM4 complex (Fig. 6A). The results verified that inhibition of Kv1.3 by PAP-1 partially restored the augmented caspase-1 cleavage and IL-1β release triggered by ATP or diazoxide in LPS-primed BV2 cells, while the NLRP3 activation was almost completely suppressed by quinine, an antagonist of K2P family (Fig. 6B−C). As expected, inhibiting kv1.3 or K2p family counteracted the obvious decline in intracellular K+ caused by ATP or diazoxide (Fig. 6D). Moreover, the BV2 cells challenged with LPS manifested the massively increased mRNA levels of representative channels such as TWIK1 (two-pore domain weak inwardly rectifying K+ channel 1), TWIK2, THIK1 (tandem pore domain halothane-inhibited K+ channel 1), and Kv1.3 (Fig. 6E). These findings prove that these representative K+ channels mediate the K+ efflux required for the NLRP3 activation.

SUR1-TRPM4 magnifies the K+ efflux via the Na+ influx-driven opening of downstream K+ channels in BV2 cells. A The schematic of the hypothesis and experiment design in this chapter. B and C Western blotting results reflecting NLRP3 activation in LPS-primed BV2 cells challenged with ATP or diazoxide, in the presence of quinine or PAP-1. D The change in intracellular K+ of BV2 cells after various stimulations and interventions. E qRT-PCR results showing the mRNA levels of representative K+ channels in LPS-primed BV2 cells. F and G Western blotting results reflecting NLRP3 activation in LPS-primed BV2 cells challenged with ATP or diazoxide, when bathed in a nominally Na+-free medium. H The change in intracellular Na+ of BV2 cells after various stimulations and interventions. I The change in membrane potential of BV2 cells after various stimulations and interventions. All data in this figure are analyzed using one-way ANOVA followed by Tukey’s post hoc test. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. n = 3 per group
Given that the opening of above K+ channels was regulated by the depolarized membrane potential [26, 27], we also addressed the mechanism by which SUR1-TRPM4 opened the downstream K+ channels. We observed that the caspase-1 activation and increased supernatant IL-1β was largely reversed, when the LPS-primed BV2 cells treated with ATP or diazoxide were bathed in a nominally Na+-free medium (Fig. 6F−G), accompanied by the inhibition of ATP- or diazoxide-induced inward Na+ current, depolarized membrane potential, and outward K+ current, which was comparable to the efficacy of silencing SUR1-TRPM4 (Fig. 6D, 6H−I). These results suggest that SUR1-TRPM4 regulates the Na+ influx and subsequent depolarization of membrane potential, thereby opening the mentioned K+ channels and amplifying K+ efflux.
NLRP3 Inflammasome Activation in RAW264.7 Cells Is Attributed to the TRPM4 Channel Alone Rather than Binding to SUR1
Given that 200 μmol/L of GLB but not glipizide entirely inhibits the NLRP3 inflammasome activation in macrophages, suggesting a SUR1-independent inhibitory effect [16], we further explored the difference in the mechanism of NLRP3 activation between microglia and macrophages. As illustrated in Fig. 7A−B, ATP-induced NLRP3 inflammasome activation in RAW264.7 cells challenged with LPS was completely abrogated by 50 μmol/L of 9-Ph, partly restored by 100 μmol/L of GLB, but not recovered by GLZ or GLM. Additionally, TRPM4 was poorly capable of binding to SUR1 in RAW264.7 cells, compared with BV2 cells (Fig. 7C). This was reminiscent of the KATP channel, and we thereby evaluated whether Kir6.x had better capacity of competitive binding to SUR1 than TRPM4 in RAW264.7 cells. The levels of SUR1 and TRPM4 were elevated in LPS-treated BV2 and RAW264.7 cells, whereas Kir6.1 level was increased in RAW264.7 but not BV2 cells (Fig. 7D−F). Consistently, SUR1-TRPM4 complex was dominant in BV2 cells, while obvious interaction between SUR1 and Kir6.1 was observed in RAW264.7 cells (Fig. 7G), demonstrating the existence of competitive relationship between TRPM4 and Kir6.1. Therefore, we suppose that the TRPM4 channel mainly exists at an unbound state in macrophages to mediate the NLRP3 inflammasome activation, and can be directly blocked by higher concentration of GLB [44].

The difference in the mechanism of NLRP3 activation between microglia and macrophages. A and B Western blotting results reflecting NLRP3 activation in LPS-primed RAW264.7 cells challenged with ATP, in the presence of SUR1 or TRPM4 inhibitors. C The association of SUR1 and TRPM4 detected by immunoprecipitation, in both BV2 and RAW264.7 cells stimulated by LPS. D–F The protein and mRNA levels of SUR1, TRPM4, Kir6.1, and Kir6.2 in LPS-primed BV2 or RAW264.7 cells. G The association between SUR1 and TRPM4, Kir6.1, or Kir6.2 detected by immunoprecipitation, in both BV2 and RAW264.7 cells stimulated by LPS. Samples from LPS-primed cells are immunoprecipitated with isotype IgG as a control. All data in this figure are analyzed using one-way ANOVA followed by Tukey’s post hoc test. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. n = 3 per group
Discussion
In the previous studies, GLB has reportedly played a neuroprotective role in several neurologic diseases such as ischemic stroke [3], subarachnoid hemorrhage [4], and traumatic brain injury [5], likely through blocking the SUR1-TRPM4 complex in neurons and astrocytes, which has been reported to mediate the dramatic influx of Na+ and water, strongly correlating with the BBB damage and cerebral edema in the above neurologic diseases [3,4,5]. However, recent studies have also found that the inhibition of SUR1-TRPM4 by GLB reduces the neuroinflammatory burden of brain damages without obvious edema [12, 13], implying a complementary neuroprotective mechanism underlying the inhibition of SUR1-TRPM4 with GLB, through restricting neuroinflammation but not reducing edema. NLRP3 inflammasome activation is a critical host defense to danger signals such as extracellular ATP released from dead cells. However, overactivity of this pathway can cause detrimental inflammatory processes and exacerbate tissue damage in various neurological disorders [49,50,51]. In terms of the findings that GLB has been reported to suppress NLRP3 activation in previous studies [16, 52, 53], we speculate that GLB prevents the activation of NLRP3 inflammasome via blocking SUR1-TRPM4.
In recent years, our team has observed that SUR1-TRPM4 is highly expressed in murine microglia after CA/CPR or ischemic stroke [40, 54]. Moreover, the critical role of microglia in providing the platform for neuroinflammation is further exemplified by our findings that the aberrant NLRP3 inflammasome activation in microglia mediates the excessive neuroinflammation in CA/CPR model [17]. In view of the finding that the expression of certain pro-inflammatory genes could be downregulated via blocking the upregulated SUR1-TRPM4 channel with GLB in LPS-activated microglia [12], we further hypothesize that GLB effectively prevents microglial NLRP3 activation through inhibiting SUR1-TRPM4. Here, we conducted in vivo experiments in the CA/CPR model which was mainly characterized by neuroinflammation. We demonstrated that the microglial NLRP3 activation after CA/CPR could be abolished by GLB. To completely exclude the interference of brain edema after CA/CPR [41], in vitro, we further explored the anti-inflammatory effect of GLB. BV2 cells were treated with LPS, and OGD/R to mimic the pathophysiologic process in CA/CPR model, further indicating that the sequential activation of SUR1-TRPM4 and NLRP3 inflammasome acts as common response to damage stress in microglia challenged with various stimuli, directly participates in the injurious neuroinflammation, and can be prevented by GLB through occluding SUR1-TRPM4. Besides, we confirmed that SUR1-TRPM4 also participated in the P2X7R-independent NLRP3 activation initiated by reduced cytoplasmic ATP in microglia, and could also be inhibited by GLB. It was noteworthy that the intracellular ATP was partially decreased in OGD/R microglia, implying that SUR1-TRPM4 may mediate the NLRP3 activation via two diverse routes simultaneously after OGD/R. Therefore, it may be a complementary route of SUR1-TRPM4-involved NLRP3 activation in vivo. Then, we further addressed whether the anti-inflammatory effects of GLB exerted complementary neuroprotection in inflammatory brain damages after CA/CPR. Inspiringly, the results showed that GLB could significantly improve neurologic outcomes, promote the recovery of cognitive function, and alleviate the neuropathologic injuries in CA/CPR model, strongly implying that GLB has beneficial effects on neurologic diseases via reducing not only edema but also inflammation.
Microglial functions are closely related to ion channels [55]. Previous study has proved that the reduction of intracellular K+ to 70–80% is sufficient to activate NLRP3 [22], suggesting the critical role of K+ channels in NLRP3 inflammasome activation. P2X7R is a non-selective cation channel which acts as an initial trigger of NLRP3 activation by sensing extracellular ATP [20], but the K+ outward current required for NLRP3 activation is mainly mediated by other K+ channels like K2P family and Kv1.3 [23,24,25]. Likewise, we observed that the mRNA levels of TWIK1, TWIK2, THIK1, and Kv1.3 were obviously upregulated in LPS-primed microglia, and the above channels collaboratively launched the ATP-induced NLRP3 activation, of which K2P family played a dominant role, as reflected by the result that quinine almost entirely counteracted the NLRP3 activation, consistent with previous studies [23, 24]. It is reported that THIK-1 is not expressed in cultured microglia [56], conflicting with our result of highly expressed THIK-1 after LPS treatment, which may be attributed to the non-modeling microglia used in the previous study, with different cell status from ours. However, the underlying mechanism by which involved channel-mediated K+ efflux is triggered after activating P2X7R remains controversial. In vitro, we found that SUR1-TRPM4 could be fueled through sensing increased intracellular Ca2+ mediated by P2X7R, and gave rise to the Na+ inward current responsible for the onset of K+ efflux. In summary, we first illustrate that SUR1-TRPM4 acts as a signal amplifier and an integrated switch of downstream K+ channels, and ultimately triggers K+ efflux-driven NLRP3 activation, through mediating Na+ influx and consequential depolarized membrane potential (Fig. 8). The opening mechanisms of related K+ channels: [1] K2P family members belong to the constitutively active K+ leak channels, and the depolarized membrane potential magnifies the driving force of K+ efflux [27]; [2] Kv1.3 belongs to voltage-gated K+ channels, and is activated by the depolarized membrane potential [26].

The regulatory mechanism of SUR1-TRPM4 in NLRP3-mediated neuroinflammation. In the current study, we have demonstrated that the environment in multiple neurological disorders can provide both the priming signal to induce the NF-κB-dependent transcriptional upregulation of SUR1-TRPM4 complex and NLRP3 inflammasome components, and the activation signal to trigger inflammasome assembly. The signal 1 (priming; left) is provided by the ischemia–reperfusion, and the activation of TLR4 with PAMPs such as LPS. After priming, the signal 2 (activation; right) is provided by DAMPs like ATP which binds to P2X7R to activate SUR1-TRPM4 by mediating Ca2+ influx. SUR1-TRPM4 acts as an integrated switch of indicated K+ channels and an amplifier of K+ efflux through mediating Na+ influx and contributing to the depolarized membrane potential, and thereby initiates the NLRP3 inflammasome activation in microglia when the decreased intracellular K.+ reaches the threshold to engage NLRP3. Formation of the inflammasome activates caspase-1, which in turn processes the precursor of IL-1β (pro-IL-1β) into its mature form. CARD, caspase recruitment domain; LRR, leucine-rich repeat; PAMPs, pathogen-associated molecular patterns; PYD, pyrin domain; TLR4, Toll-like receptor 4
High-dose GLB (200 μmol/L) can reverse the NLRP3 activation in macrophages [16], but there are major challenges towards clinical translation due to the side effect of hypoglycemia. In this study, we found that NLRP3 inflammasome activation in microglia could be entirely suppressed by 100 μmol/L of sulfonylureas including GLB. Therefore, we further provided important insights into the differences in the mechanism under NLRP3 activation between microglia and macrophages. In view of another finding that SUR1 tended to bind TRPM4 in microglia, but Kir6.1 in macrophages, we support the notion that TRPM4 in macrophages mainly participates in the NLRP3 activation at an unbound state, and is directly blocked by higher dose of GLB because of lower sensitivity to GLB compared to SUR1-TRPM4 [12, 44], whereas SUR1-TRPM4 in microglia can be inhibited by both GLB and other sulfonylureas. On account of the minimal risk of hypoglycemia [57], we hope that our research will represent a turning point in the understanding of the direct anti-inflammatory effects of GLB, and promote the translation of low-dose GLB into clinical trials for neurological disorders involving NLRP3-related inflammation.
Conclusion
This study uncovers a complementary neuroprotective mechanism of GLB, which is attributed to the inhibition of NLRP3-mediated neuroinflammation through blocking SUR1-TRPM4 in microglia, and independent of the well-documented neuroprotection through blocking SUR1-TRPM4 in neurovascular units and thus reducing the brain edema. Mechanistically, this research reveals that the SUR1-TRPM4 complex serves as a K+ efflux amplifier in activating the NLRP3 inflammasome in microglia, via mediating Na+ influx, contributing to the depolarized membrane potential, and thereby gating downstream K+ channels. In conclusion, this study will certainly be insightful for deciphering the direct anti-inflammatory effects of GLB, as well as the contribution of low-dose GLB to combating neurological disorders involving NLRP3-related inflammation.
Data Availability
The datasets used and/or analyzed during this study are available from the corresponding authors on reasonable request.
Abbreviations
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood-brain barrier
- BBG:
-
Brilliant Blue G
- CA/CPR:
-
Cardiac arrest/cardiopulmonary resuscitation
- DAMPs:
-
Danger-associated molecular patterns
- DMSO:
-
Dimethyl sulfoxide
- FBS:
-
Fetal bovine serum
- GFAP:
-
Glial fibrillary acidic protein
- GLB:
-
Glibenclamide
- GLM:
-
Glimepiride
- GLZ:
-
Gliclazide
- Iba-1:
-
Ionized calcium-binding adapter molecule-1
- IL-1β:
-
Interleukin-1β
- K2P family:
-
Two-pore domain K+ channel family
- Kv family:
-
Voltage-gated K+ channel family
- LPS:
-
Lipopolysaccharides
- MAP:
-
Mean arterial pressure
- MAP-2:
-
Microtubule-associated protein 2
- NDSs:
-
Neurologic deficit scores
- NeuN:
-
Neuronal nuclei
- NF-κB:
-
Nuclear factor kappa-B
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3
- OGD/R:
-
Oxygen-glucose deprivation/reperfusion
- PFA:
-
Paraformaldehyde
- pro-caspase-1:
-
Precursor of caspase-1
- pro-IL-1β:
-
Precursor of IL-1β
- P2X7R:
-
P2X7 receptor
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- ROSC:
-
Return of spontaneous circulation
- Sp-1:
-
Specificity protein 1
- SUR1-TRPM4:
-
Sulfonylurea receptor 1-transient receptor potential M4
- THIK1:
-
Tandem pore domain halothane-inhibited K+ channel 1
- TLR:
-
Toll-like receptor
- TWIK1:
-
Two-pore domain weak inwardly rectifying K+ channel 1
- 9-Ph:
-
9-Phenanthrol
References
Simard JM, Woo SK, Bhatta S, Gerzanich V (2008) Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol 8:42–49
Guo J, She J, Zeng W, Chen Q, Bai X, Jiang Y (2017) Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 552:205–209
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V (2006) Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat Med 12:433–440
Simard JM, Geng Z, Woo SK, Ivanova S, Tosun C, Melnichenko L, Gerzanich V (2009) Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab 29:317–330
Zweckberger K, Hackenberg K, Jung CS, Hertle DN, Kiening KL, Unterberg AW, Sakowitz OW (2014) Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 272:199–206
Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V (2007) Endothelial sulfonylurea receptor 1-regulated NCCa-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest 117:2105–2113
Kimberly WT, Bevers MB, von Kummer R, Demchuk AM, Romero JM, Elm JJ, Hinson HE, Molyneaux BJ et al (2018) Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP trial. Neurology 91:e2163–e2169
Sheth KN, Petersen NH, Cheung K, Elm JJ, Hinson HE, Molyneaux BJ, Beslow LA, Sze GK et al (2018) Long-term outcomes in patients aged ≤70 years with intravenous glyburide from the phase II GAMES-RP study of large hemispheric infarction: an exploratory analysis. Stroke 49:1457–1463
Huang K, Hu Y, Wu Y, Ji Z, Wang S, Lin Z, Xu J, Pan S (2019) Exploratory analysis of oral glibenclamide in acute ischemic stroke. Acta Neurol Scand 140:212–218
Kunte H, Busch MA, Trostdorf K, Vollnberg B, Harms L, Mehta RI, Castellani RJ, Mandava P et al (2012) Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann Neurol 72:799–806
Chang JJ, Khorchid Y, Kerro A, Burgess LG, Goyal N, Alexandrov AW, Alexandrov AV, Tsivgoulis G (2017) Sulfonylurea drug pretreatment and functional outcome in diabetic patients with acute intracerebral hemorrhage. J Neurol Sci 381:182–187
Kurland DB, Gerzanich V, Karimy JK, Woo SK, Vennekens R, Freichel M, Nilius B, Bryan J et al (2016) The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J Neuroinflammation 13:130
Makar TK, Gerzanich V, Nimmagadda VKC, Jain R, Lam K, Mubariz F, Trisler D, Ivanova S et al (2015) Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J Neuroinflammation 12:210
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411
Lamkanfi M, Dixit VM (2009) Inflammasomes: guardians of cytosolic sanctity. Immunol Rev 227:95–105
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM et al (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187:61–70
Chang Y, Zhu J, Wang D, Li H, He Y, Liu K, Wang X, Peng Y et al (2020) NLRP3 inflammasome-mediated microglial pyroptosis is critically involved in the development of post-cardiac arrest brain injury. J Neuroinflammation 17:219
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272:735–738
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S (2017) The P2X7 receptor in infection and inflammation. Immunity 47:15–31
Chen GY, Nuñez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153
Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z et al (2018) The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49:56–65
Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, Attwell D (2018) Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K+ channel THIK-1. Neuron 97:299–312
Ma D, Zhang N, Zhang Y, Chen H (2020) Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp Neurol 332:113399
Wulff H, Zhorov BS (2008) K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem Rev 108:1744–1773
Enyedi P, Czirják G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90:559–605
Huang K, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X, Xie Z, Pan S (2015) Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit Care Med 43:e341–e349
Lin Z, Huang H, Gu Y, Huang K, Hu Y, Ji Z, Wu Y, Wang S, Yang T, Pan S (2017) Glibenclamide ameliorates cerebral edema and improves outcomes in a rat model of status epilepticus. Neuropharmacology 121:1–11
Ye Y, Jin T, Zhang X, Zeng Z, Ye B, Wang J, Zhong Y, Xiong X et al (2019) Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-κB signaling pathway. Front Cell Neurosci 13:553
Zhu S, Huang J, Peng W, Wu D, Luo M, Li Q, Li Z, Feng X et al (2017) Inhibition of smoothened decreases proliferation of synoviocytes in rheumatoid arthritis. Cell Mol Immunol 14:214–222
Ding H, Zhang H, Ding H, Li D, Yi X, Ma X, Li R, Huang M et al (2017) Transplantation of placenta-derived mesenchymal stem cells reduces hypoxic-ischemic brain damage in rats by ameliorating the inflammatory response. Cell Mol Immunol 14:693–701
Geocadin RG, Ghodadra R, Kimura T, Lei H, Sherman DL, Hanley DF, Thakor NV (2000) A novel quantitative EEG injury measure of global cerebral ischemia. Clin Neurophysiol 111:1779–1787
Luo D, Ge W, Hu X, Li C, Lee C, Zhou L, Wu Z, Yu J et al (2019) Unbiased transcriptomic analyses reveal distinct effects of immune deficiency in CNS function with and without injury. Protein Cell 10:566–582
Lage SL, Dominical VM, Wong C, Sereti I (2019) Evaluation of canonical inflammasome activation in human monocytes by imaging flow cytometry. Front Immunol 10:1284
Hassan S, Eldeeb K, Millns PJ, Bennett AJ, Alexander SPH, Kendall DA (2014) Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br J Pharmacol 171:2426–2439
Seligmann B, Gallin JI (1980) Secretagogue modulation of the response of human neutrophils to chemoattractants: studies with a membrane potential sensitive cyanine dye. Mol Immunol 17:191–200
Tao Y, Yan D, Yang Q, Zeng R, Wang Y (2006) Low K+ promotes NF-kappaB/DNA binding in neuronal apoptosis induced by K+ loss. Mol Cell Biol 26:1038–1050
Yang B, Zhang L, Cao Y, Chen S, Cao J, Wu D, Chen J, Xiong H et al (2017) Overexpression of lncRNA IGFBP4-1 reprograms energy metabolism to promote lung cancer progression. Mol Cancer 16:154
Huang K, Wang Z, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X et al (2016) Glibenclamide is comparable to target temperature management in improving survival and neurological outcome after asphyxial cardiac arrest in rats. J Am Heart Assoc 5:e003465
Huang K, Wang Z, Gu Y, Ji Z, Lin Z, Wang S, Pan S, Wu Y (2018) Glibenclamide prevents water diffusion abnormality in the brain after cardiac arrest in rats. Neurocrit Care 29:128–135
Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19:477–489
Ming S, Zeng L, Guo Y, Zhang S, Li G, Ma Y, Zhai Y, Chang W et al (2020) The human-specific STING agonist G10 activates type I interferon and the NLRP3 inflammasome in porcine cells. Front Immunol 11:575818
Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM (2013) The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem 288:3655–3667
Katsnelson MA, Rucker LG, Russo HM, Dubyak GR (2015) K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194:3937–3952
Nomura J, So A, Tamura M, Busso N (2015) Intracellular ATP decrease mediates NLRP3 inflammasome activation upon nigericin and crystal stimulation. J Immunol 195:5718–5724
Nilius B, Prenen J, Voets T, Droogmans G (2004) Intracellular nucleotides and polyamines inhibit the Ca2+-activated cation channel TRPM4b. Pflugers Arch 448:70–75
Chen M, Simard JM (2001) Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci 21:6512–6521
Zhao H, Chen Y, Feng H (2018) P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol 16:1282–1295
Lee SW, de Rivero Vaccari JP, Truettner JS, Dietrich WD, Keane RW (2019) The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J Neuroinflammation 16:27
Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J, Li Y, Guo H et al (2019) Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis 10:555
Hirshman NA, Hughes FJ, Jin H, Harrison WT, White SW, Doan I, Harper SN, Leidig PD et al (2020) Cyclophosphamide-induced cystitis results in NLRP3-mediated inflammation in the hippocampus and symptoms of depression in rats. Am J Physiol Renal Physiol 318:F354–F362
Hou L, Yang J, Li S, Huang R, Zhang D, Zhao J, Wang Q (2020) Glibenclamide attenuates 2,5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol Lett 331:152–158
Wang X, Chang Y, He Y, Lyu C, Li H, Zhu J, Liu K, Hu Y et al (2020) Glimepiride and glibenclamide have comparable efficacy in treating acute ischemic stroke in mice. Neuropharmacology 162:107845
Izquierdo P, Attwell D, Madry C (2019) Ion channels and receptors as determinants of microglial function. Trends Neurosci 42:278–292
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B et al (2014) Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143
Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V (2009) Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke 40:604–609
Acknowledgements
We thank the central laboratory of Southern Medical University for technical assistance in immunofluorescence capture under the confocal microscope.
Funding
This work was supported by the National Key R&D Program of China (No. 2017YFC1307500), National Natural Science Foundation of China (No. 81701294 & 82072133 & 81871030), and Guangdong Basic and Applied Basic Research Foundation (2019A1515011446 & 2021A1515010922).
Author information
Authors and Affiliations
Contributions
Y.H. and Y.C. performed all the experiments and wrote the manuscript. Y.P., J.Z., K.L., J.C., Y.W., Z.J., Z.L., S.W., S.G., and N.Z. completed the statistical analysis. S.P. and K.H. designed, guided the experiments, and critically revised the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
All animal experiments in this study were approved by Animal Care and Use Committee of Nanfang Hospital, Southern Medical University (Guangzhou, China).
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, Y., Chang, Y., Peng, Y. et al. Glibenclamide Directly Prevents Neuroinflammation by Targeting SUR1-TRPM4-Mediated NLRP3 Inflammasome Activation In Microglia. Mol Neurobiol 59, 6590–6607 (2022). https://doi.org/10.1007/s12035-022-02998-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02998-x