
Abstract
Heart failure (HF) is one of the most critical health and economic burdens worldwide, and its prevalence is continuously increasing. HF is a disease that occurs due to a pathological change arising from the function or structure of the heart tissue and usually progresses. Numerous experimental HF models have been created to elucidate the pathophysiological mechanisms that cause HF. An understanding of the pathophysiology of HF is essential for the development of novel efficient therapies. During the past few decades, animal models have provided new insights into the complex pathogenesis of HF. Success in the pathophysiology and treatment of HF has been achieved by using animal models of HF. The development of new in vivo models is critical for evaluating treatments such as gene therapy, mechanical devices, and new surgical approaches. However, each animal model has advantages and limitations, and none of these models is suitable for studying all aspects of HF. Therefore, the researchers have to choose an appropriate experimental model that will fully reflect HF. Despite some limitations, these animal models provided a significant advance in the etiology and pathogenesis of HF. Also, experimental HF models have led to the development of new treatments. In this review, we discussed widely used experimental HF models that continue to provide critical information for HF patients and facilitate the development of new treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Definition and importance of heart failure
Heart failure (HF) is a clinical syndrome characterized by the inability to send sufficient blood to the peripheral tissue as much as its metabolic requirement due to pathologies arising from the function or structure of the heart tissue, and it usually has a progressive course [1,2,3]. The complexity of HF has been challenging the scientific world for many years. Versatile and complex processes in the pathophysiology of HF have been investigated for a long time. Also, the heterogeneous structures of the disease negatively affect the speed of scientific developments in this field. The prevalence of HF continues to increase significantly today.
Prevalence and incidence
Unfortunately, HF is a leading cause of death worldwide [4, 5]. It affects more than 26 million people worldwide, with a prevalence of more than 12% over the age of 70 [6]. Although HF is generally seen in the elderly, it occurs at all ages [7, 8]. Currently, 6.5 million people in the USA are thought to have HF, and it is predicted that 8 million people in the USA will be diagnosed with HF by 2030 [8, 9]. HF is the leading cause of death worldwide, with approximately 50% of patients dying within the first 5 years after diagnosis [10]. HF not only is affected human health but also dramatically affects the social economy. It costs several billion dollars each year and is estimated to increase by around 70 billion costs by 2030 [10]. It is essential to analyze risk factors and develop new effective treatment methods for HF patients for all these reasons.
Pathophysiology, subclassification, and symptoms
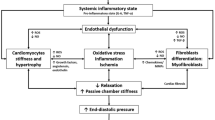
The most important defining feature of HF is the heart’s inability to pump enough blood to the body and the resulting low quality of life in these patients [11]. HF is a complex clinical picture with many different etiologies. The main feature of HF has reduced contractile force or underfilling. Systemic inflammation, hypoxic environment, cardiomyocyte damage, mechanical stress, and other profibrotic cytokines, transforming fibroblasts into myofibroblasts at the injury site, are the essential pathophysiological processes underlying heart diseases [12]. Myofibroblasts have a more contractile structure by developing stress fibers. These induce cardiomyocyte hypertrophy by causing cytokines’ secretion, such as transforming growth factor-β, tumor necrosis factor-α, and angiotensin II [9, 13,14,15,16]. As a result of myofibroblasts’ activity, it also causes the formation of extracellular connective tissue and the formation of interstitial and perivascular fibrosis [17]. This fibrotic structure adversely affects the heart tissue’s physiological processes and causes myocardial stiffening and diastolic dysfunction [18]. These changes cause left ventricular hypertrophy, inhibition of the left ventricle’s relaxation ability, and diastolic dysfunction [19]. Developments such as ischemic damage in the heart lead to cardiomyocyte death, leading to decreased contraction of heart tissue and wall thickness [20]. As a result of these damages, the heart tissue’s cytosolic functions are impaired [21]. Pathological changes such as increased stiffness of the myocardial tissue reduced left ventricular contraction and heart tissue remodeling are the main hallmarks of HF [21]. The pathophysiology of systolic and diastolic heart failure and the differences between them are shown in Fig. 1. Left HF is often the leading cause of right HF and is associated with an increased risk of sudden death [22].

The pathophysiology of systolic and diastolic heart failure and the differences between them
HF is further divided into three subgroups: (1) HF with preserved EF (HFpEF, LV EF ≥ 50%), (2) HF with midrange EF (HFmrEF, 40–49% LV EF), (3) HF with reduced EF (HFrEF, LV EF < 40%). This sub-classification has an essential place in the difference in HF treatment protocols [23]. According to the analysis of the data obtained from the Framingham Heart Study, 56.2% of the patients were diagnosed with HFpEF, 31.1% with HFrEF, and 12.8% with HfmrEF [24]. Although the relative proportion of HFrEF decreases, the absolute number of HFrEF patients is predicted to increase in the coming years. HFpEF occurs when the ventricles fail to relax correctly and are clinically defined as normal EF or HF with diastolic dysfunction. About half of HF patients suffer from HFpEF and exhibit HF symptoms, including exercise intolerance, congestion, and edema associated with cardiac hypertrophy [25, 26]. HFpEF is more common in women than men and its incidence increases in older age [27]. In addition, it can be a secondary disease that occurs as a result of various chronic diseases such as hypertension and diabetes mellitus [9, 28].
As a result of clinical studies, it is known that there is a transition between HF subtypes. According to this, including HFrEF to HFmrEF/HFpEF or HFmrEF to HFpEF, it is referred to as “HF with enhanced LVEF” (HFiEF). These patients have transitioned to the lower HF phenotype from baseline. Including HFpEF to HFmrEF/HFrEF or HFmrEF to HFrEF, it is referred to as “HF with worsened LVEF” (HFdEF). The remainder of the enrolled patients were termed “HFuEF with unchanged LVEF” (HFuEF) [29]. Age, hypertensive and in some cases diabetes-related ventricular remodeling thus creates the slowly progressive substrate upon which HFpEF is formed, and recent evidence suggests that progression of a number of abnormalities in cardiovascular function may promote the transition to overt HFpEF, including loss of contractile reserve, diastolic reserve, chronotropy, vasodilation, and endothelial function. In contrast, HFrEF most commonly develops in response to distinct pathophysiologic perturbations leading to accelerated and larger-scale myocyte loss/dysfunction, with the most common etiologies including acute myocardial infarction, genetic abnormalities, myocarditis, or toxin effects (e.g., alcohol or chemotherapy) [30].
Two-thirds of all HF patients fall under four main headings: ischemic heart disease, chronic obstructive pulmonary disease, hypertensive heart disease, or rheumatic heart disease [30]. Typical signs of HF and physical examination findings include dyspnea, weight gain, weakness, increased jugular venous pressure, pulmonary rales, and peripheral edema [31]. Symptoms of right and left-sided HF are shown in Fig. 2. These findings significantly affect the expected patient’s quality of life [32]. All these symptoms and signs occur due to increased cardiac hypertrophy, formation of fibrotic tissue, and decreased blood supply. Many cardiac problems, such as coronary artery disease, myocardial infarction, and cardiomyopathy cause HF [30]. HF causes the loss of the heart tissue’s physiological contractile ability by causing cardiomyocyte damage or death. All these pathological changes cause the insufficient pumping of blood to the systemic circulation, in other words, the emergence of systolic HF. This relationship showed that brain natriuretic peptide levels and mortality are higher in HFrEF patients than in HFpEF patients. The relative incidence of these three subgroups, a decrease in HFrEF, and an increase in HFpEF have been observed in recent years [27, 33].

Symptoms of right and left-sided HF
Possible therapeutic targets of HF
Despite significant improvements in cardiovascular mortality over the last decades, cardiovascular disease is the main reason for death in several countries. Cardiovascular therapies improved the survival of patients with cardiovascular disease but, at the same time, increased the number of subjects affected by chronic cardiovascular conditions such as HF. The development of new drugs for HF therapy must be necessarily focused on additional targets [34]. Possible therapeutic pathways and mechanisms in HF are shown in Table 1.
Experimental models of heart failure
It is well known that there are many risk factors associated with HF. Apart from risk factors, comorbidities such as obesity, hypertension, type 2 diabetes, and chronic kidney disease have revealed the necessity of developing new treatment options in HF [28, 62,63,64]. The pathogenesis of HFpEF and HFrEF is multifactorial. Therefore, it is complicated to distinguish underlying mechanisms that may be overlapping and interconnected. The complex mechanisms of all these variables make the disease difficult to understand and discover new treatment methods. One of these difficulties is the lack of ideal animal models similar to the pathophysiological features of human HF.
Various experimental animal models are used to analyze the causes of HF and to develop current treatment strategies [65]. Many small animal species are often preferred for this purpose, including mice, rats, and guinea pigs. Mice and rats are very similar to the human genome, with 30,000 protein-coding genes. The most important advantages of these experimental animal species are the short reproductive cycles and low housing costs. These models generally use genetic modifications and pharmacological and surgical approaches. Many animal models have provided significant progress in knowledge of HFrEF and HFpEF pathogenesis [26]. The HF models in small animals are shown in Fig. 3.

The HF models in small animals
Myocardial infarction (MI)
Ischemia/anoxia occurs when a part of the myocardium is not provided with adequate blood flow or if it is completely stopped due to coronary occlusion [66, 67]. Insufficient blood flow to the myocardium is one of the leading causes of HF [68,69,70].
Several animal models have been developed to mimic HF in humans with coronary artery disease [71, 72]. These models can help elucidate the pathophysiological mechanisms occurring in ischemic human hearts. The main model types used are coronary artery ligation, coronary artery embolization, hydraulic occluder or ameroid ring constrictor, and cryoinjury models. These interventions are generally used to narrow or occlude coronary vessels. This blockage can be used to trigger acute or chronic HF [71, 73]. Coronary artery ligation is the most commonly used model used to induce HF in many animals, from mice to pigs [74,75,76]. Coronary artery ligation has been performed not only in rats but also in mice [77, 78]. Since ischemic heart disease is the most important cause of HF in humans, coronary artery occlusion is the most common inducing acute myocardial injury in animal models.
Ischemic heart disease is the main cause of HF in humans [79]. Ligation of the left anterior descending (LAD) coronary artery or any of its branches is the most preferred and generally accepted method to induce HF [8, 16, 80, 81]. Coronary artery ligation is a widely used small animal HF model created by Pfeffer et al. in rats and later used by numerous groups [82]. After discovering the experimental model, Pfeffer et al. showed that the use of angiotensin-converting enzyme inhibitor (ACEI), captopril, increased the contractile function and survival of the ventricles after MI in rats [83]. These experimental studies laid the foundation for ACEIs traditionally used in MI patients today. This method also provides information about cardiac remodeling developed in the heart after MI and its pathophysiology [84]. Pathological changes occur due to the increased overload in the cardiac tissue and the increased fibrotic structure in the extracellular tissue [8, 85]. In addition, this condition is accompanied by neurohumoral activation [86]. Dilatation, necrosis, and apoptosis were observed in the cardiac tissue of the animals in which this experimental model was applied. Also, high ventricular filling pressures and decreased cardiac index were found. In addition to all these changes, the natriuretic peptide and renin-angiotensin system were activated [87, 88]. HF induced by LAD ligation is associated with myocardial hypertrophy, progressive myocardial enlargement, and late failure if 40–50% of the left ventricle is viable [14, 89]. There was no significant difference in cardiac output in rats 1 week after LAD ligation compared to the control group. However, a significant reduction in cardiac output was detected 3–5 weeks after LAD ligation [90]. A significant reduction in rats’ cardiac output, up to 25%, is observed 8 weeks after LAD ligation [91]. It is known that mice that survive progressively develop HF 4 weeks after surgery [92]. Infarction size varies between 10 and 45% and is directly related to left ventricular function damage; this ratio affects HF’s development time. In general, the infarct must affect at least 30% of the left ventricular mass to demonstrate the typical HF features and detect various biomarker levels [81].
Studies have shown that female mice undergo less ventricular remodeling. Sex hormones are thought to underlie this difference. Female mice show less extensive ventricular remodeling than males, suggesting the role of sex hormones [93]. These differences should be taken into account when designing experiments. It must be taken into attention. Due to the MI experimental model in both mice and rats, the mortality rate ranges from 35 to 50%. Ventricular fibrillation usually occurs within 1 h after MI due to severe acute HF [94]. Although progressive, non-occlusive coronary artery occlusion is usually seen in clinical patients; this model is mostly caused by a normal coronary artery’s sudden occlusion. Ischemic heart disease in humans is mostly caused by non-occlusive coronary artery narrowing. This experimental model is created by reducing the left coronary artery’s inner lumen diameter by about 60% by occluding the vessel with a probe and then removing it in rats [95]. As a result of the decrease in coronary blood flow, a reduction in left ventricular performance and deterioration in HF are observed. Ischemic HF develops more slowly than this model of total occlusion, and the overall mortality rate is approximately 43%. Although coronary artery ligation is a reliable model for inducing tissue damage that leads to HF, it does not reflect HF development in patients. In sum, the coronary artery ligation-induced coronary artery test model can be established in a robust, reproducible manner in many species [96,97,98].
Temporary LAD occlusion models have been developed to induce human ischemia–reperfusion (I/R) injury. However, results vary due to the variability observed in the left coronary anatomical structure in mice [99]. The coronary artery is ligated for 180 min and then reperfused in the I/R method. Approximately 16% infarct area and an LVEF of roughly 40% were formed in this method parallel with increased LVEDP, LVESV, and N-terminal pro-BNP after 4 weeks from MI [100]. In a different version of these models, a temporary occlusion is achieved by imitating I/R, whereby the flow in the previously occluded coronary artery bed is allowed to recover [99]. Another method used to induce myocardial ischemia/infarction is coronary artery embolization. For this purpose, intracoronary embolizations, thrombin, and fibrinogen are used [101]. Coronary microembolization has been used extensively for a while and causes a decrease in ejection fraction (EF), an increase in LVEDP, and an increase in plasma norepinephrine levels [102]. This model causes a local infarction and global ischemia as a result of the development of the aneurysm. MI is created by serial injections of 90-micron diameter polystyrene microspheres in this model’s left coronary artery. Injections are made with a catheter inserted through the femoral artery [103]. Approximately 20,000 microspheres are injected into the artery with each injection. A total of 3 injections are made at 15-min intervals and repeated every week until the desired effect is observed. It is known that a total of 4–14 injections are usually required to obtain a chronic HF model. Injections are stopped when the ejection fraction (EF) is reduced to 35%. This microembolization-induced myocardial ischemia is an irreversible model of HF [104]. A study showed that this sheep model was stable for 6 months, and the mortality rate was shown to be between 30 and 50% [103]. The microembolization model is typically only applied to large animals to cause HF [105, 106].
This model has several advantages:
-
1.
Lower risk of severe inflammatory complications than thoracotomy
-
2.
This model is similar to the clinical situation in HF
However, it is difficult to understand the exact length and location of coronary artery occlusion. Arrhythmias during the experimental model may cause difficulties in interpreting the biological response [107]. It seems that I/R models were created to overcome this limitation, and transient LAD occlusion facilitated the investigation of molecular mechanisms and tissue damage [108,109,110]. It has been shown that ischemic injury in newborn mice is triggered by LAD ligation and complete recovery occurs after 3 weeks. The regenerative potential is reduced in aged mice due to the age-related decrease in the number of healthy cardiomyocyte cells [111]. Interestingly, in another study, rats were studied 4, 8, and 16 weeks after coronary artery ligation. According to the data obtained, hemodynamic and clinical signs indicated cup failure in only 16-week-old rats [112]. Essential advantages of LAD ligation include the cost and simplicity of the process. In addition, electrophysiology, coronary anatomy, physiology, and MI developmental processes in rodents differ when compared to humans. However, this experimental model is considered a significant disadvantage since some studies have shown that the mortality associated with this experimental model can reach up to 70% [81]. Despite its widespread use, there are significant drawbacks to this intervention [107]:
-
1.
Infarctions seen in cases are relatively small (left ventricular mean ~ 21%).
-
2.
Major hemodynamic changes are not seen due to a large number of pericardial collaterals.
-
3.
Mortality rates of more than 50% are seen due to ventricular tachycardia.
Recent studies have shown that the branching pattern of coronary arteries in Lewis congenital rats is more consistent than in other strains. LAD ligation in Lewis inbred rats has been shown to have a lower mortality rate despite having a more extensive infarction than Sprague Dawley rats [95]. However, it should be kept in mind that the clinical manifestations of chronic HF and neurohumoral changes were not evaluated in this study. Therefore, this promising model needs to be studied in more detail. Studies show that recovery and termination processes begin faster in rats than in humans, which requires careful interpretation of the results. Another alternative MI model is the cryoinfarction, which causes damage to the epicardium of mice and rats [113]. However, this model has not attracted much attention from the scientific community and is no longer preferred. As a result of the study, it has been shown that myocardium wall thickness and fibrotic tissue due to infarction are less pronounced in mice than in rats [114].
Another method used to induce MI involves the use of a hydraulic occluder or ameroid ring constrictor. Procedures begin with a left anterolateral thoracotomy followed by a pericardial incision, while the left coronary artery branch is exposed and the hydraulic occluder is inserted. The occlusive is then inflated to cause partial stenosis or complete obstruction. An ultrasonic flow probe is positioned distal to the occluder to manage the degree of occlusion and record downstream flow from the left coronary artery [115]. Similarly, an ameroid ring constrictor is implanted. It will gradually shrink due to the peculiarity of the material. As a result of this method, complete or partial narrowing will occur in the coronary artery [116]. One of the essential factors in choosing a model is the variation in coronary variability and collateral vasculature between animals. Hedström et al. showed that in the region at risk, 50% infarction development time varies significantly between species [117]. Additionally, the type of anesthesia must also be considered when choosing a model affecting I/R [118]. HF model due to MI can be created in rats with the cryosurgery method. After intercostal thoracotomy, a 0.18 1.2-cm2 liquid nitrogen probe is applied to the left ventricular free wall 15 times for 20 s. Also, cryoinjury may not always induce a transmural lesion, and it should be noted that in this case, it can heal without fibrotic tissue forming and an aneurysm developing [119]. The cryoinjury model is reproducible and generally has a moderate resemblance to infarctions observed in the clinic. It has some disadvantages as with other surgical models because they are presented more acutely than clinically [120]. As a result, numerous animal models allow us to examine the effect of a drug in the field of HF. A model’s advantages must be carefully compared against disadvantages, and non-scientific factors such as cost, effort, and time must also be considered.
Chronic rapid cardiac pacing
Dilated cardiomyopathy caused by chronic tachycardia is a known clinical picture [121]. Whipple et al. showed that atrial pacing above 330 beats/min can induce physical signs of HF and cardiomyopathy due to experimental tachycardia that was first shown in 1962 [122]. The experimental model is simple and neurohumoral activation develops after about 8 weeks. Increased left ventricular filling pressures after 1–2 months of pacing are associated with pulmonary wedge pressure and right atrial pressure, ascites, pulmonary congestion, and reduced EF [123]. After pacing cessation, hemodynamic changes usually correct within 4 weeks. This is a unique feature of this model [124]. The interaction between HF and arrhythmia is well established to increase the risk of developing HF and morbidity or mortality [125]. Sudden cardiac death is one of the major causes of episodes in HF [126]. This situation can develop after arrhythmia. For example, arrhythmias such as atrial fibrillation significantly increase the risk of HF. Experimental models of arrhythmia-induced HF are characterized by chronic rapid pacing periods indicated by the pacemaker’s anatomical location [127, 128]. In tachycardia models, the pacemaker is implanted in the right or left ventricle. The right ventricle was paced at a rate ranging from 180 to 240 bpm, causing congestive HF within 3 to 4 weeks [129]. While the right atrium was paced at a rate of 400 bpm, the left and right ventricles were dilated without hypertrophy [130]. In dogs, pigs, and sheep, 128–130 rapid pacing of the atrium or ventricle for at least 3–4 weeks is known to cause progressive HF and is partially reversible upon cessation of stimulation [131].
According to experimental studies on dogs, it is known that atrial pacing above 330 beats/minute can cause symptoms of HF. In general terms, this model has been shown in creatures such as dogs, pigs, sheep, and rabbits [132]. It is known that the pacing-induced HF model is also suitable for rodents. The rapid right ventricular pacing model has been shown to cause a progressive biventricular dilatation, including decreased cardiac output and increased peripheral vascular resistance and neurohumoral activation within a few weeks [133]. Tachycardia causes a decrease in systolic, diastolic function and cardiac output. In addition, left ventricular end-diastolic pressure, mean arterial pressure, pulmonary artery pressure, and wall stress increase in this model approximately 24 h later. Ventricular dysfunction is seen after 3–5 weeks and results in HF. However, no significant changes are observed in the structure, physiology, and hypertrophy of the left ventricle. Indeed, cardiac widening is accompanied by very little cardiac hypertrophy [134, 135]. Therefore, the chronic pacing tachycardia experimental model has been used to generate dilated cardiomyopathy and chronic HF.
In the neurohumoral activity, early sympathetic activation and reduced parasympathetic activity are seen. Atrial natriuretic peptide and brain natriuretic peptide levels in plasma increase before damage to the left ventricle. This was thought to be a protective mechanism, but exogenous atrial natriuretic peptide administration did not improve hemodynamic and renal responses [136]. It has been shown that cytokines such as endothelin-1 and TNF-α increase in plasma in HF induced by this model [137]. HF created with this model develops due to left ventricular dysfunction similar to HF in humans. Accordingly, catecholamines’ plasma levels first increase in the early period and then reach the plateau [137]. Increases in plasma endothelin levels, beta-receptor density changes, and function are similar to patients with HF [138]. However, increased levels of endothelin and renin in plasma are seen in advanced stages of left ventricular dysfunction [139]. This experimental model has some essential features in terms of being similar to HF. First, major surgical traumas such as thoracotomy and pericardiectomy that may affect hemodynamics are not required. Second, HF occurs in as little as a few weeks in this experimental model, allowing sequential observations. Third, calibration can be achieved using a pacemaker to stimulate the heart rate. Fourth, rapid pacing leads to the emergence of well-defined signs of biventricular failure with cardiomegaly, hypoperfusion, pulmonary congestion, cachexia, and ascites. Finally, the HF model created by this experimental model is reversible [136, 140].
This experimental model is characterized by increasing cardiac volume with fluid retention, increasing catecholamines, atrial natriuretic peptide, renin-angiotensin, aldosterone, endothelin-1, and TNF-α [141]. Due to chronic tachycardia, calcium abnormalities, and deterioration of the extracellular matrix through the activation of myocardial matrix, metalloproteases, gelatinase, and other cytokines are seen [142,143,144,145]. After pacing for 1 week, apoptosis is seen due to myocyte damage. Both the heart’s systolic and diastolic function recovered within approximately 2–3 weeks after the cessation of stimulation [71].
As a result of all these reasons, plasma markers of neurohormonal activation in pacing models are very similar to patient populations. In terms of calcium balance, the sarcolemmal Na/Ca exchanger is known to increase, while SERCA levels are lower [141, 145, 146]. As a result of the analyses performed, it was shown that there was a significant loss in myocyte count, and this was thought to be the main component of cardiomyopathy. Increased p53 DNA binding activity to the Bax promoter, increased Bax protein expression, and decreased Bcl-2 have been shown in pacing-induced HF models [147].
The most important advantages of the model are its predictability, reproducibility, and resemblance to the hemodynamic and mechanical phenotype of HF due to dilated cardiomyopathy in humans. One of the essential advantages of this model is that the model is simple and requires simple instrumentation. This HF model closely resembles human HF due to dilated cardiomyopathy in terms of mechanical, structural, neurohormonal, and myocyte functional changes [11, 131]. However, the return of mechanical and neurohormonal changes within a few days after pacing is stopped is a limitation of this model. Limitations include the absence of myocardial hypertrophy and fibrosis and the reversibility of this myopathy [148]. Approximately 48 h after pacing is stopped, hemodynamic variables return to normal, and left ventricular EF normalizes after 1–2 weeks. During the first hours after stopping pacing, the circulating atrial natriuretic peptide level drops by 60%. Unlike hemodynamic dysfunction and neurohormonal activation, ventricular dilation continues even after pacing is terminated. Another limitation is that events due to mechanical and electrical dyshomogeneity of the heart due to the homogeneity of myocardial damage induced by rapid pacing can not be shown in this model. Unlike in humans, hypertrophy does not occur in the HF model created by chronic rapid pacing. The absence of fibrotic tissue contributes to ventricular remodeling. Therefore, it may not reflect all the features of HF [132]. The enlargement of both ventricles and mitral valve annulus shown in the experimental model has been associated with valve insufficiency [129].
Rapid pacing models have been used to evaluate intracellular and extracellular changes and develop new pharmacological treatment methods. Rapid pacing models have also been used to assess the different surgical procedures required for HF [149, 150]. It is predicted that this experimental HF model, which is used in many different animal species, may help to elucidate the mechanisms related to HF in the future.
Pressure overload
Chronic left ventricular pressure-overload is an experimental model in mice and rats that causes HF that mimics adaptations associated with hypertension and aortic valve stenosis in patients [151,152,153,154]. Although the pressure-overload model is used more in rodent studies, they are less used in large animal experiments than ischemic models [72, 155, 156]. Similar to rodents, methods such as aortic taping can also be applied to large animals [157]. Aortic constriction causes an increase in pressure in the left ventricle and hypertrophy. While banding does not initially significantly affect the contraction, gradually, the constriction’s relative intensity increases as the animal grows, resulting in cardiac hypertrophy [155].
Overpressure-induced left ventricular hypertrophy and HF have been produced in rodents by transverse aortic constriction (TAC) and abdominal aortic constriction (AAC) [12, 158, 159]. Rats exposed to pressure-overload for more than 8 weeks have impaired systolic function, fractional shortening, and EF values. Significant increases in left ventricular posterior wall thickness, left ventricular inner size, and ventricular septal wall-size indicate concentric cardiac hypertrophy [10, 143]. A decrease in E, A wave, and E/A ratio on echocardiography indicates diastolic dysfunction [160]. All results show a significant increase in left ventricular end-diastolic dimension, left ventricular end-systolic dimension, and left ventricular posterior wall thickness. Also, a 60% reduction in the maximum pressure development rate (+dP/dt) indicates diastolic dysfunction [154]. Various surgical approaches have been developed to examine HF caused by chronic overpressure on the left ventricle and to mimic the protection mechanisms associated with hypertension in patients. These experimental models cause significant macroscopic and microscopic hypertrophy, but systolic dysfunction appears to progress much more slowly than rodent models [161].
The development of animal models with preserved EF is difficult due to the complexity of the pathophysiology of the disease [162, 163]. Pressure-overload is directly related to the development of left ventricular hypertrophy seen in HF. Myocardial hypertrophy is a protective mechanism that occurs due to increased myocardial cell tension [164]. Although it is beneficial, it may cause the myocardium to decrease its elasticity and become fibrotic after a certain level. Also, it gradually surpasses all post-loading protection mechanisms. Then, despite the left ventricle’s enlargement, the load reserve is depleted, and the basal contraction becomes incompatible with the post-load level [165].
Pressure-overload usually occurs in conditions such as left ventricular outlet obstruction such as hypertension or aortic stenosis. As a result of these experimental models, it has been shown that hypertrophy and fibrotic tissue develop in the myocardium of experimental animals. Also, impaired relaxation, increased left ventricular stiffness, and left atrial remodeling were also observed [166]. According to the Framingham study, left ventricular hypertrophy is seen in many serious diseases such as sudden cardiac death, HF, myocardial infarction, and stroke. Therefore, left ventricular hypertrophy is considered to be an independent cardiac risk factor. However, reverse remodeling with normalization of the left ventricular mass can be expected after treatment. Reverse remodeling is determined clinically as a result of echocardiographic studies [167]. Despite scientific advances, cellular mechanisms in myocardial tissue are not clearly known. The experimental model of pulmonary artery banding induces right ventricle overpressure. The supravalvular aortic stenosis model is widely used in rats.
New experimental models are being developed to induce metabolic diseases in large animals to induce HF with preserved ejection fraction [157]. Transverse aortic stenosis in mice was first described by Rockman et al. [168]. Today, it has been the most used experimental model to examine HF caused by excessive left ventricular pressure. The TAC model leads to concentric hypertrophy, interstitial fibrosis, increased left ventricular stiffness, and eventually systolic HF due to increased pressure in the left ventricle [169, 170]. The TAC model causes concentric cardiac hypertrophy and subsequent systolic HF by increasing left ventricular afterload. Evaluation of the TAC procedure and comparison with the control group is done by doppler [171]. The TAC model has enabled discovering the cause of many fundamental pathological changes seen in HF, especially left ventricular remodeling [159, 172, 173]. Among this model’s advantages are the lower mortality rate (5–10%) and the occurrence of left ventricular hypertrophy [132]. Another advantage of this experimental model is that it can manipulate the degree of overpressure load by changing the contraction intensity [174]. It is also an essential advantage that thoracic aortic constrictions resemble those in humans, especially aortic stenosis [175]. The major disadvantage of the TAC procedure is that it is dependent on the experimenter. Repeating the experiment is a complicated and technically demanding model. It also has the disadvantage of large variability between individuals [171].
As a result of studies conducted in rats, it was shown that AAC initially caused an increase in contractility due to the sympathetic nervous system [176]. Only after 8 weeks of concentric hypertrophy becomes apparent, as well as systolic and diastolic dysfunction [175]. The hypertrophic response and progression to HF resulting from the TAC model depend on sex, weight, age, and genetics. Approximately 1 month after adrenal AAC in mice, cardiac hypertrophy is seen, and the degree of this narrowing causes HF after 15–21 weeks [177]. Also, as a result of the studies, the mortality rate of TAC was found to be between 6 and 45%, and it was found to be very variable [178, 179].
As a result of recent developments, the double loop-clip technique has been developed. They measured the lumen diameter of the middle aortic arch during preoperative echocardiography to calculate the inter-knot span of the suture for the double loop-clip technique modified by Merino et al. This new procedure reduced the mortality rate in experimental animals and resulted in reproducible aortic stenosis [180]. The application of this model results in marked left ventricular hypertrophy and HF associated with increased β-myosin heavy chains. Also, a decrease in EF is observed. The hemodynamic function is associated with decreased sarco(endo)plasmic reticulum calcium ATPase (SERCA) expression, glucose uptake, reduced amount of adenosine in the coronary artery, and increased cardiomyocyte microtubule density [79, 145]. A decrease in SERCA expression in the left ventricle was observed 20 weeks after the experimental model. This suggests that SERCA may be a marker in the process between hypertrophy and HF [63, 181]. In terms of changes in the neurohormonal level, when hypertrophy occurs, plasma catecholamine levels are normal, and local myocardial RAS activation is increased [182]. The number of ANP increases due to HF. Also, as a result of the studies performed, it was determined that the PTHrP/PTH bioregulatory system, which is locally expressed in the ventricular myocardium, is associated with pressure-overload hypertrophy [183]. It has been shown that not only myosin heavy chain and atrial natriuretic peptide (ANP), but also cytokines such as interleukin-1, interleukin-6, and TNF-α are increased in HF induced by this experimental model [184]. Apoptotic processes, collagen, and matrix remodeling are seen in this experimental model. Also, Ca2+ is an ion required for cell growth and the continuation of vital cell functions. In response to growth stimuli, the cytosolic Ca2+ level is increased, and calcineurin is indirectly activated. These changes also cause dephosphorylation of transcription factors, which in turn regulates the expression of specific genes. Although not well known, calcineurin is thought to be a mediator of myocardial hypertrophy [181]. Changes in the nitric oxide (NO) pathway in pressure-overloaded heart tissue are important [185]. Studies have shown that sildenafil, a phosphodiesterase-5 inhibitor, reduced the left ventricular hypertrophy in the TAC model created in mice [186, 187]. Also, exogenous administration of the NO synthase cofactor (BH4) improved this left ventricular hypertrophy in mice [188]. Aortic constriction has been demonstrated in both the infrarenal and adrenal position in rats.
The experimental model of aortic constriction causes hypoperfusion, hypertension, and left ventricular hypertrophy. The AAC model contributes to the slower development of HF [160, 189]. When aortic insufficiency is combined with aortic narrowing, HF develops more rapidly. Ezzaher et al. reported that aortic insufficiency is produced by destroying the aortic valve with a catheter introduced through the carotid artery. After 14 days, aortic narrowing was performed just below the diaphragm. HF developed approximately 1 month after the first surgical procedure [190]. Although the protein and mRNA levels of the Na+/Ca2+ exchanger increased significantly in the HF model, the Ca2+/ATPase level in the sarcoplasmic reticulum did not change significantly [79]. This model mimics the changes in myocardial function observed in the late human myocardium. Therefore, this experimental model may be very suitable for studying HF after hypertrophy. Hyperreninemia develops in less than 4 days as a result of adrenal aortic coarctation. After several weeks, ventricular ACE activity may return to typical values, which may be associated with increased hypertrophy and normalization of wall stress [191]. As a result of this experimental model, left ventricular hypertrophy and HF were associated with increased β-myosin heavy chain mRNA and atrial natriuretic factor mRNA. Interestingly, after 20 weeks of banding, a decrease in SR-Ca2+/ATPase mRNA levels was seen by the polymerase chain reaction (PCR), while not in the group that did not develop HF. These data indicate that the decrease in SR-Ca2+/ATPase mRNA levels may indicate compensatory hypertrophy’s transition to failure in these animals [181]. During compensated hypertrophy, although catecholamine levels are normal, there is a local myocardial increased renin-angiotensin system. Also, with the development of HF, catecholamine levels in plasma may increase [192]. Therefore, this model should be preferred to examine the pathophysiological changes in the transition from hypertrophy to HF at the myocardial level. It is also a significant advantage that it can elucidate the molecular mechanisms involved in reverse remodeling of left ventricular hypertrophy [193].
Volume overload
The HF model induced by volume overload can be created by many different methods such as the arteriovenous fistula, aortic valve insufficiency, or destruction of the mitral valve [154, 194, 195]. It can cause HF in hyperdynamic conditions such as hyperthyroidism, beriberi (vitamin B1 deficiency), and severe anemia [154]. A shunt is created surgically to induce HF by causing an increase in volume. The shunt is usually formed between the aorta and the vena cava, femoral artery and vein, carotid artery, and internal jugular vein. Mice and rats are generally used in the HF model induced by volume overload [26]. Although HF progression varies, these models investigate disorders in fluid balance, electrolytes, and hormones common in HF. Increasing the heart’s volume load causes an increase in left ventricular end-diastolic pressure and results in expanding all the chambers in the heart. Although the systolic function is not impaired until the last stage of HF, the heart enlarges, and eccentric hypertrophy develops [196]. A catheter creates aortic valve perforation and chronic severe aortic regurgitation in a study on rabbits. Although eccentric left ventricular hypertrophy is observed first, it is often followed by systolic dysfunction [197]. However, it is difficult to induce aortic regurgitation in an experimentally reproducible manner, so most animal models have focused on inducing mitral regurgitation [198]. While arteriovenous fistula formation is a common volume overload model in small animals, it is rarely used in large animal models [199].
Surgical or percutaneous cutting of the mitral valve chordae tendineae causes mitral regurgitation and leads to chronic HF. However, this model is known to have a high mortality rate of up to 50% [200]. The reason for high mortality is that the degree of experimental HF created is difficult to control. One method of controlling this procedure is to place a graft between the left ventricle and the left atrium [201]. Although implantation is technically difficult, clamping the graft allows the degree of HF to be controlled. Inferior vena cava filters can be placed to keep the degree of insufficiency under control in the experimental HF model created due to mitral valve insufficiency [201]. Mitral valve insufficiency can also be observed in patients with postmyocardial infarction, and this circumstance is a poor prognosis. This model is usually done in sheep and pigs by infarctions the posterior wall of the left ventricle. The prognosis of mitral valve insufficiency after ischemia is quite poor [202].
Arteriovenous fistula models are another HF model used to create volume-overload HF in large animals [203]. The degree of shunt and proximity to the heart are among the factors that determine the severity of HF. The internal jugular vein and left carotid artery are exposed in the cervical arteriovenous shunt procedure. Approximately 30 min after the opening of the shunt, a 40% increase in cardiac output is observed. The left ventricular end-diastolic diameter and left ventricular end-diastolic volume increase after 8 weeks [132]. Although the arteriovenous fistula created between the carotid artery and the jugular vein caused an increase in left ventricular end-diastolic volume by approximately 50%, it did not cause a change in the end-diastolic pressure value [204]. Pinsky et al. performed a shunt between the infrarenal aorta and the superior vena cava to induce volume overload in 1979, whereas Tessier et al. have also combined this method with doxorubicin administration with a similar approach in goats [199].
Apart from this, mitral valve regurgitation was induced in dogs causing left ventricular enlargement. While left ventricular mass increased, there was not much change in the right ventricle. Asymmetric left ventricular dilatation was observed. Septum increased its contribution to the left ventricular stroke volume [205]. Mitral valve insufficiency models do not fully reflect HF. These models do not have pathophysiological changes in the myocardial structure due to ischemia or hypertrophy and are observed in congestive HF. The most important advantage of this model is evaluating the aperture of the shunt by palpating the neck. Also, ultrasound can be used for this procedure. A simple technique, the needle technique, is used to create an aortocaval shunt [206]. Compensated hypertrophy occurs approximately 2–8 weeks after the aortocaval fistula is formed. Left ventricular end-diastolic pressure increases around five times in the first week [206]. Arteriovenous shunts cause HF due to dilated cardiomyopathy by induction of volume overload in rodents. Femoral arteriovenous fistula causes HF, although it has a high mortality rate (over 25%) [207]. Also, the HF model’s essential advantages created with an aortocaval shunt are low mortality rate, simple, rapid, and no need for thoracotomy [208]. Studies in rats showed that cardiac hypertrophy developed and left ventricular diastolic pressure increased 4 weeks after shunt induction [209].
Volume overload resulting from aortocaval fistula causes a decrease in left ventricular function. Subsequently, hypertrophy develops as a mechanism of protection, leading to near-normal function at 4 weeks [210]. The decompensated hypertrophic HF and decreased systolic and diastolic function are seen approximately 8–16 weeks after the surgical procedure [211]. After a large aortocaval fistula is opened, severe volume overload occurs. Initially, the left ventricular function is suppressed, and hypertrophy develops as a protective mechanism. Hemodynamic functions are almost the same as normal after 4 weeks [212]. It has been determined that in about 7% of cases, the shunt closes spontaneously, so the fistula opening must be confirmed at the end of the experiment. It is known that left ventricular end-diastolic pressure increase is caused only by a large shunt and develops after at least 4 weeks [213].
Aortic valve insufficiency also causes HF as a result of increased volume in rats [214]. An increase in beta-receptor density was observed in heart tissues with HF in the 16th week. Progressive neurohormonal activation derived from the chronic mitral valve regurgitation model allowed investigation of the effects of angiotensin II type 1 and beta-receptor blockade on left ventricular failure [215]. For example, Tsutsui et al. showed in a study performed by bridges that the beta-receptor blockade provides an improvement in contractile function in the left ventricle compared to the control group [216]. It was observed that basal adenylyl cyclase activity increased, and beta-adrenoceptor signal transduction was similar to hypertrophy by induced volume overload [210]. According to recent studies, it has been shown that the synthesis of ANP expression increases due to increasing volume rather than pressure [217]. Also, induced myocyte elongation, changes in myofilament structure, and decreases in myocyte contractile function were observed [218]. It has been shown that SERCA2α expression can prevent systolic and diastolic dysfunction and left ventricular remodeling [219]. Volume overload in mice causes minimal apoptosis in the absence of pathological remodeling than the TAC model [26]. Surgical-induced HF models are presented in the Table 2.
Drug-induced
Drugs induce HF model in different animals to study the etiology of HF that includes chemotoxicity, hypertension, kidney damage, and liver damage [226, 227]. Drug-induced HF models are presented in the Table 3. In summary, HF can be caused by the administration of chemical agents. However, in general, the advantages and disadvantages should be evaluated carefully, and the appropriate experimental model should be selected.
Hypertension
Hypertension is one of the essential conditions leading to diastolic HF in humans [144, 240]. Hypertension causing extensive inflammation and metabolism changes can lead to myocardial stiffness and diastolic dysfunction [241, 242]. Ventricular dysfunction develops due to the proliferation of fibroblasts, hypertrophy of vascular smooth muscle cells, and pathological accumulation of interstitial collagen [143, 243]. Hypertension-induced HF models are presented in the Table 4.
High fat diet + L-NAME
It is known that the comorbidities that increase the risk of HFpEF are diabetes, obesity, and hypertension [249]. Recently, Hill et al. proposed a “two-hit” mouse model of HFpEF that mimics the concomitant metabolic and hypertensive stress in mice [250]. In this model, a high-fat diet (HFD) induces metabolic stress (obesity, glucose intolerance, and metabolic syndrome), and a drug called Nω-nitro-l-arginine methyl ester (L-NAME), which inhibits nitric oxide synthase, also causes hypertension. This model recapitulates numerous systemic and cardiovascular features of HFpEF, including impaired cardiac filling, cardiac hypertrophy, cardiac fibrosis, decreased myocardial capillary density, pulmonary hyperemia, decreased exercise tolerance, myocardial capillary rarefaction, and increased levels of inflammatory markers [250]. Diastolic dysfunction was associated with cardiac hypertrophy and fibrosis [251]. This experimental model uses 60% kilocalories from fat (lard) and drinking water with 0.5 g/L of L-NAME [252].
Conclusion
Pre-clinical HF models are useful in the studying pathophysiology of complex cardiovascular diseases. Experimental animal models are becoming less relevant due to modern techniques to determine HF’s hemodynamics in humans. However, pre-clinical studies have great significance in the experimental HF studies currently. Also, pre-clinical studies have elucidated molecular and biochemical modifications and remodeling processes in human HF myocardium. Animal HF models may be suitable for testing the effect of new treatment modalities on hemodynamics, neurohumoral activation, and survival under preclinical conditions. Today, transgenic models provide a better understanding of HF pathophysiology at the molecular level. It enables the identification of genes that cause HF and elucidate their role in HF. It also enables it to be characterized in the molecular mechanisms responsible for HF development and progression.
Finally, various experimental animal models that mimic human HF features will play a vital role in elucidating the pathophysiology in HF and in solving molecular techniques. These studies are invaluable and provide knowledge on HF’s underlying mechanisms and contribute significantly to creating new therapeutic strategies for HF. Experimental HF models will have mechanistic perspectives that will contribute to new classes of therapy in the future. Animal HF models are now becoming excellent tools for creating new goal settings for new HF management agents.
Availability of data and materials
Not applicable.
References
Members ATF et al (2012) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 33(14):1787–1847
Kittana N (2018) Angiotensin-converting enzyme 2-Angiotensin 1–7/1-9 system: novel promising targets for heart failure treatment. Fundam Clin Pharmacol 32(1):14–25
Ozmen C et al (2020) Prognostic performance of copeptin among patients with acute decompensated heart failure. Acta Cardiol 1–10
Raghunathan S, Patel BM (2013) Therapeutic implications of small interfering RNA in cardiovascular diseases. Fundam Clin Pharmacol 27(1):1–20
Lubrano V, Balzan S (2020) Role of oxidative stress-related biomarkers in heart failure: galectin 3, α1-antitrypsin and LOX-1: new therapeutic perspective? Mol Cell Biochem 464(1–2):143–152
Pagliaro BR et al (2020) Myocardial ischemia and coronary disease in heart failure. Heart Fail Rev 25(1):53–65
Savarese G, Lund LH (2017) Global public health burden of heart failure. Card Fail Rev 3(1):7
Benjamin EJ et al (2019) Heart disease and stroke statistics-2019 update a report from the American Heart Association. Circulation
Caliskan HM et al (2020) Prognostic value of thiol/disulfide homeostasis in symptomatic patients with heart failure. Arch Physiol Biochem 1–6
Hampton C et al (2017) Early echocardiographic predictors of outcomes in the mouse transverse aortic constriction heart failure model. J Pharmacol Toxicol Methods 84:93–101
Carll AP et al (2011) Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc Toxicol 11(2):91–112
Rai V et al (2017) Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research. Mol Cell Biochem 424(1–2):123–145
Suthahar N et al (2017) From inflammation to fibrosis—molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep 14(4):235–250
Gunata M, Parlakpinar H (2020) A review of myocardial ischaemia/reperfusion injury: pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell Biochem Funct
Manea A et al (2007) Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Arch Physiol Biochem 113(4–5):163–172
Costa CRM et al (2020) Progression of heart failure is attenuated by antioxidant therapy with N-acetylcysteine in myocardial infarcted female rats. Mol Biol Rep 47(11):8645–8656
Chen W, Frangogiannis NG (2013) Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta Mol Cell Res 1833(4):945–953
Simmonds SJ et al (2020) Cellular and molecular differences between HFpEF and HFrEF: a step ahead in an improved pathological understanding. Cells 9(1):242
Ghorbanzadeh V et al (2020) The role of vasopressin V1A and oxytocin OTR receptors in protective effects of arginine vasopressin against H(2)O(2)-induced oxidative stress in H9C2 cells. Arch Physiol Biochem: 1–6
Katz MG et al (2019) Surgical and physiological challenges in the development of left and right heart failure in rat models. Heart Fail Rev 24(5):759–777
Lips DJ et al (2003) Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur Heart J 24(10):883–896
Chugh SS et al (2008) Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis 51(3):213–228
Spannbauer A et al (2019) Large animal models of heart failure with reduced ejection fraction (HFrEF): a minireview. Front Cardiovasc Med 6:117
Vasan RS et al (2018) Epidemiology of left ventricular systolic dysfunction and heart failure in the Framingham study: an echocardiographic study over 3 decades. JACC Cardiovasc Imaging 11(1):1–11
Hogg K, Swedberg K, McMurray J (2004) Heart failure with preserved left ventricular systolic function: epidemiology, clinical characteristics, and prognosis. J Am Coll Cardiol 43(3):317–327
Riehle C, Bauersachs J (2019) Small animal models of heart failure. Cardiovasc Res 115(13):1838–1849
Lejeune S et al (2020) Heart failure with preserved ejection fraction in Belgium: characteristics and outcome of a real-life cohort. Acta Cardiol: 1–10
Leonardini A, Avogaro A (2013) Abnormalities of the cardiac stem and progenitor cell compartment in experimental and human diabetes. Arch Physiol Biochem 119(4):179–187
Gu J et al (2020) Characteristics and outcomes of transitions among heart failure categories: a prospective observational cohort study. ESC Heart Fail 7(2):616–625
Borlaug BA, Redfield MM (2011) Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 123(18):2006–2014
Alpert CM et al (2017) Symptom burden in heart failure: assessment, impact on outcomes, and management. Heart Fail Rev 22(1):25–39
Ponikowski P et al (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37(27):2129–2200
Palazzuoli A et al (2018) Combined use of lung ultrasound, B-type natriuretic peptide, and echocardiography for outcome prediction in patients with acute HFrEF and HFpEF. Clin Res Cardiol 107(7):586–596
Correale M et al (2021) New targets in heart failure drug therapy. Front Cardiovasc Med 8:665797
Suematsu Y et al (2016) LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail 18(4):386–393
Habibi J et al (2017) Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol 16(1):9
Düngen HD et al (2019) Safety and tolerability of the chymase inhibitor fulacimstat in patients with left ventricular dysfunction after myocardial infarction-results of the CHIARA MIA 1 Trial. Clin Pharmacol Drug Dev 8(7):942–951
Carubelli V et al (2020) Treatment with 24 hour istaroxime infusion in patients hospitalised for acute heart failure: a randomised, placebo-controlled trial. Eur J Heart Fail 22(9):1684–1693
Tita C et al (2017) A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur J Heart Fail 19(10):1321–1332
Voors AA et al (2017) Safety and tolerability of neladenoson bialanate, a novel oral partial adenosine A1 receptor agonist, in patients with chronic heart failure. J Clin Pharmacol 57(4):440–451
Voors AA et al (2019) Safety and efficacy of the partial adenosine A1 receptor agonist neladenoson bialanate in patients with chronic heart failure with reduced ejection fraction: a phase IIb, randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 21(11):1426–1433
Vecchio EA et al (2016) The hybrid molecule, VCP746, is a potent adenosine A2B receptor agonist that stimulates anti-fibrotic signalling. Biochem Pharmacol 117:46–56
Butler J et al (2020) Effects of elamipretide on left ventricular function in patients with heart failure with reduced ejection fraction: the PROGRESS-HF phase 2 trial. J Card Fail 26(5):429–437
Filippatos G et al (2017) Patient-reported outcomes in the soluble guanylate cyclase stimulator in heart failure patients with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. Eur J Heart Fail 19(6):782–791
Gheorghiade M et al (2015) Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES-REDUCED randomized trial. JAMA 314(21):2251–2262
Goldsmith SR et al (2021) Dual vasopressin receptor antagonism to improve congestion in patients with acute heart failure: design of the AVANTI trial. J Card Fail 27(2):233–241
Patel RB, Shah SJ (2019) Drug targets for heart failure with preserved ejection fraction: a mechanistic approach and review of contemporary clinical trials. Annu Rev Pharmacol Toxicol 59:41–63
Teerlink JR et al (2016) Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC-AHF study. J Am Coll Cardiol 67(12):1444–1455
Tamaki S et al (2013) Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS ONE 8(7):e68893
Trankle CR et al (2018) Usefulness of canakinumab to improve exercise capacity in patients with long-term systolic heart failure and elevated C-reactive protein. Am J Cardiol 122(8):1366–1370
Van Tassell BW et al (2017) Interleukin-1 blockade in recently decompensated systolic heart failure: results from REDHART (recently decompensated heart failure Anakinra response trial). Circ Heart Fail 10(11)
Isakova T et al (2015) Rationale and approaches to phosphate and fibroblast growth factor 23 reduction in CKD. J Am Soc Nephrol 26(10):2328–2339
Yu L et al (2013) Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail 6(1):107–117
Gallet R et al (2016) Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 1(1–2):14–28
Goldin A et al (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114(6):597–605
Maier LS et al (2013) RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI-DHF proof-of-concept study. JACC Heart Fail 1(2):115–122
Jeong MY et al (2018) Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 10(427)
Methawasin M et al (2016) Experimentally increasing the compliance of titin through RNA binding motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 134(15):1085–1099
Yang Q et al (2011) AVE3085, an enhancer of endothelial nitric oxide synthase, restores endothelial function and reduces blood pressure in spontaneously hypertensive rats. Br J Pharmacol 163(5):1078–1085
Green EM et al (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351(6273):617–621
Spencer AG et al (2014) Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 6(227):227ra36
Kurian GA, Ansari M, Prem PN (2020) Diabetic cardiomyopathy attenuated the protective effect of ischaemic post-conditioning against ischaemia-reperfusion injury in the isolated rat heart model. Arch Physiol Biochem: 1–12
Afzal M (2021) Recent updates on novel therapeutic targets of cardiovascular diseases. Mol Cell Biochem 476(1):145–155
Dhalla NS et al (2012) Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev 17(4–5):671–681
Cops J et al (2019) Current animal models for the study of congestion in heart failure: an overview. Heart Fail Rev 24(3):387–397
Ali SS et al (2019) Anti-fibrotic actions of roselle extract in rat model of myocardial infarction. Cardiovasc Toxicol 19(1):72–81
Zhang F et al (2020) β-cryptoxanthin alleviates myocardial ischaemia/reperfusion injury by inhibiting NF-κB-mediated inflammatory signalling in rats. Arch Physiol Biochem: 1–8
Heusch G (2016) Myocardial ischemia: lack of coronary blood flow or myocardial oxygen supply/demand imbalance? Circ Res 119(2):194–196
Refaie MMM et al (2020) Cardioprotective effect of hemin in isoprenaline-induced myocardial infarction: role of ATP-sensitive potassium channel and endothelial nitric oxide synthase. Fundam Clin Pharmacol 34(3):302–312
He W, James Kang Y (2013) Ischemia-induced copper loss and suppression of angiogenesis in the pathogenesis of myocardial infarction. Cardiovasc Toxicol 13(1):1–8
Monnet E, Chachques JC (2005) Animal models of heart failure: what is new? Ann Thorac Surg 79(4):1445–1453
Janahmadi Z et al (2015) Oleuropein offers cardioprotection in rats with acute myocardial infarction. Cardiovasc Toxicol 15(1):61–68
Ou L et al (2010) Animal models of cardiac disease and stem cell therapy. Open Cardiovasc Med J 4:231
Iwanaga K et al (2004) Effects of G-CSF on cardiac remodeling after acute myocardial infarction in swine. Biochem Biophys Res Commun 325(4):1353–1359
Shettigar V et al (2016) Rationally engineered troponin C modulates in vivo cardiac function and performance in health and disease. Nat Commun 7(1):1–13
Wayman NS et al (2003) Models of coronary artery occlusion and reperfusion for the discovery of novel antiischemic and antiinflammatory drugs for the heart. Inflammation protocols. Springer, pp 199–208
Thackeray JT et al (2018) Myocardial inflammation predicts remodeling and neuroinflammation after myocardial infarction. J Am Coll Cardiol 71(3):263–275
Thackeray JT et al (2015) Molecular imaging of the chemokine receptor CXCR4 after acute myocardial infarction. JACC Cardiovasc Imaging 8(12):1417–1426
Djordjevic A et al (2018) Left ventricular remodeling after the first myocardial infarction in association with LGALS-3 neighbouring variants rs2274273 and rs17128183 and its relative mRNA expression: a prospective study. Mol Biol Rep 45(6):2227–2236
Gould KE et al (2002) Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol 282(2):H615–H621
Bayat H et al (2002) Progressive heart failure after myocardial infarction in mice. Basic Res Cardiol 97(3):206–213
Pfeffer MA et al (1979) Myocardial infarct size and ventricular function in rats. Circ Res 44(4):503–512
Pfeffer M et al (1985) Survival after an experimental myocardial infarction: beneficial effects of long-term therapy with captopril. Circulation 72(2):406–412
Patten RD, Hall-Porter MR (2009) Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail 2(2):138–144
Sheng FQ et al (2009) In rats with myocardial infarction, interference by simvastatin with the TLR4 signal pathway attenuates ventricular remodelling. Acta Cardiol 64(6):779–785
Li YY, Feldman AM (2001) Matrix metalloproteinases in the progression of heart failure. Drugs 61(9):1239–1252
Kajstura J et al (1996) Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest 74(1):86–107
Pugliese NR, Masi S, Taddei S (2020) The renin-angiotensin-aldosterone system: a crossroad from arterial hypertension to heart failure. Heart Fail Rev 25(1):31–42
Pfeffer JM et al (1979) Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 237(4):H461–H468
Ceiler DL et al (1998) Time-related adaptations in plasma neurohormone levels and hemodynamics after myocardial infarction in the rat. J Cardiac Fail 4(2):131–138
Hwang GS et al (2006) Effects of KR-31378, a novel ATP-sensitive potassium channel activator, on hypertrophy of H9c2 cells and on cardiac dysfunction in rats with congestive heart failure. Eur J Pharmacol 540(1–3):131–138
Li Z et al (2004) p38α mitogen-activated protein kinase inhibition improves cardiac function and reduces myocardial damage in isoproterenol-induced acute myocardial injury in rats. J Cardiovasc Pharmacol 44(4):486–492
Wu JC et al (2003) Influence of sex on ventricular remodeling after myocardial infarction in mice. J Am Soc Echocardiogr 16(11):1158–1162
Kuhlmann MT et al (2006) G-CSF/SCF reduces inducible arrhythmias in the infarcted heart potentially via increased connexin43 expression and arteriogenesis. J Exp Med 203(1):87–97
Liu Y et al (1997) Chronic heart failure induced by coronary artery ligation in Lewis inbred rats. Am J Physiol Heart Circ Physiol 272(2):H722–H727
Braunwald E, Kloner R (1982) The stunned myocardium: prolonged, postischemic ventricular dysfunction. Circulation 66(6):1146–1149
Mertes H et al (1995) Assessment of hibernating myocardium by dobutamine stimulation in a canine model. J Am Coll Cardiol 26(5):1348–1355
Bolukoglu H et al (1992) An animal model of chronic coronary stenosis resulting in hibernating myocardium. Am J Physiol Heart Circ Physiol 263(1):H20–H29
Michael LH et al (1995) Myocardial ischemia and reperfusion: a murine model. Am J Physiol Heart Circ Physiol 269(6):H2147–H2154
Silva KAS, Emter CA (2020) Large animal models of heart failure: a translational bridge to clinical success. JACC Basic Transl Sci 5(8):840–856
Sabbah HN et al (1994) Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation 89(6):2852–2859
Saavedra WF et al (2002) Reverse remodeling and enhancedadrenergic reserve from passive externalsupport in experimental dilated heart failure. J Am Coll Cardiol 39(12):2069–2076
Huang Y et al (1997) A stable ovine congestive heart failure model. A suitable substrate for left ventricular assist device assessment. Asaio J 43(5):M408–M413
Hedayati N et al (2002) Circulatory benefits of diastolic counterpulsation in an ischemic heart failure model after aortomyoplasty. J Thorac Cardiovasc Surg 123(6):1067–1073
Gupta RC et al (1997) SR Ca (2+)-ATPase activity and expression in ventricular myocardium of dogs with heart failure. Am J Physiol Heart Circ Physiol 273(1):H12–H18
Dixon JA, Spinale FG (2009) Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2(3):262–271
Klocke R et al (2007) Surgical animal models of heart failure related to coronary heart disease. Cardiovasc Res 74(1):29–38
Lindsey ML et al (2018) Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 314(4):H812–H838
Yeang C et al (2019) Reduction of myocardial ischaemia–reperfusion injury by inactivating oxidized phospholipids. Cardiovasc Res 115(1):179–189
Hausenloy DJ et al (2019) The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res 115(7):1143–1155
Porrello ER et al (2013) Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci 110(1):187–192
Dixon I, Lee S-L, Dhalla N (1990) Nitrendipine binding in congestive heart failure due to myocardial infarction. Circ Res 66(3):782–788
Krzemiński TF et al (2008) Wide-spread myocardial remodeling after acute myocardial infarction in rat. Features for heart failure progression. Vascul Pharmacol 48(2–3):100–108
Lutgens E et al (1999) Chronic myocardial infarction in the mouse: cardiac structural and functional change. Cardiovasc Res 41(3):586–593
Patterson RE, Kirk ES (1983) Analysis of coronary collateral structure, function, and ischemic border zones in pigs. Am J Physiol Heart Circ Physiol 244(1):H23–H31
González-Rosa JM, Mercader N (2012) Cryoinjury as a myocardial infarction model for the study of cardiac regeneration in the zebrafish. Nat Protoc 7(4):782–788
Hedström E et al (2009) Infarct evolution in man studied in patients with first-time coronary occlusion in comparison to different species-implications for assessment of myocardial salvage. J Cardiovasc Magn Reson 11(1):38
Suzuki M et al (1999) Development and evaluation of a new canine myocardial infarction model using a closed-chest injection of thrombogenic material. Jpn Circ J 63(11):900–905
Li R-K et al (1999) Smooth muscle cell transplantation into myocardial scar tissue improves heart function. J Mol Cell Cardiol 31(3):513–522
Sabbah HN et al (1991) A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol Heart Circ Physiol 260(4):H1379–H1384
Dandamudi G et al (2008) Persistent left ventricular dilatation in tachycardia-induced cardiomyopathy patients after appropriate treatment and normalization of ejection fraction. Heart Rhythm 5(8):1111–1114
Whipple G (1962) Reversible congestive heart failure due to chronic rapid stimulation of the normal heart. In Proc N Engl Cardiovasc Soc
Ohno M, Cheng C-P, Little WC (1994) Mechanism of altered patterns of left ventricular filling during the development of congestive heart failure. Circulation 89(5):2241–2250
Howard RJ et al (1988) Recovery from heart failure: structural and functional analysis in a canine model. Can J Physiol Pharmacol 66(12):1505–1512
Masarone D et al (2017) Management of arrhythmias in heart failure. J Cardiovasc Dev Dis 4(1):3
Sun J, Zhang C, Zhang Z (2019) Atorvastatin attenuates cardiac hypertrophy through AMPK/miR-143–3p/Bcl2 axis. Arch Physiol Biochem: 1–7
January CT et al (2014) 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 130(23):2071–2104
Iwasaki Y-K et al (2011) Atrial fibrillation pathophysiology: implications for management. Circulation 124(20):2264–2274
Timek TA et al (2003) Tachycardia-induced cardiomyopathy in the ovine heart: mitral annular dynamic three-dimensional geometry. J Thorac Cardiovasc Surg 125(2):315–324
Shi Y et al (2001) Remodeling of atrial dimensions and emptying function in canine models of atrial fibrillation. Cardiovasc Res 52(2):217–225
Houser SR et al (2012) Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 111(1):131–150
Halapas A et al (2008) In vivo models for heart failure research. In Vivo 22(6):767–780
Riegger GA et al (1988) Atrial natriuretic peptide in congestive heart failure in the dog: plasma levels, cyclic guanosine monophosphate, ultrastructure of atrial myoendocrine cells, and hemodynamic, hormonal, and renal effects. Circulation 77(2):398–406
Armstrong PW et al (1986) Rapid ventricular pacing in the dog: pathophysiologic studies of heart failure. Circulation 74(5):1075–1084
Wilson J et al (1987) Experimental congestive heart failure produced by rapid ventricular pacing in the dog: cardiac effects. Circulation 75(4):857–867
Moe GW, Armstrong P (1999) Pacing-induced heart failure: a model to study the mechanism of disease progression and novel therapy in heart failure. Cardiovasc Res 42(3):591–599
Margulies KB et al (1990) Increased endothelin in experimental heart failure. Circulation 82(6):2226–2230
Bristow MR (2000) β-Adrenergic receptor blockade in chronic heart failure. Circulation 101(5):558–569
Eble DM, Spinale FG (1995) Contractile and cytoskeletal content, structure, and mRNA levels with tachycardia-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 268(6):H2426–H2439
Moe G et al (1988) Early recovery from heart failure: insights into the pathogenesis of experimental chronic pacing-induced heart failure. J Lab Clin Med 112(4):426–432
Xin W et al (2011) Improved cardiac function after sarcoplasmic reticulum Ca(2+)-ATPase gene transfer in a heart failure model induced by chronic myocardial ischaemia. Acta Cardiol 66(1):57–64
Nazifova-Tasinova NF et al (2020) Circulating uncarboxylated matrix Gla protein in patients with atrial fibrillation or heart failure with preserved ejection fraction. Arch Physiol Biochem: 1–11
Givvimani S et al (2013) TIMP-2 mutant decreases MMP-2 activity and augments pressure overload induced LV dysfunction and heart failure. Arch Physiol Biochem 119(2):65–74
Givvimani S et al (2010) MMP-2/TIMP-2/TIMP-4 versus MMP-9/TIMP-3 in transition from compensatory hypertrophy and angiogenesis to decompensatory heart failure. Arch Physiol Biochem 116(2):63–72
Chakraborti S et al (2007) Calcium signaling phenomena in heart diseases: a perspective. Mol Cell Biochem 298(1–2):1–40
Hobai IA, Maack C, O’Rourke B (2004) Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res 95(3):292–299
Leri A et al (1998) Pacing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation 97(2):194–203
Shinbane JS et al (1997) Tachycardia-induced cardiomyopathy: a review of animal models and clinical studies. J Am Coll Cardiol 29(4):709–715
Oh JH et al (1998) The effects of prosthetic cardiac binding and adynamic cardiomyoplasty in a model of dilated cardiomyopathy. J Thorac Cardiovasc Surg 116(1):148–153
Lazzara RR, Trumble DR, Magovern JA (1994) Dynamic descending thoracic aortomyoplasty: comparison with intraaortic balloon pump in a model of heart failure. Ann Thorac Surg 58(2):366–371
Riehle C et al (2011) PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res 109(7):783–793
Schwarzer M et al (2013) Pressure overload differentially affects respiratory capacity in interfibrillar and subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 304(4):H529–H537
Rupp H, Benkel M, Maisch B (2000) Control of cardiomyocyte gene expression as drug target. Mol Cell Biochem 212(1–2):135–142
Balakumar P, Singh AP, Singh M (2007) Rodent models of heart failure. J Pharmacol Toxicol Methods 56(1):1–10
Xiao Y et al (2018) The association between myocardial fibrosis and depressed capillary density in rat model of left ventricular hypertrophy. Cardiovasc Toxicol 18(4):304–311
Li JM et al (2012) The therapeutic potential of G-CSF in pressure overload induced ventricular reconstruction and heart failure in mice. Mol Biol Rep 39(1):5–12
Oh JG et al (2019) Experimental models of cardiac physiology and pathology. Heart Fail Rev 24(4):601–615
Xiao C-Y et al (2005) Poly (ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther 312(3):891–898
Tannu S et al (2020) Experimental model of congestive heart failure induced by transverse aortic constriction in BALB/c mice. J Pharmacol Toxicol Methods 106:106935
Cantor EJ et al (2005) A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. J Mol Cell Cardiol 38(5):777–786
Ishikawa K et al (2015) Increased stiffness is the major early abnormality in a pig model of severe aortic stenosis and predisposes to congestive heart failure in the absence of systolic dysfunction. J Am Heart Assoc 4(5):e001925
Obokata M et al (2017) Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 136(1):6–19
Borlaug BA (2014) The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 11(9):507–515
Grossman W, Jones D, McLaurin L (1975) Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Investig 56(1):56–64
Huber D et al (1981) Determinants of ejection performance in aortic stenosis. Circulation 64(1):126–134
Zakeri R et al (2016) Left atrial remodeling and atrioventricular coupling in a canine model of early heart failure with preserved ejection fraction. Circ Heart Fail 9(10):e003238
Walther T et al (1999) Prospectively randomized evaluation of stentless versus conventional biological aortic valves: impact on early regression of left ventricular hypertrophy. Circulation 100(suppl_2):II-6–Ii−10
Rockman HA et al (1994) ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol Heart Circ Physiol 266(6):H2468–H2475
Mustonen E et al (2010) Metoprolol treatment lowers thrombospondin-4 expression in rats with myocardial infarction and left ventricular hypertrophy. Basic Clin Pharmacol Toxicol 107(3):709–717
Bosch L et al (2021) The transverse aortic constriction heart failure animal model: a systematic review and meta-analysis. Heart Fail Rev 26(6):1515–1524
Mohammed SF et al (2012) Variable phenotype in murine transverse aortic constriction. Cardiovasc Pathol 21(3):188–198
Ichinose F et al (2004) Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiol
Chen JJ et al (2019) PM25 exposure aggravates left heart failure induced pulmonary hypertension. Acta Cardiol 74(3):238–244
Moens AL et al (2009) Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the β3-adrenoreceptor. J Mol Cell Cardiol 47(5):576–585
Bramlage P et al (2004) Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens 17(10):904–910
Brede M et al (2002) Feedback inhibition of catecholamine release by two different α2-adrenoceptor subtypes prevents progression of heart failure. Circulation 106(19):2491–2496
Hara M et al (2002) Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med 195(3):375–381
deAlmeida AC, van Oort RJ, Wehrens XH (2010) Transverse aortic constriction in mice. J Vis Exp 38:1729
Hu P et al (2003) Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol 285(3):H1261–H1269
Merino D et al (2018) Experimental modelling of cardiac pressure overload hypertrophy: Modified technique for precise, reproducible, safe and easy aortic arch banding-debanding in mice. Sci Rep 8(1):3167
Feldman AM et al (1993) Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res 73(1):184–192
Weinberg EO et al (1994) Angiotensin-converting enzyme inhibition prolongs survival and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis. Circulation 90(3):1410–1422
Halapas A et al (2005) PTH-related protein and Type 1 parathyroid hormone receptor mRNA expression in rat ventricular myocardial hypertrophy. Clin Pract 2(3):415
Molina EJ et al (2009) Novel experimental model of pressure overload hypertrophy in rats. J Surg Res 153(2):287–294
Umar S, van der Laarse A (2010) Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem 333(1–2):191–201
Anand IS et al (2011) Prognostic value of baseline plasma amino-terminal pro-brain natriuretic peptide and its interactions with irbesartan treatment effects in patients with heart failure and preserved ejection fraction: findings from the I-PRESERVE trial. Cir Heart Fail 4(5):569–577
Takimoto E et al (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11(2):214–222
Moens AL et al (2008) High dose folic acid pre-treatment blunts cardiac dysfunction during ischemia coupled to maintenance of high energy phosphates and reduces post-reperfusion injury. Circulation 117(14):1810
Cui YH et al (2011) 17 beta-estradiol attenuates pressure overload-induced myocardial hypertrophy through regulating caveolin-3 protein in ovariectomized female rats. Mol Biol Rep 38(8):4885–4892
Ezzaher A et al (1991) Increased negative inotropic effect of calcium-channel blockers in hypertrophied and failing rabbit heart. J Pharmacol Exp Ther 257(1):466–471
Holtz J et al (1992) Modulation of myocardial sarcoplasmic reticulum Ca++-ATPase in cardiac hypertrophy by angiotensin converting enzyme? Cardiac Adaptation in Heart Failure. Springer, pp 191–204
Elsner D, Riegger G (1995) Characteristics and clinical relevance of animal models of heart failure. Curr Opin Cardiol 10(3):253–259
Stansfield WE et al (2007) Characterization of a model to independently study regression of ventricular hypertrophy. J Surg Res 142(2):387–393
Kleaveland JP et al (1988) Volume overload hypertrophy in a closed-chest model of mitral regurgitation. Am J Physiol Heart Circ Physiol 254(6):H1034–H1041
He ZY et al (2005) Intracardiac basic fibroblast growth factor and transforming growth factor-beta 1 mRNA and their proteins expression level in patients with pressure or volume-overload right or left ventricular hypertrophy. Acta Cardiol 60(1):21–25
Toischer K et al (2010) Differential cardiac remodeling in preload versus afterload. Circulation 122(10):993–1003
Magid NM et al (1994) Heart failure due to chronic experimental aortic regurgitation. Am J Physiol Heart Circ Physiol 267(2):H556–H562
Watanabe S et al (2018) Echocardiographic and hemodynamic assessment for predicting early clinical events in severe acute mitral regurgitation. Int J Cardiovasc Imaging 34(2):171–175
Tessier D et al (2003) Induction of chronic cardiac insufficiency by arteriovenous fistula and doxorubicin administration. J Card Surg 18(4):307–311
Watanabe S et al (2017) Protein phosphatase inhibitor-1 gene therapy in a swine model of nonischemic heart failure. J Am Coll Cardiol 70(14):1744–1756
Beaudoin J et al (2013) Late repair of ischemic mitral regurgitation does not prevent left ventricular remodeling: importance of timing for beneficial repair. Circulation 128(11 Suppl 1):S248–S252
Ishikawa K et al (2018) Reduced longitudinal contraction is associated with ischemic mitral regurgitation after posterior MI. Am J Physiol Heart Circ Physiol 314(2):H322–H329
Lu X et al (2014) Response of various conduit arteries in tachycardia-and volume overload-induced heart failure. PLoS ONE 9(8):e101645
Bolotin G et al (1999) Acute and chronic heart dilation model-induced in goats by carotid jugular AV shunt. BAM-PADOVA- 9(5):219–222
Young A et al (1996) Three-dimensional changes in left and right ventricular geometry in chronic mitral regurgitation. Am J Physiol Heart Circ Physiol 271(6):H2689–H2700
Garcia R, Diebold S (1990) Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res 24(5):430–432
Ozek C et al (1998) A new heart failure model in rat by an end-to-side femoral vessel anastomosis. Cardiovasc Res 37(1):236–238
Gomes A et al (2013) Rodent models of heart failure: an updated review. Heart Fail Rev 18(2):219–249
Scheuermann-Freestone M et al (2001) A new model of congestive heart failure in the mouse due to chronic volume overload. Eur J Heart Fail 3(5):535–543
Wang X et al (2003) Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J Appl Physiol 94(2):752–763
Wang X et al (2005) Upregulation of β-adrenergic receptors in heart failure due to volume overload. Am J Physiol Heart Circ Physiol 289(1):H151–H159
Liu Z et al (1991) Regional changes in hemodynamics and cardiac myocyte size in rats with aortocaval fistulas. 1. Developing and established hypertrophy. Circ Res 69(1):52–58
Langenickel T et al (2000) Differential regulation of cardiac ANP and BNP mRNA in different stages of experimental heart failure. Am J Physiol Heart Circ Physiol 278(5):H1500–H1506
Murakami K et al (2002) Perindopril effect on uncoupling protein and energy metabolism in failing rat hearts. Hypertension 40(3):251–255
Tallaj J et al (2003) β1-adrenergic receptor blockade attenuates angiotensin II–mediated catecholamine release into the cardiac interstitium in mitral regurgitation. Circulation 108(2):225–230
Tsutsui H et al (1994) Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Investig 93(6):2639–2648
Cavallero S et al (2007) Atrial natriuretic peptide behaviour and myocyte hypertrophic profile in combined pressure and volume-induced cardiac hypertrophy. J Hypertens 25(9):1940–1950
Spinale FG et al (1993) Structural basis for changes in left ventricular function and geometry because of chronic mitral regurgitation and after correction of volume overload. J Thorac Cardiovasc Surg 106(6):1147–1157
Kawase Y et al (2008) Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 51(11):1112–1119
Hasenfuss G (1998) Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res 39(1):60–76
Liao X et al (2007) Angiotensin-converting enzyme inhibitor improves force and Ca2 +–frequency relationships in myocytes from rats with heart failure. Acta Cardiol 62(2):157–162
Liu JG et al (2003) Effects of glucose-insulin-potassium on baroreflex sensitivity, left ventricular function and ventricular arrhythmia in the subacute phase of myocardial infarction in rats. Fundam Clin Pharmacol 17(4):443–448
Naseroleslami M et al (2020) Nesfatin-1 attenuates injury in a rat model of myocardial infarction by targeting autophagy, inflammation, and apoptosis. Arch Physiol Biochem: 1–9
Janssen PM, Elnakish MT (2019) Modeling heart failure in animal models for novel drug discovery and development. Expert Opin Drug Discov 14(4):355–363
Saura M, Zamorano JL, Zaragoza C (2022) Preclinical models of congestive heart failure, advantages, and limitations for application in clinical practice. Front Physiol 13:850301
Guo R, Ren J (2010) Alcohol dehydrogenase accentuates ethanol-induced myocardial dysfunction and mitochondrial damage in mice: role of mitochondrial death pathway. PLoS ONE 5(1):e8757
Zeiss CJ et al (2019) Doxorubicin-induced cardiotoxicity in collaborative cross (cc) mice recapitulates individual cardiotoxicity in humans. G3-Genes Genomes Genet 9(8):2637–2646
Breckenridge R (2010) Heart failure and mouse models. Dis Model Mech 3(3–4):138–143
Colak M et al (2012) Therapeutic effects of ivabradine on hemodynamic parameters and cardiotoxicity induced by doxorubicin treatment in rat. Hum Exp Toxicol 31(9):945–954
Disli O et al (2013) Effects of molsidomine against doxorubicin-induced cardiotoxicity in rats. Eur Surg Res 51(1–2):79–90
Ekinci Akdemir FN et al (2019) Protective effects of gallic acid on doxorubicin-induced cardiotoxicity; an experimantal study. Arch Physiol Biochem: 1–8
Kalyanaraman B et al (2002) Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem 234(1):119–124
Wang HL et al (2017) Synergistic effects of polydatin and vitamin C in inhibiting cardiotoxicity induced by doxorubicin in rats. Fundam Clin Pharmacol 31(3):280–291
Burdick J, Berridge B, Coatney R (2015) Strain echocardiography combined with pharmacological stress test for early detection of anthracycline induced cardiomyopathy. J Pharmacol Toxicol Methods 73:15–20
El Agaty SM (2019) Cardioprotective effect of vitamin D2 on isoproterenol-induced myocardial infarction in diabetic rats. Arch Physiol Biochem 125(3):210–219
Panda V et al (2019) Cardioprotective potential of Spinacia oleracea (Spinach) against isoproterenol-induced myocardial infarction in rats. Arch Physiol Biochem: 1–10
Rathinavel A et al (2018) Oligomeric proanthocyanidins protect myocardium by mitigating left ventricular remodeling in isoproterenol-induced postmyocardial infarction. Fundam Clin Pharmacol 32(1):51–59
Simko F et al (200) Ivabradine improves survival and attenuates cardiac remodeling in isoproterenol‐induced myocardial injury. Fundam Clin Pharmacol
Ulutas Z et al (2021) The Protective effects of compound 21 and Valsartan in isoproterenol-induced myocardial injury in rats. Cardiovasc Toxicol 21(1):17–28
Dogan MF et al (2019) Potassium channels in vascular smooth muscle: a pathophysiological and pharmacological perspective. Fundam Clin Pharmacol 33(5):504–523
Lamb HJ et al (1999) Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 99(17):2261–2267
Werida R et al (2020) Comparative effects of enalapril versus perindopril on serum levels of leptin and adiponectin in hypertensive patients. Acta Cardiol 75(6):551–556
Berenji K et al (2005) Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol 289(1):H8–H16
Ozhan O, Parlakpinar H, Acet A (2020) Comparison of the effects of losartan, captopril, angiotensin II type 2 receptor agonist compound 21, and MAS receptor agonist AVE 0991 on myocardial ischemia-reperfusion necrosis in rats. Fundam Clin Pharmacol
Valero-Muñoz M, Backman W, Sam F (2017) Murine Models of heart failure with preserved ejection fraction: a “fishing expedition.” JACC Basic Transl Sci 2(6):770–789
Noll NA, Lal H, Merryman WD (2020) Mouse models of heart failure with preserved or reduced ejection fraction. Am J Pathol 190(8):1596–1608
Conceição G et al (2016) Animal models of heart failure with preserved ejection fraction. Neth Hear J 24(4):275–286
Wei Y et al (2020) Acacetin improves endothelial dysfunction and aortic fibrosis in insulin-resistant SHR rats by estrogen receptors. Mol Biol Rep 47(9):6899–6918
Ussher JR et al (2016) The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J Am Coll Cardiol 68(25):2850–2870
Schiattarella GG et al (2019) Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568(7752):351–356
Cornuault L et al (2021) Diastolic dysfunction in high fat diet + L-NAME treated mice is associated with endothelial dysfunction. Arch Cardiovasc Dis Suppl 13(2):220
Zhang B et al (2021) Alteration of m6A RNA methylation in heart failure with preserved ejection fraction. Front Cardiovasc Med 8:647806
Author information
Authors and Affiliations
Contributions
MG was responsible for the design of figures/tables. All authors are responsible for the current information and literature research. This manuscript was written and the final manuscript is revised collaboratively by all authors.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gunata, M., Parlakpinar, H. Experimental heart failure models in small animals. Heart Fail Rev 28, 533–554 (2023). https://doi.org/10.1007/s10741-022-10286-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-022-10286-y