
Abstract
This study investigated the therapeutic potential of N-acetylcysteine (NAC) in the treatment of heart failure in female rats. Myocardial infarcted (MI) rats were given NAC (250 mg/kg/day p.o.) during 28 days after surgery (MI + NAC) or vehicle (MI + Placebo), and sham-operated rats received the same treatments (Sham + NAC and Sham + Placebo). Electrocardiographic and echocardiographic analyses were performed in the last week of treatment. Cardiac mRNA levels of types I and II superoxide dismutase (SOD), catalase, types I and III glutathione peroxidase (GPX), nerve growth factor (NGF), β1-adrenergic receptor (β1ADR), and type 2 muscarinic receptor (M2R) were assessed. Cardiac levels NADPH oxidase (NOX) activity, total content of reduced thiols, and SOD, GPX, and catalase activity were assessed. Compared to MI + Placebo group, MI + NAC group exhibited decreased NOX activity, increased content of reduced thiols, increased GPX activity, and normalized GPX III mRNA levels (p < 0.05). Heart and lung weights, left ventricular (LV) end-diastolic volume and left atrium/aorta ratio were decreased, while LV posterior wall thickness and ejection fraction were increased in MI + NAC group versus MI + Placebo rats (p < 0.05). Power density of low frequency band was decreased, while power density of high frequency and the root mean square of the successive differences were increased in MI + NAC rats versus MI + Placebo (p < 0.05). These findings indicate that NAC promotes therapeutic effects in the progression of MI-induced heart failure in female rats.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ischemic heart disease remains the leading cause of death worldwide in both men and women [1]. Prolonged periods of ischemia due to coronary artery disease are followed by irreversible myocardial damage irrespective of gender. However, women tend to exhibit suboptimal treatment pattern, and consequently worse outcomes compared to men [2]. Due to the limited healing capacity of adult mammalian hearts, most patients that survive acute myocardial infarction (AMI) eventually develop heart failure, a disease syndrome characterized by ventricular dysfunction and compensatory autonomic changes [3, 4].
So far, heart failure prognosis has been challenged by the limited efficacy of current pharmacological interventions, while organ rejection and small number of donors have imposed drastic limitations to heart transplantation [5]. This scenario has been even worse among women. Accordingly, although nearly 50% of hospital admissions due to heart failure are accounted to women, only few studies have focused on the therapeutic efficacy of treatments targeting heart failure in this population [6]. As a result, it remains questionable whether advances in heart failure therapies can indeed be extended to women. This scenario has stimulated the search for alternative heart failure treatments that might be effective for females as well.
It has been demonstrated that most pathophysiological stimuli recruited over the progression of heart failure can increase the production of reactive oxygen species (ROS) [7, 8]. Cellular antioxidant defence, on the other hand, is down-regulated [9]. This redox imbalance has been shown to impair cardiomyocyte survival and to shift cardiac hypertrophy towards decompensated phenotype in infarcted hearts [10]. Redox imbalance also contributes to deterioration of cardiac hemodynamic properties during heart failure [11, 12]. These evidences have fuelled investigations on the therapeutic efficacy of antioxidant compounds in the treatment of heart failure, including N-acetylcysteine (NAC).
NAC is a sulfhydryl-containing antioxidant compound widely used in the treatment of pulmonary diseases, psychiatric disorders, and paracetamol overdose. Clinical reports have demonstrated the efficacy of NAC therapy in the treatment of ischemic heart disease, with mild side effects [13, 14]. In patients with coronary syndrome, NAC improves antiplatelet activity of nitro-glycerine, and thereby the risk myocardial ischemia by thrombosis or vasospasm is decreased [15]. During myocardial ischemia and reperfusion, NAC administration can improve myocardial antioxidant defence and cardiomyocyte survival, preserving overall cardiac hemodynamic performance [16]. Post-MI administration of NAC has been proven effective to reduce cardiac sympathetic overactivity, arrhythmic episodes, cardiac remodelling, and infarct extension in male rats and dogs [17, 18]. These effects have also been reported in other experimental models of heart failure as well [19].
All these evidences have supported the use of NAC as an alternative therapeutic approach to attenuate heart failure progression, at least in males. However, there has few evidences on whether pathophysiological progression of heart failure can be ameliorated by NAC therapy in females, as far as we know. Here, we investigated the therapeutic efficacy of NAC therapy in the treatment of heart failure induced by acute myocardial infarction in female rats. Cardiac ROS production and antioxidant enzymatic activity were measured to estimate the impact over redox status. The pathophysiological progression of HF was assessed by cardiac morphological, hemodynamic, and autonomic changes.
Methods
Ethics
This study followed the standards and ethical guidelines of the Ethics Committee for Research of the Federal Rural University of Rio de Janeiro, and it was approved by the commission under the number 015/2015. All standards proposed by the Guide for the Care and Use of Laboratory Animals (U.S. National Institute of Health (NIH) Publication No. 85-23, revised 1996) were followed.
Experimental protocol
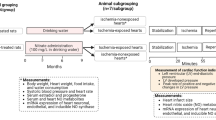
Female Wistar rats (2 months-old, n = 40) were housed in cages under controlled temperature (21 ± 2 °C), daily exposed to 12-h light-dark cycle (lights off at 7:00 pm), and water and standard chow ad libitum. Rats were randomly distributed into myocardial infarcted (MI, n = 20) or sham-operated (Sham, n = 20) groups. After confirmation of myocardial infarction by electrocardiogram (ECG), rats were treated with NAC or placebo starting 24h after surgery, totalling 4 experimental groups: Sham + Placebo (n = 10), Sham + NAC (n = 10), MI + Placebo (n = 10), and MI + NAC (n = 10). NAC was given at 250 mg/kg/day for 28 days after coronary ligation, whereas vehicle consisted of distilled water. This dose has been shown to attenuate heart failure progression post-MI in male rats [20]. Electrocardiographic (ECG) recordings were obtained 28 days post-MI to estimate cardiac autonomic modulation by heart rate variability (HRV). Part of these animals had cardiac function and morphology were assessed by echocardiography 28 days post-MI. After euthanasia, heart and lung were weighted and normalized by tibia length. Heart samples were collected and stored at − 80 °C for further analyses. All animals had their oestrous cycle regularly checked and the experimental protocols were performed when the animals were in diestrus.
Myocardial infarction model
Experimental MI was performed by permanent left coronary artery ligation, as previously described by our group [21, 22]. Animals were anesthetized with isoflurane (Biochimico, 5% for induction, 2% for maintenance). A skin incision was performed in the left parasternal level, and the pectoralis major and minor muscles were dissected to expose the left rib cage. Heart was externalized through a small incision between the fourth and fifth intercostal space. Left coronary artery was quickly ligated close to the level of coronary sinus with a 6-0 silk thread, and myocardial ischemia was confirmed by myocardial pallor. Hearts were returned to its anatomical position, and chest was then closed with continuous silk stitch, and the rats were allowed to recover. Mechanical ventilation was performed when necessary. Sham-surgery followed similar steps to MI surgery, except for coronary ligation. Myocardial infarction was further confirmed by ECG recording 24 h post-surgery. In MI rats, ECG tracings are marked by lack of Q wave at D1 lead. MI rats whose ECG tracings exhibited Q wave were excluded from the study, which totalled 20% of MI group. In addition, MI mortality trans and post-surgery totalled 40%, whereas none died in the SHAM group.
ECG recordings and HRV analysis
ECG recordings were acquired by cutaneous electrodes attached to the precordial levels. All recordings were performed in conscious, freely-moving rats for 10 min, at 1-kHz sampling rate and 12 bits amplitude resolution. Processing of ECG recordings for HRV was performed with MatLab-based algorithms using Kubios HRV 3.0.2 software (Department of Applied Physics, University of Eastern Finland, Kuopio, Finland) [23]. Series of tachograms were generated by detection of R wave peaks throughout all continuous recordings (600 seconds). Tachograms were resampled to equal intervals using cubic interpolation method at 10 Hz, and linear trend was removed for temporal (time domain) and spectral analysis (frequency domain) of HRV. Root mean square of successive differences (RMSSD) was accessed in the time domain. Power spectrum was obtained by Fast Fourier transform (Welch's periodogram: 256 points, 50% overlap and Hamming window). Peak spectral amplitudes were measured in three frequency bands: very low frequency (VLF, 0.02–0.2 Hz), low frequency (LF: 0.2–0.75 Hz), and high frequency (HF: 0.75–3 Hz) [22].
Transthoracic echodopplercardiography
Echocardiography analysis was performed in rats under anaesthesia with isoflurane (5% for induction, 2% for maintenance) with a Vevo 770® High-Resolution Imaging System (VisualSonics, Ontario, Canada) equipped with a 30 mHz electronic phased-array transductor. All analyses were performed blind and by the same operator. M-mode recordings were obtained by short-axis two-dimensional views of the left ventricle at the levels of papillary muscles. Posterior and anterior end-systolic and end-diastolic wall thickness, left ventricular, left atrial, and aortic internal dimensions, and relative wall thickness were assessed. Systolic function was expressed by the fractional shortening and ejection fraction obtained from the M-mode traces. Pulsed-wave Doppler spectra of mitral inflow was recorded from the apical four-chamber view with the guidance of the colour Doppler. All Doppler spectra [mitral flow velocity pattern: peak early diastolic filling velocity (E velocity), peak filling velocity at atrial contraction (A velocity), and their ratio (E/A)] were recorded, and morphological parameter values were measured during the echocardiographic exam. All measurements followed the guidelines of the American Society of Echocardiography and the European Association of Echocardiography Imaging [24].
NOX activity
LV tissues were homogenized in 50 mM sodium phosphate buffer, pH 7.2, 0.25 M sucrose, 0.5 mM dithiothreitol, 1 mM EGTA, supplemented with protease inhibitors: 5 µg/mL aprotinin, and 38.4 mg/mL phenylmethylsulphonyl fluoride (PMSF). Samples were centrifuged at 100 000×g, 35 min, 4 °C, and suspended in 50 mM sodium phosphate buffer, pH 7.2, containing 0.25 M sucrose, 2 mM MgCl2 and protease inhibitors. Protein content was determined by Bradford method [25]. Samples were incubated in 150 mM sodium phosphate buffer, pH 7.4, containing 100 U/mL superoxide dismutase (Sigma), 0.5 U/mL horseradish peroxidase (Roche), 50 μM Amplex red (Molecular Probes) and 1 mM EGTA. Fluorescence was measured in a microplate reader (Victor X5, PerkinElmer) at 30 °C (excitation: 530 nm; emission: 595 nm). Hydrogen peroxide production was determined using standard calibration curves. Specific enzymatic activity was expressed as nmols H2O2 h−1.mg−1.protein.
Antioxidant enzymes activity
LV tissues were homogenized in 100 mM Tris-HCl buffer, pH 7.4, supplemented with protease inhibitors (1 µg/mL aprotinine and 1 mM PMSF). Samples homogenates were centrifuged at 720×g, 4 °C, for 10 min . Protein content was determined by Bradford method [25]. Catalase, superoxide dismutase, and GPX activities were assessed as previously reported [26].
For catalase activity, samples were incubated in 50 mM phosphate buffer, 1 mM EDTA, with 15 mM hydrogen peroxide, pH 7.0. and the absorbance at 240 nm was evaluated by spectrophotometry (Hitachi U-3300) for 60 s. Catalase activity was expressed as mmol.min−1.mL−1.
For SOD activity, samples homogenates were incubated in 50 mM potassium phosphate buffer, 0.1 mM EDTA, 50 µM xanthine, 20 µM cytochrome C, and 10 µM KCN, at 37 °C and pH 7,8. Xanthine oxidase was added at 8 mU to catalyse the production of O2−. Cytochrome C is reduced by O2−, and SOD activity was estimated by measuring the rate of this reaction by spectrophotometry at 550 nm. SOD activity was expressed as U.mg−1 of protein.
For GPX activity, samples homogenates were incubated in 50 mM potassium phosphate buffer pH 7, 1 mM EDTA, 0.24 U/mL glutathione reductase, 0.5 mM GSH, and 0.15 mM nicotinamide adenine dinucleotide phosphate (NADPH). The reaction consists in the oxidation of GSH into GSSH by GPX, with tert-butyl peroxide as a substrate for GPX. GSSH is reduced into GSH by glutathione reductase, which uses NADPH as a co-factor. The reaction was started by addition of 1.2 mM tert-butyl peroxide for 5 min , and NADPH levels were measured by spectrophotometry at 340 nm. GPX activity was expressed as μmol mg–1 min–1.
Reduced thiols levels
Oxidative damage was estimated by cardiac levels of reduced thiols, as previously described [26]. Homogenized LV samples were incubated with 100 µM Dithionitrobenzoic acid (DTNB). Disulphide bonds of DTNB is converted into 2-nitro-5-tiobenzoate (NTB-) by reacting with thiol residues. NTB- is ionized into NTB2- dianion in water and neutral or alkaline pH. NTB2- levels were measure by spectrophotometry at 412 nm absorbance and was expressed as nmol.mg−1.min−1.
Quantitative real time polymerase chain reaction (qRT-PCR)
Messenger RNA (mRNA) expression levels of superoxide dismutase type 1 (SOD1), SOD2, catalase, glutathione peroxidase type 1 (GPX1) and GPX3, nerve growth factor (NGF), β1-adrenergic receptor (β1AR), and muscarinic receptor type 2 (M2R) were measured by qRT-PCR (Table S1). Total RNA was extracted from LV tissue samples using RNeasy® Fibrous Tissue Mini Kit (QIAGEN) and cDNA was prepared from 1 μg of total RNA using High-Capacity Reverse Transcription kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. mRNA levels of target genes (Table S1) were evaluated by qRT-PCR. Amplification reactions containing 1ng of cDNA were performed at 60°C during the annealing and extension cycles. The expression of chosen genes was normalized to GAPDH as an internal control. The quantification of selected mRNA was determined by 2−(ΔΔCT) method in a Viia7 Software v1.2.4 and expressed as fold change of MMZ and T4 group compared to the control group.
Statistical analysis
Data were expressed as mean ± standard error of mean (S.E.M.). Two-way ANOVA followed by Bonferroni post-hoc test was used to compare all parameters. Statistical difference was considered significant when p < 0.05 (Prism, GraphPad software inc.).
Results
Redox imbalance and oxidative damage were ameliorated by NAC therapy
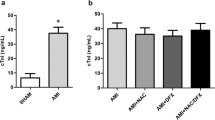
NOX activity was higher in MI + Placebo group compared to Sham + Placebo group (Fig. 1a p < 0.05), whereas it was decreased in MI + NAC versus MI + Placebo group (p = 0.05). As a result, reduced thiols levels were significantly decreased in MI + Placebo group versus Sham + Placebo (Fig. 1b p < 0.01), indicating increased level of oxidative damage, while it was decreased in MI + NAC versus MI + Placebo group (p < 0.05).

Redox imbalance of female infarcted hearts is attenuated by NAC therapy. NOX activity (A) was assessed by spectrofluorimetric assay and the total content of reduced thiols (B) was estimated by spectrophotometry in Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 5/group). *p < 0.05 versus Sham + Placebo, and #p < 0.05 versus MI + Placebo.
Cardiac antioxidant defence was enhanced by NAC therapy
Antioxidant enzymes activities are demonstrated in Fig. 2. No differences were found for SOD (Fig. 2a) and catalase (Fig. 2b) activities (p > 0.05), whereas GPX activity was found to be significantly decreased in MI + Placebo rat hearts when compared to Sham + Placebo group (Fig. 2c p < 0.01). Compared to MI + Placebo, MI + NAC exhibited higher GPX activity (p < 0.001).

GPX activity of infarcted hearts was increased by NAC therapy. SOD (a), catalase (b), and GPX (C) activities were assessed by spectrophotometry in heart samples of Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 5/group). **p < 0.01 versus MI + Placebo group; ##p < 0.01 versus Placebo group
mRNA expression levels of antioxidant enzymes are shown in Fig. 3. MI + Placebo group exhibited higher GPX3 mRNA expression levels (Fig. S1e, p < 0.001) versus Sham + Placebo group. MI + NAC group exhibited decreased GPX3 mRNA expression levels (Fig. S1e, p < 0.05) when compared to MI + Placebo group.

Cardiac remodeling and LV function of infarcted rats were improved by NAC therapy. LVEDV (a), LVPWT (b), LA/Ao (c), E/A waves (d), FS (e), and EF (f) were assessed by echocardiography and were compared between Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 4-5/group). Data are expressed as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001 versus Sham + Placebo group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus MI + Placebo group
Cardiac hypertrophy and pulmonary vascular congestion were prevented by NAC therapy
Heart weight (HW) and lung weight (LW) relative to tibia length (TBL) are demonstrated in Table 1. MI + Placebo group exhibited increased HW/TBL when compared to Sham + Placebo (p < 0.01), consistent with cardiac hypertrophy. This was prevented by NAC therapy, since HW/TBL of MI + NAC group was significantly decreased in comparison to MI + Placebo group (p < 0.05). LW/TBL was significantly increased in MI + Placebo group versus Sham + Placebo group (p < 0.01), which is indicative of pulmonary vascular congestion. In the MI + NAC group, LW/TBL was significantly decreased when compared to MI + Placebo (p < 0.05).
LV contractility and cardiac remodelling of infarcted hearts were ameliorated by NAC therapy
Echocardiographic measures are shown in Fig. 3. Compared to Sham + Placebo group, MI + Placebo group exhibited significantly increased LVEDV (Fig. 3a p < 0.05), a strong trend towards decreased LVPWT (Fig. 3b p = 0.05), and increased LA/Ao (Fig. 3c p < 0.01), characterizing dilation of left cardiac chambers. No significant changes of E/A waves maximal velocity were noted (Fig. 3d p > 0.05). Functional analyses revealed that MI + Placebo group exhibited decreased FS (Fig. 3e p < 0.05) and EF (Fig. 3f p < 0.001) when compared to Sham + Placebo group. Compared with MI + Placebo, MI + NAC group exhibited increased LVPWT (p < 0.001) and EF (p < 0.001), and decreased LA/Ao (p < 0.01).
Heart rhythm and autonomic modulation were restored by NAC therapy
MI + Placebo group exhibited increased HR when compared to Sham + Placebo group (Fig. 4a p < 0.001), whereas it was decreased in MI + NAC rats versus MI + Placebo group (p < 0.001). HRV in time domain revealed that MI + Placebo group exhibited decreased levels of the RMSSD versus Sham + Placebo group (Fig. 4b p < 0.001), consistent with decreased parasympathetic activity, whereas it was significantly increased in MI + NAC versus MI + Placebo group (p < 0.001). In the frequency domain, MI + Placebo group exhibited increased power spectral density in the LF band (Fig. 4c p < 0.05) and decreased HF density (Fig. 4d p < 0.05), resulting in increased LF/HF (Fig. 4e p < 0.001), consistent with sympathetic hyperactivity and decreased parasympathetic activity. MI + NAC group exhibited decreased LF (p < 0.01) and increased HF (p < 0.01) compared with MI + Placebo group, resulting in decreased LF/HF (p < 0.001).

Heart rhythm and cardiac autonomic activity of MI rats were restored by NAC therapy. Mean heart rate (HR, A), root mean square of successive differences (RMSSD, B), low frequency power spectral density (LF, C), high frequency power spectral density (HF, D), and LF/HF (E) were compared between Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 10/group). Data are expressed as mean ± S.E.M. *p < 0.05 and ***p < 0.001 versus Sham + Placebo group; ##p < 0.01 and ###p < 0.001 versus MI + Placebo group
NGF mRNA expression levels of infarcted female hearts were decreased by NAC therapy
mRNA expression levels of β1ADR (Fig. S2a) and M2R (Fig. S2b) were not statistically different among all experimental groups (p > 0.05). Since cardiac NGF levels have been directly associated with sympathetic nerve sprouting and activity after MI, NGF mRNA expression levels were assessed in all experimental groups. MI + Placebo group exhibited significantly higher NGF mRNA expression levels when compared to Sham + Placebo group (Fig. S2c, p < 0.001), and it was partially attenuated by NAC therapy (p = 0.05).
Discussion
The present report provides experimental evidence that chronic administration of NAC can attenuate heart failure pathophysiological progression in myocardial infarcted female rats. NAC therapy attenuated NOX activity and improved antioxidant enzymes activities, resulting in reduced oxidative damage. Cardiac remodelling, LV dysfunction, and pulmonary vascular congestion were overall improved by NAC therapy. Ultimately, autonomic activity of MI rats was restored by NAC therapy, and this was associated with decreased myocardial NGF expression levels, at least in part. As far as we know, the use of NAC therapy in the treatment of MI-induced heart failure has never been investigated in female rats.
Redox imbalance of infarcted female hearts and the effects of chronic NAC therapy
ROS production progressively increase after the onset of ischemia [12, 27]. Accumulating evidences have indicated that NOX is the major source of ROS in several cardiovascular diseases [28]. During the progression towards heart failure, ROS production by NOX can be augmented by angiotensin II, noradrenaline, hemodynamic stress, and pro-inflammatory cytokines [29]. Consistent with this, MI + Placebo rats exhibited higher levels of cardiac hydrogen peroxide when compared to Sham + Placebo group. Overtime, ROS production can reach toxic levels due to several pathophysiological mechanisms triggered after myocardial infarction, contributing to the ventricular remodeling and failure [11, 12]. Indeed, higher NOX activity of MI + Placebo rats was followed by increased oxidative damage, as evidenced by decreased total content of reduced thiols versus Sham + Placebo group. These findings suggest that myocardial infarcted female hearts undergo redox imbalance and oxidative damage during the progression towards HF, as previously demonstrated in male hearts [7, 8].
In keeping with the antioxidant properties of NAC, hydrogen peroxide production by NOX was significantly decreased in MI + NAC hearts in comparison with MI + Placebo rat hearts. Accordingly, the antioxidant effect of NAC involves direct interaction with ROS [30, 31], or up-regulation of cysteine and GSH biosynthesis [9, 32], which has been reported to be depleted in infarcted hearts in agreement with our findings [33, 34]. As a result, MI + NAC hearts exhibited increased content of reduced thiol when compared with MI + Placebo group, indicating that oxidative damage was attenuated by NAC therapy. Although previous evidences have indicated that NAC therapy can either prevent or attenuate cardiac redox imbalance following IR injury, most reports have focused in males [16, 35,36,37]. Our findings indicate that chronic NAC therapy can provide cardiac antioxidant effects in a model of chronic ischemic heart disease, and more importantly, in females.
In natural conditions, ROS levels are kept within physiological ranges by enzymatic and non-enzymatic mechanisms. Intracellular ROS are mostly neutralized by SOD, GPX, and catalase enzymes [38, 39]. In male infarcted hearts, either the expression or activity of these enzymes can be down-regulated, resulting in increased susceptibility to redox imbalance [40, 41]. Interestingly, infarcted female rats exhibited a slight, but not significantly increase in SOD1, catalase, and GPX1 mRNA levels, and significantly increased GPX3 mRNA levels when compared with the sham-operated group. These findings suggest that female hearts exhibit distinct compensatory responses when exposed to the redox imbalance developed during the progression towards HF, particularly involving GPX. Corroborating this hypothesis, SOD and catalase activities were equivalent between Sham + Placebo and MI + Placebo groups. The potential role of estrogen and nuclear factor erythroid-2 in this gender distinction has been previously highlighted [42]. GPX activity, on the other hand, was significantly down-regulated when compared to Sham + Placebo, suggesting that the redox imbalance and oxidative damage of infarcted female rat hearts is likely associated with down-regulation of GPX activity. Importantly, down-regulation of GPX activity is a robust predictor of risk during cardiovascular events, including acute coronary syndrome [43, 44]. When infarcted females were treated with NAC therapy, GPX3 mRNA levels (p < 0.05), and to a lesser extent SOD1, catalase, and GPX1 (p > 0.05) were decreased. In addition, GPX activity was kept within physiological ranges, whereas the effect on catalase activity was insignificant. Taken together, these findings indicate that the compensatory mechanism evoked during redox imbalance in infarcted female hearts are must be triggered by increased ROS levels, for it was prevented by administration of antioxidant therapy. Furthermore, these evidences indicate that the antioxidant effect of NAC therapy was associated, at least in part, with increased GPX activity.
Cardiac dysfunction of infarcted female rats and therapeutic effects of NAC therapy
Since numerous reports have emphasized the role of ROS in the pathophysiological progression of CHF, we investigated whether NAC therapy can provide therapeutic effects over the course of MI-induced HF. As expected, MI + Placebo groups exhibited increased HW/TBL and LVEDV when compared to Sham + Placebo, while LVPWT was decreased and LA/Ao was increased, consistent with cardiac hypertrophy and chamber dilation [45,46,47]. Although primarily compensatory, cardiac hypertrophy itself contributes to progression towards ventricular failure, for dilated ventricles require higher tension to develop pressure levels to the same extent of healthy ventricles, according to Laplace’s law [48]. Unsurprisingly, thus, MI + Placebo rats exhibited decreased levels of EF and FS in comparison with Sham + Placebo group. Furthermore, the increased LW/TBL of MI + Placebo rats is a classic pathognomonic sign of pulmonary vascular congestion, in this case due to LV dysfunction [49]. Future studies will be necessary to address whether NAC therapy prevents infarct expansion in females, an effect that could improve overall post-MI heart function.
As previously reported in male rats [17], NAC therapy attenuated the development of LV dilation and myocardial hypertrophy of infarcted females, evidenced by decreased LVEDV and HW/TBL when compared to MI + Placebo group. In addition, LA/Ao and LW/TBL were decreased in MI + NAC female rats in comparison with MI + Placebo group, indicating that LA dilation and pulmonary vascular congestion were decreased by this therapeutic intervention. Noteworthy, LVPWT was found to be significantly increased in MI + NAC female hearts when compared to MI + Placebo group and, somewhat surprisingly. This finding suggests that cardiac hypertrophy was shifted towards concentric phenotype, instead of dilated phenotype. Wall thinning is a hallmark of transmural myocardial infarction and has been correlated with increased systolic stress and poor prognosis [50]. Interestingly, our findings indicate that overall LV function was ameliorated by NAC therapy, for MI + NAC group exhibited increased EF levels when compared to MI + Placebo, although FS was only modestly increased. Altogether, these evidences reinforce previous reports on the role exerted by ROS signaling during cardiac remodeling following myocardial infarction, particularly in heart failure with decreased EF [51]. They also indicate that infarcted females can also benefit from NAC therapy, in addition to infarcted males.
Given the limited healing capacity of adult mammalian heart, LV dysfunction must be compensated by physiological mechanisms involving, among others, the autonomic regulation of cardiovascular system. Whereas sympathetic activity to the heart increases, parasympathetic activity decreases, contributing to the maintenance of hemodynamic homeostasis [52]. This was evidenced by the increased levels of power spectral density of LF band, the sympathetic index of HRV in the frequency domain, while indexes of parasympathetic activity of time domain (i.e., RMSSD) and frequency domain (HF) were down-regulated in MI + Placebo group. However, autonomic imbalance has also been associated with poor prognosis, and decreased HR variability is an independent predictor of mortality after myocardial infarction [53, 54]. Indeed, cardiac remodeling and arrhythmic events following myocardial infarction have been attributed, at least in part, to the autonomic imbalance [55]. Furthermore, the increased workload imposed by sympathetic overactivity to the failing heart further deteriorates LV hemodynamic performance. This has been supported by the wide benefits of sympatholytic drugs, particularly β-adrenergic receptor blockers [17], and more modestly by vagal stimulation [56], in the treatment of heart failure.
Importantly, the levels of sympathetic activity indexes of MI + NAC female rats were decreased when compared to MI + Sham group, whereas parasympathetic activity was increased, indicating that autonomic balance was kept within physiological ranges by NAC therapy. As a result, HR was kept in the same levels of Sham + Placebo rats. Our findings showing that β1AR and M2R mRNA expression levels were not affected by NAC suggest that reestablishment of autonomic activity involves other mechanisms, such as decreased release of norepinephrine and/or increased vagal activity per se. In agreement with this, previous reports have demonstrated that myocardial norepinephrine levels can be reduced by NAC therapy in myocardial infarcted male rats, while other antioxidant interventions did not promote the same effect despite similar improvements of cardiac function [17].
The mechanisms involved in the sympatholytic effect of NAC therapy remains elusive. Down-regulation of myocardial NGF levels and sympathetic innervation have been recently proposed [17]. NGF is a neurotrophic factor that plays a pivotal role in the development and maintenance of peripheral sympathetic innervation [57]. Cardiac NGF levels are substantially increased following myocardial infarction, particularly throughout infarcted site, as exhibited by MI + Placebo group [58]. Furthermore, NGF infusion has been shown to promote sympathetic nerve sprouting in myocardial infarcted dogs [59]. As a result, cardiac NGF levels can be correlated with density of sympathetic innervation. Noteworthy, we found that myocardial NGF mRNA expression levels were down-regulated by NAC therapy in MI + NAC hearts, as reported in male rat hearts [17]. Accordingly, NGF promoter can be redox regulated due to the cysteine residue within activator protein-1 molecular structure, and thus it might be a potential target of NAC, as previously hypothesized [17, 25, 60]. Furthermore, NGF receptor TrkA signaling cascade can be down-regulated by NAC in several steps that involve inhibition of mitogen-activated protein kinases, such as Ras [54, 55]. Taken together, these evidences suggest that NAC can attenuate sympathetic hyperactivity in myocardial infarcted female rats by down-regulation NGF production and signaling. Alternatively, down-regulation of NGF and thereby improvement of the initially compensatory autonomic imbalance might be as well a result of reestablishment of cardiac function and morphology by NAC.
The present report provides experimental evidence that cardiac antioxidant capacity was enhanced by chronic NAC therapy in myocardial infarcted female rats. This effect was associated with up-regulation of GPX activity. Cardiac remodeling, LV function, and pulmonary vascular congestion were ameliorated by NAC therapy, and these improvements were associated in part with reestablishment of cardiac autonomic regulation and NGF expression levels.
References
Mendis S et al. (2011) Global Atlas on Cardiovascular disease prevention and control. World Health Organization PMID:21884856
Jneid H, Fonarow GC, Cannon CP, Hernandez AF, Palacios IF, Maree AO, Wells Q, Bozkurt B, LaBresh KA, Liang L, Hong Y, Newby LK, Fletcher G, Peterson E, Wexler L (2008) Sex differences in medical care and early death after acute myocardial infarction. Circulation 118:2803–2810. https://doi.org/10.1161/CIRCULATIONAHA.108.789800
Barretto ACP, Santos AC, Munhoz R, Rondon MUPB, Franco FG, Trombetta IC, Roveda F, de Matos LNJ, Braga AMW, Middlekauff HR, Negrão CE (2009) Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 135:302–307. https://doi.org/10.1016/j.ijcard.2008.03.056
Levy D, Kannel KKL, Deedwania PC (1997) The progression from hypertension to heart failure. Am J Hypertens 10:280S-288S. https://doi.org/10.1001/jama.275.20.1557
Lund LH, Khush KK, Cherikh WS, Goldfarb S, Kucheryavaya AY, Levvey BJ, Meiser B, Rossano JW, Chambers DC, Yusen RD, Stehlik J (2017) The registry of the international society for heart and lung transplantation: thirty-fourth adult heart transplantation report—2017; focus theme: allograft ischemic time. J Hear Lung Transplant 36:1037–1046. https://doi.org/10.1016/j.healun.2017.07.019
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics Committee and Stroke Statistics Subcommittee, P (2017) Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation 135:e146–e603. https://doi.org/10.1161/CIR.0000000000000485
Junior RFR, Dabkowskia ER, Shekarb KC, O´Connella KA, Heckera PA, Murphyd MP (2018) MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med 117(2018):18–29. https://doi.org/10.1016/j.freeradbiomed.2018.01.012
Qin F, Simeone M, Patel R (2007) Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic Biol Med 43:271–281. https://doi.org/10.1016/j.freeradbiomed.2007.04.021
Hill MF, Singal PK (1996) Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 148:291–300. https://doi.org/10.1161/01.CIR.96.7.2414
Li W, Kennedy D, Shao Z, Wang X, Kamdar AK, Weber M, Mislick K, Kiefer K, Morales R, Agatisa-Boyle B, Shih DM, Reddy ST, Moravec CS, Tang WHHW (2018) Paraoxonase 2 prevents the development of heart failure. Free Radic Biol Med 121:117–126. https://doi.org/10.1016/j.freeradbiomed.2018.04.583
Brookes PS, Digerness SB, Parks DA, Darley-Usmar V (2002) Mitochondrial function in response to cardiac ischemia-reperfusion after oral treatment with quercetin. Free Radic Biol Med 32:1220–1228. https://doi.org/10.1016/S0891-5849(02)00839-0
Fiorillo C, Becatti M, Pensalfini A, Cecchi C, Lanzilao L, Donzelli G, Nassi N, Giannini L, Borchi E, Nassi P (2008) Curcumin protects cardiac cells against ischemia-reperfusion injury: effects on oxidative stress, NF-κB, and JNK pathways. Free Radic Biol Med 45:839–846. https://doi.org/10.1074/jbc.M113.496570
Ardissino D, Merlini PA, Savonitto S, Demicheli G, Zanini P, Bertocchi F, Falcone C, Ghio S, Marinoni G, Montemartini C, Mussini A (1997) Effect of transdermal nitroglycerin or N-acetylcysteine, or both, in the long-term treatment of unstable angina pectoris. J Am Coll Cardiol 29:941–947. https://doi.org/10.1016/S0735-1097(97)00005-3
Horowitz JD, Henry CA, Syrjanen ML, Louis WJ, Fish D, Smith TW, Antman EM (1988) Combined use of nitroglycerin and N-acetylcysteine in the management of unstable angina pectoris. Circulation 77:787–794. https://doi.org/10.1161/01.CIR.77.4.787
Folts JD, Stamler J, Loscalzo J (1991) Intravenous nitroglycerin infusion inhibits cyclic blood flow responses caused by periodic platelet thrombus formation in stenosed canine coronary arteries. Circulation 83:2122–2127. https://doi.org/10.1161/01.CIR.83.6.2122
Brunet J, Boily MJÉ, Cordeau S, Des Rosiers C (1995) Effects of N-acetylcysteine in the rat heart reperfused after low-flow ischemia: evidence for a direct scavenging of hydroxyl radicals and a nitric oxide-dependent increase in coronary flow. Free Radic Biol Med 19:627–638. https://doi.org/10.1016/0891-5849(95)00077-B
Lee T-MTM, Lai P-YPY, Chang N-CNC (2010) Effect of N-acetylcysteine on sympathetic hyperinnervation in post-infarcted rat hearts. Cardiovasc Res 85:137–146. https://doi.org/10.1093/cvr/cvp286
Šochman J, Kolc J, Vrána M, Fabián J (1990) Cardioprotective effects of N-acetylcysteine: the reduction in the extent of infarction and occurrence of reperfusion arrhythmias in the dog. Int J Cardiol 28:191–196. https://doi.org/10.1016/0167-5273(90)90060-I
Giam B, Chu P.-Y, Kuruppu S, Smith A I, Horlock D, Kiriazis H, Du X.-J, Kaye DM, Rajapakse NW (2016) N‐ acetylcysteine attenuates the development of cardiac fibrosis and remodeling in a mouse model of heart failure. Physiol Rep 4:e12757. https://doi.org/https://doi.org/10.14814/phy2.12757
Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT (1999) Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol 277:H2240-6. https://doi.org/10.1152/ajpheart.1999.277.6.H2240
Griendling KK, Sorescu D, Ushio-Fukai M (2000) NAD (P) H oxidase: role in cardiovascular biology and disease. Circ Res 86:494–501 (PMID: 10720409)
Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171 (PMID: 12821241)
Benrahmoune M, Thérond P, Abedinzadeh Z (2000) The reaction of superoxide radical with N-acetylcysteine. Free Radic Biol Med 29:775–782. https://doi.org/10.1016/S0891-5849(00)00380-4
Aruoma OI, Halliwell B, Hoey BM, Butler J (1989) The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 6:593–597. https://doi.org/10.1016/0891-5849(89)90066-X
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:1–2. https://doi.org/10.1016/0003-2697(76)90527-3
Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA (2007) N-Acetylcysteine-a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7:355–359. https://doi.org/10.1016/j.coph.2007.04.005
Usal A, Acarturk E, Yuregir GT, Unlukurt I, Demirci C, Kurt HI, Birand A (1996) Decreased glutathione levels in acute myocardial infarction. Jpn Hear J 37:177–182. https://doi.org/10.1536/ihj.37.177
Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Iwasaka T (2011) Ascorbic acid and N-acetyl cysteine prevent uncoupling of nitric oxide synthase and increase tolerance to ischemia/reperfusion injury in diabetic rat heart. Free Radic Res 45:1173–1183. https://doi.org/10.3109/10715762.2011.605361
Abe M, Takiguchi Y, Ichimaru S, Tsuchiya K, Wada K (2008) Comparison of the protective effect of N-acetylcysteine by different treatments on rat myocardial ischemia-reperfusion injury. J Pharmacol Sci 106:571–577. https://doi.org/10.1254/jphs.FP0071664
Kamunde C, Sharaf M, MacDonald N (2018) H2O2metabolism in liver and heart mitochondria: low emitting-high scavenging and high emitting-low scavenging systems. Free Radic Biol Med 124:135–148. https://doi.org/10.1016/j.freeradbiomed.2018.05.064
Lu J, Cheng K, Zhang B, Xu H, Cao Y, Guo F, Feng X, Xia Q (2015) Novel mechanisms for superoxide-scavenging activity of human manganese superoxide dismutase determined by the K68 key acetylation site. Free Radic Biol Med 85:114–126. https://doi.org/10.1016/j.freeradbiomed.2015.04.011
Giustarini D, Galvagni F, Dalle Donne I, Milzani A, Severi FM, Santucci A, Rossi R (2018) N-acetylcysteine ethyl ester as GSH enhancer in human primary endothelial cells: a comparative study with other drugs. Free Radic Biol Med 126:202–209. https://doi.org/10.1016/j.freeradbiomed.2018.08.013
Khaper N, Kaur K, Li T, Farahmand F, Singal PK (2003) Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol Cell Biochem 251:9–15. https://doi.org/10.1023/A:1025448908694
Luo T, Liu H, Kim JK (2016) Estrogen protects the female heart from ischemia/reperfusion injury through manganese superoxide dismutase phosphorylation by mitochondrial p38β at threonine 79 and serine 106. PLoS One 11:e0167761. https://doi.org/10.1371/journal.pone.0167761
Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, Smieja M, Cambien F, Meyer J, Lackner KJ (2003) Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med 349:1605–1613. https://doi.org/10.1056/NEJMoa030535
Holley AS, Harding SA, Sasse A, Miller JH, Larsen PD (2016) Reduced glutathione peroxidase activity predicts increased cardiovascular risk following an acute coronary syndrome. Int Cardiovasc Forum J. https://doi.org/https://doi.org/10.17987/icfj.v6i0.241
Olivetti G, Capasso JM, Sonnenblick EH, Anversa P (1990) Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. Circ Res 67:23–34 (PMID: 2364493)
Kono T, Sabbah HN, Rosman H, Alam M, Jafri S, Goldstein S (1992) Left ventricular shape is the primary determinant of functional mitral regurgitation in heart failure. J Am Coll Cardiol 20:1594–1598. https://doi.org/10.1016/0735-1097(92)90455-V
Miranda A, Costa-E-Sousa RH, Werneck-De-Castro JPS, Mattos EC, Olivares EL, Ribeiro VP, Silva MG, Goldenberg RCS, Campos-De-Carvalho AC (2007) Time course of echocardiographic and electrocardiographic parameters in myocardial infarct in rats. An Acad Bras Cienc 79:639–648. https://doi.org/10.1590/S0001-37652007000400006
Grossman W (1980) Cardiac hypertrophy: Useful adaptation or pathologic process? Am J Med 69:576–584. https://doi.org/10.1016/0002-9343(80)90471-4
Joachim H, Riede UN, Mittermayer C (1978) Das Lungengewicht A diagnostisches Kriterium (Abgrenzung der Schocklungen von Normallungen durch histologische, morphometrische and biochemische Untersuchungen). Pathol Res Pract 162:24–40. https://doi.org/10.1016/S0344-0338(78)80129-0
McKay RG, Pfeffer MA, Pasternak RC, Markis JE, Come PC, Nakao S, Alderman JD, Ferguson JJ, Safian RD, Grossman W (1986) Left ventricular remodeling after myocardial infarction: A corollary to infarct expansion. Circulation 74:693–702. https://doi.org/10.1161/01.CIR.74.4.693
Francis J, Weiss RM, Wei SG, Johnson AK, Felder RB (2001) Progression of heart failure after myocardial infarction in the rat. Am J Physiol Integr Comp Physiol 281:R1734–R1745. https://doi.org/10.1152/ajpregu.2001.281.5.R1734
Münzel T, Gori T, Keaney JF, Maack C, Daiber A (2015) Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 36:2555–2564. https://doi.org/10.1093/eurheartj/ehv305
La Rovere MT, Bigger JT, Marcus FI, Mortara A, Schwartz PJ (1998) Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. Lancet 351:478–484. https://doi.org/10.1016/S0140-6736(97)11144-8
Opthof T, Misier ARR, Coronel R, Vermeulen JT, Verberne HJ, Frank RGJ, Moulijn AC, Van Capelle FJL, Janse MJ (1991) Dispersion of refractoriness in canine ventricular myocardium: effects of sympathetic stimulation. Circ Res 68:1204–1215. https://doi.org/10.1161/01.RES.68.5.1204
Vaishnav S, Stevenson R, Marchant B, Lagi K, Ranjadayalan K, Timmis AD (1994) Relation between heart rate variability early after acute myocardial infarction and long-term mortality. Am J Cardiol 73:653–657. https://doi.org/10.1016/0002-9149(94)90928-8
Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH (1996) The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. carvedilol heart failure study group. N Engl J Med 334:1349–55. https://doi.org/10.1056/NEJM199605233342101
Zannad F, De Ferrari GM, Tuinenburg AE, Wright D, Brugada J, Butter C, Klein H, Stolen C, Meyer S, Stein KM, Ramuzat A, Schubert B, Daum D, Neuzil P, Botman C, Caste MA, D’Onofrio A, Solomon SD, Wold N, Ruble SB (2014) Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the neural cardiac therapy for heart failure (NECTAR-HF) randomized controlled trial. Eur Heart J 36:425–433. https://doi.org/10.1093/eurheartj/ehu345
Levi-Montalcini R, Angeletti PU (1963) Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol 7:653–659. https://doi.org/10.1016/0012-1606(63)90149-0
Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS (2004) Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res 95:76–83. https://doi.org/10.1161/01.RES.0000133678.22968.e3
Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, Lai WW, Karagueuzian HS, Wolf PL, Fishbein MC, Chen PS (2000) Nerve sprouting and sudden cardiac death. Circ Res 86:816–821. https://doi.org/10.1161/01.RES.86.7.816
Colangelo AM, Johnson PF, Mocchetti I (1998) β-Adrenergic receptor-induced activation of nerve growth factor gene transcription in rat cerebral cortex involves CCAAT/enhancer-binding protein δ. Proc Natl Acad Sci 95:10920–10925. https://doi.org/10.1073/pnas.95.18.10920
Vinson C, Sigler P, McKnight S, Curran T (1989) Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science 246:911–916. https://doi.org/10.1126/science.2683088
Kamata H, Oka SI, Shibukawa Y, Kakuta J, Hirata H (2005) Redox regulation of nerve growth factor-induced neuronal differentiation of PC12 cells through modulation of the nerve growth factor receptor, TrkA. Arch Biochem Biophys 434:16–25. https://doi.org/10.1016/j.abb.2004.07.036
Olivares EL, Costa-e-Sousa RH, Werneck-de-Castro JPS, Pinho-Ribeiro V, Silva MG, Ribeiro KC, Mattos EC, Goldenberg RCS, Campos de Carvalho AC, Masuda MO (2007) Cellular cardiomyoplasty in large myocardial infarction: can the beneficial effect be enhanced by ACE-inhibitor therapy? Eur J Heart Fail 9:558–567. https://doi.org/10.1016/j.ejheart.2007.02.003
Souza N, Dos-Santos R, da Silveira ALB, Gantus MAV, Fortes F, Olivares EL (2016) Effects of autonomic balance and fluid and electrolyte changes on cardiac function in infarcted rats: a serial study of sexual dimorphism. Clin Exp Pharmacol Physiol 43:476–483. https://doi.org/10.1111/1440-1681.12543
Tarvainen MP, Niskanen JP, Lipponen JA, Ranta-aho PO, Karjalainen PA (2014) Kubios HRV - Heart rate variability analysis software. Comput Methods Programs Biomed 113:210–220. https://doi.org/10.1016/j.cmpb.2013.07.024
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt J-U (2015) afbeelding 5. J Am Soc Echocardiogr 28:1-39.e14. https://doi.org/10.1016/j.echo.2014.10.003
Frankenfeld SSP, de Oliveira L, Ortenzi VH, Rego-Monteiro ICC, Chaves EEA, Ferreira AC, Leitão AC, Carvalho DP, Fortunato RSR (2014) The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male wistar rats. PLoS One 9:e102699. https://doi.org/10.1371/journal.pone.0102699
Acknowledgements
This study was supported by the Department of Science and Technology - Brazilian Ministry of Health (DECIT/SCTIE/MS), the Brazilian Council for Scientific and Technological Development (CNPq), the Rio de Janeiro State Research Foundation (FAPERJ), the Coordination for the Improvement of Higher Education Personnel (CAPES).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2020_5907_MOESM1_ESM.tif
Fig. S1 Antioxidant enzymes mRNA expression levels were restored by NAC therapy in infarcted hearts. mRNA expression levels of SOD1 (a), SOD2 (b), catalase (c), GPX1 (d), and GPX3 (e) were compared between Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 5-7/group). Data are expressed as mean ± S.E.M. ***p < 0.001 versus Sham + Placebo group; #p < 0.05 versus MI + Placebo group (TIF 102 kb)
11033_2020_5907_MOESM2_ESM.tif
Fig. S2 NGF mRNA expression levels were attenuated by NAC therapy. mRNA expression levels of β1-adrenergic receptor (a), type 2 muscarinic receptor (b), and NGF (c) were compared between Sham-operated (white bars) and MI rats (black bars) treated with placebo or NAC therapy (n = 6/group). ***p < 0.001 versus Sham + Placebo group (TIF 95 kb)
Rights and permissions
About this article

Cite this article
Costa, C.R.M., Seara, F.A.C., Peixoto, M.S. et al. Progression of heart failure is attenuated by antioxidant therapy with N-acetylcysteine in myocardial infarcted female rats. Mol Biol Rep 47, 8645–8656 (2020). https://doi.org/10.1007/s11033-020-05907-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05907-4