
Abstract
Heart failure (HF) is characterized as a limitation to cardiac output that prevents the heart from supplying tissues with adequate oxygen and predisposes individuals to pulmonary edema. Impaired cardiac function is secondary to either decreased contractility reducing ejection (systolic failure), diminished ventricular compliance preventing filling (diastolic failure), or both. To study HF etiology, many different techniques have been developed to elicit this condition in experimental animals, with varying degrees of success. Among rats, surgically induced HF models are the most prevalent, but they bear several shortcomings, including high mortality rates and limited recapitulation of the pathophysiology, etiology, and progression of human HF. Alternatively, a number of non-invasive HF induction methods avoid many of these pitfalls, and their merits in technical simplicity, reliability, survivability, and comparability to the pathophysiologic and pathogenic characteristics of HF are reviewed herein. In particular, this review focuses on the primary pathogenic mechanisms common to genetic strains (spontaneously hypertensive and spontaneously hypertensive heart failure), pharmacological models of toxic cardiomyopathy (doxorubicin and isoproterenol), and dietary salt models, all of which have been shown to induce left ventricular HF in the rat. Additional non-invasive techniques that may potentially enable the development of new HF models are also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is a global public health problem imposing considerable disease and economic burdens on individuals and societies. Cardiac disease in general leads to more hospitalizations (4.4 million annually) and deaths (a third of annual fatalities) in the United States than any other single cause [1, 2]. Heart failure (HF) is the end-phase syndrome of chronic cardiac disease that occurs when the left ventricle becomes too weak to provide adequate tissue perfusion [3, 4]. According to the U.S. Centers for Disease Control, HF specifically causes more hospitalizations (1.1 million annually) than any single type of cardiac disease [1]. Roughly 6 million Americans live with HF, and hospital discharges for HF, including deaths, nearly tripled from 1979 to 2005 [3, 5]. Because HF is difficult for practitioners to define, and its symptomology is exceptionally variable [4], currently reported morbidity and mortality rates may grossly underestimate the impact of HF due to the difficulty of classifying the disease [6].
The staggering health and economic burdens imposed by HF (60,000 deaths and $37 billion annually in the United States [5]) demand an improved understanding of how potential therapies and toxins may specifically affect this syndrome. Investigations into the mechanisms of exacerbation and treatment of HF are often limited by the inability of animal models to reliably mimic the pathogenesis of human HF. Several left ventricular HF models have been developed in rats using surgical, pharmacological, dietary, and genetic manipulations, all with notable shortcomings and advantages. In this review, several of the surgically induced models are described briefly, but primarily as counterpoints to the main emphasis on non-invasively induced models. In order to focus on a few major non-invasive models and expose lesser known emerging models, this review omits several models reviewed previously, including those induced by myocarditis, alcohol, streptozotocin, and hyperhomocysteinemia, as well as models of right ventricular heart failure (cor pulmonale) [7].
In particular, the merits of genetic strains (spontaneously hypertensive and spontaneously hypertensive heart failure), pharmacological models of toxic cardiomyopathy (doxorubicin and isoproterenol), and dietary salt models in the rat are described in detail. Additionally, less well-established non-invasive techniques that may enable the development of new HF models are also described.
Pathogenesis
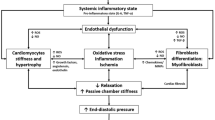
The pathogenesis of left ventricular HF has been reviewed extensively [8–11]. Briefly, the hallmark of HF is the heart’s inability to provide adequate tissue perfusion [4]. This state of cardiac insufficiency usually results after an initial “index event” (e.g., acute myocardial infarct, gradual onset of systemic hypertension, myocardial inflammation, valvular insufficiency, a genetic mutation, coronary heart disease, diabetes mellitus, and/or cardiomyopathy) alters ventricular wall stress at end-diastole (preload) and/or at ejection (afterload), eventually impairing cardiac output [10]. The decline in cardiac function activates the release of neurohormones (e.g., catecholamines, angiotensin II, or endothelin) and cytokines, increasing water retention and cardiac workload in an effort to preserve blood pressure and kidney perfusion [10]. If persistent, the neurohormonal response may sequentially elicit the myocardial fetal gene program, cardiomyocyte hypertrophy, structural remodeling of the heart, cardiac dilatation, cardiac insufficiency, and circulatory congestion (Fig. 1) [3, 8, 10–12]. As well, the type of index event often dictates the course and features of HF pathogenesis. For instance, aortic regurgitation and ventricular septal defect stimulate water retention to increase blood volume (hypervolemia), augmenting stroke volume through increased preload, ultimately inducing ventricular volume overload and cardiac dilatation but typically not ventricular wall thickening. Conversely, unlike physiologic hypertrophy, the innocuous and temporary consequence of physical exertion, prolonged pathologic cardiac hypertrophy is an adaptation to increased afterload that is characterized by several features of cardiac remodeling, including increased cardiomyocyte size, protein synthesis, and sarcomere number and organization [10–12]. With cardiomyocyte hypertrophy, left ventricular (LV) volume usually decreases with the thickening of ventricular walls; however, chamber dilatation can suppress these two gross features despite severe cardiomyocyte hypertrophy [13]. Ultimately, decompensation ensues when LV dilatation (indicated by increased chamber volume, and the thinning, weakening, and fibrosis of the LV wall) and neurohormonal compensatory mechanisms fail to maintain sufficient cardiac output for the body’s needs [8, 11, 14]. At or near decompensation, the myocardium becomes ATP deficient due to impaired ATP generation. Consequently, metabolic function often shifts from fatty acid oxidation to glucose utilization within the myocardium of rodent HF models; however, fatty acid uptake remains unchanged within the failing human heart [15]. Notably, other important pathophysiological differences in HF progression exist between rodent models and humans, including a shift from α-myosin heavy chain (MHC) protein expression to βMHC in the rodent myocardium and the opposite response in humans. Although congestion (systemic or pulmonary) is not required for HF, it frequently occurs along with dyspnea (labored breathing) at rest or upon mild physical exertion. Other HF symptoms can include lethargy, dizziness, angina pectoris, weight loss and/or gain, swelling of the limbs, syncope (fainting), and cyanosis. The modes of fatality in HF include sudden death (usually from arrhythmia), congestion (especially pulmonary), pump failure, insufficient perfusion of the heart (ischemia and infarction), and inadequate cardiac output [16].

Development and progression of decompensated left ventricular hypertrophy. Compensated left ventricular hypertrophy manifests as a decrease in chamber volume and an increase in LV wall thickness characterized by cardiomyocyte growth in response to hemodynamic stress and/or myocardial injury. Neurohormonal and cytokine activation induces development of LV hypertrophy, often with enhancements in LV contractile performance maintaining cardiac output despite decreased chamber volume. Cardiomyocyte death provokes transition to cardiomyopathic dilation and wall thinning, corresponding with decreased LV contractile performance. Along with a host of molecular changes, these events are termed ‘decompensation’ and usually confer a decline in cardiac output
Functional, Structural, and Hemodynamic Indicators
Although this review focuses especially on non-invasive HF models, it must be noted that confirming HF in even these models often requires invasive techniques such as implantable telemetry or ventricular catheterization to obtain arterial and cardiac pressures as well as ECG measurements. Conversely, echocardiography provides a non-invasive means for confirming HF. Heart failure is typically verified by changes in cardiac performance, dimension, and pressure. When the heart’s compensatory mechanisms fail and decompensation ensues, LV end-diastolic volume and pressure both increase, while LV end-systolic pressure (LVESP) and cardiomyocyte contractile performance usually decline [10, 11, 17]. Elevated LV end-diastolic pressure (LVEDP) impedes chamber filling and reduces stroke volume (SV), thereby serving as a major indicator of HF—particularly, diastolic failure [18–21]. Four-fifths of humans with HF symptoms have diastolic failure, while less than half have systolic failure [5]. In these instances, elevations in the mitral valve ratio of early-to-late inflow velocities (E/A, an echocardiographic parameter) parallels elevated LVEDP to indicate diastolic dysfunction and failure [22]. Although susceptible to preload and afterload conditions in general, the peak rate of increase and decrease in LV pressure (LV dP/dt max and dP/dt min) represents contractile and diastolic performance, respectively [9]. Additionally, significant declines in fractional shortening (FS) and ejection fraction (EF)—both expressed in percentage values—indicate systolic dysfunction; progressive decreases are prognostic of decompensation and death from HF [23]. For instance, in a large human cohort study, every incremental 5% decrease in EF below 45% corresponded with a 15% increase in risk of death within 2 years [24]. EF is estimated from the difference in LV volume between systole and diastole (stroke volume) as a fraction of diastolic volume. LV volumes typically are calculated from ultrasound measurements of LV dimension, using well-established geometric assumptions of LV shape. In healthy experimental animals, EF typically ranges from 45% to 70%, depending on anesthesia, heart rate, and the procedures used [9]. Notably, anesthetics differentially affect cardiac output and hemodynamics as well. A comparison on the effects of isoflurane, urethane, sodium pentobarbital, and ketamine–xylazine in mice demonstrated that—in ascending order and with many significant differences between agents—all depressed mean arterial pressure and cardiac output relative to conscious mice [25].
Invasive Models of HF in the Rat
Models Requiring Thoracotomy
A discussion of the limitations and merits of surgical models of heart failure is required to convey the relative advantages of non-invasively induced models of heart failure. Surgical induction of HF often requires opening the thoracic cavity. One example of such a procedure is coronary artery ligation, which causes myocardial ischemia and subsequent necrosis that decreases the number of viable cardiomyocytes, impairs electrical conduction, and results in HF accompanied by structural remodeling of the lung by 2 weeks post-surgery [26–28]. Following a hypervolemic response to decreased contractility and stroke volume, elevated preload is primarily responsible for the progression to HF in this model, which significantly and dramatically impairs both systolic and diastolic function [19, 26–28]. In one study, diastolic function was significantly impaired at 2 weeks post-surgery, but systolic function (dP/dt max) was enhanced—likely due to a neurohormonal augmentation of contractility (Table 1) [28]. In another study, cardiac performance data and gross clinical signs of HF indicated that systolic and diastolic failure were achieved concomitantly at 5 weeks post-surgery [19]. The authors used a threshold LVEDP value to delineate ligated rats as either “non-failing” (LVEDP < 15 mmHg) and “failing” (LVEDP ≥ 15 mmHg). They demonstrated that when MI led to LVEDP ≥ 15 mmHg (62% of ligated rats), multiple indicators of decompensated HF were evident, including pleural effusion, ascites, tachypnea, increased diameters of the left atrium and LV at end diastole, and reductions in weight gain, heart rate, cardiac output (CO), fractional shortening, and contractility (LV dP/dt max) [19]. Nevertheless, the average cardiac performance parameters for all ligated rats (regardless of LVEDP) still differed dramatically from rats with sham surgery. Survival was not reported in this study, but other investigators demonstrated similar cardiac dysfunction at 9 weeks post-surgery with only 10% premature mortality [29]. An additional study lacking cardiac performance data achieved even lower mortality rates (<5%) by specifically ligating the LAD 1–2 mm below the junction of the pulmonary conus and the left atrial appendage [30]. Despite the low mortality rates reported previously, coronary artery ligation in rats demands surgical expertise and precision that, if lacking, can rapidly lead to premature mortality of 30–62% [27, 30]. Furthermore, all coronary artery ligation procedures require invasive and potentially traumatic thoracotomy.
A second invasive model involves constrictive banding or suturing of the thoracic aorta (often called “transverse aortic constriction” [TAC] or aortic stenosis), which induces myocardial “pressure overload” and leads to heart failure after about 15–27 weeks, resulting in about 30% premature mortality (Table 1) [31–36]. Initially, dramatic increases in afterload cause hypertrophy and thickening of the posterior wall and interventricular septum, as well as increased neurohormones [37]. Aortic constriction impairs cardiac performance in part by causing dysfunction of the 2a isoform of the sarcoplasmic reticulum Ca2+ ATPase pump (SERCA2a)—a pathology common to HF [33, 38–40]. However, thoracic aortic stenosis, like coronary artery ligation, can carry high mortality rates and typically requires surgical opening of the thoracic cavity (sternotomy or thoracotomy), which may elicit chronic pain, local and systemic inflammation, epicardial inflammatory cell infiltration, fibrous adhesions, and extensive thoracic scarring, and may thus interfere with experimentation long after surgery [39, 41, 42].
Invasive Models Not Requiring Thoracotomy
Importantly, del Monte and colleagues have developed a less invasive technique of transverse aortic constriction that circumvents thoracotomy via a suprasternal incision and the cutting of a clavicle to access the aorta equally as proximal to the heart as methods requiring a thoracotomy [39, 43]. Only a limited number of studies have successfully applied the supraclavicular approach in rats to elicit cardiac dilation and dysfunction comparable to the models requiring thoracotomy or sternotomy (Table 1) [33, 39, 43, 44]. A similar technique requiring a suprasternal notch has been developed for the mouse and is used much more commonly [45, 46]. Abdominal aortic constriction provides an additional alternative approach to stenotic-increased afterload that is less demanding of surgical precision, requires a less traumatic and invasive surgery (laparotomy does not require mechanically assisted ventilation, pneumothorax, or cutting of bone or cartilage), and eventually leads to HF. At 4–5 weeks after this surgery, researchers have observed significant evidence of cardiac hypertrophy with marked increases in LV wall thicknesses but no significant change in EF or FS [37, 47]. In contrast, Kobayashi and colleagues witnessed significantly enhanced LV systolic function and pressure at this same time-point with no impairment in diastolic pressure [48]. Importantly, these values paralleled those of thoracic aortic constricted rats in both time and magnitude, suggesting strong homology between thoracic and abdominal aortic stenosis in the early compensatory stages of HF progression. In another study, Wistar rats were shown at 7 weeks post-surgery to have strong indications of systolic failure, including significantly decreased ejection fraction and sharply increased end-systolic volume (Table 1) [49]. Less dramatic effects were observed at 8 weeks post-surgery in another study using SD rats [37]. Others have noted that 8 weeks of abdominal aortic constriction caused 15% mortality, increased systolic pressure, and did not alter LV dP/dt max relative to control [50, 51]. Declines in FS, marked myocardial fibrosis, and pulmonary edema have been demonstrated at 20 weeks post-surgery in 44% of the rats surviving abdominal aortic constriction; however, survival rate was not reported [52]. At 24 weeks post-surgery, investigators in another study observed significantly increased LVEDP and depressed systolic and diastolic performance with no significant pulmonary edema, but survival data were not reported [53]. Others observed that 28 weeks of abdominal aortic banding failed to induce significant pulmonary congestion or ascites [37].
Pressure overload, resulting from chronically elevated afterload, causes concentric hypertrophy, with thickening of the posterior and septal walls through addition of sarcomeres in parallel [37]. Because 75% of HF cases in the United States have antecedent hypertension [3], the pressure overload-induced model of aortic constriction simulates the most common natural pathogenesis to HF; nevertheless, it remains limited by premature mortality, a 6- to 7-month delay for HF onset, difficult surgical procedures, and complications from invasive surgeries.
Additional surgical models of heart failure in the rat that do not require thoracotomy and pericardiectomy include rapid ventricular pacing, aortocaval shunt (sometimes termed ‘fistula’), arteriovenous fistula, and induction of mitral valve regurgitation [54]. In contrast to pressure overload, rat models of volume overload hypertrophy simulate the cardiac effects of anemia, heart block, atrial or ventricular septal defects, mitral or aortic valve regurgitation, or other congenital diseases that lead to dramatic elevations in preload. Volume overload causes eccentric hypertrophy (myocyte elongation by series replication of sarcomeres), with LV dilatation [37]. Aortocaval shunt (or fistula) has been used in rats to induce volume overload. A syringe needle (often 18-gauge) is used to create a puncture that links the aorta and vena cava, leading to the death of roughly 10% of male rats at surgery, 25% by 8 weeks, a highly variable fraction (13–60%) by 16 weeks, and up to 80% by 21 weeks post-surgery [37, 55–58]. At least two groups have demonstrated that at 4–8 weeks after shunt surgery, LVEDP elevates significantly and peaks at 16-24 weeks post-surgery [53, 55]. In contrast, others have noted peak LVEDP elevations at 3 weeks, with lesser elevations at 5 and 8 weeks [59]. A handful of studies have noted pulmonary edema, lethargy, pitted edema, pleural effusion, and/or ascites in a large portion of rats after anywhere from 0–32 weeks of shunt surgery [37, 53, 55, 56, 59]. Despite the effective induction of HF in rats via aortocaval shunt, the variable timing of both HF onset and subsequent death limits the utility of this model [37, 56].
Arteriovenous fistula is similar to aortacaval shunt, but instead of a puncture between the vena cava and the aorta, the fistula is made in the wall between the carotid artery and jugular vein [54]; however, this application has also been mostly limited to large animals such as dogs, goats, and sheep. Rapid ventricular pacing via chronic electrode stimulation of the LV for 4–6 weeks increases heart rate to elicit HF by “cardiac overdrive” with minimal premature mortality [60–63]. Although this model facilitates the study of the sequential progression of HF over several weeks, it fails to induce myocardial ischemia and hypertrophy, and animals tend to recover from treatment with a reversal of the induced dilated cardiomyopathy [64, 65]. Moreover, the model has been limited to larger animals such as dogs, sheep, and rabbits and is thus likely not suited for rats.
Surgical models bear several advantages, such as dramatically impaired cardiac function (particularly for TAC and coronary ligation) through the simulation or induction of key stimuli of HF progression (i.e., hypertension-induced increases in afterload, myocardial infarction–induced increases in preload). Ultimately, however, surgical models of HF present several challenges to laboratory research, including long-term complications from highly invasive surgeries, difficult surgeries requiring expertise, high premature mortality, ethical concerns, and variable timing in HF onset within treatment groups.
Non-Invasive Models of HF in the Rat
Genetic Models
Selectively bred and genetically engineered rodent models of HF often replicate the natural etiologies of and progression to HF while avoiding potentially confounding surgical procedures and toxic treatments. Several genetically engineered mouse strains have been developed that closely mimic specific aspects of HF, often through altered protein expression [66–68]. Among rats, however, genetically engineered HF models have scarcely been researched [69, 70], while selectively bred HF-prone rat strains have been extensively investigated and incorporated into toxicologic and therapeutic HF research. Chief among these are the spontaneously hypertensive (SH) and spontaneously hypertensive heart failure (SHHF) rat strains, which mimic several key aspects of human HF pathogenesis while presenting fewer challenges to research than invasive models.
Derived from the inbreeding of hypertensive Wistar Kyoto rats, SH rats (SHRs) are predisposed to systemic hypertension, concentric hypertrophy, and thus afterload-driven HF. At 11 and 27 weeks of age, unrestrained unanesthetized male SHRs have a 24-h mean arterial pressure (MAP) of 135 and 150 mmHg, respectively, in contrast to 100 and 105 mmHg for the normal WKY at these two ages [71–73]. At 18–24 months of age, 57% of male SHRs have been shown to progress from hypertension-induced compensated hypertrophy to decompensated HF, with 13% surviving without HF and 30% dying or euthanized due to non-cardiac reasons (e.g., stroke, tumor, or debilitation) [74]. Several features common to human HF emerge during the SHR’s transition to decompensated HF, including marked increases in myocardial non-cross-linked collagen, fibronectin mRNA, LV necrosis, LV fibrosis, and βMHC protein expression, as well as the complete loss of αMHC [75, 76]. The transition to decompensation corresponds with tachypnea and shallow rapid breathing as well as profoundly decreased EF, heart rate, and LVESP, and increased LVEDP, end-diastolic and end-systolic volumes relative to age-matched non-failing SHRs (Table 2) [74]. Within 1 week of onset of labored breathing and large drops in echocardiographic measures, most of these rats have pericardial/pleural effusions, atrial thrombi, and right ventricular hypertrophy [74, 75]. In one study, systolic failure was less demonstrable in male SHRs than diastolic failure. At 20 months of age, these rats lacked declines in FS or EF despite a doubling of E/A ratio, 27% fibrosis, pleural or pericardial effusions, and thickening of the posterior wall and interventricular septum relative to 12-month-old SHRs [77].
The SHHF/Mcc-fa cp strain originates from the seventh backcross of the normal SHR with “Koletsky obese” rats (inbred from the hypertensive offspring of a SD/SH cross [78, 79]). SHHF rats possess characteristics similar to the SHR, except 100% eventually acquire dilated cardiomyopathy and HF preceded by Type II diabetes mellitus and consequent diabetic nephropathy accentuated in obese and male rats [79, 80]. The SHHF’s additional pathology is attributed to a nonsense mutation, fa, which encodes a premature stop codon in the leptin receptor [80, 81]. “Lean” and “obese” SHHF rats differ in disease severity and progression primarily by their responsiveness to leptin—a hormone released upon eating that inhibits appetite, provokes a sense of satiation, stimulates the sympathetic nervous system, and increases energy expenditure in a receptor-dependent manner [82]. The autosomal recessive corpulence trait (cp) manifests as obesity in rats homozygous for the fa mutation (fa cp/fa cp), while homozygous wild-type (+/+) or heterozygous (+/fa cp) SHHFs are lean [81, 83, 84]. Among lean males, heterozygotes develop congestive HF and die sooner than the homozygous wild types [83, 85]. Heterozygosity confers mild hyperleptinemia and insulin resistance, with marked effects in homozygous mutant (fa cp/fa cp) rats [83, 86]. Notably, leptin administration has been shown to induce eccentric dilatation of the left ventricle [87], while leptin receptor polymorphisms and circulating leptin associate with human HF [88]. In contrast to 10- to 12-month-old SHRs with concentric hypertrophy, age-matched lean male SHHF rats develop eccentric hypertrophy and lack ventricular wall thickening [89]. Although leptin stimulates sympathetic nerve activity and may increase arterial pressure, leptin-induced sympathetic excitation is absent in the obese phenotype of another rat strain (Zucker) homozygous for the same mutated leptin receptor gene [82]. Thus, heart failure in the SHHF is not entirely a result of hypertension-induced increases in afterload and may result partly from preload-driven volume overload. Unanesthetized, unrestrained lean male SHHF rats have hypertension exceeding the SHR (24-h MAP: 161 mmHg at 10 weeks and 145 mmHg at 15 weeks) [90, 91], while several studies suggest that unanesthetized, unrestrained obese male SHHFs have less severe hypertension (MAP: 119 mmHg at 18–26 weeks, 133 mmHg at 40 weeks, and 127 mmHg at 54 weeks) [92, 93]. Some studies have reported higher systolic pressure in the obese relative to the lean; however, pressure measurements in these studies use anesthesia or restraint (e.g., tail-cuff), which may differentially affect the two phenotypes [81, 83, 85].
SHHFs express LV hypertrophy at 3 months regardless of gender or obesity. Decompensated HF with gross symptoms occurs at 10–13 months in obese males [85, 92, 94], 15 months in obese females [95], 18 months in lean males [96–100], and 24 months in lean females [85, 98, 101, 102]. The overt signs of decompensated HF found in the SHHF often include subcutaneous edema, tachypnea and shallow rapid breathing, cold tails, cyanosis, lethargy, piloerection, pulmonary edema, pleural effusion, ascites, cardiomegaly, left and right atrial dilatation, and hepatomegaly [85]. Death typically occurs at 18 months in obese females [103] and 19 months in lean males [86, 96] as HF severity increases with decompensation. Although HF onset is more rapid in obese SHHFs, lean males compare well to the hypertrophic qualities of obese SHHFs and also compare closely to human HF pathogenesis [81]. In 18- to 20-month-old lean male SHHFs, a reduction in αMHC and β-adrenergic receptor (βAR) density as well as increases in ventilatory rate (>200 breaths/min), βMHC, circulating TNF-α, IL-6, natriuretic peptides, and leptin suggests a profound comparability between HF pathogenesis in SHHF rats and humans [86, 96, 97, 104]. Furthermore, exceptional homology has been demonstrated between lean and obese male SHHFs in increases in neurohormonal, apoptotic, fibrotic, inflammatory, metabolic, hypertrophic, and structural gene expression at 10 months relative to 4 months of age [81]. Thus, the pathogenesis of HF in lean and obese males is strikingly similar with exception to rate of progression.
Among SHHFs, the lean male has been the most thoroughly studied for cardiac dysfunction. Yet, timing of systolic and diastolic dysfunction has been inconsistent between several of these studies (Table 2) [85, 96, 97]. The variability in observations may stem from different anesthetics used during physiologic measurements; however, similar variability has not been observed in studies using other models and different anesthetics. Female SHHFs differ from males in timing to progression and gross signs of HF. A few studies have noted the absence of several common HF traits in 24-month-old lean female SHHFs [98, 101] despite marked declines in LV systolic and diastolic performance and significant cardiomegaly [98, 102] (Table 2).
In addition to the SHR and SHHF, normotensive male Fischer 344 (F344) rats at 25-29 months acquire the symptoms, myocardial histopathology, and cardiac dysfunction inherent to HF [105, 106]. Notably, investigators observed 50% mortality among F344 rats between 24 and 26 months of age [105]. The F344 strain is best suited as a model of aging-related HF independent of hypertension. Given that 75% of HF cases have antecedent hypertension [3], the etiology of HF in the normotensive F344 strain may render it less pathophysiologically relevant to the human condition than the SH and SHHF strains. Furthermore, the F344’s duration to HF onset is highly variable and prolonged relative to the SHHF and SH strains. Nevertheless, with improved health care management of hypertension in society, age-related HF that is independent of hypertension may increase in prevalence and thus become more relevant. Additional strains with potential as HF models exist, including the Zucker diabetic fatty (ZDF) rat, which acquires diabetes- and obesity-associated decrements in cardiac function; nevertheless, some common traits of HF, including hypertension and decreased SERCA2a, are not seen in this strain [107].
The utility of the SHR and SHHF strains extends beyond their abilities to mimic the etiology of cardiac failure in humans. These strains have minimal premature mortality and, along with other strain-based models, lack the major side effects common to surgical, pharmacologic, or dietary HF models. Moreover, the steady progression to HF in all three aforementioned strains (1–2.25 years in SHR, SHHF, and F344 rats) enables long-term therapeutic treatments or chronic toxic exposures that are more comparable to human conditions. Yet, in contrast with many other models, the relatively slow progression to HF and often variable time to HF onset in these strains demands extensive time, husbandry, and resources. Finally, the incorporation of other methods of inducing hypertrophy or HF in the SH, SHHF, and F344 strains may expedite their otherwise slow progression to HF at the risk of compromising their pathophysiologic relevance.
Pharmacological Models
Models of pharmacologically induced cardiomyopathy and ensuing HF often involve simpler, easier, and less traumatic methods than surgically invasive models of HF. Doxorubicin (DOX) and isoproterenol (ISO; either acute or subchronic) have been shown to significantly impair cardiac function in rats to the point of HF. Other agents with promise for yielding models of HF through chronic or subchronic administration include propylthiouracil, angiotensin II, and TNF-α.
Doxorubicin. Doxorubicin (DOX; adriamycin) is an anthracycline antibiotic and antineoplastic agent used in cancer chemotherapy that has been shown to induce free radical formation and extensive lipid peroxidation leading to myocardial inflammation, morphological disorganization of myofibrils, necrosis, apoptosis, interstitial fibrosis, and cardiac dilatation and failure that is both progressive and irreversible [54, 64, 108–115]. Assisted by the enzyme NADPH cytochrome P450 reductase, the redox cycling of the quinone structure of DOX and semiquinone generates electrons that interact with molecular oxygen to produce reactive oxygen species (ROS) [110, 116]. Free radicals are also generated non-enzymatically when DOX reacts with either iron or nitric oxide [110]. Although DOX elicits cardiotoxicity primarily through oxidative stress, especially in mitochondria [111], it causes additional cardiac injury by inducing proteolysis through activation of matrix metalloproteinases [110] as well as hyperactivation of the ubiquitin–proteasome system (UPS) [117, 118]. Despite that UPS hyperactivation and oxidative stress promote hypertrophy, evidence that DOX elicits hypertrophy remains scarce [119]. Instead, DNA lesions in cardiomyocytes, cardiomyocyte apoptosis, intracellular Ca2+ overload via SERCA2a dysfunction, depletion of endogenous antioxidants, and disruption of mitochondrial structure and bioenergetic metabolism are all believed to contribute to DOX-induced HF [110, 115, 120].
The severity of DOX-induced HF in both patients and experimental animals is highly dependent upon the cumulative dose [110]. In humans, the likelihood of developing HF with bolus intravenous DOX delivered once every 3 weeks was 3% after a cumulative dose of 430 mg/m2, 7% after 575 mg/m2, and 21% after 728 mg/m2, equivalent to 12, 16, and 20.25 mg/kg cumulative doses in the rat, respectively [121, 122]. In experimental models, serial daily or weekly doses of DOX may elicit HF in a more gradual and survivable manner despite similar decrements in systolic function relative to a single bolus dose [123]. Liu and colleagues administered 3.3 mg/kg DOX per week intravenously to SD rats for 4 weeks to cause major declines in LV systolic and diastolic performance and 14% mortality at completion of the regimen (Table 3) [113]. Among the DOX-treated rats that survived the 4-wk regimen, half of them died over the following 4 weeks. Slightly lower doses may have dramatically improved survival; no deaths were observed following this same regimen at a dose of 3 mg/kg/week DOX, i.v. [124]. Intraperitoneal injection of 2.5 mg/kg DOX six times over 2 weeks in Wistar rats resulted in 100% survival 2 weeks afterward accompanied by significant declines in FS, dP/dt max, and LV end-systolic pressure as well as increases in LVEDP and LV end-diastolic and end-systolic diameters relative to the control group [125]. At 3 weeks after completion of this same regimen in 8-week-old SD rats, investigators noted 25% mortality, marked accumulation of ascites, and marked declines in FS and heart weight [126]. A 5-week tail vein administration of 3 mg/kg/week DOX in SD rats achieved similar reductions in FS as well as significant ventricular wall thinning 4 weeks after dosing, but survival was not reported [127]. Therefore, at specific doses, DOX can elicit systolic failure more effectively than the aforementioned surgical procedures while minimizing premature mortality.
Despite evidence that serial administration can be less lethal yet achieve substantially greater decrements in FS than a bolus injection at an equal cumulative dose, at least one study has provided contradictory evidence. Serial administration of DOX (1 mg/kg DOX for 10 days) caused greater mortality than a 10 mg/kg bolus injection (40 vs. 20%) and took longer (32 vs. 14 days post-treatment) to reduce FS by 15% [123]. Serial injection impaired FS the greatest at 67 days post-treatment (−25%), but this effect resulted in 70% mortality. Ultimately, the utility of bolus vs. serial DOX administration, as well as other HF models, depends on the goals of the study (e.g., short vs. long monitoring period, level of systolic dysfunction, and survival rate).
The pathophysiologic relevance of the DOX model to human HF is limited because it results in HF primarily through eliciting systolic dysfunction and subsequent preload-driven volume overload while only mildly impairing diastolic function. Furthermore, the frequent occurrence of ascites and the scarcity of pulmonary edema in the DOX-HF model indicate that, relative to the left ventricle, the right ventricle is disproportionately impaired. Finally, DOX can have major gastrointestinal, renal, hepatic, and bone marrow toxicities that may be unsuitable for a model of heart failure [64, 112, 128].
Isoproterenol (also, isoprenaline or isuprel). Isoproterenol is a synthetic catecholamine and non-selective βAR agonist formerly prescribed as a bronchodilator for asthmatics as well as a treatment for cardiac arrest, heart block, and bronchospasm. By stimulating the β1AR, ISO increases chronotropy (heart rate), inotropy (contractile force), dromotropy (electrical conduction rate of the atrio-ventricular node), and lusitropy (cardiac relaxation), while it causes vasodilation and bronchodilation through β2AR stimulation [129]. As such, ISO bears competing short-term effects on cardiac afterload (increasing it as a positive inotrope but decreasing it as a vasodilator and positive lusitrope), while it decreases preload via tachycardia-impaired diastole and reduced central venous pressure—the latter a natural adaptation to increased contractility.
As evidenced by the profound success of beta-blockers in HF treatment, βAR stimulation by catecholamines is crucial to HF pathogenesis [130]. Regulating cardiac function, myocyte growth, and apoptosis, βARs are seven transmembrane-spanning receptors coupled intracellularly with guanine-nucleotide-binding regulatory proteins (G proteins). The heart has three βAR subtypes (β1-3), among which β1 and β2ARs have the most prominent effects on cardiac function. The binding of the endogenous catecholamines norepinephrine and epinephrine to βARs enables G proteins to stimulate adenylyl cyclase, which opens L-type Ca2+ channels, enabling Ca2+ to bind to ryanodine receptors, thereby triggering the release of stored Ca2+ from the sarcoplasmic reticulum into the contractile apparatus to provoke cardiomyocyte contraction while increasing the synthesis of cyclic AMP, PKA, and—with β2AR-activation only—MAPK [55, 130]. The consequent elevation in cyclic AMP increases transport of myocardial Ca2+ into the cytosol—enhancing myocardial filament contractility—and activates sarcoplasmic reticulum calcium-dependent ATPases (SERCA), thereby increasing Ca2+ efflux and myofilament relaxation. In addition to enhancing contractility, βAR stimulation by catecholamines expedites chronotropic depolarization of the sinoatrial node, thereby increasing heart rate, cardiac output, and workload [9, 131]. In this manner, long-term elevations in catecholamines can deplete high-energy phosphate stores as well as SERCA [132]. Elevated myocardial Ca2+ may also increase arrhythmias and risk for sudden cardiac death [9, 133]. Likewise, increased plasma catecholamine levels occur in response to overload hypertrophy [132] and myocardial infarction [134] while correlating with the likelihood of sudden death in HF patients [135] and promoting HF pathogenesis. Acute catecholamine elevations are the putative cause of Takotsubo stress cardiomyopathy, characterized by cyclic AMP-mediated Ca2+ overload, decreased myocyte viability, contraction band necrosis, angina, and gross myocardial ballooning [136, 137]. Meanwhile, chronic β1AR stimulation (from elevated norepinephrine) advances cardiac remodeling via cardiomyocyte hypertrophy, necrosis, apoptosis, and fibrosis, and further impairs cardiac performance through depletion of βARs—especially β1ARs—attendant to increased βAR kinases [130, 138–141].
ISO has been used extensively to induce toxic cardiomyopathy and HF. Although ISO administration increases contractility through Ca2+ accumulation in myocytes, Ca2+ accumulation inevitably reduces the heart’s ability to relax and stretch, thereby inhibiting ventricular filling and stroke volume and increasing preload [133]. ISO has been shown in several studies to cause tachycardia, hypotension, Ca2+ overload, free radical generation, coronary vasospasm, high-energy phosphate exhaustion, and impaired myocardial glucose metabolization in the short term, as well as decreased SERCA expression, ryanodine receptor PKA hyperphosphorylation, and βAR down-regulation and uncoupling in the long term that lead to a progression from myocardial hypoxia to ischemia, inflammation, necrosis, apoptosis, and fibrosis accompanied by HF-like symptoms [90, 134, 142–149]. Ultimately, these alterations in the myocardium decrease contractility and heart rate while increasing water and Na+ retention, thereby decreasing afterload while increasing preload. In contrast to the DOX-HF model, the predominance of pulmonary edema and scarcity of ascites suggest that ISO-induced decrements in cardiac function occur primarily in the left ventricle.
Traditionally, the model of ISO-induced HF has incorporated acute administration (i.e. subcutaneous or intraperitoneal injections) of high bolus doses to achieve cardiac necrosis and associated declines in cardiac function. Zhang and colleagues subcutaneously injected male SD rats twice with 340 mg/kg ISO. Four months later, heart failure was strongly indicated by pulmonary congestion, edema, anorexia, and depressed dP/dt max [150]. This dose also significantly reduced responsiveness of the L-type Ca2+ receptor, for which declines in function and expression are known to impair contractility in human HF [151]. Unfortunately, high mortality (~50% after 48 h) [150] and hepatic and renal necrosis (at ISO doses ≥ 200 mg/kg) [152] may limit the utility of this model of HF.
Lower ISO doses can yield more stable models of HF while more closely approximating catecholamine elevations common to HF pathogenesis. For example, a single 150 mg/kg sc injection in male SD rats impaired systolic function at 1 week post-injection [153], while in female SD rats, it led to 25% mortality and caused significant LV diastolic dysfunction and hypertrophy after 2 weeks that persisted after 4 months [152, 154]. Other studies with similar dose regimens show variations in survival, cardiotoxicity, symptomatology, and strain dependence (e.g., pre-existing hypertension appears to enhance the cardiotoxicity of ISO administration). Importantly, chronic and subchronic administration of very low doses of ISO may mimic natural HF pathogenesis more accurately than large acute doses because they can elicit necrosis and fibrotic cardiomyopathy through free radical-induced extracellular matrix biosynthesis [150, 155–157]. SD rats injected with 0.04–0.1 mg/kg/day ISO for 3–7 months had very limited sudden cardiac death and several strong similarities to a subgroup of aortic banded rats showing overt signs of HF: impaired myocardial collagen cross-linking, LV chamber dilatation, an equivalent reduction in FS, and impaired LV developed pressure–volume relations [52] as well as marked apoptosis and β1 and β2AR inotropic downregulation [158]. Moreover, long-term exposure of cardiomyocytes to beta agonists leads to functional “uncoupling” of βARs and adenylate cyclase [159], characteristic of human HF. Nevertheless, the stress inflicted on rats by daily handling and injections over several weeks and the protracted length of such regimens limit this model’s utility.
Alternatively, osmotic pumps provide a convenient means to circumvent these issues while maintaining levels more comparable to endogenous neurohormones. A host of cardiac alterations consistent with the pathogenesis of heart failure have been revealed following continuous ISO infusion with subcutaneously implanted osmotic pumps [76, 90, 132, 139, 142, 144, 146, 157, 160–167]. Infusion of 2.4 mg/kg/day ISO in Wistar rats caused less than 5% mortality while it altered gene expression in a strikingly similar magnitude and time-course as aortic constriction through day 8 of infusion; however, most of these changes reverted after 26 days of infusion [132]. Although the myocardial challenge presented by long-term ISO infusion may mimic the neurohormonal pathogenesis of HF and enables substantial reductions in premature mortality relative to other HF models, only a small handful of rat studies have examined the effects of long-term ISO infusion on in vivo cardiac function [76, 164, 167]. ISO infusion over 3–7 days in Wistar rats caused significant declines in LV dP/dt max [164], CO [167], and SV [167], but did not change LVESP [164, 167] or EF [167] (Table 3)—consistent with a decrease in contractility and a hypertrophy-induced decrease in chamber volume. Nevertheless, even several hours after treatment cessation, temporary myocardial adaptations to ISO (e.g., hypertrophy) may transiently mask underlying cardiac pathophysiology. For instance, LVESP remained unchanged and LV volume was decreased at 2 h after a 7-day infusion in one of these models, but it decreased 2 days post-infusion when LV volume rebounded to normal [167]. Prolonged infusion may bear more dramatic and persistent effects; 6-16 weeks of ISO significantly reduced EF and FS and markedly increased LV systolic diameter, indicative of systolic failure accompanied by cardiac hypertrophy [167]. Infusion of 0.02 mg/kg/day ISO for 5 months caused no fatalities and induced a phenotype in 12-month-old SH rats that compared closely to the LV dilatation, fibrosis, and systolic and diastolic dysfunction seen in untreated 22-month-old SH rats in decompensated HF [76]. While these extended low-dose periods may appear prohibitively long to some researchers, the induction of HF with pathophysiologically relevant levels of circulating catecholamines may render a model with exceptional comparability to human HF pathogenesis and low premature mortality.
Angiotensin II (ANG II). In both human HF and animal models of HF, pro-hypertrophic factors typically induce a progression from hypertrophy to dilated cardiomyopathy and subsequent HF. By prolonging stimuli of concentric cardiac hypertrophy, investigators may render new models of HF if they can induce sufficient fibrosis, apoptosis, and/or autophagy in the myocardium. ANG II infusion is one well-established induction method for cardiac hypertrophy that bears promise for yielding a new HF model. A major component of the renin-aldosterone-angiotensin system (RAAS), ANG II originates from angiotensin I via its converting enzyme (ACE). ANG II causes vasoconstriction and sodium retention (leading to afterload-dependent pressure overload and preloaded volume overload, respectively) as well as myocardial inflammation, oxidative stress, and fibrosis [168]. As evidenced by the benefits of ACE inhibitors in humans with HF and of angiotensin receptor blockade in a hypertensive rat HF model, ANG II is known to play a key pathogenic role in HF [169]. Several investigators have administered exogenous ANG II to rodents by osmotic pump to induce hypertrophic cardiomyopathy, but very few have examined the effects of long-term ANG II administration on cardiac function [67, 161, 168, 170–172]. Freund and colleagues elicited significant cardiac hypertrophy in mice via infusion of ANG II (2.0 mg/kg/day for 14 days) [171]. Despite perivascular infiltration of inflammatory cells and increased LV wall thickness, echocardiographic and histological analysis indicated no cardiac dilatation, myocyte apoptosis, or declines in cardiac function [171]. Notably, this regimen risks vascular complications; a lower infusion rate of ANG II (1.44 mg/kg/day) for 4 weeks has been shown to induce aortic aneurysm in 63% and aortic rupture in 25% of apolipoprotein E-deficient mice [173]. Infusion of lower concentrations (0.29 mg/kg/day) in mice for 8 weeks did not significantly change cardiac function or dimension despite causing cardiac fibrosis [170]. While work on models of Ang II-induced HF has thus far yielded limited results, a modified regimen that focuses on prolonged Ang II exposure at higher concentrations may elicit a more pronounced fibrosis and eventually lead to dilated cardiomyopathy and/or HF symptomatology.
Propylthiouracil (PTU). Although hypertrophic models such as that achieved by ANG II may hold promise for future models of HF, cardiac dysfunction and HF can occur independent of hypertrophy in less common cases [174]. Hypothyroidism is a common condition affecting 10% of women and 6% of men >65 that in some cases can lead to dilated cardiomyopathy and HF without hypertrophy [175]. Hypothyroidism occurs when the thyroid fails to secrete sufficient levels of the thyroid hormones thyroxine (T4) and/or triiodothyronine (T3), and often manifests clinically as developmental retardation in children (cretinism) and depressed mental and physical activity in adults (myxedema) [176]. Experimental induction of hypothyroidism by thyroidectomy has been shown to decrease cardiac output, blood volume, LVESP, heart rate, aortic pressure, and LV dP/dtmin—a measure of cardiac relaxation [177]. Hypothyroidism induces cardiac unloading (decreasing both afterload and preload), which if sustained can lead to cardiac atrophy and dilatation [178]. PTU is an antithyroid medication prescribed for hyperthyroidism that blocks T3 synthesis and has been shown to induce hypothyroidism in rats with similar effects on cardiac output as thyroidectomy [178–180]. Like the HF seen in other models, PTU treatment represses SERCA and the more efficient alpha isoform of myosin heavy chain (MHC) while dramatically increasing expression of βMHC in the rat heart [68, 75, 181]. Tang and associates demonstrated in female SD rats that 0.025% PTU in drinking water for 6 weeks and 1 year was associated with dramatic loss of myocardial arterioles, major declines in FS, EF, LVESP, and body weight, but no changes in LVEDP (Table 3) [178]. Another study examined the effects of this same dose of PTU for 6 months in female SHHFs to determine if hypothyroidism could accelerate the progression to heart disease in the context of hypertensive hypertrophy [180]. The treatment elicited traits similar to those seen in SD rats, including cardiac dilatation, unchanged LVEDP, and decreased FS, EF, and LV dP/dtmax; however, in contrast to SD rats, myocardial arteriolar number was not affected by PTU in SHHF rats. Ultimately, PTU hypothyroidism parallels HF pathogenesis by impairing myocardial blood flow while inducing cardiomyocyte atrophy and cardiac dilatation with series addition of sarcomeres, but it differs from common HF etiologies by failing to cause hypertrophy and diastolic dysfunction [178].
Tumor Necrosis Factor-α (TNF-α) is elevated in HF and believed to play a major role in HF pathogenesis and mortality [10, 182]. By impairing contractility, decreasing stroke volume, and causing LV dilatation [183], one would predict that TNF-α promotes increased preload and a progression to HF consistent with volume overload. Although evidence of TNF-α-induced HF is scarce in rats, studies of TNF-α overexpression in mice provide further evidence of the promise for a rat model of TNF-α-induced HF. Specifically, in genetically modified mice overexpressing cardiac TNF-α, declines in FS have been observed along with increased cardiomyocyte apoptosis, mislocalization of desmin and intercalated disc proteins, and desmin aggregation [184]. TNF-α administration has been shown to impair rat cardiomyocyte βAR responsiveness to catecholamines [185] while causing coronary vasoconstriction in canine hearts and severe intracellular and mitochondrial oxidative stress in the rat myocardium, significantly impairing systolic and diastolic function by hindering energy utilization for excitation–contraction coupling [186] (Table 3). In SD rats, subchronic infusion of 3.6 mg/kg/day of TNF-α (sufficient to maintain levels comparable to HF patients) for 15 days caused fibrillar collagen deterioration, cardiomyocyte hypertrophy, cardiac dilatation, and impaired FS without contraction band necrosis or fibrosis) [187]. Further characterization of this HF model may enhance its utility in therapeutic and toxicological studies.
Dietary Salt Models
Salt Alone. Consumption of dietary salt stimulates water retention and, potentially, hypertension, increasing both afterload and preload. A high salt diet increases sympathetic activity and circulating norepinephrine in humans with salt-sensitive hypertension while having the opposite effect on normal individuals [188]. Similarly, administration of 1% NaCl in drinking water for 4 weeks substantially increases mean arterial pressure, noradrenaline (in urine, plasma, and the myocardium), adrenaline, sympathetic activity, and vasopressin in the SHR but not the WKY rat [189–191]. Studies have indicated that the administration of a high salt diet (salt loading) is a feasible and simple means of inducing heart failure in SH [192, 193], SHHF [194], and Dahl salt-sensitive (DS) rats [195]. The combination of increased afterload from salt-sensitive hypertension and increased preload from hypervolemia is critical to the development of HF in salt-fed rats. An 8% NaCl diet fed to 2-month-old SH and WKY rats for 8 weeks elicits marked ventricular fibrosis [192, 193, 196] and hypervolemia [196]. In one such study, 13% fatality occurred in SHRs, as well as signs of congestive HF (labored breathing and lethargy with decreased FS and a 59% increase in right ventricular mass) among a third of the surviving SHRs [192]. Unexpectedly, cardiac output and SV declined only in the non-congestive SHRs, while congestive SHRs had cardiac dilatation and systolic hypotension. In a separate study, this regimen affected neither contractile function nor LVEDP, but it impaired the rate of diastolic relaxation for both ventricles and caused premature mortality in SHRs with lesser effects in WKY rats [193].
The effects of dietary salt on the SHHF strain are not as well characterized. Mediated by leptin resistance and increased renal endothelin production, salt sensitivity has been confirmed in obese—but not lean—SHHF males by the dramatic exacerbation of hypertension and cardiac hypertrophy from a 7-day 8% NaCl diet [83]. Notably, one study demonstrated that salt enhances cardiac hypertrophy in obese SHHFs independent of hypertension and endothelin, potentially by decreasing nitric oxide production [197]. Despite the obese SHHF’s salt sensitivity, reports of salt-accelerated HF in the SHHF strain are scarce and limited to the lean phenotype [194]. Although no cardiac function data were provided, one laboratory has reported eliciting HF in 6-week-old lean male SHHF rats fed an 8% NaCl diet for 5 months. Similar to aortic banding of SHHF rats, salt significantly increased pro-apoptotic signaling in SHHF cardiomyocytes [194].
Salt loading in the DS strain has been shown to rapidly elicit HF with a swift progression from decompensation to death (with 8% salt diet, death < 2 weeks after decompensation) [195, 198]. Collectively, two studies have shown that 6-week-old DS rats fed an 8% NaCl diet develop cardiac hypertrophy after 5–6 weeks, transition to systolic failure on the 9th week, have full-blown dilatation and decompensated HF with 60-70% mortality by the 11th week, and have 100% mortality by the 13th week of salt diet (Table 4) [198, 199]. Unfortunately, the effects of this regimen on cardiac function are not entirely consistent. In another study, the same regimen unexpectedly increased FS, LV thickness, systolic blood pressure, and molecular markers of cardiac hypertrophy while causing high fatality (56%) by the 12th week of salt diet [195]. Thus, the DS and SH strains are both variable in timing and physiologic manifestation of salt-induced HF. Survivability may be improved by either a lower salt dose over a longer period of time or removal of the elevated salt diet prior to anticipated decompensation.
Salt with Other Agents. While salt loading often enhances cardiac fibrosis, acute injections of ISO typically elicit infarct-like cardiac necrosis. Therefore, the combination of these two treatments may more readily mimic the HF pathogenesis found among survivors of myocardial infarction. To date, only one publication appears to have addressed the effects of concomitant chronic salt loading and ISO treatment. Treatment with salt (1% NaCl in drinking water for 2 weeks) did not exacerbate ISO-induced LV hypertrophy in male Wistar rats [166]; nevertheless, data on cardiac function after ISO salt co-treatment are scarce. Other methods may enhance the pathologic effects of salt while simulating the human diet. Co-treatment of SHRs with 1% NaCl and 5% sucrose in water induced hypertension, tachycardia, renal excretion of noradrenaline, and responsiveness to pressor substances exceeding either treatment alone [190]; however, the effects of this regimen on cardiac pathology were not examined. Overall, the potential of such combination treatments to induce HF remains largely unexplored. Conversely, the co-treatment of rats with salt and deoxycorticosterone acetate (DOCA) is an emerging model of HF. DOCA increases salt retention and thus enables salt-induced hypertension in non-salt-sensitive strains. The combination of DOCA with salt after surgical unilateral nephrectomy has been shown in rats to significantly impair LV diastole and, in some cases, LV systole. In one study, 1% NaCl in water combined with bi-weekly 15 mg/kg DOCA injections for 5 weeks impaired diastolic function (Table 4) [200]. In SD rats, 1% NaCl drinking water and 100 mg/kg/week DOCA (by subcutaneous injection) for 6 weeks significantly impaired both systolic and diastolic function [201]. Similar to human HF, these changes were accompanied by increased plasma markers of cardiac remodeling, including matrix metalloproteinase-2, tissue inhibitor of metalloproteinase-1, and osteopontin. Despite these findings, there remain only a few rat studies demonstrating reduced cardiac function following DOCA and salt administration. Moreover, this model typically requires invasive surgery for the removal of a single kidney. Ultimately, the limited literature covering the effects of DOCA + salt on cardiac function, survival, and gross HF signs limit the reliability of this model for HF studies.
Conclusions: The Application of Rat Models of Non-invasively Induced HF
As noted, most of the previously described models of non-invasively induced HF have shown exceptional comparability to more invasive models. Furthermore, they often involve routine techniques that require less expertise while reducing the physical stress and premature mortality commonly inflicted on animals through surgical techniques. Finally, many of these models have been used to bolster evidence toward the benefits of therapies and/or the susceptibilities to toxins in humans with cardiac failure. For instance, aged SHHF rats and salt-loaded DS rats have been used to demonstrate that exercise enhances survival and reduces adverse symptoms in HF [86, 194, 195]. Additionally, the aged SHHF has been used to show the benefits of inhibition of matrix metalloproteinase and blockade of the A1 receptor [84, 94]. Meanwhile, the model of PTU-induced hypothyroidism has been compared with surgical models of HF to demonstrate mechanisms of micro-RNA function in cardiac remodeling [68]. DOX and ISO have each been used to demonstrate the cardioprotective effects of a number of antioxidants [124, 125, 147, 202]. The DOX model has also been used to show improved survival and cardiac function with administration of a neuregulin-1/erbB-activating agent [113]. Also, an endothelin receptor antagonist has been shown capable of reversing ISO-induced HF [203]. Collectively, animal models have enabled deep mechanistic insight into the multifactorial condition of HF. A majority of the non-invasive methods described herein enable faster turnaround of simpler and more efficient HF models while remaining pathophysiologically relevant to human HF.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ANG II:
-
Angiotensin II
- βAR:
-
Beta adrenergic receptor
- CO:
-
Cardiac output
- DM:
-
Diabetes mellitus
- DOX:
-
Doxorubicin
- dP/dt max :
-
Peak rate of increase in LV pressure
- dP/dt min :
-
Peak rate of decrease in LV pressure
- DOCA:
-
Deoxycorticosterone acetate
- DS:
-
Dahl salt sensitive
- E/A:
-
Ratio of early-to-late inflow velocities
- EF:
-
Ejection fraction
- ESV:
-
End-systolic volume
- FS:
-
Fractional shortening
- HF:
-
Heart failure
- HR:
-
Heart rate
- i.v. :
-
Intravenous
- ISO:
-
Isoproterenol
- LAD:
-
Left anterior descending coronary artery
- LV:
-
Left ventricular
- LVP:
-
LV pressure
- LVEDP:
-
LV end-diastolic pressure
- LVESP:
-
LV end-systolic pressure
- LVOT:
-
LV outflow tract
- MAP:
-
Mean arterial pressure
- MHC:
-
Myosin heavy chain
- MI:
-
Myocardial infarction
- PO:
-
Pressure overload
- PTU:
-
Propylthiouracil
- s.c. :
-
Subcutaneous
- SD:
-
Sprauge Dawley
- SERCA:
-
Sarcoplasmic reticulum Ca2+ ATPase pump
- SERCA2a:
-
SERCA type “2”, isoform ‘a’
- SH:
-
Spontaneously hypertensive
- SHHF:
-
Spontaneously hypertensive heart failure
- SHR:
-
Spontaneously hypertensive rat
- SV:
-
Stroke volume
- T3 :
-
Triiodothyronine
- T4 :
-
Thyroxine
- TAC:
-
Transverse aortic constriction
- TNF-α:
-
Tumor necrosis factor-α
- UPS:
-
Ubiquitin–proteasome system
- VO:
-
Volume overload
References
DeFrances, C. J. & Podgornik, M. N. (2006). 2004 National hospital discharge survey. In Advance data. United States, pp. 1–19.
Minino, A. M., Heron, M. P., & Smith, B. L. (2006). Deaths: preliminary data for 2004. National vital statistics reports: From the centers for disease control and prevention, National center for health statistics. National Vital Statistics System, 54(19), 1–49.
Rosamond, W., et al. (2008). Heart disease and stroke statistics—2008 update: A report from the American heart association statistics committee and stroke statistics subcommittee. Circulation, 117(4), e25–e146.
Coronel, R., de Groot, J. R., & van Lieshout, J. J. (2001). Defining heart failure. Cardiovascular Research, 50(3), 419–422.
Lloyd-Jones, D., et al. (2009). Heart disease and stroke statistics—2009 update: A report from the American heart association statistics committee and stroke statistics subcommittee. Circulation, 119(3), e21–e181.
Goff, D. C., Jr., et al. (2000). Congestive heart failure in the United States: Is there more than meets the I(CD code)? The corpus christi heart project. In Archives of internal medicine, USA, pp. 197–202.
Balakumar, P., Singh, A. P., & Singh, M. (2007). Rodent models of heart failure. Journal of Pharmacological and Toxicological Methods, 56(1), 1–10.
Francis, G. S. (2001). Pathophysiology of chronic heart failure. American Journal of Medicine, 110(Suppl 7A), 37S–46S.
Braunwald, E., Ross, J. & Sonnenblick, E. (1976). Mechanisms of contraction of the normal and failing heart, vol. 2, p. 417. Boston: Little, Brown and Company.
Mann, D. L., & Bristow, M. R. (2005). Mechanisms and models in heart failure: The biomechanical model and beyond. Circulation, 111(21), 2837–2849.
Diwan, A. & Dorn, G. W. II (2007). Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda), 22, 56–64.
Frey, N. & Olson, E. N. (2003). Cardiac hypertrophy: The good, the bad, and the ugly. Annual Review of Physiology, 65, 45–79.
Schoen, F. J. (2005). In V. Kumar, A. K. Abbas, & N. Fausto (Eds.), The heart, in robbins and cotran pathologic basis of disease. Elsevier Saunders: Philadelphia, PA, pp. 555–618.
Kang, Y. J. (2006). Cardiac hypertrophy: A risk factor for QT-prolongation and cardiac sudden death. Toxicologic Pathology, 34(1), 58–66.
Funada, J., et al. (2009). Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PLoS One, 4(10), p. e7533.
Zile, M. R., et al. (2010). Mode of death in patients with heart failure and a preserved ejection fraction: Results from the irbesartan in heart failure with preserved ejection fraction study (I-Preserve) trial. Circulation, 121(12), 1393–1405.
Jackson, G., et al. (2000). ABC of heart failure. Pathophysiology. BMJ, 320(7228), 167–170.
Tonnessen, T., et al. (1997). Increased cardiac expression of endothelin-1 mRNA in ischemic heart failure in rats. Cardiovascular Research, 33(3), 601–610.
Sjaastad, I., et al. (2000). Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. Journal of Applied Physiology, 89(4), 1445–1454.
Morgan, E. E., et al. (2004). Validation of echocardiographic methods for assessing left ventricular dysfunction in rats with myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 287(5), H2049–H2053.
Holt, E., et al. (1998). Mechanisms of cardiomyocyte dysfunction in heart failure following myocardial infarction in rats. Journal of Molecular and Cellular Cardiology, 30(8), 1581–1593.
Wake, R., et al. (2005). Beneficial effect of candesartan on rat diastolic heart failure. Journal of Pharmacological Science, 98(4), 372–379.
Itter, G., et al. (2004). A model of chronic heart failure in spontaneous hypertensive rats (SHR). Laboratory Animals, 38(2), 138–148.
Pocock, S. J., et al. (2006). Predictors of mortality and morbidity in patients with chronic heart failure. European Heart Journal, 27(1), 65–75.
Janssen, B. J., et al. (2004). Effects of anesthetics on systemic hemodynamics in mice. American Journal of Physiology. Heart and Circulatory Physiology, 287(4), H1618–H1624.
Pfeffer, M. A., et al. (1979). Myocardial infarct size and ventricular function in rats. Circulation Research, 44(4), 503–512.
Opitz, C. F., et al. (1995). Arrhythmias and death after coronary artery occlusion in the rat. Continuous telemetric ECG monitoring in conscious, untethered rats. Circulation, 92(2), 253–261.
Lefebvre, F., et al. (2006). Modification of the pulmonary renin-angiotensin system and lung structural remodelling in congestive heart failure. Clinical Science (London), 111(3), pp. 217–224.
Mulder, P., et al. (1997). Role of endogenous endothelin in chronic heart failure: Effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation, 96(6), 1976–1982.
Samsamshariat, S. A., Samsamshariat, Z. A., & Movahed, M. R. (2005). A novel method for safe and accurate left anterior descending coronary artery ligation for research in rats. Cardiovascular Revascularization Medicine, 6(3), 121–123.
Molina, E. J., et al. (2008). Novel experimental model of pressure overload hypertrophy in rats. Journal of Surgical Research.
Rockman, H. A., et al. (1991). Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proceedings of National Academic Science of USA, 88(18), 8277–8281.
Suckau, L., et al. (2009). Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation, 119(9), 1241–1252.
Weinberg, E. O., et al. (1994). Angiotensin-converting enzyme inhibition prolongs survival and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis. Circulation, 90(3), 1410–1422.
Gupta, D., et al. (2008). Adenoviral beta-adrenergic receptor kinase inhibitor gene transfer improves exercise capacity, cardiac contractility, and systemic inflammation in a model of pressure overload hypertrophy. Cardiovascular Drugs and Therapy, 22(5), 373–381.
Schwarzer, M., et al. (2009). The metabolic modulators, Etomoxir and NVP-LAB121, fail to reverse pressure overload induced heart failure in vivo. Basic Research in Cardiology, 104(5), 547–557.
Cantor, E. J., et al. (2005). A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. Journal of Molecular and Cellular Cardiology, 38(5), 777–786.
Rivera, D. M., & Lowes, B. D. (2005). Molecular remodeling in the failing human heart. Current Heart Failure Reports, 2(1), 5–9.
del Monte, F., et al. (2001). Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2 +)-ATPase in a rat model of heart failure. Circulation, 104(12), 1424–1429.
Gianni, D., et al. (2005). SERCA2a in heart failure: Role and therapeutic prospects. Journal of Bioenergetics and Biomembranes, 37(6), 375–380.
Tsukioka, T., et al. (2007). Local and systemic impacts of pleural oxygen exposure in thoracotomy. BioFactors, 30(2), 117–128.
Buvanendran, A., et al. (2004). Characterization of a new animal model for evaluation of persistent postthoracotomy pain. Anesthesia and Analgesia, 99(5), 1453–1460; table of contents.
del Monte, F., et al. (2002). Novel technique of aortic banding followed by gene transfer during hypertrophy and heart failure. Physiol Genomics, 9(1), 49–56.
Lebeche, D., et al. (2004). In vivo cardiac gene transfer of Kv4.3 abrogates the hypertrophic response in rats after aortic stenosis. Circulation, 110(22), 3435–3443.
Hu, P., et al. (2003). Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. American Journal of Physiology. Heart and Circulatory Physiology, 285(3), H1261–H1269.
Stansfield, W. E., et al. (2007). Characterization of a model to independently study regression of ventricular hypertrophy. Journal of Surgical Research, 142(2), 387–393.
Pawlush, D. G., et al. (1993). Echocardiographic evaluation of size, function, and mass of normal and hypertrophied rat ventricles. Journal of Applied Physiology, 74(5), 2598–2605.
Kobayashi, S., et al. (1996). Influence of aortic impedance on the development of pressure-overload left ventricular hypertrophy in rats. Circulation, 94(12), 3362–3368.
Shah, K. B., et al. (2009). The cardioprotective effects of fish oil during pressure overload are blocked by high fat intake: Role of cardiac phospholipid remodeling. Hypertension, 54(3), 605–611.
Linz, W., et al. (1996). ACE inhibition decreases postoperative mortality in rats with left ventricular hypertrophy and myocardial infarction. Clinical and Experimental Hypertension, 18(5), 691–712.
Luo, J. D., et al. (1999). Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clinical and Experimental Pharmacology and Physiology, 26(11), 903–908.
Woodiwiss, A. J., et al. (2001). Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation, 103(1), 155–160.
Sethi, R., et al. (2007). Dependence of changes in beta-adrenoceptor signal transduction on type and stage of cardiac hypertrophy. Journal of Applied Physiology, 102(3), 978–984.
Monnet, E., & Orton, E. C. (1999). A canine model of heart failure by intracoronary adriamycin injection: Hemodynamic and energetic results. Journal of Cardiac Failure, 5(3), 255–264.
Wang, X., et al. (2003). Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. Journal of Applied Physiology, 94(2), 752–763.
Brower, G. L., & Janicki, J. S. (2001). Contribution of ventricular remodeling to pathogenesis of heart failure in rats. American Journal of Physiology. Heart and Circulatory Physiology, 280(2), H674–H683.
Janicki, J. S., et al. (2006). Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovascular Research, 69(3), 657–665.
Gardner, J. D., Brower, G. L., & Janicki, J. S. (2002). Gender differences in cardiac remodeling secondary to chronic volume overload. Journal of Cardiac Failure, 8(2), 101–107.
Brower, G. L., Henegar, J. R. & Janicki, J. S. (1996). Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. American Journal of Physiology. Heart and Circulatory Physiology, 271(5 Pt 2), pp. H2071–H2078.
Moreno, J., et al. (2005). Effect of remodelling, stretch and ischaemia on ventricular fibrillation frequency and dynamics in a heart failure model. Cardiovascular Research, 65(1), 158–166.
Hanna, N., et al. (2004). Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovascular Research, 63(2), 236–244.
Cardin, S., et al. (2003). Evolution of the atrial fibrillation substrate in experimental congestive heart failure: Angiotensin-dependent and -independent pathways. Cardiovascular Research, 60(2), 315–325.
Takagaki, M., et al. (2002). Induction and maintenance of an experimental model of severe cardiomyopathy with a novel protocol of rapid ventricular pacing. Journal of Thoracic and Cardiovascular Surgery, 123(3), 544–549.
Monnet, E., & Chachques, J. C. (2005). Animal models of heart failure: What is new? Annals of Thoracic Surgery, 79(4), 1445–1453.
Chekanov, V. S., et al. (2000). Effects of electrical stimulation postcardiomyoplasty in a model of chronic heart failure: Hemodynamic results after daily 12-hour cessation versus a nonstop regimen. Pacing and Clinical Electrophysiology, 23(7), 1094–1102.
Hasenfuss, G. (1998). Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovascular Research, 39(1), 60–76.
Maass, A. H., et al. (2004). Hypertrophy, fibrosis, and sudden cardiac death in response to pathological stimuli in mice with mutations in cardiac troponin T. Circulation, 110(15), 2102–2109.
van Rooij, E., et al. (2007). Control of stress-dependent cardiac growth and gene expression by a microRNA. Science, 316(5824), 575–579.
de Resende, M. M., Kriegel, A. J., & Greene, A. S. (2006). Combined effects of low-dose spironolactone and captopril therapy in a rat model of genetic hypertrophic cardiomyopathy. Journal of Cardiovascular Pharmacology, 48(6), 265–273.
Muller, D. N., Derer, W., & Dechend, R. (2008). Aliskiren—Mode of action and preclinical data. Journal of Molecular Medicine, 86(6), 659–662.
Wichers, L. B., et al. (2004). Effects of instilled combustion-derived particles in spontaneously hypertensive rats. Part I: Cardiovascular responses. Inhalation Toxicology, 16(6–7), 391–405.
Calhoun, D. A., et al. (1994). Diurnal blood pressure variation and dietary salt in spontaneously hypertensive rats. Hypertension, 24(1), 1–7.
El-Mas, M. M., & Abdel-Rahman, A. A. (2005). Longitudinal studies on the effect of hypertension on circadian hemodynamic and autonomic rhythms in telemetered rats. Life Science, 76(8), 901–915.
Bing, O. H., et al. (1995). The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. Journal of Molecular and Cellular Cardiology, 27(1), 383–396.
Boluyt, M. O., Bing, O. H. & Lakatta, E. G. (1995). The ageing spontaneously hypertensive rat as a model of the transition from stable compensated hypertrophy to heart failure. European Heart Journal, 16(Suppl N), pp. 19–30.
Badenhorst, D., et al. (2003). Beta-adrenergic activation initiates chamber dilatation in concentric hypertrophy. Hypertension, 41(3), 499–504.
Kuoppala, A., et al. (2003). Expression of bradykinin receptors in the left ventricles of rats with pressure overload hypertrophy and heart failure. Journal of Hypertension, 21(9), 1729–1736.
Koletsky, S. (1975). Pathologic findings and laboratory data in a new strain of obese hypertensive rats. American Journal of Pathology, 80(1), 129–142.
McCune, S., Baker, P. & Stills, H. Jr. (1990). SHHF/Mcc-cp rat: Model of obesity, non-insulin-dependent diabetes, and congestive heart failure. ILAR (Institute for Laboratory Animal Research) Journal, 32.
Muders, F., & Elsner, D. (2000). Animal models of chronic heart failure. Pharmacological Research, 41(6), 605–612.
Roncalli, J., et al. (2007). NMR and cDNA array analysis prior to heart failure reveals an increase of unsaturated lipids, a glutamine/glutamate ratio decrease and a specific transcriptome adaptation in obese rat heart. Journal of Molecular and Cellular Cardiology, 42(3), 526–539.
Mark, A. L., et al. (2003). A leptin-sympathetic-leptin feedback loop: Potential implications for regulation of arterial pressure and body fat. Acta Physiologica Scandinavica, 177(3), 345–349.
Radin, M. J., et al. (2003). Increased salt sensitivity secondary to leptin resistance in SHHF rats is mediated by endothelin. Molecular and Cellular Biochemistry, 242(1–2), 57–63.
Jackson, E. K., et al. (2001). A(1) receptor blockade induces natriuresis with a favorable renal hemodynamic profile in SHHF/Mcc-fa(cp) rats chronically treated with salt and furosemide. Journal of Pharmacology and Experimental Therapeutics, 299(3), 978–987.
McCune, S. A., et al. (1995). SHHF/Mcc-facp rat model: effects of gender and genotype on age of expression of metabolic complications and congestive heart failure and on response to drug therapy. In E. Shafir (Ed.), Lessons from animal diabetes V. Smith-Gordon: London, pp. 255–270.
Emter, C. A., et al. (2005). Low-intensity exercise training delays onset of decompensated heart failure in spontaneously hypertensive heart failure rats. American Journal of Physiology. Heart and Circulatory Physiology, 289(5), H2030–H2038.
Abe, Y., et al. (2007). Leptin induces elongation of cardiac myocytes and causes eccentric left ventricular dilatation with compensation. American Journal of Physiology. Heart and Circulatory Physiology, 292(5), H2387–H2396.
Bienertova-Vasku, J. A., et al. (2009). Association between variants in the genes for leptin, leptin receptor, and proopiomelanocortin with chronic heart failure in the Czech population. Heart and Vessels, 24(2), 131–137.
Haas, G. J., et al. (1995). Echocardiographic characterization of left ventricular adaptation in a genetically determined heart failure rat model. American Heart Journal, 130(4), 806–811.
Carll, A. P., et al. (2010). Particulate matter inhalation exacerbates cardiopulmonary injury in a rat model of isoproterenol-induced cardiomyopathy. Inhalation Toxicology, 22(5), 355–368.
Carll, A. P. (2010). Unpublished data.
Schlenker, E. H., Kost, C. K., Jr., & Likness, M. M. (2004). Effects of long-term captopril and l-arginine treatment on ventilation and blood pressure in obese male SHHF rats. Journal of Applied Physiology, 97(3), 1032–1039.
Poornima, I., et al. (2008). Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circulation. Heart failure, 1(3), 153–160.
Peterson, J. T., et al. (2001). Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation, 103(18), 2303–2309.
Hohl, C. M., et al. (1993). Effects of obesity and hypertension on ventricular myocytes: Comparison of cells from adult SHHF/Mcc-cp and JCR:LA-cp rats. Cardiovascular Research, 27(2), 238–242.
Heyen, J. R., et al. (2002). Structural, functional, and molecular characterization of the SHHF model of heart failure. American Journal of Physiology. Heart and Circulatory Physiology, 283(5), H1775–H1784.
Anderson, K. M., et al. (1999). The myocardial beta-adrenergic system in spontaneously hypertensive heart failure (SHHF) rats. Hypertension, 33(1 Pt 2), pp. 402–407.
Tamura, T., Said, S., & Gerdes, A. M. (1999). Gender-related differences in myocyte remodeling in progression to heart failure. Hypertension, 33(2), 676–680.
Reffelmann, T., & Kloner, R. A. (2003). Transthoracic echocardiography in rats. Evaluation of commonly used indices of left ventricular dimensions, contractile performance, and hypertrophy in a genetic model of hypertrophic heart failure (SHHF-Mcc-facp-Rats) in comparison with Wistar rats during aging. Basic Research in Cardiology, 98(5), 275–284.
Janssen, P. M., et al. (2003). Selective contractile dysfunction of left, not right, ventricular myocardium in the SHHF rat. American Journal of Physiology. Heart and Circulatory Physiology, 284(3), H772–H778.
Onodera, T., et al. (1998). Maladaptive remodeling of cardiac myocyte shape begins long before failure in hypertension. Hypertension, 32(4), 753–757.
Gerdes, A. M., et al. (1996). Myocyte remodeling during the progression to failure in rats with hypertension. Hypertension, 28(4), 609–614.
Park, S., et al. (1997). Verapamil accelerates the transition to heart failure in obese, hypertensive, female SHHF/Mcc-fa(cp) rats. Journal of Cardiovascular Pharmacology, 29(6), 726–733.
Ferrara, C. M., et al. (1996). Exercise training and the glucose transport system in obese SHHF/Mcc-fa(cp) rats. Journal of Applied Physiology, 81(4), 1670–1676.
Pacher, P., et al. (2004). Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. American Journal of Physiology. Heart and Circulatory Physiology, 287(5), H2132–H2137.
Anversa, P., et al. (1994). Effects of aging on quantitative structural properties of coronary vasculature and microvasculature in rats. American Journal of Physiology. Heart and Circulatory Physiology, 267(3 Pt 2), pp. H1062–H1073.
Bugger, H., & Abel, E. D. (2009). Rodent models of diabetic cardiomyopathy. Disease Models & Mechanisms, 2(9–10), 454–466.
Bristow, M. R., et al. (1980). Acute and chronic cardiovascular effects of doxorubicin in the dog: The cardiovascular pharmacology of drug-induced histamine release. Journal of Cardiovascular Pharmacology, 2(5), 487–515.
Djelmami-Hani, M., et al. (2007). Induction of heart failure: Haemodynamic comparison of three different canine models. Laboratory Animals, 41(1), 63–70.
Ferreira, A. L., Matsubara, L. S., & Matsubara, B. B. (2008). Anthracycline-induced cardiotoxicity. Cardiovascular & Hematological Agents In Medicinal Chemistry, 6(4), 278–281.
Gille, L., & Nohl, H. (1997). Analyses of the molecular mechanism of adriamycin-induced cardiotoxicity. Free Radical Biology and Medicine, 23(5), 775–782.
Herman, E. H., et al. (1998). Comparison of the chronic toxicity of piroxantrone, losoxantrone and doxorubicin in spontaneously hypertensive rats. Toxicology, 128(1), 35–52.
Liu, X., et al. (2006). Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. Journal of the American College of Cardiology, 48(7), 1438–1447.
Saad, S. Y., Najjar, T. A., & Al-Rikabi, A. C. (2001). The preventive role of deferoxamine against acute doxorubicin-induced cardiac, renal and hepatic toxicity in rats. Pharmacological Research, 43(3), 211–218.
Arola, O. J., et al. (2000). Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Research, 60(7), 1789–1792.
Li, T., Danelisen, I., & Singal, P. K. (2002). Early changes in myocardial antioxidant enzymes in rats treated with adriamycin. Molecular and Cellular Biochemistry, 232(1–2), 19–26.
Liu, J., et al. (2008). A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. American Journal of Physiology. Heart and Circulatory Physiology, 295(6), H2541–H2550.
Kumarapeli, A. R., et al. (2005). A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB Journal, 19(14), 2051–2053.
Tu, V. C., Bahl, J. J., & Chen, Q. M. (2002). Signals of oxidant-induced cardiomyocyte hypertrophy: Key activation of p70 S6 kinase-1 and phosphoinositide 3-kinase. Journal of Pharmacology and Experimental Therapeutics, 300(3), 1101–1110.
Olson, R. D., et al. (2005). Doxorubicin cardiac dysfunction: Effects on calcium regulatory proteins, sarcoplasmic reticulum, and triiodothyronine. Cardiovascular Toxicology, 5(3), 269–283.
Pfizer Inc. (2010). Doxorubicin Hydrochloride for Injection, USP. Available from: http://www.pfizer.com/files/products/uspi_adriamycin.pdf.
U.S. Food and Drug Administration. (2010). Oncology Tools: Dose Calculator. Available from: http://www.accessdata.fda.gov/scripts/cder/onctools/animalresults.cfm.
Hayward, R., & Hydock, D. S. (2007). Doxorubicin cardiotoxicity in the rat: An in vivo characterization. Journal of American Association of Laboratory in Animal Science, 46(4), 20–32.
Kozluca, O., et al. (1996). Prevention of doxorubicin induced cardiotoxicity by catechin. Cancer Letters, 99(1), 1–6.
Chen, X., et al. (2007). Preventive cardioprotection of erythropoietin against doxorubicin-induced cardiomyopathy. Cardiovascular Drugs and Therapy, 21(5), 367–374.
Matsui, H., et al. (1999). Protective effects of carvedilol against doxorubicin-induced cardiomyopathy in rats. Life Science, 65(12), 1265–1274.
Ueno, M., et al. (2006). Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. Journal of Pharmacological Science, 101(2), 151–158.
Deepa, P. R., & Varalakshmi, P. (2003). Protective effect of low molecular weight heparin on oxidative injury and cellular abnormalities in adriamycin-induced cardiac and hepatic toxicity. Chemico-biological Interactions, 146(2), 201–210.
Zheng, M., Han, Q. D., & Xiao, R. P. (2004). Distinct beta-adrenergic receptor subtype signaling in the heart and their pathophysiological relevance. Sheng li xue bao, 56(1), 1–15.
Rockman, H. A., Koch, W. J., & Lefkowitz, R. J. (2002). Seven-transmembrane-spanning receptors and heart function. Nature, 415(6868), 206–212.
Wu, Y., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proceedings of National Academic Science USA, 106(14), 5972–5977.
Boluyt, M.O., et al., Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. American Journal of Physiology. Heart and Circulatory Physiology, 1995. 269(2 Pt 2): p. H638-47.
Zevitz, M. E. & October (2006). Heart Failure. eMedicine from WebMD.
Rona, G. (1985). Catecholamine cardiotoxicity. Journal of Molecular and Cellular Cardiology, 17(4), 291–306.
Peng, Y., et al. (2003). Effects of catecholamine-beta-adrenoceptor-cAMP system on severe patients with heart failure. Chinese Medical Journal, 116(10), 1459–1463.
Abraham, J., et al. (2009). Stress cardiomyopathy after intravenous administration of catecholamines and beta-receptor agonists. Journal of the American College of Cardiology, 53(15), 1320–1325.
Wittstein, I. S. (2008). Acute stress cardiomyopathy. Current Heart Failure Reports, 5(2), 61–68.
Iaccarino, G., et al. (2005). Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. European Heart Journal, 26(17), 1752–1758.
Iaccarino, G., et al. (1999). Bbeta-adrenergic receptor kinase-1 levels in catecholamine-induced myocardial hypertrophy: Regulation by beta- but not alpha1-adrenergic stimulation. Hypertension, 33(1 Pt 2), 396–401.
Lamba, S., & Abraham, W. T. (2000). Alterations in adrenergic receptor signaling in heart failure. Heart Failure Reviews, 5(1), 7–16.
Keys, J. R. & Koch, W. J. (2004). The adrenergic pathway and heart failure. Recent progress in hormone research, 59, 13–30.
Nishikawa, M., et al. (1993). Differential down-regulation of pulmonary beta 1- and beta 2-adrenoceptor messenger RNA with prolonged in vivo infusion of isoprenaline. European Journal of Pharmacology, 247(2), 131–138.
Yeager, J. C., & Iams, S. G. (1981). The hemodynamics of isoproterenol-induced cardiac failure in the rat. Circulatory Shock, 8(2), 151–163.
Maisel, A. S., et al. (1989). Regulation of cardiac beta-adrenergic receptors by captopril. Implications for congestive heart failure. Circulation, 80(3), 669–675.
Murray, D. R., Prabhu, S. D., & Chandrasekar, B. (2000). Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation, 101(20), 2338–2341.
Nerme, V., Abrahamsson, T., & Vauquelin, G. (1990). Chronic isoproterenol administration causes altered beta adrenoceptor-Gs-coupling in guinea pig lung. Journal of Pharmacology and Experimental Therapeutics, 252(3), 1341–1346.
Jia, Y. X., et al. (2006). Apelin protects myocardial injury induced by isoproterenol in rats. Regulatory Peptides, 133(1–3), 147–154.
Montgomery, R. L., et al. (2008). Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. Journal of Clinical Investigation, 118(11), 3588–3597.
Reiken, S., et al. (2003). Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. Journal of Biological Chemistry, 278(1), 444–453.
Zhang, Z. S., et al. (2005). Enhanced inhibition of L-type Ca2 + current by beta3-adrenergic stimulation in failing rat heart. Journal of Pharmacology and Experimental Therapeutics, 315(3), 1203–1211.
Borlak, J., & Thum, T. (2003). Hallmarks of ion channel gene expression in end-stage heart failure. FASEB Journal, 17(12), 1592–1608.
Grimm, D., et al. (1998). Development of heart failure following isoproterenol administration in the rat: Role of the renin-angiotensin system. Cardiovascular Research, 37(1), 91–100.
Yan, Y. H., et al. (2008). Effects of diesel exhaust particles on left ventricular function in isoproterenol-induced myocardial injury and healthy rats. Inhalation Toxicology, 20(2), 199–203.
Grimm, D., et al. (1999). Effects of beta-receptor blockade and angiotensin II type I receptor antagonism in isoproterenol-induced heart failure in the rat. Cardiovascular Pathology, 8(6), 315–323.
Brouri, F., et al. (2002). Toxic cardiac effects of catecholamines: Role of beta-adrenoceptor downregulation. European Journal of Pharmacology, 456(1–3), 69–75.
Brouri, F., et al. (2004). Blockade of beta 1- and desensitization of beta 2-adrenoceptors reduce isoprenaline-induced cardiac fibrosis. European Journal of Pharmacology, 485(1–3), 227–234.
Zhang, G. X., et al. (2005). Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovascular Research, 65(1), 230–238.
Osadchii, O. E., et al. (2007). Cardiac dilatation and pump dysfunction without intrinsic myocardial systolic failure following chronic beta-adrenoreceptor activation. American Journal of Physiology. Heart and Circulatory Physiology, 292(4), H1898–H1905.
Harden, T. K., Su, Y. F., & Perkins, J. P. (1979). Catecholamine-induced desensitization involves an uncoupling of beta-adrenergic receptors and adenylate cyclase. Journal of Cyclic Nucleotide Research, 5(2), 99–106.
Benjamin, I. J., et al. (1989). Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circulation Research, 65(3), 657–670.
Friddle, C. J., et al. (2000). Expression profiling reveals distinct sets of genes altered during induction and regression of cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America, 97(12), 6745–6750.
Leenen, F. H., White, R., & Yuan, B. (2001). Isoproterenol-induced cardiac hypertrophy: Role of circulatory versus cardiac renin-angiotensin system. American Journal of Physiology. Heart and Circulatory Physiology, 281(6), H2410–H2416.
Gengo, P., et al. (1988). Regulation by chronic drug administration of neuronal and cardiac calcium channel, beta-adrenoceptor and muscarinic receptor levels. Biochemical Pharmacology, 37(4), 627–633.
Hayes, J. S., Pollock, G. D., & Fuller, R. W. (1984). In vivo cardiovascular responses to isoproterenol, dopamine and tyramine after prolonged infusion of isoproterenol. Journal of Pharmacology and Experimental Therapeutics, 231(3), 633–639.
Bos, R., et al. (2005). Inhibition of catecholamine-induced cardiac fibrosis by an aldosterone antagonist. Journal of Cardiovascular Pharmacology, 45(1), 8–13.
Oliveira, E. M., & Krieger, J. E. (2005). Chronic beta-adrenoceptor stimulation and cardiac hypertrophy with no induction of circulating renin. European Journal of Pharmacology, 520(1–3), 135–141.
Takeshita, D., et al. (2008). Isoproterenol-induced hypertrophied rat hearts: Does short-term treatment correspond to long-term treatment? The Journal of Physiological Sciences, 58(3), 179–188.
Johar, S., et al. (2006). Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB Journal, 20(9), 1546–1548.
Sakata, Y., et al. (2003). Angiotensin II type 1 receptor blockade prevents diastolic heart failure through modulation of Ca(2 +) regulatory proteins and extracellular matrix. Journal of Hypertension, 21(9), 1737–1745.
Di Zhang, A., et al. (2008). Cross-talk between mineralocorticoid and angiotensin II signaling for cardiac remodeling. Hypertension, 52(6), 1060–1067.
Freund, C., et al. (2005). Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation, 111(18), 2319–2325.
Beck, A., et al. (1985). Angiotensin-induced hypertension in conscious dogs: Biochemical parameters and baroreceptor reflex. Cardiovascular Research, 19(11), 721–726.
Cao, R. Y., et al. (2010). The murine angiotensin II-induced abdominal aortic aneurysm model: Rupture risk and inflammatory progression patterns. Frontiers in Pharmacology, 1(9), 1–7.
Perrino, C., et al. (2006). Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. Journal of Clinical Investigation, 116(6), 1547–1560.
Hershman, J. M. & March (2008). Hypothyroidism: Thyroid Disorders: Merck Manual Professional. Merck.
Maitra, A. & Abbas, A. K. (2005). The endocrine system. In V. Kumar, A. K. Abbas, & N. Fausto (Eds.), Robbins and Cotran pathologic basis of disease. Philadelphia, PA: Elsevier Saunders, pp. 1155–1226.
Gay, R. G., et al. (1988). Effects of thyroid state on venous compliance and left ventricular performance in rats. American Journal of Physiology, 254(1 Pt 2), pp. H81–H88.
Tang, Y. D., et al. (2005). Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation, 112(20), 3122–3130.
Liu, Z., & Gerdes, A. M. (1990). Influence of hypothyroidism and the reversal of hypothyroidism on hemodynamics and cell size in the adult rat heart. Journal of Molecular and Cellular Cardiology, 22(12), 1339–1348.
Kisso, B., et al. (2008). Effect of low thyroid function on cardiac structure and function in spontaneously hypertensive heart failure rats. Journal of Cardiac Failure, 14(2), 167–171.
Schuyler, G. T., & Yarbrough, L. R. (1990). Changes in myosin and creatine kinase mRNA levels with cardiac hypertrophy and hypothyroidism. Basic Research in Cardiology, 85(5), 481–494.
Seta, Y., et al. (1996). Basic mechanisms in heart failure: The cytokine hypothesis. Journal of Cardiac Failure, 2(3), 243–249.
Bryant, D., et al. (1998). Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation, 97(14), 1375–1381.
Panagopoulou, P., et al. (2008). Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. Journal of Cell Biology, 181(5), 761–775.
Prabhu, S. D. (2004). Cytokine-induced modulation of cardiac function. Circulation Research, 95(12), 1140–1153.
Mariappan, N., et al. (2007). TNF-alpha-induced mitochondrial oxidative stress and cardiac dysfunction: Restoration by superoxide dismutase mimetic Tempol. American Journal of Physiology. Heart and Circulatory Physiology, 293(5), H2726–H2737.
Bozkurt, B., et al. (1998). Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation, 97(14), 1382–1391.
Biaggioni, I. (2007). The sympathetic nervous system and blood volume regulation: Lessons from autonomic failure patients. American Journal of the Medical Sciences, 334(1), 61–64.
Gradin, K., Elam, M. & Persson, B. (1985). Chronic salt loading and central adrenergic mechanisms in the spontaneously hypertensive rat. Acta Pharmacologica et Toxicologica (Copenh), 56(3), 204–213.
Gradin, K., et al. (1988). Adrenergic mechanisms during hypertension induced by sucrose and/or salt in the spontaneously hypertensive rat. Naunyn-Schmiedebergs Arch Pharmacol, 337(1), 47–52.
Takata, Y., et al. (1988). Central and peripheral mechanisms of the enhanced hypertension following long-term salt loading in spontaneously hypertensive rats. Japanese Circulation Journal, 52(11), 1317–1322.
Ahn, J., et al. (2004). Cardiac structural and functional responses to salt loading in SHR. American Journal of Physiology. Heart and Circulatory Physiology, 287(2), H767–H772.
Varagic, J., et al. (2006). Myocardial fibrosis, impaired coronary hemodynamics, and biventricular dysfunction in salt-loaded SHR. American Journal of Physiology. Heart and Circulatory Physiology, 290(4), H1503–H1509.
Watson, P. A., et al. (2007). Restoration of CREB function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. American Journal of Physiology. Heart and Circulatory Physiology, 293(1), H246–H259.
Miyachi, M., et al. (2009). Exercise training alters left ventricular geometry and attenuates heart failure in dahl salt-sensitive hypertensive rats. Hypertension, 53(4), 701–707.
Yu, H. C., et al. (1998). Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation, 98(23), 2621–2628.
Radin, M. J., et al. (2008). Salt-induced cardiac hypertrophy is independent of blood pressure and endothelin in obese, heart failure-prone SHHF rats. Clinical and Experimental Hypertension, 30(7), 541–552.
Iwanaga, Y., et al. (2001). Differential effects of angiotensin II versus endothelin-1 inhibitions in hypertrophic left ventricular myocardium during transition to heart failure. Circulation, 104(5), 606–612.
Takenaka, H., et al. (2006). Angiotensin II, oxidative stress, and extracellular matrix degradation during transition to LV failure in rats with hypertension. Journal of Molecular and Cellular Cardiology, 41(6), 989–997.
Ogata, T., et al. (2004). Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. Journal of the American College of Cardiology, 43(8), 1481–1488.
Kramer, F., et al. (2008). Plasma concentrations of matrix metalloproteinase-2, tissue inhibitor of metalloproteinase-1 and osteopontin reflect severity of heart failure in DOCA-salt hypertensive rat. Biomarkers, 13(3), 270–281.
Bjelogrlic, S. K., et al. (2007). Effects of dexrazoxane and amifostine on evolution of Doxorubicin cardiomyopathy in vivo. Experimental Biology and Medicine, 232(11), 1414–1424.
Xu, M., et al. (2008). Protective effect of the endothelin antagonist CPU0213 against isoprenaline-induced heart failure by suppressing abnormal expression of leptin, calcineurin and SERCA2a in rats. Journal of Pharmacy and Pharmacology, 60(6), 739–745.
Suzuki, M., et al. (1998). Altered inotropic response of endothelin-1 in cardiomyocytes from rats with isoproterenol-induced cardiomyopathy. Cardiovascular Research, 39(3), 589–599.
Meszaros, J., & Levai, G. (1990). Ultrastructural and electrophysiological alterations during the development of catecholamine-induced cardiac hypertrophy and failure. Acta Biologica Hungarica, 41(4), 289–307.
Teerlink, J. R., Pfeffer, J. M., & Pfeffer, M. A. (1994). Progressive ventricular remodeling in response to diffuse isoproterenol-induced myocardial necrosis in rats. Circulation Research, 75(1), 105–113.
Bruch, C., et al. (2000). Tei-index in patients with mild-to-moderate congestive heart failure. European Heart Journal, 21(22), 1888–1895.
Kim-Mitsuyama, S., et al. (2004). Additive beneficial effects of the combination of a calcium channel blocker and an angiotensin blocker on a hypertensive rat-heart failure model. Hypertension Research, 27(10), 771–779.
Acknowledgments
The authors thank and acknowledge Drs. Urmila Kodavanti of the U.S. EPA and David Kurtz of Experimental Pathology Laboratories for their reviews of this manuscript.
Disclaimer
This paper has been reviewed and approved for release by the National Health and Environmental Effects Research Laboratory, U.S. EPA. Approval does not signify that the contents necessarily reflect the views and policies of the U.S. EPA, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Declaration of interest
Alex Carll is supported by UNC/EPA CR83323601.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carll, A.P., Willis, M.S., Lust, R.M. et al. Merits of Non-Invasive Rat Models of Left Ventricular Heart Failure. Cardiovasc Toxicol 11, 91–112 (2011). https://doi.org/10.1007/s12012-011-9103-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-011-9103-5