
Abstract
Around three out of one hundred thousand people are diagnosed with glioblastoma multiforme, simply called glioblastoma, which is the most common primary brain tumor in adults. With a dismal prognosis of a little over a year, receiving a glioblastoma diagnosis is oftentimes fatal. A major advancement in its treatment was made almost two decades ago when the alkylating chemotherapeutic agent temozolomide (TMZ) was combined with radiotherapy (RT). Little progress has been made since then. Therapies that focus on the modulation of autophagy, a key process that regulates cellular homeostasis, have been developed to curb the progression of glioblastoma. The dual role of autophagy (cell survival or cell death) in glioblastoma has led to the development of autophagy inhibitors and promoters that either work as monotherapies or as part of a combination therapy to induce cell death, cellular senescence, and counteract the ability of glioblastoma stem cells (GSCs) for initiating tumor recurrence. The myriad of cellular pathways that act upon the modulation of autophagy have created contention between two groups: those who use autophagy inhibition versus those who use promotion of autophagy to control glioblastoma growth. We discuss rationale for using current major therapeutics, their molecular mechanisms for modulation of autophagy in glioblastoma and GSCs, their potentials for making strides in combating glioblastoma progression, and their possible shortcomings. These shortcomings may fuel the innovation of novel delivery systems and therapies involving TMZ in conjunction with another agent to pave the way towards a new gold standard of glioblastoma treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme, commonly called glioblastoma, is the most common and aggressive primary brain tumor in adults. With an occurrence of about 3.19 cases per 100,000 people and a median overall survival (OS) rate of 14.6 months, the prognosis of glioblastoma patients undergoing the current standard of treatment is grim [1, 2]. Glioblastoma is histologically characterized by anaplasia, high levels of mitotic activity, cellular pleomorphism, nuclear atypia, and coagulation necrosis. They are high grade, infiltrative astrocytomas with extensive vascular proliferation. The diffuse and infiltrative nature of these tumors prevents complete surgical resection, leaving malignant cells that can prompt recurrence of the disease. Radiotherapy (RT) and concomitant and adjuvant chemotherapy follow surgical resection to prevent recurrence and eliminate remaining tumorigenic cells. RT is typically administered in fractions of 1.8 to 2.0 Gy amounting to a total of 58 to 60 Gy at the site of tumor resection, while concomitant chemotherapy with the alkylating agent temozolomide (TMZ) is given daily at a dosage of 75 mg/m2 at the site of tumor resection. After a four-week break, TMZ is administered as adjuvant chemotherapy at about 150 to 200 mg/m2 for a maximum of six cycles [5]. The drug TMZ remains a landmark achievement since it is the only chemotherapeutic agent that has shown survival benefit in Phase III clinical trials; however, limitations in its efficacy prevent it from substantially improving the OS of all glioblastoma patients.
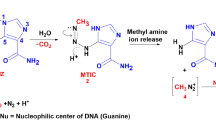
A major advantage of the DNA-alkylating chemotherapy TMZ is that it has the ability to penetrate the blood–brain barrier (BBB). When the drug is orally administered, it converts into the active metabolite 5-(3-methyltriazen-1-yl)-imidazole-4-carboxamide (MTIC) at physiologic pH. MTIC is an unstable compound that readily degrades into the highly reactive methyldiazonium cation, a powerful methylating agent. The most common DNA lesions that are produced by the methyldiazonium ion are at the N7 position of guanine, N3 position of adenine, and the O6 position of guanine (O6-MeG). Although methylation of the O6 guanine is the least frequent, it is directly involved in induction of apoptosis in methylguanine-DNA methyltransferase (MGMT)-deficient cancer cells. MGMT is a mismatch repair (MMR) enzyme that readily repairs O6-MeG in normal cells by removing the methyl adduct. However, errors occur in DNA replication in MGMT-deficient cells, activating the DNA MMR pathway. Since the MMR pathway cannot repair the O6-MeG, which remains in the template strand, repeated cycles of reinsertion and excision carried out by the pathway lead to extensive DNA resection and induction of apoptosis in the cells [3].
A limiting factor of TMZ chemotherapy is that only 45% of patients have MGMT-promoter methylation, which inhibits the activation of MGMT. The other 55% of patients experience only little to no clinical benefits from TMZ chemotherapy [4]. Another limiting factor includes the acquired resistance of the tumor cells to TMZ. In a landmark study, it was found that patients that had undergone prolonged chemotherapy had tumors consisting of hypermutated clones with mutations in the mutator S homolog 6 (MSH6) DNA-repair enzyme [6]. This shows that the current method of treatment can still cause recurrent, increasing growth of glioblastoma even in patients that have MGMT-promoter methylation.
The poor prognosis that glioblastoma carries has spurred the investigation of alternative therapeutic options that can address the limitations of this gold standard of therapy and increase patient OS and progression free survival (PFS). Examples of alternative therapeutic options include anti-angiogenic therapy, targeted molecular therapy, tumor-treating fields (TTFields, otherwise known as alternating electric fields or AEFs), and immunotherapy. However, therapeutic barriers like treatment resistant glioblastoma stem cells (GSCs), intratumoral and intertumoral heterogeneity, signaling pathway redundancy, and the BBB make creating an effective therapy difficult [7,8,9,10].
Alterations in three signaling pathways are known to be the main drivers for the emergence of glioblastoma and its cell proliferation. These drivers are the retinoblastoma (CDK4/6-p16-Rb1-E2F), the p53 (p14-MDM2-MDM4-p53), and the phosphoinositide 3-kinase (RTK-Ras-PI3K) pathways [13,14,15]. Specifically, around 90% of high-grade gliomas have PI3K pathway alteration, with the overexpression of a variety of cell surface tyrosine kinase receptors (TKRs)—like epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor-alpha (PDGFRα)—and the mutations in tumor suppressor genes such as the phosphatase and tensin homolog or PTEN gene [15]. Suppression of PTEN gene leads to activation of protein kinase B (also known as Akt), which promotes cell survival and proliferation in addition to altering the regulation of intracellular homeostasis processes like autophagy. The p53 protein encoded by the TP53 gene also plays essential roles in tumor suppression, impacting DNA repair, cell cycle arrest, and apoptosis [14]. Dysregulation of these pathways leads to the formation of tumorigenic properties that enforce therapeutic barriers.
GSCs are particularly challenging when devising therapies for glioblastoma. Not only are they resistant to standard RT and chemotherapy, but they also have an increased propensity for invading brain parenchyma, evading and suppressing the immune system, and promoting angiogenesis [11]. Autophagy, the process where cellular components and organelles are recycled to provide metabolic substrates for cellular metabolism, is a main driver in maintaining GSCs’ self-renewal, pluripotency, and tumorigenesis capabilities [12].
In normal cells, autophagy plays an important role in homeostasis, degrading long-lived proteins and damaged organelles [16] (Fig. 1). Within the context of the tumor microenvironment, autophagy plays a protective role, maintaining glioblastoma cell survival even in hypoxic and nutrient-deficient conditions and conferring resistance to glioblastoma cells even in the presence of chemotherapeutic agents like TMZ that can induce cytotoxicity [17] (Fig. 2). Autophagy also maintains glioblastoma survival before vascular proliferation is in full effect [16]. In late stages of tumor growth, autophagy increases metastasis, survival during therapy, and resistance to apoptosis [12]. By recycling damaged organelles, autophagy provides a source of metabolic plasticity for the tumor cells, aiding their survival, increasing chances of tumor recurrence, and conferring therapeutic resistance. Only in rare cases are the tumor cells overwhelmed with the quantities of metabolic and genotoxic stress to the level that leads to type II programmed cell death or autophagic cell death [149] (Fig. 3). In these cases, the entire cell is digested through the action of autolysosomes, and this only occurs in tumorigenic cells lacking the pro-apoptotic proteins Bax and Bak, preventing the utilization of type I programmed cell death [19]. However, most of the time, autophagy promotes tumor proliferation, and it is relatively uncommon for tumor masses to have mutations in autophagy-related genes [20].

Under normal physiological conditions, autophagy promotes cellular homeostasis by removing damaged organelles and protein aggregates, which result from typical sources of stress that a cell may experience

In the context of the tumor microenvironment, additional sources of stress such as hypoxia and nutrient starvation trigger autophagy, which acts cytoprotectively. Chemotherapy, radiotherapy, and targeted therapy can also induce autophagy by triggering metabolic and genotoxic stress. Autophagy lets tumor cells survive, increase chances of tumor recurrence, and confer therapeutic resistance

In rare cases, the tumor cells overwhelmed with metabolic and genotoxic stress succumb to type II programmed cell death or autophagic cell death via the digestion of the entire cell by autolysosomes. This pathway is a last resort in the event that pro-apoptotic proteins are not present in the cell, preventing type I programmed cell death or apoptosis
In the midst of therapeutic options that have had limited success in glioblastoma, modulation of autophagy regulation has become a new avenue of investigation. Although the process of autophagy itself has been well studied, its application to glioblastoma treatment has been controversial, with proposed interventions that either upregulate autophagy to cause autophagic cell death or downregulate autophagy to promote apoptosis. The divergence in approaches suggests that the usage of autophagy is context-dependent [18]. The focus of this article is to consider both conventional inhibition of autophagy and unconventional promotion of autophagy to predict what the future of autophagy-targeted treatments in glioblastoma will look like.
Autophagy and its importance in promoting and inhibiting glioblastoma growth
Autophagy plays an essential role in maintaining cellular homeostasis, and thus, is also implicated in cancer progression. Its involvement in the degradation of intracellular components to produce high-energy macromolecules that can maintain intracellular metabolism is what makes autophagy, specifically macroautophagy, a key target in developing novel therapies for glioblastoma.
There are three types of autophagy: macroautophagy, chaperone-mediated autophagy, and microautophagy. Macroautophagy is characterized by the formation of an autophagosome that engulfs complete regions of the cytosol and then fuses with a lysosome to digest its contents using hydrolases. Microautophagy does away with the autophagosome and involves the formation of invaginations in the lysosomal membrane that allows the movement of cytosolic regions into the lysosome for degradation. Lastly, chaperone-mediated autophagy is somewhat similar to microautophagy in that it does not involve the usage of an autophagosome; however, it is much more specific in that chaperones in the cytosol selectively recognize proteins that are lysosomal targets and then those proteins are able to cross the lysosomal membrane through interactions with receptor proteins at the surface of the lysosomal membrane [21]. Since macroautophagy, which we will refer to as autophagy from here on out, consists of the degradation and “recycling” of large quantities of intracellular materials, it is most heavily involved in tumor progression when the tumor microenvironment causes cellular stress.
Autophagy occurs at basal levels in most tissues, and it serves as a mechanism of quality control for cellular macromolecules and organelles. It is stimulated by several signals, including nutrient and growth factor deficiency, decreased energy status, increased oxidative or endoplasmic reticulum (ER) stress, and pathogenic infection, with the most well-studied being nutrient starvation [20, 22]. At tumor initiation phase, autophagy promotes tumor suppression by providing an anti-inflammatory function, preserving genetic and genomic stability, degrading potentially oncogenic proteins, participating in anti-cancer immunosurveillance, maintaining a normal metabolism with optimal bioenergetics, and having anti-viral and anti-bacterial effects [23]. However, in progressive and established tumors, autophagy can play a different role that enhances tumor progression. The tumor microenvironment is conducive to the upregulation of autophagy since the lack of expression of tumor suppression genes and the overexpression of oncogenes lead to high levels of cell proliferation that result in hypoxia, nutrient deficiency, and growth factor deprivation [24]. In response to these stressors, autophagy can take a cytoprotective role by preventing the toxic accumulation of the damaged proteins and organelles, reducing the sensitivity of tumor cells to microenvironmental factors that normally result in type I programmed cell death or apoptosis [23]. Not only does autophagy protect the tumor mass from death-inducing factors like hypoxia and nutrient starvation, but it can also promote survival in the presence of RT, chemotherapy, and targeted agents, ultimately promoting therapeutic resistance [24]. In well-established tumors, autophagy confers resistance to starvation, endothelial-to-mesenchymal transition, hypoxia, therapy-induced cell death, the maintenance of cancer stem cells, and the survival of senescent cancer cells that can support disease relapse [23, 25] (Fig. 4).

Repeated and chronic stress can lead to the activation of multiple pathways to mediate DNA damage. Cellular senescence is one mechanism that provides “time” for cells to undergo DNA repair and to evade apoptosis, and usually occurs during the G1/S or G2/M interfaces of the cell cycle [159]. However, it can be a mechanism used by cancer cells and the tumor microenvironment (TME) to escape cell death and promote disease relapse. In case of cancer-associated fibroblasts (CAFs) and glioblastoma cells, TMZ-induced DNA damage led to induction of autophagy and mitophagy in a transient manner, leading to mitochondrial dysfunction and ultimately senescence [43, 160]. Induction of senescence in CAFs promoted tumor growth and metastasis, potentiating tumorigenesis [160]. On the other hand, in the context of stem cells, senescence impedes their ability to regenerate and proliferate, so activation of autophagy acts as a mechanism of evading induction of senescence to maintain characteristics of stemness [37, 159]
The molecular machinery of autophagy is quite complex as it is tightly regulated in the cells. Mechanistic target of rapamycin complex 1 (mTORC1) is the best understood major regulator of autophagy [26]. Aa a part of the PI3K-family, mTORC1 is regulated by the cell-surface tyrosine kinase receptors (TKRs) that are commonly dysregulated in malignant tumors. An upstream negative regulator of mTORC1 is adenosine 5′ monophosphate-activated protein kinase (AMPK), which is upregulated in hypoxic environments and in the presence of genotoxic stress, but AMPK is down regulated during high energy availability [26, 27]. mTORC1 is an upstream regulator and inhibitor of Unc-51 like autophagy activating kinase 1 (ULK1), autophagy-related gene 13 (Atg13), and focal adhesion kinase family-interacting protein of 200 kDa (FIP200), ultimately leading to the repression of autophagy [27]. During nutrient deficiency, mTORC1 expression is inhibited via AMPK, leading to an increase in autophagy [28].
Two pathways, which are commonly altered in glioblastoma and connected to the molecular machinery of autophagy include the p53 pathway and the RTK-Ras-PI3K pathway. Most glioblastomas have dysregulation of the p53 pathway either through the mutation or deletion of the TP53 gene that occurs in about 28% of glioblastomas, the amplification of mouse double minute 2/4 homolog (MDM2/4) that occurs in about 15% of glioblastomas, or the deletion of cyclin-dependent kinase inhibitor 2A (CDKN2A) that occurs in about 58% of glioblastomas [14]. Alterations in TP53 are generally mutually exclusive with MDM2/4 mutations and are more common in secondary glioblastomas, which usually occur in younger patients and are less malignant [29]. In the RTK-Ras-PI3K pathway, about 90% of high-grade gliomas have PI3K pathway alterations, and about 50% of glioblastomas show epidermal growth factor receptor (EGFR) amplification, with the most common EGFR mutation being the intragenic rearrangement and in-frame deletion of exons 2–7 that produces the EGFRvIII allele [15, 30].
The dual role of autophagy, promoting tumor suppression during initial stages of tumor development and enhancing tumor progression in later stages, is reflected in the molecular pathways that glioblastoma progression and autophagy regulation share. In early stages of tumorigenesis, hyperactivation of p53 due to genotoxic stress causes the activation of AMPK, which suppresses the formation of mTORC1 and indirectly promotes autophagy, counteracting tumor formation. However, gain-of-function mutations in p53 protein suppressing autophagy have been observed, leading to an accumulation of cellular damage that promotes tumorigenesis [31]. Autophagy-defective cells have shown an accumulation of sequestosome 1 (SQSTM1, also known as p62), ER chaperones, damaged mitochondria, reactive oxygen species (ROS), and genome damage, leading to alterations in nuclear factor-kappa B (NF-κB) regulation, and promoting tumor growth and the formation of tumor cells with giant nuclei exhibiting aberrations in ploidy [33].
During later phases of tumor progression, at the time when multiple genetic mutations have already accrued, autophagy can promote cancer growth and survival and confer resistance to therapeutic agents. Dysregulated EGFR signaling causes changes in gene expression and cytoskeletal rearrangement, inhibits apoptosis, and increases cell proliferation. EGFR-dysregulated tumors are more dependent on autophagy for survival and use autophagy to increase resistance against EGFR-targeting agents [32]. The usage of autophagy against current therapeutic agents renders them largely ineffective even though many cancerous tumors present EGFRvIII mutations. Moreover, DNA damage can activate ataxia telangiectasia mutated (ATM), a serine/threonine kinase, via double strand breaks leading to phosphorylation of PTEN, a tumor suppressor. Phosphorylation of PTEN leads to nuclear translocation, which in turn, activates autophagy through the p-Jun-SESN2-AMPK pathway, promoting survival even in the presence of DNA-damaging treatment [34]. Furthermore, increased expression of autophagic markers Beclin-1 and light chain-3 II (LC3II) correlates with decreased progression-free survival (PFS) and high-grade gliomas. Although autophagy was functionally non-defective in both low-grade and high-grade gliomas, high-grade gliomas showed increased turnover of autophagy [35].
Autophagy also plays a major role in maintaining the stemness of GSCs and in promoting their migration and invasion. Cancer stem cells are critical in conferring therapeutic resistance and in tumor initiation due to their capacity for self-renewal and pluripotency [11, 36]. In glioblastoma, not only can GSCs give rise to a variety of neural stem cell types, but they can also transdifferentiate into tumor endothelial cells, which promote angiogenesis [39]. Autophagy maintains the stemness of GSCs by preventing cellular senescence, protecting GSCs against irradiation injury, and by producing metabolites that correlate with gene profiles normally associated with stemness. In ageing cells, autophagy failure leads to increases in mitochondrial dysfunction and oxidative stress, which trigger senescence via a loss of organelle and protein homeostasis, while the regenerative properties of stem cells can be restored through the re-establishment of autophagy [37]. Moreover, when autophagy was promoted via starvation or rapamycin (an mTORC1 inhibitor), ROS accumulation induced by irradiation was decreased, maintaining the stemness of mesenchymal stem cells (MSCs). On the other hand, when autophagy was inhibited, ROS accumulation and DNA damage was increased, ultimately leading to the loss of stemness of MSCs [36]. In another study, cancer cells that were cultured with lactates and ketones, the high energy metabolites often produced by aerobic glycolysis and fibroblastic production resulting from autophagy, demonstrated genetic profiles that correlated with stemness and poor clinical outcome [38]. Stressors like nutrient starvation, hypoxia, and RT and chemotherapy activate the metabolic autophagy pathway in the established tumors. This causes the expression of high levels of autophagy-associated factors like the DNA damage-regulated autophagy modulator 1 (DRAM1) and the SQSTM1 that are correlated with shorter OS in glioblastoma patients and with greater potential of the tumor for migration and invasion. Along similar lines, autophagy is induced at greater levels in GSCs that are actively migrating and invading [40].
Autophagy maintains cellular homeostasis through the complex mechanisms, as we schematically outlined (Fig. 5). The mechanistic pathway of autophagy induction overlaps with pathways that are commonly altered in cancers and specifically in glioblastoma. The complexity of autophagy and its dual roles as a tumor suppressor and a tumor promoter yield multiple avenues of investigation in finding novel therapies to combat glioblastoma growth.

Pathways involved in autophagy regulation and autolysosome formation. Initiation of autophagy is caused by genotoxic or metabolic stress; examples include deletion of TP53 and mutation in EGFR. Stressors then modulate the mTORC1 or the Bcl-2 family of proteins through intermediates such as AMPK, MDM2, Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway, or PI3K. Inhibition of members of the mTORC1 promotes the formation of a complex (containing ULK1, ATG101, ATG13, and FIP200), which is necessary for the initiation of autophagy. Induction of anti-apoptotic members of the Bcl-2 family of proteins can cause inhibition of Beclin-1 and autophagy, while activation of the Raf-mitogen activated protein kinase (MAPK) or extracellular signal regulated kinase 1/2 (ERK1/2) kinase-ERK1/2 (Raf-MEK-ERK1/2) pathway promotes Beclin-1 and, in turn, autophagy. Modulation of the Bcl-2 family also impacts apoptosis progression; for example, p53 may upregulate Puma and other pro-apoptotic proteins of the Bcl-2 family to trigger mitochondrial release of cytochrome c, leading to activation of downstream caspase cascade and ultimately induction of apoptosis [156,157,158]. The relationship between autophagy and apoptosis becomes even more complex with the consideration of mitophagy. Mitophagy, the selective degradation of dysfunctional mitochondria, can relieve the cell of pro-apoptotic signals, preventing induction of apoptosis [162]. Other proteins of note are LC3I and LC3II, which are important in the lengthening of the autophagosome and its fusion with the lysosome to form the autolysosome. Modulation of various key elements in these pathways is the basis for autophagy inhibition and promotion for therapies
Autophagy modulates efficacy of TMZ in glioblastoma
As mentioned earlier, TMZ is a DNA-alkylating chemotherapeutic agent that is part of the gold standard of therapy currently administered to patients diagnosed with glioblastoma [2]. Its ability to methylate the O6 position of guanine in DNA is cytotoxic and causes cancer cells to undergo programmed cell death or apoptosis. Even though TMZ is a breakthrough therapy in that it is one of the only chemotherapies that have shown consistent increase in OS and PFS even in Phase III clinical trials, it is still largely ineffective due to the intracellular presence of mismatch-repair enzymes like MGMT and processes like autophagy that confer resistance to RT and chemotherapy [4, 6, 41].
Administration of TMZ has been correlated with an induction of autophagy in glioblastoma. Immunohistochemical analyses of the proteins LC3B, lysosome-associated membrane protein (LAMP)-1, and LAMP-2 implicated in autophagy progression reflect an increase in autophagy in surgical glioblastoma tissues after TMZ treatment [42]. The induction of autophagy due to TMZ administration is thought to occur through the sustained inhibition of the Akt/mTOR pathway, which negatively regulates autophagy. In addition to causing an uptick in autophagy, Akt/mTOR inhibition suppresses apoptosis. A transient induction of autophagy is thought to confer resistance to TMZ therapy in glioblastoma cells [43]. Following TMZ treatment, an increase in autophagy in glioblastoma cells has been associated with a concomitant surge in ATP production. The surge in ATP indicates that autophagy has a cytoprotective effect against TMZ therapy [41]. It has been suggested that TMZ mainly exerts its cytotoxic activity through pro-autophagic processes [44].
Increased autophagy has an inverse effect on TMZ sensitization of glioblastoma cells. The autophagy-associated ATP surge recorded in the tumor cells treated with TMZ protects the cells from drug-induced cell death by providing the energy required to meet intracellular metabolic requirements, ultimately contributing to drug resistance [41]. In addition, to compensate for a loss of nutrient uptake, autophagy can provide growth-factor deprived cells with substrates to maintain catabolism and ATP production [45]. Processes like autophagy strengthen and potentiate genomic instability by circumventing programmed cell death [43].
Even more disturbing is TMZ's effect on GSCs. Even after therapeutic treatment of glioblastoma involving surgical resection, RT, and chemotherapy with TMZ, disease relapse is almost impossible to prevent due to the presence of endogenous tumor cells that have cancer stem cell properties, giving them the ability to reproduce highly proliferative tumor cell populations [46]. When treated with TMZ, members of the non-GSC population started to exhibit GSC markers like CD133 + , Sox2, Oct4, and Nestin. These newly formed GSCs acquired the phenotypic and functional characteristics of the native GSCs of the original tumor [47]. Similarly, hypoxia-inducible factors (HIFs) can induce conversion between non-CSC and CSC cell types, as HIF-1α induced autophagy has the ability of converting non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells [48]. The acquisition of these characteristics gives a way for the replenishment of the original tumor population even after surgical resection, RT, and chemotherapy. Moreover, the novel tumor population is increasingly infiltrative and chemoresistant, leading to decreased response to later rounds of treatment [47].
Since TMZ is an autophagy-inducer, multiple therapeutic avenues are under investigation in determining which type of cell death will best counteract glioblastoma progression. Currently, both autophagy inhibitors and promoters are under consideration. Autophagy inhibitors can be broken down by the point of autophagy progression that is under examination. Early-stage blockers or inhibitors of autophagy are often combined with adjuvant RT and chemotherapy with TMZ to activate apoptosis pathways resulting in cell death. These inhibitors prevent the formation of autophagosomes, block an important source of energy for tumor cells, and eliminate the autophagy’s cytoprotective effect [49]. A common early-stage autophagy inhibitor is 3-methyladenine (3-MA), which suppresses class III PI3Ks that positively regulate autophagy [50]. In addition, the ATP surge produced by TMZ-associated autophagy could also be blocked via the small interfering RNA (siRNA) technology targeting of Beclin-1 [41]. However, early-stage autophagy inhibitors can have some negative effects on tumor suppression. For example, in one study, when autophagy was inhibited at an early stage during TMZ treatment, the anti-tumor effect of TMZ was suppressed [51].
The ineffectiveness of using solely early-stage autophagy inhibitors has called for the usage of late-stage autophagy inhibitors and possibly even autophagy promoters. Although late-stage inhibitors allow the formation of the autophagosome, they prevent the fusion of the autophagosome with lysosomes, impeding their ability to recycle cellular components to produce energy. Not only do late-stage inhibitors repress the tumor cells' acquisition of energy, but they also load tumor cells with non-degradable autophagic vesicles and cause waste loading that ultimately leads to cell death [43, 49]. Bafilomycin A1, which is a compound commonly used to inhibit autolysosome formation, has been observed to enhance apoptosis through the release of pro-apoptotic agents like cathepsin B from lysosomes and through mitochondrial membrane permeabilization (MMP) [51]. A proposed model of treatment using late-stage inhibitors includes starting treatment with PI3K/Akt/mTOR suppressors to promote autophagy, then using late-stage inhibitors in conjunction with TMZ and RT to induce high levels of autophagosome accumulation and prevent functional autophagy to lead to cell death [49]. Autophagy promoters can also cause autophagic cell death via induction of autophagy through the usage of continued RT, TMZ, and the suppression of the PI3K/Akt/mTOR pathway to cause uncontrolled intracellular digestion that ultimately leads to cell death [49].
TMZ induces autophagy, while autophagy increases tumor resistance to TMZ by maintaining optimal intracellular bioenergetics and inducing stemness in non-GSC populations. This leads to the genesis of increasingly resistant tumor populations and poor patient outlook. Given the integral role of autophagy in glioblastoma progression, its utilization has been approached from different angles. This array of autophagy-targeted therapeutics shows varying levels of success, leading to controversy and begging for additional investigation.
Targeting autophagy as a therapeutic strategy in glioblastoma
Due to the limited efficacy of the current gold standard of treatment for glioblastoma, combination therapies are becoming an increasingly favorable option to increase the number of potential targets and improve the survival of patients diagnosed with glioblastoma. Combination therapies that target and inhibit autophagy to increase chemo- and radio-sensitivity or those that enhance autophagy to induce type II programmed cell death are constantly being developed.
Currently, the one of the only United States Food and Drug Administration (FDA)-approved drugs that have been used in the context of combination treatment to combat glioblastoma is Bevacizumab (BEV, Avastin®), which is a recombinant humanized vascular endothelial growth factor-A (VEGF-A)-specific monoclonal antibody that inhibits tumor angiogenesis and normalizes mature tumor vasculature. BEV has been shown to increase PFS of glioblastoma patients in a variety of clinical trials [52,53,54]. Although BEV has shown promise as a monotherapy, it greatly improved the PFS of patients when combined with Irinotecan, a topoisomerase I inhibitor (29% PFS in monotherapy versus 46% PFS for combination therapy) [53, 55]. The improved efficacy of BEV when combined with Irinotecan can be attributed to increased uptake of Irinotecan into the central nervous system (CNS) and/or the tendency for BEV to target GSCs and for Irinotecan to target differentiated cells [56, 57]. However, cytoprotective autophagy can confer resistance to BEV and glioblastoma xenografts treated with BEV have shown increased hypoxia-associated autophagy, which ultimately promoted resistance to chemotherapy [58, 59]. This reinforces that even though BEV may be able to increase the duration of PFS in glioblastoma patients, the OS of glioblastoma patients does not increase because any recurring tumors become more biologically aggressive than the original tumor, which is reflected in the decreased post-operative survival rates in patients treated pre-operatively with BEV [60].
Another drug that has traditionally been used as a late-stage autophagy inhibitor for glioblastoma is chloroquine (CQ) and its less toxic metabolite, hydroxychloroquine (HCQ). CQ's anti-cancer properties include autophagy inhibition, DNA intercalation, and enhanced penetration of chemotherapeutic drugs in tumor masses [61]. CQ inhibits autophagy by increasing the vacuolar pH of acidic organelles, like lysosomes, due to its properties as a weak base. This prevents the fusion of autophagosomes with lysosomes and the ultimate formation of autolysosomes [62]. In Phase I/II clinical trials, it has been seen that LC3II accumulation in peripheral blood mononuclear cells (PBMCs), which indicates inhibition of autophagy, is increased as HCQ exposure is increased. However, at the maximum tolerated dose (MTD) of 600 mg/day, autophagy inhibition was inconsistent and there was no notable improvement in OS. At higher doses of HCQ (of around 800 mg/day), with RT and concurrent and adjuvant TMZ, all subjects experienced high grades of neutropenia and thrombocytopenia, with one patient going into sepsis. Thus, although HCQ and CQ have potent autophagy-inhibiting and anti-cancer properties, the toxicity of the drugs can make them rather prohibitive for clinical use [63].
To recruit drugs that have anti-cancer properties and minimal toxicity, researchers are turning to the use of phytochemicals, which are natural plant compounds that can be found in everyday foods or concentrated in dietary supplements [64]. The benefit of phytochemicals is that they have high efficacy and can impact multiple cellular pathways, have little side effects and toxicity, and they are relatively cheap to produce. Not only can phytochemicals enhance the efficacy of chemotherapeutic treatment, but they can also ameliorate adverse effects of RT and chemotherapy and decrease chemoresistance. However, certain herb–drug interactions can lead to adverse effects like liver injury, so these need to be investigated before putting certain phytochemical combination therapies into practice [65]. Phytochemicals can activate both pro-autophagic and pro-apoptotic pathways [66, 67]. Examples of phytochemicals that are apoptosis promoters and autophagy inhibitors are quercetin, silibinin, and luteolin when combined with the tumor suppressor microRNA (miR)-7–1-3p; proanthocyanidin (PAC) when combined with miR-30e (an autophagy inhibitor); and thymoquinone (TQ), resveratrol, and celastrol when combined with a heat-shock protein 90 (HSP90) inhibitor [68,69,70,71,72,73]. Specifically, celastrol is a late-stage autophagy inhibitor much like CQ; however, this phytochemical can increase lysosomal content at minimally toxic doses, unlike CQ, showing the therapeutic potential of using autophagy-inhibiting phytochemicals [74]. Some phytochemicals are both autophagy and apoptosis inducers, like cucurbitacin and evodiamine. Pharmacological inhibition of autophagy in combination with these phytochemicals has led to increased frequency of apoptosis and decreased tumor cell viability [75, 76]. Other phytochemicals that induce type II programmed cell death through autophagy induction are berberine, curcumin, and ursolic acid [77,78,79]. Phytochemicals target a variety of intracellular pathways, they relatively lack side effects when used in treatment, and they have lower cost of production, all of which make phytochemicals potentially desirable anti-cancer agents.
Long non-coding RNAs (lncRNAs) and microRNAs (miRs) have also been investigated to target the epigenetic regulation of autophagy in combating glioblastoma. These RNAs play an important role in epigenetic modification in almost every single cellular process, and when it comes to autophagy, they can regulate some ATG genes at specific stages of autophagy, including induction, vesicle nucleation, vesicle elongation, maturation, and lysosomal fusion [80]. An example of a lncRNA that has the ability to suppress autophagy and reduce chemoresistance in glioblastoma through the induction of cisplatin, a platinum-based drug, is AC023115.3. Studies show that AC023115.3 competes with miR-26a and acts as a “sponge” reducing the inhibitory effect of miR-26a on glycogen synthase kinase 3 beta (GSK3β), a serine-threonine kinase that causes a decrease in cytoprotective autophagy, which ultimately increases the chemosensitivity of glioblastoma cells [81]. In addition, as seen above with phytochemicals, miRs can be used in combination treatment to have an enhanced positive effect on decreasing tumor viability. For example, combining an miR that is a tumor suppressor and autophagy inhibitor, like miR-30e, with an apoptosis-inducer like PAC (a plant-derived polyphenol), can lead to increased apoptosis in GSCs [70]. Also, the use of miR-7-1-3p as a tumor suppressor allowed for the enhancement of the anti-tumor effects of luteolin and silibinin, with a focus on inducing apoptotic cell death rather than the induction of autophagy, which could play a cytoprotective role [69]. Using epigenetic modifiers like lncRNAs and miRs can provide more direct and efficient treatment when used in conjunction with other compounds like phytochemicals.
Modulating the patient immune system has been another avenue of treatment development in an attempt to counteract the immunosuppressive effects of glioblastoma. The brain was classically considered less immunologically active due to constraints placed by the BBB, the postulated lack of CNS lymphatic drainage, the limited number of resident cells expressing major histocompatibility complex (MHC) molecules, and the lack of antigen-presenting cells (APCs) [82, 83]. However, recent developments argue against this idea as it has been found that the BBB is not impenetrable when it comes to the recruitment of peripheral immune cells, T cells and antigens in the CNS are still able to drain into cervical lymphatics, and that resident macrophages of the CNS, otherwise known as microglia, can present MHCs [82]. The realized complexity of the immune system of the brain and CNS has allowed for newfound approaches for targeting autophagy and stimulating glioblastoma cell immunogenicity. One group used polyglycerol-functionalized nanodiamonds bearing doxorubicin (Nano-DOX) to stimulate glioblastoma cells to emit antigens and damage-associated molecular patterns (DAMPs) that resulted in the activation of dendritic cells (DCs), enhancing the tumor's ability to elicit an immune response [84]. The same group developed polyglycerol-functionalized nanodiamonds (dND-PG) as a drug delivery system that avoided macrophages and exhibited selective uptake in cancer cells to potentiate the cytotoxic effects of DOX [85]. Although DOX primarily could induce apoptosis via intercalation with DNA and inhibition of topoisomerase-II, it was seen that Nano-DOX exerted its ability to increase immunogenicity through the induction of autophagy [84]. Another study focused on the late-stage inhibition of autophagy to enhance anti-tumor immunity via the use of a histone deacetylase (HDAC) inhibitor. In the study, it was found that inhibition of glioblastoma cell proliferation via the HDAC6 inhibitor, J22352, correlated with the dosage of J22352 that was administered, and that J22352 enhanced anti-tumor immunity by decreasing programmed death-ligand 1 (PD-L1) expression, and promoting CD8 + T cell activation [86]. Overall, autophagy plays an important role in the regulation of intrinsic anti-tumor responses and using it as a target in the context of immunotherapy may promote tumor regression.
Another novel therapy that has shown promise in combating glioblastoma and improving the PFS and OS of glioblastoma patients is TTFields, as mentioned earlier. TTFields are alternating electrical fields of a low intensity that can disrupt mitosis in replicating cells, leading to cancer cell death [87]. In a clinical trial, concomitant administration of TTFields in conjunction with TMZ has been shown to increase PFS by about three months, and OS by around five months as opposed to the sole use of TMZ therapy [88]. Another study investigated the relationship between TTFields and autophagy. In this study, it was found that autophagy mediated TTFields-induced cell death, and that blocking autophagy limited the anti-tumor efficacy of TTFields treatment [89]. The promise TTFields show reflects how the convergence of multiple scientific fields can lead to innovative, game-changing treatments.
The complexity of glioblastoma and autophagy limits the efficacy of the gold standard of treatment; however, it paves the way for the discovery of new combination therapies. From using anti-angiogenesis drugs, to recruiting phytochemicals, and to using electrical fields, it seems that both autophagy promotion and inhibition can have anti-tumor properties and play a role in improving PFS and OS. Further investigation into these different avenues of treatment is required to fully understand the role of autophagy in glioblastoma progression, its implications in combination therapy, and whether the modulation of autophagy will ultimately be based on a patient’s individual physiology or the pathological characteristics of their specific case of glioblastoma.
Inhibition of autophagy as a conventional therapeutic strategy in glioblastoma
Since the discovery that autophagy plays a cytoprotective role in late stages of tumor progression, increasing focus has been placed on finding efficient autophagy inhibitors that sensitize glioblastoma cells to inducers of cell death and that are tolerable in terms of toxicity. The result has been the development of both early-stage and late-stage autophagy inhibitors that attack various parts of autolysosome formation. This section will describe these inhibitors, their efficacy, and evaluate the progress made starting from long-standing to recently developed therapeutics.
Early-stage autophagy inhibitors
Early-stage autophagy inhibitors target various substrates required in the initiation of autophagy, including PI3Ks and Unc-51-like autophagy activating kinase 1 (ULK1) (Table 1). PI3Ks are intracellular lipid kinases that phosphorylate the 3′-hydroxyl group of phosphatidylinositols, and they are split into three classes (classes I, II, and III). As master regulators, they are implicated in a variety of crucial cellular pathways, including those for cellular metabolism, survival and polarity, vesicle trafficking, autophagy, and more [90]. Although much research has gone into classifying the role of classes I and III PI3Ks, not as much is known about class II PI3Ks. Class I PI3Ks are important in glucose metabolism and can play a role in autophagy suppression through the activation of Akt (protein kinase B), which is a serine/threonine kinase implicated in mTOR regulation. Unlike class I PI3Ks, class III PI3Ks are implicated in vesicular trafficking and are positive regulators of autophagy [91]. Manipulation of class I and class III PI3Ks is the basic mechanism of PI3K-specific early-stage autophagy inhibitors. Examples of PI3K-specific early-stage autophagy inhibitors include 3-methyladenine (3-MA), wortmannin, LY294002, and pyrvinium.
One of the most common autophagy inhibitors used, other than CQ, is 3-MA. A classic PI3K inhibitor, 3-MA is mainly employed for its ability to inhibit class III PI3Ks and is often used in combination therapy to enhance the efficacy of other chemotherapeutic agents [92,93,94]. During nutrient starvation, which often occurs in the tumor microenvironment due to rapid cellular proliferation and lagging neovascularization, 3-MA persistently blocks both class I and class III PI3Ks, ultimately leading to inhibition of autophagy. However, another study reported that in nutrient-rich media, 3-MA could act similar to rapamycin, an autophagy promoter, due to its persistent blockage of class I PI3Ks and transient blockage of class III PI3Ks, raising possible concerns as to its efficacy as an autophagy inhibitor. In addition, it seems that 3-MA acts on multiple cellular pathways, affecting other metabolic processes such as proteolysis in Atg5-deficient cells [91]. As this is so, although 3-MA has been used in combination therapy as an autophagy inhibitor in a myriad of investigations, other PI3K autophagy inhibitors should be investigated as well.
Another common PI3K-specific early-stage autophagy inhibitor is LY294002, otherwise known as 2-(4-morpholinyl)-8-phenylchromone. Using a mechanism similar to 3-MA, LY294002 also exerts a transient effect on class III PI3K for its inhibition. This is most likely due to the structural similarity of LY294002 to adenosine triphosphate (ATP), limiting the efficacy of the drug due to competition between the two molecules [96]. The result is that a greater concentration of LY294002 is required in order to have an inhibitory effect similar to wortmannin, which is another PI3K inhibitor. However, even though LY294002 only transiently inhibits PI3Ks, it has been shown to have a similar effect on increasing TMZ cytotoxicity as CQ [99]. In the clinical context, when 44 patients with B-cell malignancies were treated with SF1126 (an RGDS peptide conjugate of LY294002), around 58% of the patients that could be evaluated had stable disease [97]. The peptide conjugate could provide an opportunity to circumvent some of the downsides of LY294002—such as limited bioavailability—to make clinical application more feasible.
The last major PI3K-specific early-stage autophagy inhibitor is wortmannin, which is an antifungal antibiotic [95]. Wortmannin, like 3-MA, affects both class I and class III PI3Ks; however, wortmannin persistently inhibits class III PI3Ks, while transiently inhibiting class I PI3Ks (the inverse of 3-MA), which means that wortmannin is still effective in high-nutrient conditions [91]. It is thought that the persistent inhibition of class III PI3Ks is due to the permanence of the covalent bonding wortmannin forms with the kinase [96]. In addition to abrogating the formation of autophagosomes, when wortmannin was used in combination treatment with RT, it reduced the size of spheroids of the M059J cell line (derived from a human glioblastoma) by 68%, indicating that wortmannin induced radio-sensitization in tumor cell populations [98]. However, it is possible that wortmannin may also promote tumor progression, as shown by a study focused on thrombin-induced migration. The necrotic lesions formed during the development of glioblastoma are vulnerable to thrombin exposure, which is a regulator of matrix metalloproteinase-9 (MMP-9) expression and cancer cell migration. When rat glioblastoma C6 cells were treated with wortmannin, thrombin-induced migration increased, which showed that it was possible that the PI3K-inhibiting effects of wortmannin acted to potentiate the migratory properties of tumor cells [95]. These results indicate that care needs to be taken when using PI3K inhibitors, as the expansiveness of cellular pathways regulated by PI3Ks can lead to undesirable results.
Pyrvinium pamoate, an FDA-approved anti-parasitic drug mainly used for treating pinworms, has also been shown to exert early-stage inhibitory effects on autophagy progression. Rather than acting directly on PI3Ks, pyrvinium has been shown to reduce the levels of Vps34, Beclin-1, p150, and Atg14 in a dose-dependent manner, ultimately leading to the inhibition of starvation and stimulation of autophagy flux [100]. Its low toxicity also makes it an attractive early-stage autophagy inhibitor. Moreover, pyrvinium has been shown to have inhibitory effects on the proliferation of GSCs. At low doses, pyrvinium targets the self-renewal and proliferative capacity of CD133+ cell lines, initiating cell death even in TMZ-resistant cell lines [101]. Pyrvinium's effect on tumor cell lines, and specifically on GSCs, makes it a promising contender for an effective treatment for glioblastoma.
A variety of phytochemicals and bioflavonoids inhibit early-stage autophagy. Quercetin, a bioflavonoid that is commonly found in foods like kale and red onions, has anti-carcinogenic, anti-inflammatory, and anti-viral activities; however, its usage in combination therapy has not been thoroughly investigated in the context of glioblastoma [102]. Due to quercetin's limited bioavailability during monotherapy, combination therapy with the HDAC inhibitor sodium butyrate led to a synergistic increase in apoptosis in glioblastoma cells via autophagy inhibition [68]. The undetected cleavage of LCB I to LCB II indicates that quercetin acts as an early-stage autophagy inhibitor and prevents the maturation of the autophagosome [68]. Although other research shows that quercetin may also act as an autophagy promoter [103], the difference in results may be attributed to whether the environment was nutrient rich or nutrient deficient. Much like 3-MA, when quercetin is used to combat glioblastoma proliferation in a nutrient-deprived environment, quercetin inhibits autophagy, and vice versa in a nutrient rich environment [68]. These studies show that the low toxicity of naturally derived flavonoids provides potential for their use in combination treatment.
Moreover, a plant-derived polyphenol, PAC, has the capability of acting synergistically with the targeted miRs to increase apoptosis in glioblastoma cell lines. In one study, PAC, a potent apoptosis promoter, is combined with miR-30e, which is an inhibitor of both autophagy promoters and apoptosis inhibitors, including Beclin-1, AVEN, and BIRC6 [70]. When used in combination therapy to combat hypoxic-induced autophagy, PAC and miR-30e exhibited synergistic inhibition of autophagy and induction of apoptosis [70]. This was confirmed by decreased expression of LCB II, increased mTOR signaling, and the presence of commonly known morphological features of apoptosis. Since this combination therapy was tested on both glioblastoma SNB19 cells and GSCs, this treatment showed promise for combating recurrent tumor formation.
The last early-stage inhibitor of autophagy described in this section is a polyphenolic small molecule, the ULK1 inhibitor known as SBI-0206965, which will be referred to as 6965. This drug could down regulate autophagy originally induced by mTOR inhibition, and after 18 h of amino acid deprivation, around 42% of cells treated with 6965 demonstrated late-stage apoptotic markers as opposed to only 15% of ULK1/ULK2 knockout mouse embryonic fibroblasts [104]. In addition, it only required 10 µM 6965 to elicit a pro-apoptotic response as opposed to 20 µM CQ, showing that it could be used to reduce treatment toxicity. ULK1 inhibitors show promise for use in combination therapy with mTOR inhibitors.
Late-stage autophagy inhibitors
Late-stage autophagy inhibitors rely on the accumulation of proteotoxic stress to trigger mitochondrial apoptotic pathways, and ultimately apoptotic cell death (Table 2). Instead of inhibiting the formation of autophagosomes, late-stage inhibitors prevent the fusion between the autophagosome and the lysosome, causing the accumulation of autophagosomes and other non-essential proteins in the cell that cannot be removed via autophagy. The mechanism by which fusion between the autophagosome and lysosome is prevented depends on the type of late-stage inhibitor. Fusion can be prevented via disorganization of the endo-lysosomal system, de-acidification of the lysosome, or increase in lysosomal permeability [51, 71, 105].
Although not FDA-approved for use as an autophagy inhibitor, Bafilomycin A1 (BafA1) is one of the most studied late-stage autophagy inhibitors. Vacuolar H+ ATPase (V-ATPase) is a lysosomal proton pump that increases the acidification of cellular microenvironments and confers therapeutic resistance to tumors via cytoprotective autophagy [106]. BafA1 is a selective inhibitor of V-ATPase, which prevents the acidification of lysosomes, thus, preventing the progression of autophagy. In addition to being a selective inhibitor of V-ATPase, BafA1 can reduce the stemness of glioblastoma cell lines, suppressing the expression of stem cell markers like nestin and CD133+ [106]. When used in combination treatment with TMZ, BafA1 synergistically decreases glioblastoma cell viability by inhibiting autophagy and activating caspase-3, an executioner of apoptosis. When compared to the early-stage autophagy inhibitor 3-MA, viability of cells treated with 3-MA combination treatment after three days was at 52%, while viability of cells treated with BafA1 combination treatment after the same period was only 19%, showing that the proteotoxic stress that resulted from autophagosome accumulation might hasten the activation of apoptotic pathways [51].
The only FDA-approved autophagy inhibitor, CQ and its derivative HCQ, can inhibit autophagy at a late-stage by disorganizing the endo-lysosomal system, rather than inhibiting lysosomal degradation capacity [105]. When tested for its ability to decrease lysosomal acidification, CQ did not impact LysoTracker Red staining in cell lines, while BafA1 did decrease LysoTracker Red staining, showing that CQ most likely did not prevent autophagy via lysosomal de-acidification. Moreover, the same group found that when cells were treated with CQ versus BafA1 and other lysosomal inhibitors, the cellular degradative compartments (DGCs) for the cells treated with BafA1 contained cytoplasmic components indicative of obstruction of degradation, while those treated with CQ did not have intact cytoplasmic materials and reflected the morphology of untreated cells, showing that CQ utilized a different mechanism to inhibit late-stage autophagy than BafA1 and other lysosomal inhibitors. As this is so, the suggested mechanism for CQ-mediated autophagy inhibition involved the redistribution and depletion of COPI subunits (the specific coat protein complex that initiates the budding process on the cis-Golgi membrane), causing defects in endosomal function and disorganization of the Golgi apparatus, ultimately preventing the fusion between autophagosomes and components of the endo-lysosomal system [105]. The discrepancy between the pathways acted upon by BafA1 and CQ raises a red flag, since CQ seems to have a more wide-spread effect on cell lines. In addition to adversely impacting endo-lysosomal function, CQ does increase lysosomal membrane permeability in glioblastoma cell lines [58]. In the context of combination therapy, when glioblastoma cells were treated with HCQ and BEV, HCQ potentiated the anti-cancer effects of BEV at a relatively low concentration [107]. Although CQ and HCQ are widely used autophagy inhibitors, the pathways they act on are still not entirely clear and merit additional investigation.
The success that CQ and HCQ has had in preventing autophagy has earned these drugs FDA-approval; however, their toxicity can make clinical applications daunting. Oftentimes, HCQ is as an alternative to CQ, but HCQ is also toxic when used in combination with other therapeutics such as TMZ. In a Phase II trial where HCQ was administered with RT and concomitant and adjuvant TMZ, when a continuous low-dosage schedule of TMZ was used, even low doses of HCQ (at around 800 mg) caused neutropenia and/or thrombocytopenia in all three glioblastoma patients in the trial [63]. This contrasts other clinical trials, where patients were able to tolerate doses of even 1200 mg HCQ; however, the discrepancy was most likely a result of a continuous dosing schedule in comparison to an intermittent dosing schedule. These results indicate that the toxicity of HCQ makes its clinical applications limited [63]. In another study focused on effects of CQ and HCQ on the retina, the investigators found that even at doses much lower than the recommended maximum, patients suffered from retinopathy that lasted even after CQ and HCQ treatment stopped, indicating that CQ and its derivative could cause retinal damage [108]. The widespread toxic effects of CQ and HCQ show that other late-stage autophagy inhibitors with less toxicity will need to be developed in order to have maximal use in clinical applications.
Flavonoids are a source of potential late-stage autophagy inhibitors that exhibit low toxicity. Two examples are celastrol, a triterpene derived from the Thunder God Vine, and TQ, the primary bioactive component of black seed oil [71, 74]. Celastrol promotes apoptosis by increasing p62 accumulation and proteotoxic stress due to autophagy inhibition. When glioblastoma cells were treated with cycloheximide (CHX), an inhibitor of de novo protein synthesis, cell death was greatly delayed, confirming that celastrol-induced cell death was dependent on excess p62 accumulation [74]. Unlike CQ, celastrol can inhibit autophagy flux at minimally toxic low doses while indirectly overburdening lysosomal degradation pathways [74]. Evidently, the autophagy-inhibiting and pro-apoptotic effects of celastrol in conjunction with its low toxicity make it an attractive and potential therapeutic agent to combat glioblastoma.
TQ primarily causes late-stage autophagy inhibition by increasing lysosomal membrane permeabilization, resulting in the translocation of lysosomal hydrolases to the cytosol. In addition, TQ affects lysosomal localization, negatively impacting autophagosome–lysosomal fusion [71]. Moreover, TQ can cause greater cytoplasmic vacuolization than CQ, making TQ a more effective autophagy inhibitor. The investigators also found that TQ concomitantly triggers apoptosis in a caspase-independent manner, which was reflected by a dose-dependent increase in propidium iodide and Annexin-V double positive cells. Contrary to the action of CQ, TQ is able to cause cell death in glioblastoma cell lines at low doses while not impacting normal astrocytes even at high doses. TQ at a dose of 2 µM affected glioblastoma cell proliferation, while doses of even up to 16 µM did not impact normal astrocyte growth [71]. TQ is also effective in combination therapy. In one study, when TQ was used in combination therapy with TMZ, 66% of the cells were apoptotic, while only 19% and 36% of cells treated with TMZ and TQ monotherapies, respectively, were apoptotic. They also noticed that the combination therapy reduced the colony-formation ability of glioblastoma cell lines [72]. The low toxicity of TQ, high efficacy at low doses, and promise in combination therapy opens the door for its use in glioblastoma treatment.
The cytoprotective effect of autophagy in the context of cancer has spurred the development of autophagy inhibitors with a variety of targets. These early-stage and late-stage inhibitors have varying levels of efficacy and toxicity, which impact their applicability to the clinical setting. As the mechanisms for long-standing early-stage and late-stage autophagy inhibitors are deduced, novel autophagy inhibitors should continue to be investigated to determine which therapeutic option has the highest efficacy and lowest toxicity in glioblastoma patients. Greater understanding of current therapeutics and the development of novel therapeutics will pave the way for creating safe and effective treatments for combating glioblastoma.
Promotion of autophagy as an unconventional therapeutic strategy in glioblastoma
Rather than inhibiting the already-produced side effects of conventional therapies, autophagy-inducers can capitalize on cytoprotective processes to cause type II programmed cell death, leading to decreased tumor growth and improving patient outlook. This unconventional approach to modulating autophagy in cancer cell lines has led to the development of novel treatments that can be classified by their corresponding targets in key pathways required for autolysosome formation. Although some of these treatments show promise, one of their major impediments is limited bioavailability, which has either been counteracted by developing variations of the therapeutic or through combination treatment. This section will mainly focus on three groups of treatments: tyrosine kinase inhibitors, mTOR inhibitors, and activators of the JNK pathway and inhibitors of the Bcl-2 family of proteins (Table 3).
Tyrosine kinase inhibitors
Two commonly overexpressed receptor tyrosine kinases are EGFR and the PDGFR-A, which lead to the overactivation of the Ras/Raf, MAPK, ERK, and PI3K/Akt/mTOR pathways in glioblastoma. Since these pathways are inherently implicated in important cellular processes such as cell proliferation, angiogenesis, migration, and invasion, their dysregulation leads to the overexpression of anti-apoptotic and cell survival proteins and the decreased expression of pro-apoptotic proteins [109]. The most common EGFR mutant is EGFRvIII, which involves the increase in transcriptions that do not contain exons 2–7 or alternative mRNA splicing that lacks amino acids 6–273. Approximately 50% of high-grade and low-grade astrocytomas contain this mutant, and it is a defining genetic characteristic for the classic glioblastoma gene expression subtype [110, 111]. On the other hand, PDGFR, with the most common isoform being PDGFR-A, is a defining genetic characteristic for the proneural glioblastoma gene expression subtype, and it is amplified in around 23% of glioblastomas [112].
Gefitinib, which is a small molecule and selective receptor tyrosine kinase inhibitor of EGFR, promotes autophagy via AMPK activation [113]. Although it has been previously shown that gefitinib promotes apoptosis via Bad phosphorylation, at lower concentrations, gefitinib also produces cytotoxic autophagy that causes growth inhibition and reduction in long-term clonogenic survival [113, 114]. The exact mechanism that gefitinib uses to upregulate the LKB1/AMPK pathway is still unknown; however, it is hypothesized that its induction of endoplastic reticulum (ER) stress and oxidative stress lead to the activation of tumor suppressor LKB1, calcium/calmodulin-dependent protein kinase kinase, and AMPK. Since gefitinib does not affect the expression of other pro-autophagic proteins such as Beclin-1, Atg5, Atg12, or Vps34, it is thought that the promotion of autophagy is a downstream effect of its upregulation of AMPK. The concentrations of gefitinib required to induce autophagy are higher than those required to inhibit EGFR, suggesting that autophagy promotion is an off-target effect of its action, and as a monotherapy it is not clinically practical due to the possibility that the concentrations required may lead to adverse effects on healthy tissues [113]. As a result, the pro-autophagic effects of gefitinib have been investigated in combination therapy as well. When combined with valproic acid, a short-chain fatty acid that is an established treatment for epilepsy, the two synergistically inhibited cell growth and long-term clonogenic survival via induction of autophagy [115]. Their combined action was independent of the Akt/mTOR pathway and acted upon the LKB1/AMPK pathway yet again, and the synergistic effect was due to greatly elevated levels of oxidative stress. Even though non-toxic levels of valproic acid were required, concentrations of 10 µM gefitinib or more were required for the combinatory effect, which could make clinical applications less practical [115].
Imatinib, which is an ATP analog that can bind to the receptors of PDGFR-A, breakpoint cluster region protein (BCR), stem cell factor (SCF), and tyrosine-protein kinase KIT (c-KIT), also promotes autophagy. Like gefitinib, as a monotherapy, its efficacy is rather limited; however, it is due to its inability to reach proper brain tissue distribution because of the action of p-glycoprotein [116]. This is why the action of imatinib has been investigated with a variety of combination therapies, such as with chlorimipramine, an anti-depressant, and with carvedilol, a treatment for chronic heart failure. Both of these drugs have shown anti-neoplastic characteristics, leading to synergistic effects when combined with imatinib to combat glioblastoma. In one study, the level of autophagy produced by imatinib only led to chemoresistance; however, when it was combined with chlorimipramine, levels of autophagy increased to the point of induction of autophagic cell death. This was also confirmed by increased autophagy and membrane fusion in the combination treatment group [116]. Carvedilol, in addition to acting upon PDGFR, also inhibited the action of p-glycoprotein, which led to the synergistic decrease in cell number, spheroid volume, percentage of cells in S-phase, and cAMP levels when combined with imatinib [117]. Both drugs have shown promise in vitro; however, further studies are required to determine their efficacy in vivo. Another possible explanation for limited action of imatinib as a monotherapy is its multi-target mechanism of action. Although it may act as an upstream promoter of autophagy, a study reports that in the context of hepatocellular carcinoma, imatinib can inhibit late stages of autophagy, prohibiting lysosomal fusion to the autophagosome. The additional ability of imatinib to counteract the pro-autophagic properties of sorafenib makes the usage of imatinib solely in the context of autophagy promotion questionable [161]. To use imatinib in this fashion, more molecular insights would have to be understood about its targets and mechanisms of action.
Overall, although tyrosine kinase inhibitors are an example of targeted therapies that take advantage of key identified characteristics of glioblastoma, monotherapies have only shown limited efficacy due to the refractory response of the tumor. Combination therapies show some promise, but may be clinically inapplicable, or require further exploration.
mTOR inhibitors
Of the modern mTOR inhibitors, rapamycin, otherwise known as sirolimus, was approved by the FDA long ago. Rapamycin was derived from the bacterium Streptomyces hygroscopicus, which was found in soil samples of Easter Island, a special territory of Chile in the southeastern Pacific Ocean. Found to have antibiotic properties, it was approved by the FDA in 1999 to help prevent kidney transplant rejection. It was not until Blommaart and colleagues analyzed the effects of rapamycin on S6 phosphorylation that the link between rapamycin and its ability to induce autophagy was discovered [118]. After rapamycin or sirolimus was determined to have anti-neoplastic qualities, other derivatives such as temsirolimus, everolimus (or RAD001), and ridaforolimus were developed for determining their therapeutic values [119]. Everolimus has been approved by the FDA in 2009 for advanced kidney cancer and it differs from rapamycin in its solubility and terminal half-life. This means that it has higher bioavailability, faster steady state levels once treatment starts, and it is eliminated from the body faster after the treatment has ended. The mechanism upon which rapamycin and its derivatives acts is that rather than directly binding to the mTOR protein, it binds to immunophilin FK506 binding protein (FKBP), which then forms a complex with mTORC1, leading to its inhibition. Newer mTOR inhibitors directly inhibit mTOR (such as by targeting its ATP-binding domain), possibly making them more effective; however, none are at the point of receiving FDA approval yet [119].
Rapamycin has been shown to promote the differentiation of stem cells in glioblastomas and increase radio-sensitization via autophagy. When SU-2 cells were treated with rapamycin or combination of rapamycin and 3-MA, the cells that were part of the rapamycin only group demonstrated decreased stem cell markers, while increased expression of differentiation markers [120]. The ability of rapamycin to cause differentiation of the stem cells was counteracted by 3-MA, an early-stage autophagy inhibitor, in the combination treatment group indicating that differentiation was the result of pro-autophagic processes. The same results were observed in rapamycin-treated tumor xenografts [120]. Around the same time, another group found that rapamycin increased the radiosensitivity of GSCs, leading to a synergistic effect that resulted in high levels of differentiation and cell death. When treated with monotherapy (just rapamycin), double therapy (rapamycin and irradiation), and triple therapy (rapamycin, irradiation, and 3-MA), the monotherapy and triple therapy led to the formation of large tumors with small satellite tumors while the double therapy led to the formation of small, confined lesions [121]. The monotherapy and triple therapy also led to greater toxicity, while the double therapy resulted in levels of apoptosis twenty to thirty times greater than the control [121].
Combination treatments consisting of EGFR inhibitors, PI3K and Akt inhibitors, and oncolytic adenoviruses potentiate the activity of rapamycin. When rapamycin was combined with erlotinib, an EGFR inhibitor, the ability of erlotinib to promote growth arrest and cell death increased even in PTEN-deficient cell lines, which are commonly resistant to the action of EGFR inhibitors [122]. Similar results were shown in rapamycin-resistant cell lines when rapamycin was combined with the PI3K inhibitor LY294002 and the Akt inhibitor UCN-01 (7-hydroxystaurosporine). Although LY294002 commonly is used as an autophagy inhibitor, when combined with rapamycin it is observed that it synergistically increases autophagy levels, reinforcing that its mechanism of action depends on the cell conditions [123]. Rapamycin was also combined with oncolytic adenovirus OBP-405, which was derived from human adenovirus stereotype 5. OBP-405 was determined to be more effective than other oncolytic adenoviruses at causing cell death regardless of the coxsackievirus and adenovirus receptor (CAR) expression level, and combination treatment led to an increased PFS in the mouse model of around 9.3 days when compared to monotherapy [124]. Clinical trials have been conducted with combination therapy of rapamycin and EGFR and VEGFR2 inhibitor vandetanib. Only two out of the nineteen selected patients showed response, and the PFS at 6 months was 15.8%; however, the clinical trial was limited in that participants were not pre-selected by the levels of activation of VEGFR, EGFR, and PI3K/Akt/mTOR pathways [125]. More clinical trials have to be conducted to determine the efficacy of these combination treatments and their applications to the current standard of glioblastoma treatment.
Although not as well studied as rapamycin, everolimus has been studied in combinations with other treatments such as the current standard of care, other oncolytic adenoviruses, and VEGFR and EGFR inhibitors. In a Phase I clinical trial, everolimus and TMZ were combined to determine the efficacy of treating newly diagnosed and progressive glioblastoma. Even though the full dose of TMZ could not be used in the combination treatment, out of the 28 patients in the study, 3 had partial responses while 16 had stable disease [126]. In addition, when everolimus was combined with oncolytic adenovirus Delta-24-RGD, long-term survival was observed in 80% of experimental animals while only 10% and 50% was observed in everolimus and Delta-24-RGD monotherapies, respectively. The cell cycle arrest and anti-angiogenic effect caused by everolimus potentiated the efficacy of the adenovirus, leading to significantly increased median survival [127]. Delayed tumor growth was also observed when everolimus was combined with the EGFR inhibitor AEE788. When combined, the drugs delayed tumor growth by 31.7 days rather than by 5.2 and 7.3 days with everolimus and AEE788 monotherapies, respectively [128]. Although they have shown promise in tumor xenografts and early stages of clinical trials, it is not until further clinical investigation is conducted that the efficacy of the combination therapies can be determined.
So far, sirolimus or rapamycin and everolimus have already been approved by the FDA for usage. Both treatments as monotherapies show efficacy in combating glioblastoma, and they show even greater potential when used in combination therapies. Since they are well-tolerated for the most part, further clinical investigation of combination therapies is required to determine the applicability of these therapies to enhance the efficacy of mTOR inhibitors.
Flavonoids have also proven their efficacy in modulating cellular pathways when it comes to mTOR inhibition. Cucurmin, a yellow pigment extracted from turmeric and commonly used in South Asian medicine, is a flavonoid that has been shown to modulate the Akt/mTOR pathway in both glioblastoma and endothelial cell lines [129, 130]. When curcumin was combined with nimustine hydrochloride (ACNU), a chemotherapeutic agent commonly used in Central Europe and Asia, it suppressed the Akt/mTOR pathway, synergistically inhibited cell proliferation, and promoted cell-cycle arrest [131]. Although it has shown promise, one of the major drawbacks of curcumin is its low bioavailability. As a result, one research group created solid lipid curcumin particles to increase its solubility. The treatment with this novel method of preparation led to elevated levels of mTOR pathway inhibition, as indicated by increased levels of Beclin-1, Atg5, and Atg7. In addition to promoting autophagy, this preparation of curcumin also led to the decrease in expression of pro-mitophagic markers Bcl-2 and adenovirus E1B 19-kDa-interactive protein 3-like (BNIP3L), indicating promotion of mitochondrial dysfunction leading to increase in cell death [129]. Perhaps the development of more efficient methods of curcumin distribution could lead to the formation of a low-toxicity polyphenol-based therapy that capitalizes on mTOR inhibition and autophagy promotion for inhibition of glioblastoma progression.
JNK activators and Bcl-2 inhibitors
JNK (c-Jun N-terminal kinase) is a member of a subfamily of MAPK (mitogen-activated protein kinase) and it has three variants: JNK1, JNK2, and JNK3. JNK1 and JNK2 are commonly found throughout the body while JNK3 is mainly found in the brain, heart, and testis. c-Jun is a phosphorylation-activated transcription factor that is a downstream effector of JNK. When activated, it regulates expression of the pro-apoptotic and anti-apoptotic and autophagic genes such as Bax (Bcl-2 associated X-protein) and Bcl-2 (B-cell lymphoma 2). Bcl-2 is a protein that binds to Beclin-1, preventing formation of the Beclin-1-hVps34-PI3K complex and inhibition of autophagy, and it also acts in preventing apoptosis. Activated JNK1 results in Bcl-2 phosphorylation, causing it to dissociate from Beclin-1 and resulting in induction of autophagy [133]. Manipulating this pathway has been a source of interest in promotion of autophagy, as it can lead to autophagic cell death while also indirectly enhancing pro-apoptotic pathways.
A polyphenol that affects Bcl-2 expression is gossypol, which is derived from cottonseeds. In the past, gossypol has demonstrated pro-apoptotic effects through the inhibition of the anti-apoptotic proteins Bcl-2, Bcl-xL, Mcl-1, and Bcl-W. When used in combination therapy with TMZ in both MGMT-negative and MGMT-expressing cells, (−)-gossypol synergistically increased cell death via autophagy, as confirmed by the formation of autophagosomes without lysosomal damage or caspase activation [134]. The decrease in levels of Mcl-1, down regulation of which has been determined as specifically important in causing cell death, has been hypothesized to contribute to the polyphenol's ability to cause stress-triggered degradation through upregulation of JNK1. Although the effect of gossypol was reduced in the MGMT-expressing cell line, the ability of gossypol to enhance the activity of TMZ as a pan-Bcl-2 inhibitor shows some hope. When further analyzed by another group, it was found that the responsiveness of cell lines to a gossypol-TMZ combination treatment depended on their individual Mcl-1 and MGMT expression statuses, which mean that there may be some applicability in the limited cases [135]. In clinical trials, gossypol can be orally ingested and is well-tolerated; however, on its own it does not do much to improve patient's PFS and OS [134, 136].
Cucurbitacin, another flavonoid, also acts upon JNK and the action of Bcl-2 family proteins to induce autophagy and suppress growth of glioblastoma. Commonly used in traditional medicine in Brazil, China, India, and Peru, cucurbitacin can be found in a variety of plant families. When glioblastoma cell lines were treated with this flavonoid, levels of phosphorylated p38, JNK, and c-JUN increased, while significant G2/M phase arrest followed. The action of cucurbitacin modulated the cytoskeleton and affected the JNK pathway [137]. In another study, cucurbitacin decreased the expression of anti-apoptotic Bcl-2 and Bcl-xL and increased the expression of pro-apoptotic Bax. The mechanism of action found in relation to a decrease in expression of Bcl-2 was the down regulation of the JAK/STAT3 pathway, which led to decrease in HIF-1α expression and decrease in interaction between Bcl-2 and Beclin-1 [75]. Overall, it shows some promise; however, it is possible that its action is relatively limited in inducing type II programmed cell death. Additional in vivo investigation for mechanistic studies and preliminary clinical trials are required.
Other flavonoids such as ursolic acid and evodiamine have shown similar ability to cause the upregulation of autophagy through the JNK pathway; however, their ability to cause levels of autophagy that lead to type II programmed cell death has not been thoroughly investigated [76, 79]. In general, the role of the JNK pathway in autophagy regulation is largely undiscovered and the low toxicity of many flavonoids may provide an opportunity to utilize the action of the JNK pathway to develop novel therapies, but this is still in its infancy.
Autophagy promoters have shown varying levels of success. Oftentimes, as a monotherapy they are largely ineffective, but when combined with other inducers of autophagy, they have demonstrated the ability to induce senescence, increased levels of apoptosis, anti-angiogenic properties, and increased PFS. Increased efficacy as a monotherapy and in combination therapy could possibly be attained through development of strategies to increase bioavailability of the treatment, such as how everolimus was developed. In vitro efficacy of these therapies has been deduced; however, there is limited research conducted on certain groups of autophagy promoters in vivo and in clinical trials. As new avenues of investigation are pursued, it is possible that the promotion of autophagy will be a therapeutic option to combat glioblastoma, but its relative novelty places the question of whether it will be incorporated in mainstream care in the near future.
Future of autophagy in glioblastoma therapy
Autophagy plays a critical role in glioblastoma progression because of its ability to either promote resistance to radiation and chemotherapy when it is activated in a cytoprotective fashion, or to cause cell death and tumor suppression when it is activated in a cytotoxic mode. Its dual role has been addressed through the development of novel treatments that either cause inhibition or promotion of autophagy. Autophagy inhibitors are long-standing and can be split into either early-stage or late-stage inhibitors. Whether they are PI3K inhibitors or they prevent lysosome and autophagosome fusion, autophagy inhibitors have shown promise in reducing tumor cell viability, combating recurrent tumor formation, and increasing activation of apoptotic pathways. However, problems still exist with the efficacy and the toxicity of certain therapies that involve autophagy inhibition. These problems also translate into therapies that promote autophagy. Although these inhibitors have shown great strides in promoting cell death, reducing the formation of large tumors, and delaying tumor growth, many therapies focused on autophagy promotion have shown little efficacy or ability to improve PFS or OS of the patients suffering from glioblastoma. Limited efficacy in both scenarios can be due to a lack of full understanding of the pathways involved in action of certain therapeutics, or they can also be due to difficulties in transporting therapeutics to the desired site of action [91]. Toxicity is yet another concern that is being resolved by finding new therapies that have lower toxicity, such as those that are derived from bioflavonoids and polyphenols.
The BBB is another limiting factor in obtaining optimal therapeutic bioavailability. Playing an important role in brain homeostasis, the BBB provides biomechanical and cellular resistance to a variety of elements including systematically circulating hormones, cytokines, waste and toxins, and items that can cause drastic change in ion gradients [151]. Although it is semi-permeable, passive diffusion only occurs for molecules that are under 400–500 Daltons (Da) in size and that are lipid-soluble. Therapeutics such as TMZ do fit the bill and are at a size of around 192 Da and have lipophilic properties; however, their relative penetration across the BBB is still only a mere 20%. Moreover, changes in the tumor microenvironment due to upregulation of HIF-1α and VEGF causes the formation of leaky vasculature that results in the reflux of chemotherapies back into the vasculature even after they have reached the parenchyma [151]. As this is so, a variety of methods have been developed in an attempt to overcome this challenge, such as biochemical drug modification, intra-arterial injection, hyperosmotic disruption, other surgical approaches, and thermotherapy.
Specifically, the development of novel treatments in the past meant creating a derivative of the ‘parent’ treatment (biochemical drug modification), such as in the case of everolimus and HCQ. However, this does not address the transport of current treatments such as those that involve bioflavonoids and can take quite a bit of time to develop. Recently, nanoparticles have been used to deliver treatments to their desired site. Common types of nanoparticles include polymeric nanoparticles, metallic nanoparticles, dendrimers, and carbon nanomaterials. Some carbon nanomaterials have shown promise, with Fullerene C60 being a potent autophagy inducer; however, problems with bioaccumulation leading to obstruction of autophagy flux still remain [138]. Nanomaterials have also been combined with the late-stage autophagy inhibitor CQ to induce apoptotic cell death. When albumin nanoparticles were modified with folic acid and loaded with paclitaxel and CQ, investigators noticed a synergistic increase in cellular apoptosis and a reduced quantity of expression of stemness-associating genes [139]. Ultimately, nanomaterials show promise in increasing the bioavailability of current treatments and are the next step to possibly creating great strides in producing therapies for glioblastoma since they can greatly augment the anti-tumorigenic action of current treatments without needing to use them at toxic levels.
Another method of delivery that has shown promise is the use of thermotherapy, specifically magnetic resonance-guided focused ultrasound (FUS). Considered relatively safe compared to other types of therapeutics, FUS can cause BBB disruption without irreversibly damaging neighboring tissues via emittance of low intensity ultrasound energy coupled with circulating microbubbles, focusing the effect of the ultrasound energy on vasculature [152]. FUS has been able to increase permeability of the BBB for molecules that are up to the sizes of 2000 kDa, providing the framework for the transport of nanoparticles containing chemotherapeutics like TMZ across the BBB to improve TMZ half-life [151, 152]. In one study using a rat model, investigators noticed that TMZ levels (measured as the cerebrospinal fluid-to-plasma ratio) increased from 22.7 to 38.6% in the presence of ultrasound therapy [151]. The high efficacy and minimal toxicity of FUS make it an attractive therapy for application to clinical trials and current standard of therapy.
In addition to finding new combination treatments or monotherapies that have lower toxicity and greater efficacy, the role of autophagy needs to be further investigated. Due to the great intra-tumoral heterogeneity that exists in glioblastoma, it is possible that different roles of autophagy can exist within one tumor [140]. As a result, which role of autophagy predominates within a particular cancer needs to be determined in addition to when autophagy should be targeted during tumor progression [141]. The long-term effects of modulating autophagy also have to be considered. For example, autophagy is essential for the development of memory T cells, and thus prolonged inhibition of autophagy could lead to even further immunosuppression [142]. By understanding the role of autophagy within a particular cancer, and how modulating autophagy affects the body in the long term, more targeted treatments can be developed.
The future of autophagy in glioblastoma is using precision medicine to give targeted therapy that is most effective in the individual patient being treated. Through greater understanding of the role of autophagy in glioblastoma, and the development of more efficient methods of delivery, current treatments can lead to greater tumor remission and improved OS for people who are diagnosed with glioblastoma.
Conclusions
As the most prominent primary malignant brain tumor in adults, glioblastoma makes up about 80% of all malignant brain tumors [1, 2]. Although TMZ, the current gold standard of treatment, has increased the life expectancy of patients diagnosed with glioblastoma by about two months, the prognosis is still grim. Oftentimes, patients hardly live to see another year after diagnosed with the disease, with a median survival still around 14.6 months after diagnosis [143]. The current standard of treatment relies heavily on MGMT-promoter methylation, which only exists in about 45% of patients diagnosed with glioblastoma [4]. As a result, alternative therapies need to be developed to consider the patients that do not have MGMT-promoter methylation and that capitalize on other pathways that are commonly dysregulated in glioblastoma [15].
Autophagy is a process that occurs at basal levels in most tissues and serves as a form of quality control. Although at incipient stages of tumor progression autophagy acts as a tumor repressor, at later stages, autophagy enhances tumorigenesis by conferring resistance to starvation, hypoxia, and therapy-induced cell death and maintaining cancer stem cells, which ultimately support relapse of the malignancy [23, 25]. The widespread role of autophagy and its implication in tumor progression make it a desirable target for novel therapeutics. TMZ, the current standard of treatment, upregulates autophagy that confers resistance to chemotherapeutics and RT, giving two possible avenues of investigation: either continued upregulation of autophagy leading to type II programmed cell death, also called autophagic cell death (unconventional), or inhibition of autophagy to upregulate type I programmed cell death, also called apoptotic cell death (conventional).
Much research has been conducted on both fronts. From the use of flavonoids to oncolytic adenoviruses, the progression of autophagy has been modulated to increase cell death. Some therapies have existed for over a decade and have been approved by the FDA for use in the clinical setting, while others are novel and have yet to be tested in larger clinical trials. Although almost all avenues seem to have a positive effect on controlling tumor progression in vitro, they fall short in vivo when it comes to bioavailability or their efficacy as a monotherapy. Since the development of TMZ and RT concomitant treatment, no other therapy has shown as great of advancement in the increase in PFS and OS. As a result, either novel treatments with lower toxicity and higher bioavailability or more efficient methods of distributing treatments that have already shown some promise need to be developed.
It is clear that certain questions need to be answered before advancements can be made and a singular direction can be taken on the development of treatments that modulate autophagy. These questions concern the intra-tumoral and intertumoral heterogeneity of autophagy activity, how autophagic activity impacts the efficacy of certain drugs (especially in the context of combination therapy), and how modulation of autophagy can impact other cellular processes, such as the immune system. As current therapies are further investigated in higher levels of clinical trials, the practicality of each therapeutic option can be determined and a decision can be made about whether early-stage autophagy inhibitors, late-stage autophagy inhibitors combined with an autophagy promoter, or autophagy promoters are the most effective at curbing glioblastoma progression. The wide variety of therapies that have been developed in addition to the use of nature-derived flavonoids and polyphenols shows great promise for glioblastoma treatments of the future. However, now is the time for increased understanding of autophagy function and the development of improved delivery methods to determine which therapeutics need to be left behind and which ones are worth pursuing further.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Witthayanuwat S, Montien P, Supaadirek C et al (2018) Survival analysis of glioblastoma multiforme. Asian Pac J Cancer Prev 19:2613–2617. https://doi.org/10.22034/APJCP.2018.19.9.2613
Stupp R, Mason W, Bent M et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. https://doi.org/10.1056/NEJMoa043330
Thomas A, Tanaka M, Trepel J et al (2017) Temozolomide in the era of precision medicine. Cancer Res 77:823–826. https://doi.org/10.1158/0008-5472.CAN-16-2983
Hegi M, Diserens A, Gorlia T et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. https://doi.org/10.1056/NEJMoa043331
Hottinger A, Stupp R, Homicscko K (2014) Standards of care and novel approaches in the management of glioblastoma multiforme. Chin J Cancer 33:32–39. https://doi.org/10.5732/cjc.013.10207
Hunter C, Smith R, Cahill D et al (2006) A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66:3987–3991. https://doi.org/10.1158/0008-5472.CAN-06-0127
Singh S, Hawkins C, Clarke I et al (2004) Identification of human brain tumor initiating cells. Nature 432:396–401. https://doi.org/10.1038/nature03128
Patel A, Tirosh I, Trombetta J et al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344:1396–1401. https://doi.org/10.1126/science.1254257
Furnari F, Cloughesy T, Cavenee W et al (2015) Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 15:302–310. https://doi.org/10.1038/nrc3918
Oberoi R, Parrish K, Sio T et al (2016) Strategies to improve delivery of anticancer drugs across the blood–brain barrier to treat glioblastoma. Neuro Oncol 18:27–36. https://doi.org/10.1093/neuonc/nov164
Lathia J, Mack S, Mulkearns-Hubert E et al (2015) Cancer stem cells in glioblastoma. Genes Dev 29:1203–1217. https://doi.org/10.1101/gad.261982.115
Ciechomska I (2018) The role of autophagy in neoplastic cells: characteristics of the interdependencies between autophagy and apoptosis; modulation of autophagy as a new therapeutic strategy in the treatment of gliomas. Postep Biochem 64:2. https://doi.org/10.18388/pb.2018_121
Goldhoff P, Clarke J, Smirnov I et al (2012) Clinical stratification of glioblastoma based on alterations in retinoblastoma tumor suppressor protein (RB1) and association with the proneural subtype. J Neuropathol Exp Neurol 71:83–89. https://doi.org/10.1097/NEN.0b013e31823fe8f1
Brennan C, Verhaak R, McKenna A et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Aldape K, Zadeh G, Mansouri S et al (2015) Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:829–848. https://doi.org/10.1007/s00401-015-1432-1
Danial N, Hockenbery D (2018) Cell death. In: Hoffman R (ed) Hematology: basic principles and practice, vol 18, 7th edn. Elsevier, Mumbai, pp 186–196
Huang Z, Zhou L, Chen Z et al (2016) Stress management by autophagy: implications for chemoresistance. Int J Cancer 139:23–32. https://doi.org/10.1002/ijc.29990
Yang K, Niu L, Bai Y et al (2019) Glioblastoma: targeting the autophagy in tumorigenesis. Brain Res Bull 153:334–340. https://doi.org/10.1016/j.brainresbull.2019.09.012
Shimizu S, Yoshida T, Tsujioka M et al (2014) Autophagic cell death and cancer. Int J Mol Sci 15:3145–3153. https://doi.org/10.3390/ijms15023145
Kimmelman AC, White E (2017) Autophagy and tumor metabolism. Cell Metab 25:1037–1043. https://doi.org/10.1016/j.cmet.2017.04.004
Martinez-Vicente M, Cuervo A (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet 6:352–361. https://doi.org/10.1016/S1474-4422(07)70076-5
Colella B, Faienza F, Bartolomeo S (2019) EMT regulation by autophagy: a new perspective in glioblastoma biology. Cancers (Basel) 11:312. https://doi.org/10.3390/cancers11030312
Galluzzi L, Pietrocola F, Bravo-San Pedro J et al (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34:856–880. https://doi.org/10.15252/embj.201490784
Amaravadi R, Lippincott-Schwartz J, Yin X et al (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17:654–666. https://doi.org/10.1158/1078-0432.CCR-10-2634
López-Otín C, Blasco M, Partridge L et al (2013) The hallmarks of aging. Cell 153:1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15:713–720. https://doi.org/10.1038/ncb2788
Laplante M, Sabatini D (2009) mTOR signaling at a glance. J Cell Sci 122:3589–3594. https://doi.org/10.1242/jcs.051011
Wong P, Puente C, Ganley I et al (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9:124–137. https://doi.org/10.4161/auto.23323
Ohgaki H, Kleihues P (2011) Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol 28:177–183. https://doi.org/10.1007/s10014-011-0029-1
Frattini V, Trifonov V, Chan J et al (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45:1141–1149. https://doi.org/10.1038/ng.2734
Cordani M, Butera G, Pacchiana R et al (2017) Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim Biophys Acta Rev Cancer 1867:19–28. https://doi.org/10.1016/j.bbcan.2016.11.003
Jutten B, Rouschop K (2014) EGFR signaling and autophagy dependence of growth, survival, and therapy resistance. Cell Cycle 13:42–51. https://doi.org/10.4161/cc.27518
Mathew R, Karp C, Beaudoin B et al (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075. https://doi.org/10.1016/j.cell.2009.03.048
Chen J, Zhang P, Chen W et al (2015) ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 11:239–252. https://doi.org/10.1080/15548627.2015.1009767
Padmakrishnan CJ, Easwer HV, Vijayakurup V et al (2019) High LC3/Beclin expression correlates with poor survival in glioma: a definitive role for autophagy as evidenced by in vitro autophagic flux. Pathol Oncol Res 25:137–148. https://doi.org/10.1007/s12253-017-0310-7
Hou J, Han Z, Jing Y et al (2013) Autophagy prevents irradiation injury and maintains stemness through decreasing ROS generation in mesenchymal stem cells. Cell Death Dis 4:884. https://doi.org/10.1038/cddis.2013.338
García-Prat L, Martínez-Vicente M, Periguero E et al (2016) Autophagy maintains stemness by preventing senescence. Nature 529:37–42. https://doi.org/10.1038/nature16187
Martinez-Outschoorn U, Prisco M, Ertel A et al (2011) Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via metabolo-genomics. Cell Cycle 10:1271–1286. https://doi.org/10.4161/cc.10.8.15330
Ricci-Vitiani L, Pallini R, Biffoni M et al (2010) Tumor vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468:824–828. https://doi.org/10.1038/nature09557
Galavotti S, Bartesaghi S, Faccenda D et al (2013) The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 32:699–712. https://doi.org/10.1038/onc.2012.111
Katayama M, Kawaguchi T, Berger M et al (2007) DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 14:548–558. https://doi.org/10.1038/sj.cdd.4402030
Natsumeda M, Aoki H, Miyahara H et al (2011) Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 31:486–493. https://doi.org/10.1111/j.1440-1789.2010.01197.x
Filippi-Chiela E, Bueno e Silva M, Thomé M, et al (2015) Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 11:1099–1113. https://doi.org/10.1080/15548627.2015.1009795
Lefranc F, Kiss R (2006) Autophagy, the Trojan horse to combat glioblastomas. Neurosurg Focus 20:E7. https://doi.org/10.3171/foc.2006.20.4.4
Lum J, Bauer D, Kong M et al (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120:237–248. https://doi.org/10.1016/j.cell.2004.11.046
Chen J, Li Y, Yu T et al (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488:522–526. https://doi.org/10.1038/nature11287
Auffinger B, Tobias A, Han Y et al (2014) Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ 21:1119–1131. https://doi.org/10.1038/cdd.2014.31
Zhu H, Wang D, Liu Y et al (2013) Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of the non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int 13:119. https://doi.org/10.1186/1475-2867-13-119
Koukourakis MI, Mitrakas AG, Giatromanolakis A (2016) Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: evidence and issues to resolve. Br J Cancer 114:485–496. https://doi.org/10.1038/bjc.2016.19
Ogier-Denis E, Codogno P (2003) Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 1603:113–128. https://doi.org/10.1016/s0304-419x(03)00004-0
Kanzawa T, Germano IM, Komata T et al (2004) Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 11:448–457. https://doi.org/10.1038/sj.cdd.4401359
Friedman HS, Prados MD, Wen PY et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733–4740. https://doi.org/10.1200/JCO.2008.19.8721
Kreisl TN, Kim L, Moore K et al (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27:740–745. https://doi.org/10.1200/JCO.2008.16.3055
Nagane M, Nishikawa R, Narita Y et al (2012) Phase II study of single-agent bevacizumab in Japanese patients with recurrent malignant glioma. Jpn J Clin Oncol 42:887–895. https://doi.org/10.1093/jjco/hys121
Vredenburgh J, Desjardins A, Herndon J II et al (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25:4722–4729. https://doi.org/10.1200/JCO.2007.12.2440
Bao S, Wu Q, Sathornsumetee S et al (2006) Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66:7843–7848. https://doi.org/10.1158/0008-5472.CAN-06-1010
Jahangiri A, Flanigan P, Aghi MK (2016) Antiangiogenic therapy for glioblastoma. In: Brem S, Abdullah KG (eds) Glioblastoma, vol 10. Elsevier, Philadelphia, pp 143–146
Hombach-Klonisch S, Mehrpour M, Shojaei S et al (2018) Glioblastoma and chemoresistance to alkylating agents: involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol Ther 184:13–41. https://doi.org/10.1016/j.pharmthera.2017.10.017
Hu Y-L, DeLay M, Jahangiri A et al (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72:1773–1783. https://doi.org/10.1158/0008-5472.CAN-11-3831
Clark AJ, Lamborn KR, Butowski NA et al (2012) Neurosurgical management and prognosis of patients with glioblastoma that progresses during bevacizumab treatment. Neurosurgery 70:361–370. https://doi.org/10.1227/NEU.0b013e3182314f9d
Pascolo S (2016) Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 771:139–144. https://doi.org/10.1016/j.ejphar.2015.12.017
Lee SW, Kim H-K, Lee N-H et al (2015) The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett 360:195–204. https://doi.org/10.1016/j.canlet.2015.02.012
Rosenfeld MR, Ye X, Supko JG et al (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10:1359–1368. https://doi.org/10.4161/auto.28984
Braicu C, Zanoaga O, Zimta A-A et al (2020) Natural compounds modulate the crosstalk between apoptosis- and autophagy-regulated signaling pathways: controlling the uncontrolled expansion of tumor cells. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2020.05.015
Lin S-R, Change C-H, Hsu C-F et al (2019) Natural compounds as potential adjuvants to cancer therapy: preclinical evidence. Br J Pharmacol 177:1409–1423. https://doi.org/10.1111/bph.14816
Hasima N, Ozpolat B (2014) Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis 5:e1509. https://doi.org/10.1038/cddis.2014.467
Zhou J, Li G, Zheng Y et al (2015) A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 8:1259–1279. https://doi.org/10.1080/15548627.2015.1056970
Taylor MA, Khathayer F, Ray SK (2019) Quercetin and sodium butyrate synergistically increase apoptosis in rat C6 and human T98G glioblastoma cells through inhibition of autophagy. Neurochem Res 44:1715–1725. https://doi.org/10.1007/s11064-019-02802-8
Chakrabarti M, Ray SK (2015) Anti-tumor activities of luteolin and silibinin in glioblastoma cells: overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis 21:312–328. https://doi.org/10.1007/s10495-015-1198-x
Chakrabarti M, Klionsky DJ, Ray SK (2016) miR-30e blocks autophagy and acts synergistically with proanthocyanidin for inhibition of AVEN and BIRC6 to increase apoptosis in glioblastoma stem cells and glioblastoma SNB19 cells. PLoS ONE 11:e0158537. https://doi.org/10.1371/journal.pone.0158537
Racoma IO, Meisen WH, Wang Q-E et al (2013) Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS ONE 8:e72882. https://doi.org/10.1371/journal.pone.0072882
Pazhouhi M, Sariri R, Rabzia A et al (2016) Thymoquinone synergistically potentiates temozolomide cytotoxicity through the inhibition of autophagy in U87MG cell line. Iran J Basic Med Sci 19:890–898. https://doi.org/10.22038/IJBMS.2016.7472
Lin C-J, Lee C-C, Shih Y-L et al (2012) Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med 52:377–391. https://doi.org/10.1016/j.freeradbiomed.2011.10.487
Boridy S, Le PU, Petrecca K et al (2014) Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis 5:e1216. https://doi.org/10.1038/cddis.2014.182
Yuan G, Yan S-F, Xue H et al (2014) Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J Biol Chem 289:10607–10619. https://doi.org/10.1074/jbc.M113.528760
Liu A-J, Wang S-H, Chen K-C et al (2013) Evodiamine, a plant alkaloid, induces calcium/JNK-mediated autophagy and calcium-mitochondria-mediated apoptosis in human glioblastoma cells. Chemico-biol Interact 205:20–28. https://doi.org/10.1016/j.cbi.2013.06.004
Wang J, Qi Q, Feng Z et al (2016) Berberine induces autophagy in glioblastoma by targeting the AMPK/mTOR/ULK1-pathway. Oncotarget 7:66944–66958. https://doi.org/10.18632/oncotarget.11396
Singletary K, Milner J (2008) Diet, autophagy, and cancer: a review. Cancer Epidemiol Biomark Prev 17:1596–1610. https://doi.org/10.1158/1055-9965.EPI-07-2917
Shen S, Zhang Y, Zhang R et al (2014) Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chemico-biol Interact 218:28–41. https://doi.org/10.1016/j.cbi.2014.04.017
Jing Z, Han W, Sui X et al (2015) Interaction of autophagy with microRNAs and their potential therapeutic implications in human cancers. Cancer Lett 356:332–338. https://doi.org/10.1016/j.canlet.2014.09.039
Ma B, Yuan Z, Zhang L et al (2017) Long non-coding RNA AC023115.3 suppresses chemoresistance of glioblastoma by reducing autophagy. Biochim Biophys Acta 1864:1393–1404. https://doi.org/10.1016/j.bbamcr.2017.05.008
Yang AI, Maus MV, O’Rourke DM (2016) General principles of immunotherapy for glioblastoma. In: Brem S, Abdullah KG (eds) Glioblastoma, vol 19. Elsevier, Philadelphia, pp 237–246
Kaminska B, Ciechomska IA, Cyranowski S (2020) Chapter 3—autophagy in brain tumor immune evasion and responses to immunotherapy. In: Chouaib S (ed) Autophagy in immune response: impact on cancer immunotherapy. Academic, London, pp 29–52
Li T-F, Xu Y-H, Li K et al (2019) Doxorubicin–polyglycerol–nanodiamond composites stimulate glioblastoma cell immunogenicity through activation of autophagy. Acta Biomater 86:381–394. https://doi.org/10.1016/j.actbio.2019.01.020
Zhao L, Xu Y-H, Akasaka T et al (2014) Polyglycerol-coated nanodiamond as a macrophage-evading platform for selective drug delivery in cancer cells. Biomaterials 35:5393–5406. https://doi.org/10.1016/j.biomaterials.2014.03.041
Liu J-R, Yu C-W, Hung P-Y et al (2019) High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhances antitumor immunity in glioblastoma. Biochem Pharmacol 163:458–471. https://doi.org/10.1016/j.bcp.2019.03.023
Hottinger AF, Abdullah KG, Stupp R (2016) Current standards of care in glioblastoma therapy. In: Brem S, Abdullah KG (eds) Glioblastoma, vol 6. Elsevier, Philadelphia, pp 73–80
Stupp R, Taillibert S, Kanner AA et al (2015) Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA 314:2535–2543. https://doi.org/10.1001/jama.2015.16669
Silginer M, Weller M, Stupp R et al (2017) Biological activity of tumor-treating fields in preclinical glioma models. Cell Death Dis 8:e2753. https://doi.org/10.1038/cddis.2017.171
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619. https://doi.org/10.1038/nrg1879
Wu Y-T, Tan H-L, Shui G et al (2010) Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861. https://doi.org/10.1074/jbc.M109.080796
Zhang X, Li W, Wang C et al (2014) Inhibition of autophagy enhances apoptosis induced by proteasome inhibitor bortezomib in human glioblastoma U87 and U251 cells. Mol Cell Biochem 385:265–275. https://doi.org/10.1007/s11010-013-1835-z
Wang J, Qi Q, Zhou Q et al (2018) Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 14:2007–2022. https://doi.org/10.1080/15548627.2018.1501133
Jin X, Liu Y, Liu X et al (2014) Role of autophagy in high linear energy transfer radiation-induced cytotoxicity to tumor cells. Cancer Sci 105:770–778. https://doi.org/10.1111/cas.12422
Kim J, Lee J-W, Kim S-I et al (2011) Thrombin-induced migration and matrix metalloproteinase-9 expression are regulated by MAPK and PI3K pathways in C6 glioma cells. Korean J Physiol Pharmacol 15:211–216. https://doi.org/10.4196/kjpp.2011.15.4.211
Blommaart EF, Krause U, Schellens JP et al (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243:240–246. https://doi.org/10.1111/j.1432-1033.1997.0240a.x
Mahadevan D, Chiorean EG, Harris WB et al (2012) Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. https://doi.org/10.1016/j.ejca.2012.06.027
Fedrigo CA, Grivicich I, Schunemann DP et al (2011) Radioresistance of human glioma spheroids and expression of HSP70, p53, and EGFR. Radiat Oncol 6:156. https://doi.org/10.1186/1748-717X-6-156
Ryabaya OO, Inshakov AN, Egorova AV et al (2017) Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anticancer Drugs 28:307–315. https://doi.org/10.1097/CAD.0000000000000463
Deng L, Lei Y, Liu R et al (2013) Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis 4:e614. https://doi.org/10.1038/cddis.2013.142
Venugopal C, Hallett R, Vora P et al (2015) Pyrvinium targets CD133 in human glioblastoma brain tumor-initiating cells. Clin Cancer Res 21:5324–5337. https://doi.org/10.1158/1078-0432.CCR-14-3147
Li Y, Yao J, Han C et al (2016) Quercetin, inflammation, and immunity. Nutrients 8:167. https://doi.org/10.3390/nu8030167
Kim H, Moon JY, Ahn KS et al (2013) Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxid Med Cell Longev. https://doi.org/10.1155/2013/596496
Egan DF, Chun MG, Vamos M et al (2015) Small molecule inhibition of autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59:285–297. https://doi.org/10.1016/j.molcel.2015.05.031
Mauthe M, Orhon I, Rocchi C et al (2018) Chloroquine inhibits autophagy flux by decreasing autophagosome–lysosome fusion. Autophagy 14:1435–1455. https://doi.org/10.1080/15548627.2018.1474314
Cristofori AD, Ferrero S, Bertolini I et al (2015) The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 6:17514–17531. https://doi.org/10.18632/oncotarget.4239
Liu L-Q, Wang S-B, Shao Y-F et al (2019) Hydroxychloroquine potentiates the anti-cancer effect of bevacizumab on glioblastoma via the inhibition of autophagy. Biomed Pharmacother 118:109339. https://doi.org/10.1016/j.biopha.2019.109339
Michaelides M, Stover N, Francis P et al (2011) Retinal toxicity associated with hydroxychloroquine and chloroquine. Arch Ophthalmol 129:30–39. https://doi.org/10.1001/archophthalmol.2010.321
Trejo-Solís C, Serrano-Garcia N, Escamilla-Ramírez Á et al (2018) Autophagic and apoptotic pathways as targets for chemotherapy in glioblastoma. Int J Mol Sci 19:3773. https://doi.org/10.3390/ijms19123773
Voldborg BR, Damstrup L, Spang-Thomsen M et al (1997) Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol 8:1197–1206. https://doi.org/10.1023/a:1008209720526
Ostrom QT, Liao P, Stetson LC et al (2016) Epidemiology of glioblastoma and trends in glioblastoma survivorship. In: Brem S, Abdullah KG (eds) Glioblastoma, vol 2. Elsevier, Philadelphia, pp 11–19
Abdullah KG, Adamson C, Brem S (2016) The molecular pathogenesis of glioblastoma. In: Brem S, Abdullah KG (eds) Glioblastoma, vol 3. Elsevier, Philadelphia, pp 21–31
Chang C-Y, Kuan Y-H, Ou Y-C et al (2014) Autophagy contributes to gefitinib-induced glioma cell growth inhibition. Exp Cell Res 327:102–112. https://doi.org/10.1016/j.yexcr.2014.05.011
Chang C-Y, Shen C-C, Su H-L et al (2011) Gefitinib induces apoptosis in human glioma cells by targeting Bad phosphorylation. J Neuro-oncol 105:507–522. https://doi.org/10.1007/s11060-011-0632-3
Chang C-Y, Li J-R, Wu C-C et al (2015) Valproic acid sensitizes human glioma cells to gefitinib-induced autophagy. IUBMB Life 67:869–879. https://doi.org/10.1002/iub.1445
Bilir A, Erguven M, Oktem G et al (2008) Potentiation of cytotoxicity by combination of imatinib and chlorimipramine in glioma. Int J Oncol 32:829–839. https://doi.org/10.3892/ijo.32.4.829
Erguven M, Yazihan N, Aktas E et al (2010) Carvedilol in glioma treatment alone and with imatinib in vitro. Int J Oncol 36:857–866. https://doi.org/10.3892/ijo_00000563
Blommaart EF, Luiken JJ, Blommaart PJ et al (1995) Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rate hepatocytes. J Biol Chem 270:2320–2326. https://doi.org/10.1074/jbc.270.5.2320
MacKeigan JP, Krueger DA (2015) Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol 17:1550–1559. https://doi.org/10.1093/neuonc/nov152
Zhuang W-Z, Long L-M, Ji W-J et al (2011) Rapamycin induces differentiation of glioma stem/progenitor cells by activating autophagy. Chin J Cancer 30:712–720. https://doi.org/10.5732/cjc.011.10234
Zhuang W, Li B, Long L et al (2011) Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer 129:2720–2731. https://doi.org/10.1002/ijc.25975
Wang M, Lu KV, Zhu S et al (2006) Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res 66:7864–7869. https://doi.org/10.1158/0008-5472.CAN-04-4392
Takeuchi H, Kondo Y, Fujiwara K et al (2005) Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 65:3336–3346. https://doi.org/10.1158/0008-5472.CAN-04-3640
Yokoyama T, Iwado E, Kondo Y et al (2008) Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther 15:1233–1239. https://doi.org/10.1038/gt.2008.98
Chheda MG, Wen PY, Hochberg FH et al (2015) Vandetanib plus sirolimus in adults with recurrent glioblastoma: results of a phase I and dose expansion cohort study. J Neurooncol 121:627–634. https://doi.org/10.1007/s11060-014-1680-2
Mason WP, MacNeil M, Kavan P et al (2012) A phase I study of temozolomide and everolimus (RAD001) in patients with newly diagnosed and progressive glioblastoma either receiving or not receiving enzyme-inducing anticonvulsants: an NCIC CTG study. Investig N Drugs 30:2344–2351. https://doi.org/10.1007/s10637-011-9775-5
Alonso MM, Jiang H, Yokoyama T et al (2008) Delta-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol Ther 16:487–493. https://doi.org/10.1038/sj.mt.6300400
Goudar RK, Shi Q, Hjelmeland MD et al (2005) Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther 4:101–112
Maiti P, Scott J, Sengupta D et al (2019) Curcumin and solid lipid curcumin particles induce autophagy, but inhibit mitophagy and the PI3K-Akt/mTOR pathways in cultured glioblastoma cells. Int J Mol Sci 20:399. https://doi.org/10.3390/ijms20020399
Guo S, Long M, Li X et al (2016) Curcumin activates autophagy and attenuates oxidative damage in EA.hy926 cells via the Akt/mTOR pathway. Mol Med Rep 13:2187–1293. https://doi.org/10.3892/mmr.2016.4796
Zhao J, Zhu J, Lv X et al (2017) Curcumin potentiates the potent antitumor activity of ACNU against glioblastoma by suppressing the PI3K/AKT and NF-κB/COX-2 signaling pathways. Oncotargets Ther 10:5471–5482. https://doi.org/10.2147/OTT.S149708
Karmakar S, Banik NL, Patel SJ et al (2006) Curcumin activated both receptor-mediated and mitochondria-mediated proteolytic pathways for apoptosis in human glioblastoma T98G cells. Neurosci Lett 407:53–58. https://doi.org/10.1016/j.neulet.2006.08.013
Zhou Y-Y, Li Y, Jiang W-Q et al (2015) MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep 35:e00199. https://doi.org/10.1042/BSR20140141
Jarzabek MA, Amberger-Murphy V, Callanan JJ et al (2014) Interrogation of gossypol therapy in glioblastoma implementing cell line and patient-derived tumour models. Br J Cancer 111:2275–2286. https://doi.org/10.1038/bjc.2014.529
Fiveash J (January 2008–June 2012). Gossypol in treating patients with progressive or recurrent glioblastoma multiforme. Identifier NCT00540722. https://clinicaltrials.gov/ct2/show/NCT00540722
Voss V, Senft C, Lang V et al (2010) The pan-Bcl-2 inhibitor (−)-Gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res 8:1002–1016. https://doi.org/10.1158/1541-7786.MCR-09-0562
Yin D, Wakimoto N, Xing H et al (2008) Cucurbitacin B markedly inhibits growth and rapidly affects the cytoskeleton in glioblastoma multiforme. Int J Cancer 123:1364–1375. https://doi.org/10.1002/ijc.23648
Agarwal S, Maekawa T (2020) Nano delivery of natural substances as prospective autophagy modulators in glioblastoma. Nanomed Nanotechnol Biol Med 19:102270. https://doi.org/10.1016/j.nano.2020.102270
Lu L, Shen X, Tao B et al (2019) The nanoparticle-facilitated autophagy inhibition of cancer stem cells for improved chemotherapeutic effects on glioblastomas. J Mater Chem B 12:2054–2062. https://doi.org/10.1039/C8TB03165G
Ulasov I, Fares J, Timashev P et al (2020) Editing cytoprotective autophagy in glioma: an unfulfilled potential for therapy. Trends Mol Biol 26:252–262. https://doi.org/10.1016/j.molmed.2019.11.001
Ishaq M, Ojha R, Sharma AP et al (2020) Autophagy in cancer: recent advances and future directions. Semin Cancer Biol 66:171–181. https://doi.org/10.1016/j.semcancer.2020.03.010
Ishimwe N, Zhang W, Qian J et al (2020) Autophagy regulation as a promising approach for improving cancer immunotherapy. Cancer Lett 475:34–42. https://doi.org/10.1016/j.canlet.2020.01.034
Kelly C, Majewska P, Ioannidis S et al (2017) Estimating progression-free survival in patients with glioblastoma using routinely collected data. J Neurooncol 135:621–627. https://doi.org/10.1007/s11060-017-2619-1
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79:1889–1892. https://doi.org/10.1073/pnas.79.6.1889
Vlahos CJ, Matter WF, Hui KY et al (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269:5241–5248. https://doi.org/10.1016/S0021-9258(17)37680-9
Oppermann H, Faust H, Yamanishi U et al (2019) Carnosine inhibits glioblastoma growth independent from PI3K/Akt/mTOR signaling. PLoS ONE 14:e0218972. https://doi.org/10.1371/journal.pone.0218972
Yamamoto A, Tagawa Y, Yoshimori T et al (1998) Bafilomycin A1 prevents maturation of autophagy vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42. https://doi.org/10.1247/csf.23.33
Hori YS, Hosoda R, Akiyama Y et al (2014) Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J Neuro-oncol 122:11–20. https://doi.org/10.1007/s11060-014-1686-9
Bursch W (2006) Multiple cell death programs: Charon’s lifts to Hades. FEMS Yeast Res 5:101–110. https://doi.org/10.1016/j.femsyr.2004.07.006
Coyle T, Levante S, Shetler M et al (1994) In vitro and in vivo cytotoxicity of gossypol against central nervous system tumor cell lines. J Neurooncol 19:25–35. https://doi.org/10.1007/BF01051046
Hendricks BK, Cohen-Gadol AA, Miller JC (2015) Novel delivery methods bypassing the blood–brain and blood–tumor barriers. Neurosurg Focus 38:E10. https://doi.org/10.3171/2015.1.FOCUS14767
Bunevicius A, McDannold NJ, Golby AJ (2020) Focused ultrasound strategies for brain tumor therapy. Oper Neurosurg (Hagerstown) 19:9–18. https://doi.org/10.1093/ons/opz374
Han C, Gu H, Wang J et al (2013) Regulation of l-threonine dehydrogenase in somatic cell reprogramming. Stem Cells 31:953–965. https://doi.org/10.1002/stem.1335
Morsi RZ, Hage-Sleiman R, Koveissy H (2018) Noxa: role in cancer pathogenesis and treatment. Curr Cancer Drug Targets 18:914–928. https://doi.org/10.2174/1568009618666180308105048
Ciuffreda L, Di Sanza C, Incani UC et al (2012) The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J Mol Med 90:667–679. https://doi.org/10.1007/s00109-011-0844-1
Hammouda MB, Ford AE, Liu Y et al (2020) The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 9:857. https://doi.org/10.3390/cells9040857
D’Arcy MS (2019) Cell death: a review of the major forms of apoptosis, necrosis, and autophagy. Cell Biol Int 43:582–592. https://doi.org/10.1002/cbin.11137
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516. https://doi.org/10.1080/01926230701320337
Pawlowska E, Szczepanska J, Szatkowska M et al (2018) An interplay between senescence, apoptosis and autophagy in glioblastoma multiforme-role in pathogenesis and therapeutic perspective. Int J Mol Sci 18:889. https://doi.org/10.3390/ijms19030889
Capparelli C, Guido C, Whitaker-Menezes D et al (2012) Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 11:2285–2302. https://doi.org/10.4161/cc.20718
Xiao M-C, Qian H, Huang C-K et al (2021) Imatinib inhibits the malignancy of hepatocellular carcinoma by suppressing autophagy. Eur J Pharmacol 906:174217. https://doi.org/10.1016/j.ejphar.2021.174217
Ray SK (2020) Modulation of autophagy for neuroprotection and functional recovery in traumatic spinal cord injury. Neural Regen Res 15:1601–1612. https://doi.org/10.4103/1673-5374.276322
Acknowledgements
The work was supported in part by the earlier R01 grants (CA-091460 and NS-057811) from the National Institutes of Health (Bethesda, MD, USA) and by the current awards from the South Carolina Honors College Research Program (Columbia, SC, USA) and the University of South Carolina Magellan Scholar Research Program (Columbia, SC, USA).
Author information
Authors and Affiliations
Contributions
All authors wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manea, A.J., Ray, S.K. Regulation of autophagy as a therapeutic option in glioblastoma. Apoptosis 26, 574–599 (2021). https://doi.org/10.1007/s10495-021-01691-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-021-01691-z