
Abstract
Prokaryotic diversity was investigated in a Tunisian salt lake, Chott El Jerid, by quantitative real-time PCR, denaturing gradient gel electrophoresis (DGGE) fingerprinting methods targeting the 16S rRNA gene and culture-dependent methods. Two different samples S1-10 and S2-10 were taken from under the salt crust of Chott El Jerid in the dry season. DGGE analysis revealed that bacterial sequences were related to Firmicutes, Proteobacteria, unclassified bacteria, and Deinococcus-Thermus phyla. Anaerobic fermentative and sulfate-reducing bacteria were also detected in this ecosystem. Within the domain archaea, all sequences were affiliated to Euryarchaeota phylum. Quantitative real-time PCR showed that 16S rRNA gene copy numbers of bacteria was 5 × 106 DNA copies g−1 whereas archaea varied between 5 × 105 and 106 DNA copies g−1 in these samples. Eight anaerobic halophilic fermentative bacterial strains were isolated and affiliated with the species Halanaerobium alcaliphilum, Halanaerobium saccharolyticum, and Sporohalobacter salinus. These data showed an abundant and diverse microbial community detected in the hypersaline thalassohaline environment of Chott El Jerid.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hypersaline environments are abundant worldwide and are characterized by salt concentrations higher than that of seawater (Ventosa et al. 2008). The microbial community of hypersaline environments close to saturation has been studied by both culture (Emmerich et al. 2012) and molecular methods (Maturrano et al. 2006; Foti et al. 2008) regarding the potential applications of halophiles in biotechnology (Oren 2010). These environments are generally limited to arid and semi-arid areas. In North Africa, salt lakes are called sebkhat or chott. In Tunisia, shallow salt lakes extend from the south coast of Tunisia to the Atlas Mountains (Algeria). Chott el Jerid is the largest saline depression, located in the south-west of Tunisia. It covers an area of approximately 5000 km2. During the dry season, salt-covered depression where salt crusts are precipitated covers most of this area. Sometimes in winter when a lot of rain falls, the area may be largely flooded and it acquires the aspect of lake during some months (Ori et al. 2009). Recent investigations have shown that solid salts in this lake come from the geological formations of the surrounding mountains. Differently to other continental sebkhats and according to its physicochemical properties, this shallow lake contains similar ions to seawater with a salt concentration varying between 250 and 330 g L−1, although it does not communicate with the Mediterranean Sea (Kbir-Ariguib et al. 2001). The major elements are sodium and chloride followed in abundance by SO4 2− obtained in previous studies in this ecosystem (Kbir-Ariguib et al. 2001). Chott El Jerid is a terrestrial evaporitic environment that has been the subject of sedimentological and hydrochemical studies but its microbial diversity has never been studied using molecular approaches. This is the first report describing the microbial populations present in the water and sediment of Chott El Jerid using molecular methods [denaturing gradient gel electrophoresis (DGGE) and quantitative real-time PCR]. Few ecological studies of this environment based on the cultivation approach have been investigated (Ben Abdallah et al. 2015; Mezghani et al. 2012). The main purpose of this research is to obtain an overview of the abundance, the structure, and microbial diversity of both archaeal and bacterial communities in water and sediment samples of Chott El Jerid using culture-dependent and culture-independent techniques. It is also aimed at comparing the results obtained with other studied hypersaline ecosystems.
Materials and methods
Site description, samples collection
Sampling was done from a continental salt lake: Chott El Jerid located in the south of Tunisia. Two samples S1-10 (33°54′42.21″N and 8°31′7.98″E) and S2-10 (33°54′44.15″N and 8°31′9.01″E) were taken from Chott El Jerid in the dry season (October 2010). S1-10 and S2-10 samples, a mixture of saturated salt waters and sediments, were collected 10 cm below the salt crust surface in sterile bottles and kept aseptically at −80 and 4 °C until further analysis. The sediments of S1-10 and S2-10 samples have a black and sandy color, respectively. Temperature, salinity, and pH of water samples were determined. The water at the samples site was at 23 °C. The pH of the water samples S1-10 and S2-10 were 6.61 and 6.9, respectively. The total salt concentration in the water was 346 g L−1.
Isolation of anaerobic halophilic fermentative bacteria
In an attempt to isolate anaerobic halophilic fermentative bacteria; the following medium was used: (per liter distilled water): NH4Cl, 1.0 g; K2HPO4, 0.5 g; MgSO4·7H2O, 20 g; CaCl2·2H2O, 0.1 g; NaCl, 200 g; tryptone, 0.5 g; KCl, 1 g; yeast extract, 0.5 g; cysteine hydrochloride, 0.5 g; 10 ml trace mineral element solution (Balch et al. 1979). The pH was adjusted to 6.8 with 10 M KOH. The medium was boiled under a stream of O2− free N2 gas and cooled to room temperature. 5 mL aliquots or 25 mL were then dispensed into Hungate tubes or serum bottles, respectively, under a stream of N2/CO2 (80:20, v/v) gas and autoclaved for 20 min at 120 °C. Prior to inoculation of the medium in each bottle, 0.5 mL 2 % (w/v) Na2S·9H2O; 0.5 mL 10 % (w/v) NaHCO3; 0.5 mL (1 M) glucose or 0.625 mL (20 %) tryptone or 0.625 mL (20 %) yeast extract were injected from sterile stock solutions. The medium in each Hungate tube was similarly supplemented, using 0.1 mL of each stock solution. For enrichment cultures in serum bottles, 10 % of sample was inoculated into growth medium followed by incubation at 37 °C for 1 week. The culture was purified by repeated use of the Hungate roll tube method in medium with 2 % (w/v) agar added (Hungate 1969). Cellular morphology and purity of the cultures were assessed under an Optiphot (Nikon) phase-contrast microscope.
DNA extraction
DNA was extracted from 250 mg of sediment of S1-10 and S2-10 samples using Ultra Clean Soil DNA kit (MO BIO) according to the protocol of the manufacturer. Fermentative bacterial genomic DNA was extracted by Wizard Genomic DNA Purification kit (Promega) according to the manufacturer’s recommendations.
PCR amplification of bacterial and archaeal 16S rRNA genes
For DGGE analysis, PCR amplifications of the variable region V3–V5 of bacterial and archaeal 16S rRNA genes were performed by Nested PCR. Bacterial 16S rRNA genes were amplified from all the samples using the following primer sets (Table 1): FD1/RD1 tested in the first round to amplify the entire DNA. In the second round of amplification, PCR products obtained were amplified with a second pair of primers: 341F-GC/907R. Within the domain archaea, DNA was amplified with the primer pairs 21F/1492R in a first amplification. Amplified archaeal 16S rRNA gene fragments were used as the template for a nested PCR with archaeal specific primers 344F-GC/915R (Table 1). PCR reactions were performed using a thermocycler (Applied Biosystems).
PCR amplification was performed in a total volume of 50 µL mixtures containing 1× Taq buffer, 0.2 mM each dNTP, 0.2 µM of each primer, 0.1 mg mL−1 BSA (Sigma), 50 ng DNA template and 1.25 U of Taq DNA polymerase (Fermentas). The first round of amplification program for bacteria was 94 °C for 2 min, and 30 cycles of 94 °C for 30 s, 55 °C for 45 s, 72 °C for 1 min 45 s. For the archaea, the following protocol was used for the first round of amplification: 94 °C for 4 min, and 30 cycles of 94 °C for 1 min, 58 °C for 1 min, and 72 °C for 1 min 45 s. The final extension step was at 72 °C for 10 min for both bacteria and archaea. The nested PCR program was 94 °C for 4 min; followed by 30 cycles of 94 °C for 1 min, 63 °C for 1 min (for archaea) or 58 °C for 45 s (for bacteria), and 72 °C for 45 s; ending with 10 min at 72 °C. Negative controls were included with no addition of template DNA. Molecular identification of fermentative bacterial isolates was performed. The 16S rRNA genes were amplified with specific bacterial primers FD1/RD1 (Table 1). The PCR condition was as follows: after initial denaturation (95 °C for 1 min), 30 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 1 min 30 s were performed, followed by a final extension (10 min, 72 °C).
DGGE analysis of bacterial and archaeal diversity
For each sample, 300 ng of archaeal and bacterial amplicons were subjected to DGGE analysis. DGGE was performed as previously described by using the D-Code multiple system (BioRad, USA). PCR products obtained were electrophoresed on a 6 % (w/v) polyacrylamide gel (acrylamide: bis-acrylamide, 37.5:1) with 40–80 % (archaeal) and 30–50 % (bacterial) denaturing gradient (100 % of denaturing agents were 7 M urea and 40 % (v/v) deionized formamide) in 1× TAE buffer (40 mM tris, pH 8.5; 20 mM acetic acid; 1 mM EDTA) at 60 °C. Electrophoresis conditions were 150 V for 5 h for both domains. After 30 min of ethidium bromide staining, DGGE gels were photographed under UV light and photographed with a Gel doc XR Imaging system (BioRad). Selected bands of each sample (in terms of intensity and frequency of appearance) were cut from the gel, resuspended overnight in 35 µL of MilliQ water, reamplified (using the appropriate corresponding bacteria or archaea primers devoid of the GC clamps), cloned, and sequenced. PCR products were ligated into the vector pGEM-T easy vector (Promega Corporation, Madison, WI) following the manufacturer’s recommendations. Recombinant plasmids were isolated from overnight cultures using EZ-10 Spin Column Plasmid DNA kit and screened by restriction analysis with EcoRI. Positive transformants were selected for sequencing.
Sequencing and phylogeny analysis
DGGE bands were sequenced using the Big Dye® Terminator cycle Sequencing kit and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The primers used for sequencing were T7 and SP6 presented in pGEM-T easy vector (Table 1). PCR products from each isolate were sequenced using FD1 and RD1 primers (Table 1). Sequence similarity searches were performed using the online sequence analysis resources “BLAST” (Basic Local Alignment Search Tool) (Altschul et al. 1997) and “Seqmatch” (Ribosomal Database project II; Release 10) (Cole et al. 2009). Multiple alignments were generated by the MUSCLE program (Edgar 2004) and phylogenetic tree was constructed with MEGA program version 6 (Tamura et al. 2013) based on evolutionary distances that were calculated using the Neighbour-Joining method with Juke-Cantor distance model. We performed bootstrap resampling analysis for 1000 replicates to estimate degrees of confidence in tree topologies. Then, chimerical sequences were checked using chimera check on DECIPHER database (http://decipher.cee.wisc.edu/FindChimeras.html) (Wright et al. 2012).
Quantitative real-time PCR (qPCR)
To evaluate the abundance of archaea and bacteria in all samples, qPCR was used by targeting 16S rRNA genes. The archaeal and bacterial amplification reactions were performed in triplicate on a Bio-Rad CFX-96 real-time system (Bio-Rad). Archaeal and bacterial 16S rRNA genes were quantified using the primer sets 344F/519R and 331F/797R, respectively (Table 1). The same reaction components and protocol were used for the bacterial and archaeal real-time PCR. Each qPCR mixture contained the following components: 1× SsoAdvanced SYBR Green Supermix (Bio-Rad), 250 nM of each primer, 50 ng of DNA template (tenfold dilution series of standard PCR product or environmental DNA sample) or distilled water (negative control), and RNase/DNase-free water to a final volume of 20 µL. The qPCR reactions were carried out as follows: Initial denaturation at 98 °C for 2 min, followed by 39 cycles of a 2-step PCR protocol with a 5 s denaturation phase at 98 °C and a 30 s annealing/elongation phase at 56 °C for the archaeal real-time PCR.
The bacterial qPCR reactions had an initial denaturing step of 10 min at 95 °C, followed by 35 cycles of 30 s of denaturing at 95 °C, 30 s of annealing phase at 60 °C, and 30 s of primer extension at 72 °C. Fluorescence was measured at the end of each cycle. To assess the specificity of the primers, a post-PCR melting curve analysis was carried out, in which the temperature varied between 65 and 95 °C in 0.5 °C increments. qPCR standard curves were constructed from serial tenfold dilutions of DNA standards of known concentration. For bacterial 16S rRNA gene qPCR, standard DNA fragments were amplified from Halomonas alimentaria YKJ-16 T using the primer sets 27F-907R (Table 1). For archaeal 16S rRNA gene qPCR, standard DNA fragments were amplified from Halorubrum chaoviator DSM 19316 using the primer sets 109F-958R (Table 1). Triplicate PCR products were pooled and purified with Wizard® SV and PCR Clean-up System (Promega), according to the manufacturer’s instructions. Purified PCR products were subsequently quantified using the BioSpec-nano Spectrophotometer (Shimadzu) and used as DNA standards. Measured concentrations of purified PCR products were then converted to copies per microliter as described and the concentration was adjusted to 1 × 109 copies μL−1 prior to performing serial dilutions from 108 to 101 copies per μL of DNA. For all standard curves, the coefficients of determination (R2) were higher than 99.0 %. Cell numbers per g sediment were calculated from the qPCR 16S rRNA gene copy numbers according to the average 16S rRNA copy number in bacterial (3.82) and archaeal genomes (1.62).
Nucleotide sequence accession numbers
The 16S rRNA gene sequences from bands DGGE determined in this study were deposited in the GenBank database under accession numbers KT363000 to KT363010 (bacteria) and KT363011 to KT363036 (archaea). The 16S rRNA gene sequences of the fermentative isolates have the accession numbers KU180220 to KU180227.
Results
DGGE profiles
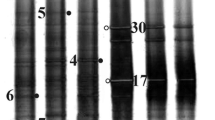
The microbial community structure was investigated in Chott El Jerid through PCR-DGGE analysis. Bacterial and archaeal community profiles elucidated by DGGE are shown in Fig. 1. Similar DGGE profiles are obtained for the S1-10 and S2-10 samples from Chott El Jerid for bacteria or archaea. Numerous predominant bands are presented in all profiles (archaeal or bacterial) indicating diverse microbial community present in S1-10 and S2-10 samples. For bacteria, 19 bands were excised. Only seven of them (B1, B4, B6, B7, B9, B10, B18) were successfully reamplified, cloned and sequenced. For archaea, 19 excised DGGE bands (A1–A19) were reamplified, cloned, and sequenced. Nucleotide sequences were compared to the GenBank and RDP databases using BLASTN and SeqMatch, respectively. Sequencing results on bacterial and archaeal 16S rRNA gene sequences from DGGE bands are reported in Tables 2 and 3. Phylogenetic analyses based on the bacterial and archaeal 16S rRNA gene sequences were performed (Figs. 2, 3).

Denaturing gradient gel electrophoresis profiles of PCR-amplified archaeal (a) and bacterial (b) 16S rRNA gene fragments obtained from samples S1-10 and S2-10

Phylogenetic tree based on 16S rRNA gene of excised DGGE bands compared to representatives of the domain bacteria and environmental clones. The tree is based on Neighbour-Joining method. The scale bar represents 5 % estimated sequence divergence. The sequence of Methanothermobacter thermophilus was used as the outgroup

Phylogenetic tree based on a comparison of 16S rRNA sequences from excised DGGE bands to those of representative archaeal species and environmental clones. The tree is based on the Neighbour-Joining method. The scale bar represents 5 % estimated sequence divergence. The sequence of Desulfococcus multivorans was used as the outgroup
Phylogenetic affiliations of bacterial community members
Phylogenetic groups within the bacterial domain were regrouped in the Firmicutes, Proteobacteria (Alphaproteobacteria, Gammaproteobacteria and Deltaproteobacteria), Deinococcus-Thermus phyla and unclassified bacteria.
Within the Proteobacteria Phylum, the sequences were clustered with the Rhodobacteraceae, Aeromonadaceae, and Desulfobacteraceae family’s bacteria belonging to Alphaproteobacteria, Gammaproteobacteria and Deltaproteobacteria classes, respectively. Band B1.C23 sequence was related to uncultured bacterium clone HSS104 (97 % similarity), detected in coastal saline soils in India and presented 86 % similarity with anaerobic halophilic sulfate-reducing Deltaproteobacteria belonging to Desulfatibacillum and Desulfosalsimonas genus. The sequences B4.C15 and B7.C13 were related to a facultative anaerobic bacteria strain Aeromonas sharmana DSM 17445, isolated from water collected from a warm spring in India. Band B10.C72 and B18.C17 sequences showed 97 % similarity with Roseovarius pacificus, isolated from deep-sea sediment and 98 % similarity with Oceanicola marinus, isolated from seawater collected from the coast of Taiwan, respectively. Band B18.C26 sequence was affiliated to uncultured bacterium clone, which was isolated from urban stormwater sediments. This band showed 85 % similarity with sulfate-reducing bacterium Desulfofrigus oceanense, isolated in marine Arctic sediments.
The second major phylum was the Firmicutes and the sequences were grouped with members of Clostridia and Bacilli classes. Band B1.C1, detected only in the S2-10 sample, presented 99 % similarity with uncultured bacterium clone kasin-B2-F08 recovered from iron-rich salt lake sediments lake Kasin, southern Russia. Band B1.C1 sequence was also related to anaerobic halophilic fermentative genera Halanaerobium and Halocella species belonging to Halanaerobiaceae family. The sequence of B6.C6 showed 97 % similarity with denitrifying bacterium Bacillus azotoformans LMG 9581.
Within the Deinococcus-Thermus phylum, Band DGGE B10.C14 sequence, detected only on S1-10 DGGE profile, was closely related to Truepera radiovictrix DSM 17093 isolated from hot spring runoffs on the Island of São Miguel in the Azores. This species was extremely ionizing radiation resistant (60 % of the cells survive 5.0 kGy). Two sequences from band B9.C16 and B4.C18 were affiliated with uncultured clones of bacteria retrieved from a hypersaline lake in Iran.
Phylogenetic affiliations of archaeal community members
Within the domain archaea, all sequences were grouped with members of Euryarchaeota phylum including Halobacteriaceae, Natrialbaceae, Haloferacaceae, Methanosarcinaceae and Methanocaldococcaceae families. Bands A2, A4, A13, A15, A17, A18 and A19 sequences were related to the genus Halorubrum spp. (Haloferacaceae family) which were widely found in diverse hypersaline environments such as solar salterns, salt lakes, coastal sabkhas, soda lakes. Band A3.C18 is related to Halorubrum ejinorense isolated from lake Ejinor, Inner Mongolia, China. It also appears that band A5.C51 and A6.C63 showed 91 % of similarity with Halococcus hamelinensis and Halococcus dombrowskii isolated from stromatolites in Shark Bay, Australia and from a Permian alpine salt deposit, respectively. Band A7.C73 is affiliated to Natrinema versiforme (Natrialbaceae family) found in Aibi salt lake in China, whereas, band A12.C24 was closely related to uncultured bacterium clone described in hypersalin sediments of lake Kasin in Russia. Band A12.C12 sequence was related to species of Natronomonas (97 % of similarity), isolated from salt-saturated lakes, from a marine solar saltern crystallizer, and from a solar saltern in Korea. Band A14.C36 and A14.C44 sequences were affiliated to Haloarcula marismortui isolated from Dead Sea. Band A10.C14 exhibited 93 % of sequence similarity with Halosimplex pelagicum isolated from salted brown alga Laminaria China. The sequence of Band A8.C86 was related to an extremely halophilic archaeon Halapricum salinum (96 % similarity) identified from non-purified solar salt. Furthermore, sequences from bands A11.C101 and A9.C13 showed high similarity to the uncultured archaeal clones previously described in hypersaline environments. Within the Methanococci class, bands A1.C10 and A11.C6 sequences were clearly affiliated to the family Methanocaldococcaceae and Methanosarcinaceae, respectively. Sequences of band A11.C6 and A1.C10 were related to Methanohalobium evestigatum isolated from a salt lagoon in Crimea, Russia and Methanotorris igneus isolated from a coastal hydrothermal environment in Iceland with sequence similarity of 99 and 83 %, respectively.
Quantification of 16S rRNA genes
The abundance of bacterial and archaeal groups in all samples, determined by quantitative real-time PCR, was reported as DNA copy numbers of 16S rRNA genes per gram of wet sediment. The abundance of archaea was 5 × 105 to 106, whereas bacteria abundance was 5 × 106 DNA copies g−1.
Identification of fermentative bacteria
Eight fermentative anaerobic halophilic bacterial strains obtained in pure culture were selected based on their morphological characteristics of colonies and were characterized by 16S rRNA gene analysis. Cells of strictly anaerobic isolates were rods occurring singly or in pairs. All strains were motile. The isolates are designated by letters indicating first the origin of strains from Chott El Jerid (CEJ), Fermentative (F) followed by type of substrate Tryptone (T), Yeast extract (YE) or Glucose (G), then a number indicating the sampling site 1 (S1-10) or 2 (S2-10).
PCR amplification of 16S rRNA gene from strains CEJFG1, CEJFG2, CEJFT1A, CEJFT1C, CEJFYE1A, CEJFYE1B, CEJFYE2C and CEJFYE2D resulted in sequences of 1058, 1545, 1543, 1546, 1048, 1544, 1524, 1008 bp, respectively.
Phylogenetic analysis revealed that those isolates were belonged to the order of Halanaerobiales placed in the phylum Firmicutes (Fig. 4). Six strains were affiliated to Halanaerobiaceae family and two strains to Halobacteroidaceae family. CEJFT1A, CEJFT1C, CEJFYE1B, CEJFG1, CEJFYE1A were related (98–99 % of similarity) to species H. saccharolyticum. The strain CEJFG2 exhibited 99 % of sequence similarity with Halanaerobium alcaliphilum. Strains CEJFYE2C, CEJFYE2D were affiliated (98 % of similarity) to species Sporohalobacter salinus.

Phylogenetic tree based on similarities of 16S rRNA sequences of isolates and its relatives. The tree is based on the Juke-Cantor model and the Neighbour-Joining method. The sequence of Bacteroides nordii was used as the outgroup. Bootstrap values based on 1000 replicates are shown
Discussion
In the present study, the microbial diversity present in the largest Tunisian salt lake, Chott El Jerid, was investigated for the first time by culture-independent methods. Denaturing gradient gel electrophoresis (DGGE) and quantitative real-time PCR (qPCR) were performed to assess the bacterial and archaeal communities’ structure and to evaluate their abundance. A remarkable microbial diversity was obtained in the extremely halophilic ecosystem, Chott El Jerid, among both the bacteria and archaea domains. Predominant phylogenetic groups were represented by Proteobacteria and Firmicutes of the bacteria domain. These taxonomic groups were also detected previously in hypersaline lakes (Demergasso et al. 2008; Makhdoumi-Kakhki et al. 2012; Emmerich et al. 2012), salterns, and hypersaline sediments (Mouné et al. 2003; Baati et al. 2010).
What is notable in our study is the absence of sequences representing the Bacteroidetes group. This group is reported to dominate with Proteobacteria in many different saline environments including hypersaline lakes such as the Tebenquiche in the Salar de Atacama (Demergasso et al. 2008), the largest hypersaline playa Aran-Bidgol in Iran (Makhdoumi-Kakhki et al. 2012), and Tunisian multipond solar saltern (Baati et al. 2008).
In this work, eight obligately halophilic anaerobic fermentative bacteria were isolated from the same samples (S1-10) and (S2-10) used for DGGE analysis. Those strains were affiliated with three species H. alcaliphilum, H. saccharolyticum and S. salinus. The species H. alcaliphilum was a strictly anaerobic, moderately halophilic, Gram-negative bacterium and was isolated from the sediments of Great Salt Lake (Tsai et al. 1995). H. saccharolyticum was retrieved from sediments of hypersaline lakes (Cayol et al. 1994). The species S. salinus was recently isolated from Chott El Jerid (Ben Abdallah et al. 2015). These results are consistent with those obtained from DGGE. In fact, in our study, a number of DGGE band sequences were related to groups of anaerobic bacteria, principally fermentative bacteria. Previous studies were established by the isolation of anaerobic halophilic fermentative bacterial strains from surface sediments of Chott El Jerid (Hedi et al. 2009; Mezghani et al. 2012).
Moreover, a number of DGGE band sequences related to sulfate-reducing bacteria were detected in our study, but no sulfate-reducing strains have been isolated in pure cultures from this lake up to now.
Within the bacterial clone libraries, the most abundant Deltaproteobacterial representatives related to halophilic sulfate-reducing bacteria and members of the phylum Firmicutes related to the strictly anaerobic fermentative bacteria were found inhabiting the hypersaline ponds in Mediterranean salterns and in salt lakes (Mouné et al. 2003; Makhdoumi-Kakhki et al. 2012).
Interesting was the detection of Deinococcus-Thermus in Chott El Jerid. It is a small group of bacteria that are highly resistant to radiation. The presence of representatives of this group was scarce in hypersaline ecosystems studied using rRNA gene clones libraries. Members of Deinococcus-Thermus were also detected from the RNA-derived DGGE profiles in Cock Lake, a soda lake in Russia at low salinity 60 g L−1 and this group was absent at high salinities in other investigated soda lakes (Foti et al. 2008).
With regard to the archaea population on the earth, halophilic archaea are the dominant microbial flora in hypersaline environments such as solar salterns, hypersaline lakes, hypersaline microbial mats, and the Dead Sea (Oren 2002). In this work, it could be noticed that all the 16S rRNA gene sequences obtained were affiliated with the orders Halobacteriales and Haloferacales positioned in the phylum Euryarchaeota, with most representatives belonging to the Halobacteria class, extreme aerobic halophiles growing at high salinity close to saturation.
In this study, no sequences affiliated with the Crenarchaeota group were detected. However, clones related to this phylum were recovered in anoxic sediments of hypersaline ponds in Salin-de-Giraud salterns from the station of lower salinity (150–200 g L−1) (Mouné et al. 2003).
The same results were obtained in other studies assessing the diversity of Haloarchaea in salt lakes which they have shown that most sequences were affiliated to Halobacteria (Makhdoumi-Kakhki et al. 2012). Here, interestingly, we detected via DGGE method two sequences, retrieved from each sample, related to members of the orders Methanosarcinales and Methanococcales. Methanogenic orders seem to be in low rates of all archaeal 16S rRNA gene sequences from clone’s libraries from hypersaline lakes, and hypersaline ponds in Mediterranean salterns (Mouné et al. 2003). It is well known that in hypersaline environments, characterized by high concentrations of sulfate, sulfate-reducing bacteria outcompete methanogens for the substrate such as H2, acetate and formate.
According to qPCR results, Chott El Jerid shelters an important number of bacterial and archaeal populations (106 cells per g wet sediment) in comparison with previous studies performed in other hypersaline systems using quantitative PCR. In contrast, higher numbers of cells 107 to 109 per g wet sediment were detected in saline Qinghai Lake, China (Dong et al. 2006). Most studies demonstrated that the microbial communities in many hypersaline environments were dominated by halophilic archaea numbers. Spatial and temporal effects on the structure of microbial communities should be further investigated. Other samples will be taken in the rainy season in order to analyze the spatial and seasonal effects on the prokaryotic community in the Chott El Jerid.
Our results showed that Chott El Jerid presents an important diversity of bacterial and archaeal types found in thalassohaline hypersaline environments from different geographic regions (Baati et al. 2010; Makhdoumi-Kakhki et al. 2012).
In conclusion, culture-independent study reports on the microbial diversity, using 16S rRNA gene sequences analysis within two samples from Chott El Jerid. The diversity of bacteria and archaea found in Chott El Jerid was specified. Bacteria domain was represented exclusively by Proteobacteria and Firmicutes. Archaea domain was closely related to the Euryarchaeota phylum. More importantly, Firmicutes were affiliated with the anaerobic genera Halanaerobium, Halocella and the group Proteobacteria with anaerobic halophilic sulfur reducing Deltaproteobacteria. Additionally, the DGGE analysis confirmed the presence of microorganisms previously described through clone libraries in these ecosystems, such as methanogen archaea and Deinococcus-Thermus phylum. Our results suggest that microorganisms as well as uncultured clones detected in Chott El Jerid were found in hypersaline thalassohaline environments, despite the continental origin of our ecosystem. The cultivation approach led us to isolate anaerobic fermentative bacteria related to three species H. alcaliphilum, H. saccharolyticum and S. salinus grouped within the phylum Firmicutes. The culture-dependent and culture-independent are complementary methods and give a good description of the prokaryotic diversity in hypersaline environments. However, using novel molecular strategies such as metagenomic approaches is an essential step to gaining a better understanding of microbial ecology in hypersaline environments.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi:10.1093/nar/25.17.3389
Baati H, Guermazi S, Amdouni R, Gharsallah N, Sghir A, Ammar E (2008) Prokaryotic diversity of a Tunisian multipond solar saltern. Extremophiles 12:505–518. doi:10.1007/s00792-008-0154-x
Baati H, Guermazi S, Gharsallah N, Sghir A, Ammar E (2010) Novel prokaryotic diversity in sediments of Tunisian multipond solar saltern. Res Microbiol 161:573–582. doi:10.1016/j.resmic.2010.05.009
Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS (1979) Methanogens: reevaluation of a unique biological group. Microbiol Rev 43:260–296
Ben Abdallah M, Karray F, Mhiri N, Cayol JL, Tholozan JL, Alazard D, Sayadi S (2015) Characterization of Sporohalobacter salinus sp. nov., an anaerobic, halophilic, fermentative bacterium isolated from a hypersaline lake. Int J Syst Evol Microbiol 65:543–548. doi:10.1099/ijs.0.066845-0
Brosius J, Dull TJ, Sleeter DD, Noller HF (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J Mol Biol 148:107–127. doi:10.1016/0022-2836(81)90508-8
Casamayor EO, Massana R, Benlloch S, Øvreås L, Díez B, Goddard VJ, Gasol JM, Joint I, Rodríguez-Valera F, Pedrós-Alió C (2002) Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4:338–348. doi:10.1046/j.1462-2920.2002.00297.x
Cayol JL, Ollivier B, Lawson A, Soh ALS, Fardeau ML, Ageron E, Grimont PAD, Prensier G, Guezennec J, Magot M, Garcia JL (1994) Haloincola saccharolytica subsp. senegalensis subsp. nov., isolated from the sediments of a hypersaline lake, and emended description of Haloincola saccharolytica. Int J Syst Bacteriol 44:805–811. doi:10.1099/00207713-44-4-805
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal Database Project: improved alignements and new tools for rRNA analysis. Nucleic Acids Res 37:141–145. doi:10.1093/nar/gkn879
DeLong EF (1992) Archaea in costal marine environments. Proc Natl Acad Sci USA 89:5685–5689. doi:10.1073/pnas.89.12.5685
Demergasso C, Escudero L, Casamayor EO, Chong G, Balagué V, Pedrós-Alió C (2008) Novelty and spatio–temporal heterogeneity in the bacterial diversity of hypersaline Lake Tebenquiche (Salar de Atacama). Extremophiles 12:491–504. doi:10.1007/s00792-008-0153-y
Dong H, Zhang G, Jiang H, Yu B, Chapman LR, Lucas CR, Fields MW (2006) Microbial diversity in sediments of saline Qinghai Lake, China: linking geochemical controls to microbial ecology. Microb Ecol 51:65–82. doi:10.1007/s00248-005-0228-6
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi:10.1093/nar/gkh340
Emmerich M, Bhansali A, Lösekann-Behrens T, Schröder C, Kappler A, Behrens S (2012) Abundance, distribution, and activity of Fe(II)-oxidizing and Fe(III)-reducing microorganisms in hypersaline sediments of Lake Kasin, southern Russia. Appl Environ Microbiol 78:4386–4399. doi:10.1128/AEM.07637-11
Foti MJ, Sorokin DY, Zacharova EE, Pimenov NV, Kuenen JG, Muyzer G (2008) Bacterial diversity and activity along a salinity gradient in soda lakes of the Kulunda Steppe (Altai, Russia). Extremophiles 12:133–145. doi:10.1007/s00792-007-0117-7
Großkopf R, Janssen PH, Liesack W (1998) Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl Environ Microbiol 64:960–969
Hedi A, Fardeau ML, Sadfi N, Boudabous A, Ollivier B, Cayol JL (2009) Characterization of Halanaerobaculum tunisiense gen. nov., sp. nov., a new halophilic fermentative, strictly anaerobic bacterium isolated from a hypersaline lake in Tunisia. Extremophiles 13:313–319. doi:10.1007/s00792-008-0218-y
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes. Methods Microbiol 3B:117–132
Kbir-Ariguib N, Ben Hassan Chehimi D, Zayani L (2001) Treatment of Tunisian salt lakes using solubility phase diagrams. Pure Appl Chem 73:761–770
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acids techniques in bacterial systematics. Wiley, Chichester, pp 115–147
Makhdoumi-Kakhki A, Amoozegar MA, Kazemi B, Pašić L, Ventosa A (2012) Prokaryotic diversity in Aran-Bidgol salt lake, the largest hypersaline playa in Iran. Microbes Environ 27:87–93. doi:10.1264/jsme2.ME11267
Maturrano L, Santos F, Rosselló-Mora R, Antón J (2006) Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol 72:3887–3895. doi:10.1128/AEM.02214-05
Mezghani M, Alazard D, Karray F, Cayol JL, Joseph M, Postec A, Fardeau ML, Tholozan JL, Sayadi S (2012) Halanaerobacter jeridensis sp. nov., isolated from a hypersaline lake. Int J Syst Evol Microbiol 62:1970–1973. doi:10.1099/ijs.0.036301-0
Mouné S, Caumette P, Matheron R, Willison JC (2003) Molecular sequence analysis of prokaryotic diversity in the anoxic sediments underlying cyanobacterial mats of two hypersaline ponds in Mediterranean salterns. FEMS Microbiol Ecol 44:117–130. doi:10.1016/S0168-6496(03)00017-5
Muyzer G, De Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nadkarni MA, Elizabeth Martin F, Jacques NA, Hunter N (2002) Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiol 148:257–266. doi:10.1099/00221287-148-1-257
Oren A (2002) Diversity of halophilic microorganisms: environments, phylogeny, physiology and applications. J Ind Microbiol Bitechnol 28:56–63. doi:10.1038/sj/jim/7000176
Oren A (2010) Industrial and environmental applications of halophilic microorganisms. Environ Technol 31:825–834. doi:10.1080/09593330903370026
Ori GG, Pascucci V, Gasmi N, Barbieri R (2009) Tunisian desert: a perfect place to simulate the landing on mars. In: 27th IAS meeting of sedimentology- field trips guide book, SASSARI, EDES, pp 315–342
Ruff-Roberts AL, Kuenen JG, Ward DM (1994) Distribution of cultivated and uncultivated cyanobacteria and chloroflexus-like bacteria in hot spring microbial mats. Appl Environ Microbiol 60:697–704
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Tsai CR, Garcia JL, Patel BKC, Cayol JL, Baresi L, Mah RA (1995) Haloanaerobium alcaliphilum sp. nov., an anaerobic moderate halophile from the sediments of Great Salt Lake, Utah. Int J Syst Evol Microbiol 45:301–307. doi:10.1099/00207713-45-2-301
Ventosa A, Mellado E, Sanchez-Porro C, Marquez MC (2008) Halophilic and halotolerant micro-organisms from soils. In: Dion P, Nautiyal CS (eds) Microbiology of extreme soils. Springer, Berlin, pp 87–115. doi: 10.1007/978-3-540-74231-9_5
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wright ES, Safak Yilmaz L, Noguera DR (2012) DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol 78:717–725. doi:10.1128/AEM.06516-11
Acknowledgments
MBA was supported by the Tunisian Ministry of Higher Education, Scientific Research and Technology fellowship. This work was published with the support of AIRD (JEAI HALOBIOTECH project “Traitement anaérobie des effluents industriels salins et hypersalins par des bioréacteurs membranaires”).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by A. Oren.
Rights and permissions
About this article

Cite this article
Abdallah, M.B., Karray, F., Mhiri, N. et al. Prokaryotic diversity in a Tunisian hypersaline lake, Chott El Jerid. Extremophiles 20, 125–138 (2016). https://doi.org/10.1007/s00792-015-0805-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-015-0805-7