
Abstract
Saline lakes at high altitudes represent an important and extreme microbial ecosystem, yet little is known about microbial diversity in such environments. The objective of this study was to examine the change of microbial diversity from the bottom of the lake to sediments of 40 cm in depth in a core from Qinghai Lake. The lake is saline (12.5 g/L salinity) and alkaline (pH 9.4) and is located on the Qinghai–Tibetan Plateau at an altitude of 3196 m above sea level. Pore water chemistry of the core revealed low concentrations of sulfate and iron (<1 mM), but high concentrations of acetate (40–70 mM) and dissolved organic carbon (1596–5443 mg/L). Total organic carbon and total nitrogen contents in the sediments were ∼2 and <0.5%, respectively. Acridine orange direct count data indicated that cell numbers decreased from 4 × 109 cells/g at the water–sediment interface to 6× 107 cells/g wet sediment at the 40-cm depth. This change in biomass was positively correlated with acetate concentration in pore water. Phospholipid fatty acid (PLFA) community structure analyses determined decrease in the proportion of the Proteobacteria and increase in the Firmicutes with increased depth. Characterization of small subunit (SSU) rRNA genes amplified from the sediments indicated a shift in the bacterial community with depth. Whereas the α-, β-, and γ-Proteobacteria and the Cytophaga/Flavobacterium/Bacteroides (CFB) were dominant at the water–sediment interface, low G + C gram-positive bacteria (a subgroup of Firmicutes) became the predominant group in the anoxic sediments. Both PLFA and the sequence data showed similar trend. The Proteobacteria, CFB, and gram-positive bacteria are present in other saline lakes, but thepresence of Actinobacteria and Acidobacteria/Holophaga in significant proportions in the Qinghai Lake sediments appears to be unique. The archaeal diversity was much lower, and clone sequences could be grouped inthe Euryarchaeota and Crenarchaeota domains. The archaeal clones were not related to any known cultures but to sequences previously found in methane-rich sediments. Acetate-utilizing methanogens were isolated from sediment incubations, and α- and γ-proteobacterial isolates were obtained from a water sample from the lakebottom (23 m). Our data collectively showed that the observed diversity and shift in the community structure with depth was correlated with geochemical parameters (the redox state and availability of electron acceptor and donor). Heterotrophic methanogenesis is possibly adominant metabolic process in the Qinghai Lake sediments. These results reinforce the importance of geochemical controls on microbial ecology in saline and alkaline lake environments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Saline and alkaline lakes are interesting model systems for studies on microbial diversity and ecosystem functions in extreme environments. Recent studies have shown that such environments are highly productive [30] and have been a valuable source of novel microorganisms [39]. Both cultivation and molecular-based approaches (SSU rRNA gene analysis) have been employed to reveal diverse bacterial and archaeal communities over a range of salinity, pH, and temperature. Various lakes from around the world have been studied, including Mono Lake in California [28], saline meromictic Lake Kaiike in Japan [34], soda lakes in Mongolia [60] and Inner Mongolia of China [40], soda lakes in the Kenyan–Tanzanian Rify Valley [54], saline and alkaline lakes of the Wadi el Natrun in the Libyan Desert, Egypt, [22], athalassohaline lakes of the Atacama Desert, Chile [12], and lakes in Antarctic [62, 70]. Many of these studies were conducted to understand cycling of carbon, sulfur, and nitrogen under oxic and anoxic conditions in diverse environments.
Despite the diverse physical and chemical propertiesof the lakes studied, only a limited number of bacterial groups predominate. The Proteobacteria and theCytophaga–Flexibacter–Bacteroides (CFB) group are commonly dominant in lake waters [12, 22, 28, 54]. Allsubdivisions of the Proteobacteria (α, β, γ, δ, and ɛ subdivisions) have been observed. CFB is recently recognized as the main bacterial inhabitants of some hypersaline environments [12]. Demergasso et al. [12] observed that the relative abundance of the CFB increased with salinity and concluded that hypersaline waters apparently constituted an important environment for CFB and a great natural laboratory for studying the ecology of this group of bacteria.
Compared to the recent interest in microbial ecology in the water column of many lakes, the number of investigations related to lake sediments is low. In this regard, Antarctic lakes probably represent one of the best-studied environments. Stackbrandt and Brambilla [62] compiled the major bacterial groups represented in the clone library of SSU rRNA genes isolated from sediments and mat materials of nine Antarctic lakes and reported that the Proteobacteria and gram-positives were the two most abundant groups. Duckworth et al. [14] and Rees et al. [54] reported gram-positive isolates and clone sequences from waters and soil/sediments in soda lakes of the Kenyan–Tanzanian Rift Valley. Thus, it appears that gram-positives may be an important group in anoxic lake sediments.
Despite these recent findings, microbial ecology in saline and alkaline lakes at high altitude has not been investigated except for one study in Chile [12]. The authors of that paper studied prokaryotic diversity in waters of athalassohaline lakes of the Atacama Desert, northern Chile. The lakes are saline (salinity 1–36%), alkaline (pH 7–9), and at high altitude (800–4140 m). The authors observed a trend of increasing contribution (abundance) of CFB with higher salinity and altitude. The authors, however, did not investigate microbial diversity in the lake sediments.
Qinghai Lake is a saline (12.5 g/L salinity) and alkaline (pH 9.4) lake on the Tibetan Plateau. The altitude of the lake is 3196 m above sea level. Qinghai Lake distinguishes itself from other lakes by its unique location. It is located on the northwest margin of the Tibetan Plateau in the juncture of three climatic systems. The objective of this investigation was to study microbial diversity and abundance in such a unique environment. We performed geochemical and microbiological analyses on a sediment core (40 cm in length) from Qinghai Lake and correlated microbial characteristics to geochemistry. Water geochemistry, sediment mineralogy, and geochemistry were analyzed. Culture-dependent and -independent approaches were employed to characterize microbial diversity. The results indicated that microbial community structure changed with sediment depth in the core, and these changes were correlated with the geochemical characteristics.
Methods
Introduction to Qinghai Lake
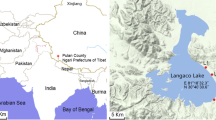
Qinghai Lake (36°32′–37°15′N, 99°36′–100°47′E) is a perennial lake located in a structural intermontane depression at the northeastern corner of the Qinghai–Tibetan Plateau (Fig. 1). It is located in a juncture in the confluence of East Asian monsoon moisture that originates from low latitudes and the cold, dry polar airflows from the Siberian high pressure. The lake has an area of 4300 km2 and lies within a catchment of limestones, sandstones, and shales [25]. The average water depth is 19.2 m, and the maximum is 28.7 m. The altitude of the lake is 3196 m above sea level (masl), and the surrounding mountains rise to above 5200 masl. The evaporation of the lake (∼1400 mm/year) is in excess of mean annual precipitation (∼400 mm/year), resulting in the development of a saline lake with salinity of 12.5 g/L [58]. Qinghai Lake is the largest inland saline lake in China and possesses a continuous sedimentary sequence at least to the Pliocene period (5million years). It is a natural archive of environmental changes and stores abundant information about the history of climatic change, especially climatic variation, vegetation succession, tectonic activity, glacial effect, and their connection with global change.

Location map of Qinghai lake in northwest China. Qinghai Lake is a perennial lake located in a structural intermontane depression on the northeastern corner of the Qinghai–Tibetan Plateau. Courtesy of Chinese Academy of Sciences.
Study Site and Sampling Rationale
Recent seismic survey [1] suggests that Qinghai Lake consists of northern and southern subbasins separated by a normalfaulting horst in the middle of the lake. The northern subbasin is apparently disturbed by a few active faults, whereas the southern one shows relatively simple structure and continuous stratigraphic sequences implying a stable depositional environment. Approximately 700-m-thick unconsolidated sediments were detected.
The separation of Qinghai Lake into two subbasins is also reflected in mixing dynamics of water column and resulting vertical distribution of geochemistry. Overall, the northern subbasin, because of input of a major river in the northwest and strong northwest wind, is more dynamic, and the entire water column is well mixed in summer. The southern subbasin is relatively quiet, and water stratification occurs seasonally. A previous study shows stratification of dissolved oxygen in the water column in the eastern depression of the southern subbasin with a relatively long water column (∼23 m) [58]. We selected our sampling site at approximately 15 km north of a fishing station called Erlangjian for easy access (Fig. 1) and potential of stratified water column. This site was one of the sites selected for drilling 700-m cores by International Continental Drilling Program in the summer of 2005.
Field Measurements and Sampling of Lake Water
Field measurements and sampling of lake water were conducted in August of 2003 on a large boat. The water depth was 23 m at the sampling location. A submersible pump was used to continuously pump lake water from a given depth (every 2 m from the lake surface to the bottom) to a large bucket to facilitate measurement and sampling. The following parameters were measured with YSI DO, pH, and conductivity probes: pH, temperature, conductivity, salinity, and dissolved oxygen. Field colorometric Hach kits were used to measure soluble Fe (Fe2+), sulfide, sulfate, phosphate, nitrite, and nitrate concentrations. Because of homogeneity of the measured parameters along the vertical water column, only four depths at 0, 4, 10, and 23 m were sampled for further analyses back in the laboratory. The four water samples were used for cultivation, cations, anions, and dissolved and total organic carbon (DOC and TOC) analyses in the laboratory following published protocols [44].
Sediment Core Collection and Processing
Four sediment cores of 50 cm in length and 8 cm in diameter were collected from the same sampling site (Fig. 1) with a gravity drop-coring device. The cores were cased inside polyvinyl chloride tubing that had been sterilized with alcohol. Once the cores were brought up to the boat, they were immediately capped. Cores for cultivation were kept at 4°C, and those for SSU rRNA gene analysis were frozen in dry ice. In less than 1 week, the cores were taken to China University of Geosciences in Beijing and were stored in a −20°C freezer (for those originally stored in dry ice) and a refrigerator (for those originally stored at 4°C). Within 1 month, the cores were shipped to Miami University. Because all four collected cores were the same in color, grain size, and mineralogy, only one core was selected for geochemistry and microbiology work. The selected core was dissected inside a glove box (Coy Laboratory Products, Ann Arbor, MI), and the external layers were carefully removed using sterile tools to avoid possible contamination. Subsamples were taken every 2 cm starting from the top of the core to a depth of 40 cm. Because of the loss of some material, only 19 subsamples were recovered. The bottom sample was labeled as QLS40 (i.e., Qinghai Lake sediment from depth 40 cm). The sediment subsamples were stored inside airtight Balsch tubes.
Pore water was collected for cultivation and geochemistry by centrifugation of the individual sediment subsamples. Upon return to the glove box, pore water samples were separated from the sediments and preserved for geochemical analyses using protocols similar to those used for lake water samples [44]. The sediment subsamples were stored anaerobically in airtight Balsch tubes until the time of analyses. The sediment subsamples and their corresponding pore water were subjected to geochemical and microbiological analyses. Geochemical analyses included pore water chemistry (dissolved oxygen, pH, cation, anion, dissolved organic carbon, and acetate), sediment mineralogy, sediment organic carbon and organic compound, sediment total nitrogen, and bioavailable phosphorous. Microbiological analyses included direct and colony-forming unit (CFU) counting, enrichment and cultivation with various media, phospholipid fatty acid (PLFA) analysis, and sequence determination of the SSU rRNA gene sequences.
Geochemical Analyses of Lake and Pore Water Samples in the Laboratory
Cations were analyzed by direct current plasma emission spectrometry (DCP). Common anions (F, Cl, Br, NO3, PO4, SO4) were analyzed by high-performance liquid chromatography (HPLC) on a Dionex (Sunnyvale, CA) DX500 ion chromatograph using an IonPac AS14 analytical column (250 × 4 mm). Acetate concentration was determined with an IonPac AS11-HC analytical column using 0.3 mM NaOH as the eluent. Samples for DOC were first filtered through 0.45-μm filters and then acidified to pH 4 to remove inorganic carbon. DOC concentration was measured as CO2 generated by catalytic combustion (EPA Method #514.1) using an infrared detector (Shimadzu TOC5000A, Columbia, MD) in the nonpurgable organic carbon mode.
Geochemical Analyses of Sediments
To identify mineralogy in the sediments, powder X-ray diffraction (XRD) patterns were obtained with a Philips PW3040/00 X'Pert MPD system, using CuKα radiation with a variable divergent slit and a solid-state detector. The routine power was 700 W (35 kV, 20 mA). Low-background quartz XRD slides (Gem Dugout, Inc., Pittsburgh, PA) were used. For analysis, powder samples were tightly packed into the well of the slides (1/4-in. diameter).
Sediment samples of 0.01–1.0 g in weight were also analyzed for total nitrogen, total and inorganic carbon, and bioavailable phosphorous in the STAR lab of the Ohio State University. Total nitrogen was analyzed with the combustion method (Dumas method). Total organic carbon was analyzed with the dry combustion method and total inorganic carbon with the US EPA method 9060A. These methods are available on-line at http://www.oardc.ohio-state.edu/starlab/references.htm. Available phosphorus was analyzed following a previously published method [36]. Organic compound analyses were performed at China University of Petroleum following the method of Xie et al. [76].
PLFA Analyses
The analysis of PLFAs is useful for evaluating biomass, community structure, and physiological changes in environmental samples. Because phospholipids break down rapidly upon cell death [72, 74], the PLFA biomass does not contain “fossil” lipids of dead cells. The sum of the PLFA, expressed as picomoles, is proportional to the number of cells. The proportion typically used, 20,000 cells/pmol, is taken from cells grown in laboratory media (Microbial Insights, Inc.).
The assessment of community structure using the PLFA method is based on the fact that broad phylogenic groups of microbes have different fatty acid profiles, making it possible to distinguish among them [13, 16, 73, 75, 79–81]. The method is also valuable for estimating physiological status. Toxic compounds or environmental stress conditions that disrupt the membrane cause some bacteria to make trans fatty acids from the usual cis fatty acids [24]. Thus, the ratio of trans/cis fatty acids is a measure of environmentally induced stress on microorganisms. Many Proteobacteria and others respond to starvation by making cyclopropyl [24] or mid-chain-branched fatty acids [71]. Therefore, the ratio of cy/cis fatty acids is a reflection of the amount of starvation that microorganisms experienced.
Because of an insufficient amount of sediment samples, sediment subsamples from 4-cm-depth intervals were combined for PLFA analyses (i.e., QLS40–QLS38, QLS30–QLS32, QLS20–QLS22, QLS10–QLS12, and QLS0–QLS2). PLFAs were analyzed by extraction of the total lipid [72] and then separation of the polar lipids by column chromatography [23]. The polar lipid fatty acids were derivatized to fatty acid methyl esters, which were quantified using gas chromatography [55]. Fatty acid structures were verified by chromatography/mass spectrometry and equivalent chain length analysis.
Cultivation of Bacterial Isolates from Qinghai Lake Water
One oxic water sample (dissolved oxygen, ∼3 mg/L) from the depth of 23 m was used as an inoculum for the cultivation of general heterotrophs and/or nitrate reducers. Bacteria were isolated using the following medium that was approximately 0.5% sodium chloride and pH 9.5. For possible nitrate reducers, diluted MR2A (DMR2A) was made as previously described, except that all final concentrations were diluted 5-fold [19]. In addition to DMR2A, for general heterotrophs, a modified Middlebrook 7H9 broth (MMB) was used and contained per liter the following: 0.5 g sodium acetate; 0.5 g yeast extract; 4.7 g Middlebrook 7H9 (Difco); 0.5 g casamino acids; 0.5 g sodium thiosulfate; 10 mL mineral solution; 5 g NaCl. The mineral solution was the same as that used for DMR2A.
Total and CFU Counts in Sediments
To determine the total number of cells in the sediments, fresh subsamples (0.5 g) were prepared and enumerated by the acridine orange direct count [20] following cell detachment from sediments [7]. To determine the proportion of cultivable aerobic cells from the total direct counts, different aliquots of the same sediment subsamples were diluted with 1.3% NaCl solution (salinity in the lake) and were aerobically plated onto plates of LB agar, a general medium with similar salinity as that in the lake. CFUs were visually counted.
Bacterial Enrichments from Sediments
Anaerobic enrichments were prepared for the growth of methanogens, sulfate-reducing bacteria (SRB), and Fe(III) reducers. The medium for the methanogen enrichment was based on the medium for Anaerobranca gottschalkii [52]. Cultures were incubated at 37°C and were provided with 0.5% (w/v) starch or xylan as possible carbon sources. The headspace was monitored for methane production by gas chromatography. The quantification limit was 5 ppm. In those enrichments where methane was detected, we speculated the presence of starch- or xylan-utilizing fermenters to produce acetate that was then used by methanogens. Indeed, the presence of fermentation products acetate and formate was confirmed by HPLC. To confirm the presence of acetate-utilizing methanogens, new enrichments were set up where acetate was the sole carbon source in the new medium and neither starch nor xylan was added. A minimal culture medium M1 [35] was used for the enrichment of iron-reducing bacteria with hydrous ferric oxide as the electron acceptor and acetate as the carbon source. The medium for SRB was based on a previously published recipe for halophilic SRB [48], and the carbon source was sodium acetate.
PCR and Sequence Determination of Isolates from the Lake Water
Cell lysates were made of the isolated microorganisms from the water sample (23-m depth) for the polymerase chain reaction (PCR) amplification of the SSU rRNA genes by boiling cells suspended in Tris–EDTA buffer for 5 min at 100°C. PCR reactions were treated with SeqMix (Q-Biogene, Irvine, CA) prior to sequencing reactions according to the manufacturer's instructions. The SSU rRNA gene was amplified with the universal primers FD1 and 1540r as previously described [77]. The primer 529r (Escherichia coli number designation) was used for sequence determination with ET Dye chemistry (Amersham Pharmacia Biotech Inc., Piscataway, NJ), and the 529r primer allows for the sequence determination of approximately the first 500 bp that contain the V2–V6 region of the SSU rRNA gene sequence. Sequence determination was performed with a 3100 DNA analyzer, and sequences were compared with sequences in GenBank. The sequences were checked for chimeras with Chimera Check and were aligned with ClustalW. Phylogenetic analyses of partial SSU rRNA gene sequences were conducted using molecular evolutionary genetics analysis software (MEGA) version 2.1. Neighbor-joining phylogenies were constructed from dissimilatory distances and pairwise comparisons with a Jukes–Cantor distance model.
DNA Isolation, Amplification, Cloning, and Sequence Analyses of Sediment Samples
Genomic DNA was extracted from 0.5- to 0.7-g samples of frozen sediment (−80°C). DNA extraction was accomplished with an UltraClean Soil DNA Isolation Kit, according to the recommended procedure by the manufacturer (Mo Bio Laboratory Inc., Solana Beach, CA). All stocks, working solutions, and water used for reagents were filtered through a 0.2-μL filter and were then autoclaved. Plastic wares were autoclaved (for example, Fisherbrand 1.5-mL microcentrifuge tubes are autoclavable). The extracted nucleic acid was then purified with a QIAGEN® RNA/DNA Midi Kit (Qiagen Inc., Chatsworth, CA).
Purified DNA was used as template for the amplification of SSU rRNA genes via PCR according to the procedure of the Failsafe Kit (Epicenter Communications Inc., Sausalito, CA). Typical PCR reactions consisted of 10 mM Tris pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 200 μm each deoxyribonucleotide triphosphate, 0.2 μm each primer, and 1.25-U FailSafe PCR Enzyme Mix in 50-μL reaction volume. Bacteria-specific primer sequences wereBac27F: 5′-AGAGTTTGATCMTGGCTCAG and Univ1492R: 5′-CGGTTACCTTGTTACGACTT. The archaea-specific primer sequences were 21F: TTCYGG TTGATCCYGCCRGA and Univ1492R: 5′-CGGTTA CCTTGTTACGACTT. The following conditions were used for bacterial SSU rRNA gene amplification: 30 cycles, denaturing at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 2 min. The standard conditions for archaeal SSU rRNA gene amplification were as follows: 45 cycles, denaturing at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 2 min. Amplified SSU rRNA gene fragments were ligated into pGEM®-T vector (Promega Inc., Madison, WI). The resulting ligation products were used to transform into E. coli DH5α competent cells. Environmental clone libraries were constructed, and randomly chosen colonies per sample were analyzed for insert SSU rRNA gene sequences. Plasmid DNA that contained SSU rRNA gene inserts was prepared with a Qiagen kit. Sequencing reactions were carried out with a DYEnamic ET terminator cycle sequencing ready reaction kit (Amersham Pharmacia Biotech Inc.) using primers Bac27F and Arch21F for bacteria and archaea, respectively. The SSU rRNA genes were sequenced with an ABI 3100 sequencer (approximately 600–700 bp). Forty clones were sequenced for the top (QLS0) and bottom (QLS40) subsamples, and approximately 25 clones were sequenced for the other subsamples.
Phylogenetic Analysis
Clone sequences were manually checked with the sequencer program, and secondary structure diagrams and the Chimera Check program were utilized to identify potential chimeras formed during PCR. The sequences obtained were compared to the Ribosomal Database Project (RDP-II) and GenBank for the identification of closely related sequences. The sequences were aligned with the ClustalW multiple sequence alignment program (http://clustalw.genome.jp/) [68]. The aligned sequences were then manually checked with the MacClade software (version 3.0; Sinauer Associates, Inc., Sunderland, MA). Phylogenetic analyses were performed with the MEGA (http://www.megasoftware.net/). Trees generated with neighbor-joining and minimum evolution methods were not significantly different. Phylogenetic inference and evolutionary distance calculations were made with the distance Jukes–Cantor model (gamma parameter equal to 2.0). Bootstrap analysis (500 replicates) was used to obtain confidence estimates for the phylogenetic trees.
Nucleotide Sequence Accession Numbers
The sequences determined in this study have been deposited in the GenBank database under accession numbers AY937389–AY937396 for the isolate sequences, AY940493–AY940576 for the bacterial clone sequences, and AY940577–AY940613 for the archaeal clone sequences.
Results
Water Chemistry of Qinghai Lake
During our sampling periods (summer 2003), Qinghai Lake did not show any stratification in salinity (1.3% throughout the water column), pH (9.4), or conductivity (18,000–21,000 μS/cm), but water temperature slightly decreased from 17°C at the surface to 14°C at the bottom (23-m water depth). Dissolved oxygen also showed a slight gradient, from 6 ppm at the surface to 3 ppm at the bottom. Water chemistry measured by Hach kits in the field indicated that reduced chemical species [iron(II), sulfide, nitrite, ammonium] were nondetectable. Laboratory DCP analyses determined concentrations of major cations (mg/L): Na+ (6018 ± 195), Mg2+ (701 ± 27), K+ (138 ± 12), Ca2+ (9 ± 1), and total Fe (2 ± 1). The standard deviations represented averages of four samples throughout the entire water column and reflected vertical variations of the cations. HPLC analyses determined concentrations of major anions (mg/L) as Cl− (19,472 ± 584), SO4 2− (2258 ± 68), F− (43 ± 2), NO3 − (17 ± 1), and Br− (6 ± 1).
Pore water was anoxic from 2.5 cm below the water–sediment interface to 50-cm depth (Fig. 2A) with nearly constant pH of 9.3 and conductivity of 16,000–18,000 μS/cm. Concentrations of potential electron acceptors such as sulfate and iron were low except for the sample at the 14-cm depth where sulfate concentration was 3.5 mM (Fig. 2A). Phosphate was not detectable. DOC concentration was high, ranging from 1596 to 5443 mg/L, and it showed an increasing trend with depth (Fig. 2A). Acetate concentration was high, ranging from 40 to 67 mM and showed a decreasing trend with depth. Lactate was not detected. The major cations were as follows (concentration in mg/L): Na+ (2404 ± 42), Mg2+ (769 ± 129), K+ (190 ± 19), Ca2+ (66 ± 41), and total Fe (1.4 ± 1). The major anion (mg/L) was Cl− (5301 ± 322). The standard deviation represented vertical variations across the length of the core (50 cm).

Depth distribution of selected physicochemical and biological variables in the pore water of the Qinghai lake sediment. (A) Dissolved oxygen, total Fe, sulfate, acetate, and dissolved organic carbon. (B) Total nitrogen and organic carbon. (C) Bacterial abundance as measured by acridine orange direct count (AODC) and colony-forming unit (CFU). Note that some variable values are scaled as indicated (by the numbers following slashes) to fit axis ranges.
Sediment Properties
X-ray diffraction results indicated that the sediment samples were dominated with quartz, feldspars, illite, kaolinite, calcite, dolomite, and aragonite in decreasing order of abundance. TOC content ranged from 1.8 to 2.4% with no particular trend with depth (Fig. 2B). Total nitrogen concentration ranged from 0.23 to 0.32%. Organic compound analyses indicated that saturated hydrocarbons ranged from 1.6 to 6.2%, aromatic hydrocarbons from 0.1 to 0.6%, resin (nonhydrocarbons) from 68.7 to 79.0%, and asphaltene from 16.8 to 28.4%. Available phosphorus was approximately 1 μg/g.
Quantitation of Biomass by Acridine Orange Direct Count, CFU, and PLFA
Both acridine orange direct count and CFU data indicated that the number of total and cultivable aerobic cells decreased with increased depth (Fig. 2C). The total cells ranged from approximately 109 cells/g wet sediment at the water–sediment interface to 107 cells/g at the 50-cm depth. Cultivable aerobic biomass was much lower, ranging from 107 cells/g at the lake bottom to 103 g at the 50-cm depth. Estimates of viable biomass as determined by the total concentration of PLFA and a conversion factor of 20,000 cells/pmol ranged from 109 cells/g at the lake bottom to 108 cells/g at the 40-cm depth.
PLFA Community Profiling
Phospholipid fatty acid profiles indicated that all samples contained relatively diverse microbial communities in that all fivecategories of PLFA were present in each sample (Table 1). The PLFA profiles of the bottom four sediment subsamples, i.e., QLS36–QLS40, QLS30–QLS32, QLS20–QLS22, and QLS10–QLS12, were quite similar but different from the interface sample (QLS0–QLS2). Normal saturated PLFA was the predominant PLFA present in all samples. Monoenoic PLFA, indicative of the Proteobacteria, was the second most prevalent type of PLFA observed. In the interface sample, monoenoic PLFA represented approximately 33% of the total PLFA, but in the other samples, the proportion of monoenoic PLFA was approximately 15–19%. Terminally branched saturated lipids are indicative of anaerobic gram-negative bacteria and Firmicutes, and they were observed in an inverse proportion to the monoenoic PLFA. The interface sample contained approximately 5% terminally branched PLFA, but in all other samples, the proportion was at least double this amount. These results suggested that the deeper samples contained anaerobic microorganisms in accordance with the geochemical data. Other anaerobic biomarkers (mid-chain-branched saturates and branched monoenoics) were also present in lower proportions in the interface sample than in the deeper samples. Analyses of particular biomarkers (10me16:0, 10me18:0, and i17:1w7c) indicated that SRB were possibly present in the bottom four samples but in small proportions. Eukaryotic biomarkers (polyenoic PLFA) were observed in all samples and were highest in the interface sample, which indicates that the sediment at the water–sediment interface represented a more oxic environment. This was consistent with the geochemical measurement of lake water in that dissolved oxygen was high (∼3 ppm) at the lake bottom, but pore water at the 1.25-cm depth was anoxic. Physiological status biomarkers indicated that all of the samples were undergoing a relatively low amount of starvation (i.e., cy/cis < 1). The amount of starvation observed was inverse to the patterns observed in the biomass levels, and these results may suggest that samples with low biomass were limited for a nutrient(s). The PFLA data also suggest that environmentally induced stresses were not present (Table 1).
Detection of Methanogenic Microorganisms in Sediments
Methane was produced in all of the incubated microcosms for methanogens when starch was used as a carbon source, and visible growth was observed. Methane was detected in the headspace gas as measured by gas chromatography, ranging from 58 to 98 ppm. This range represented 90–93% of all gasses in the headspace. When acetate was used as the sole carbon source in the methanogen growth medium, a similar amount of methane production occurred and growth was clearly visible, suggesting enrichment of acetate-utilizing methanogens. Growth was not observed in enrichment tubes that contained either sulfate or iron hydroxides.
Bacterial Diversity in Sediments
Partial sequences of the SSU rRNA genes could be classified into the following groups.
α-Proteobacteria
Sequences most closely related to the α-Proteobacteria were from isolates obtained from the water sample at the bottom of the lake (23 m) and the water–sediment interface sample (QLS0; Fig. 3). One sequence from QLS30 and two sequences from QLS40 also belonged to this group. Three isolates from the water sample (23-m water depth, bottom of the lake) were closely related to (>98% similarity) Caulobacter MBIC1405 (AB016847). Two sequences from QLS0 were 96% similar to Paracoccus sp. RA19 isolated from a Polynesian lagoon (AJ507806, GenBank description). Two others from the same sample were 97% similar to Haplosporidium sp. endosymbiont. Of the two sequences from the deepest sediment sample (QLS40), one was 92% similar to a clone sequence from a soda lake (GenBank description, AF507004).

Neighbor-joining tree (partial sequences, ∼600 bp) showing phylogenetic relationships of bacterial 16S rRNA gene sequences cloned from the Qinghai Lake sediments to closely related sequences from GenBank. Clone sequences from this study are coded as follows, with QLS30-B23 as an example: QLS, Qinghai Lake sediment; 30, sample depth in cm; B, bacteria; 23, clone number. Therefore, it reads bacterial clone 23 from Qinghai Lake sediment of 30 cm in depth. The isolates were obtained from a water sample from 23-m water depth (lake bottom). They are coded as follows: QLW23, Qinghai Lake water from 23 m depth, isolateQ. Scale bars indicate Jukes–Cantor distances. Bootstrap values of >50% (for 500 iterations) are shown. Aquifex pyrophilus is used as an outer group, and a single tree showing all bacterial sequences is created. Because of its large size, it is divided into two subtrees. This figure is the first bacterial subtree showing the α-, β-, and γ-Proteobacteria, Planctomycetes, and Cytophaga–Flexibacter–Bacteroides (CFB) groups.
β-Proteobacteria
Similar to the α-Proteobacteria, the sequences in this group were primarily observed in the clonelibrary of QLS0, with one sequence each from 10-, 20-, and 40-cm depth (out of 25 clones from each respective library; Fig. 3). The sequences from the QLS0 clone library were related to a diverse group of bacteria, including Comamonas sp. TK41 from an activated sludge-derived microbial community (>98% similarity; GenBank description), a β-proteobacterium OS-ac-16 from a Yellowstone National Park hot-spring microbial mat [46], and an uncultured bacterium from a phosphorus-removal ecosystem (88% similarity; AF314424) [11]. The sequence from QLS10 was 95–96% similar to autotrophic ammonia-oxidizing bacterium Nitrosospira briensis. The sequence from QLS20 was >98% similar to uncultured Nitrosomonas sp. (AJ431350) from an alkaline, cold ecological niche in Greenland [64].
γ-Proteobacteria
Nine clonal sequences and five bacterial isolates from the water sample clustered with the γ-Proteobacteria (Fig. 3). Three of the isolates (QLW23-isolateQ, QLW23-isolateC, and QLW23-isolateM) clustered together and were 96–97% similar to Pseudomonas anguilliseptica. P. anguilliseptica has been observed from sediments at uranium waste piles [59] and is also an opportunistic fish pathogen that canbe isolated from diseased fish [3]. QLW23-isolateX and QLW23-isolateZ had approximately 92% sequence similarity to Rheinheimera baltica BA 33. Rheinheimera is a recently named bacterial genus, and R. baltica is a novel, blue-pigmented bacterium that was isolated from the Baltic Sea [9]. However, on the tested medium, the two isolates did not produce blue pigmentation. Other species of Rheinheimera have been isolated from the deep Pacific Ocean [56].
Two of the four clonal sequences from QLS0 were closely related to Pseudomonas fulva and Stenotrophomonas maltophilia LMG 20578, respectively. P. fulva was previously isolated from strawberry plants, and LMG 20578 is capable of reducing selenate and selenite to elemental selenium under microaerophilic conditions [15]. One sequence was related to (94% similarity) a sulfur-oxidizing endosymbiont of Ridgeia piscesae (AY129120) found in tubeworms from hydrothermal vent systems [45]. The sequence from QLS10 clustered with one sequence from QLS0 and was related to (98% similarity) P. fulva or Pseudomonas sp. Fa27. One sequence from QLS30 clustered with the two sequences from QLS40, and they were 88–90% similar to an uncultured sponge symbiont (AF186447) from nutrient-limited environments such as deep-sea sediment [38].
δ-Proteobacteria
Sequences in this group were dominated by those from the clone library of QLS30 (6out of 16) with one from QLS0, two from QLS10, and two from QLS40 (Fig. 4). Two sequences from QLS30 and one each from QLS0 and QLS40 formed a cluster and had approximately 95% similarity to a clone sequence recovered from benzene-contaminated groundwater/uranium mining waste piles and mill tailings (AJ534628) (GenBank description). One sequence from QLS10 and two sequences from QLS30 formed a small cluster and had 98% identity with an uncultured hydrocarbon seep bacterium (AF154102; GenBank description) and were moderately related (90–92% similarity) toa benzene-mineralizing, sulfate-reducing consortium and Desulfobacterium anilini. D. anilini is capable of degrading the aromatic compound aniline [57]. Two sequences (one each from QLS30 and QLS40) had 94% identity with an uncultured delta proteobacterium (AJ535249) in sediments above gas hydrates from Cascadia Margin, Oregon [32]. One sequence from QLS30 was closely related to (96% similarity) uncultured Desulfosarcina sp. or Desulfosarcina variabilis. Members of Desulfosarcina sp. have commonly been isolated in methane gas hydrates [32].

This figure is the second bacterial subtree showing Low G + C gram-positive, gram-positive, Firmicutes, Actinobacteria, δ-Proteobacteria, Acidobacteria/Holophaga, and green nonsulfur bacteria.
Cytophaga–Flexibacter–Bacteroides Group
Eight sequences (four from QLS0, three from QLS10, and one from QLS20) could be classified into the CFB clade (Fig. 3). Two sequences in QLS0 were 88% similar to an uncultured bacterium clone from Lake Sapgyo in Korea (AY135907, GenBank description). One sequence from QLS0 was distantly related to (88% similarity) Cytophaga sp. EPR 14 from a deep-sea hydrothermal vent (GenBank description). Of the three sequences from QLS10, two distantly (85% similarity) formed a cluster with Cytophaga sp. or Cytophaga fermentans. Sequences belonging to the Cytophaga group have been isolated from deep-sea sediments [38]. One sequence in the Cytophaga group was closely related to a benzene-mineralizing, sulfate-reducing consortium clone. The other sequence from QLS10 was distantly related to (∼90%) an uncultured bacterium from a phosphorus-removal ecosystem (AF314435) [11].
Planctomycetes
Three sequences clustered with Planctomycetes. One sequence from QLS0 was distantly related to (90% similarity) an uncultured Planctomycete clone recovered from Mono Lake, CA (AF507874) [28]. One sequence from QLS20 was distantly related to (89%) Planctomycete GMD14H10 isolated from the Sargasso Sea (in the middle of the Atlantic Ocean, between the West Indies and the Azores) at a depth of 3 m [78], and the other QLS20 sequence was remotely related to (∼90%) an uncultured bacterial clone retrieved in gas hydrates from Nankai Trough, Japan (AY093482) [53].
Low G + C Gram-Positive Bacteria (Firmicutes)
Twenty-seven sequences could be classified as the low G + C gram-positive group (Fig. 4). Low G + C gram-positive bacterial sequences dominated the clone library for the deepest sample. These sequences were also abundant in the libraries from QLS10, QLS20, and QLS30, but not in the library from QLS0. Twenty-six sequences formed a separate lineage and had between 90 and 92% identity with uncultured low G + C gram-positive (AF507875) and Bacillus sp. clone sequences previously recovered from the anoxic zone of a soda lake in California (Mono Lake) [28]. Whereas most sequences were closely related to uncultured low G + C gram-positive bacterial sequences from Mono Lake, some sequences from QLS40 formed a deep branch and were only distantly related to (81–90% similarity) the clones from the lake. Mono Lake is an alkaline (pH 9.8) and hypersaline (84- to 94-g/L salinity) soda lake located east of the Sierra Nevada mountains, approximately 160 km south of Lake Tahoe, CA, USA.
Gram-Positive Group (Firmicutes)
Nine sequences (six from QLS20 and three from QLS10) formed a tight cluster and were closely related to clone sequences retrieved from gas hydrate-bearing sediments from the Gulf of Mexico (AY211667, AY053496) [37, 43], the Sea of Okhotsk (AB094958) [29], Nankai Trough (AY093459) [53], and Cascadia Margin, Oregon [32]. The sediments from the Gulf of Mexico were at 550- to 575-m water depth and had visible oil and gas seepage and methane-rich hydrates. The bottom pressure and temperature were at 50 atm and 7°C. Some of the sequences in this group were also related to a benzene-mineralizing, sulfate-reducing bacterial consortium (AF029050) [51].
Firmicutes
One sequence from QLS0 belonged to the Firmicutes that was not low G + C gram-positive or gram-positive subgroup. It was closely related to Planococcus sp. isolated from desert dust in Mali, West Africa (GenBank description).
High G + C Gram-Positive Bacteria (Acinobacteria)
Seven sequences clustered with this group. Three sequences (two from QLS10 and one from QLS20) formed a small cluster and were moderately related to (92–94%) uncultured bacterial clones from the intertidal flat of Ganghwa Island (AY568849) (GenBank description) and gas hydrate-bearing deep-sea sediments (AB015540) [38]. Three other sequences (each from QLS0, QLS10, and QLS20) were not related to any previously documented sequences.
Acidobacteria
Clonal sequences classified in this group were predominantly from the clone library QLS40. A group of clone sequences from QLS40 formed a cluster and were related to (86–91% similarity) uncultivated Holophaga sp. (AJ535239) recovered from sulfate-reducing and methane-oxidizing sediments above gas hydrates in Cascadia Margin, Oregon [32] and an uncultured bacterium from cold-seep sediments (AY279049; GenBank description). Holophaga/Acidobacterium have been shown to be common in freshwater lakes [61] and are often observed with α-Proteobacteria. One sequence from QLS40 was remotely related to (83%) an uncultured Verrucomicrobia bacterium (AY114317) from anoxic, ammonium-oxidizing marine sediments [18].
Green Nonsulfur Bacteria
Two sequences from QLS30 and QLS40 each formed a small cluster and were related to clone sequences recovered from hydrothermal sediments in the Guaymas Basin (AF419665) [67] and bacterial community in the intertidal flat of Ganghwa Island (AY568796; GenBank description).
Archaeal Diversity in Sediments
Archaeal diversity was low, and the clonal sequences were not closely related to any known isolates. Sequences from different libraries (QLS0 through QLS40) clustered together, and sequences from the different depths were not distinct. Two clusters were observed within the Euryarchaeota. The sequences in the first cluster, which contained the majority of archaeal SSU rRNA gene sequences (74 out of 108), were closely related to (97–99% similarity) an uncultured archaeon clone recovered from methanogenic communities in eutrophic and pristine areas of the Florida Everglades (AY457656) [10] and uncultured archaeon clone sequences from Lake Kinneret sediments (AJ310856) [47]. The related sequences from the Lake Kinneret sediments formed a cluster with clone sequences Eel-TA1e6 [26] and VAL2 within the Euryarchaeotal group III named by Jurgens et al. [31]. Clone sequences Eel-TA1e6 and VAL2 were obtained from marine and forest lake sediments [26, 31]. The sequences in the second cluster were related to (∼94% similarity) an uncultured archaeon (AY341270) from a low-salt, sulfide-, and sulfur-rich spring [17] or to uncultured Euryarchaeote (AJ131274 or AJ131275) in bacterioplankton of a boreal forest lake [31].
There were three small clusters in the Crenarchaeota (Fig. 5). Five sequences formed the first, and they were related to (96–99%) anaerobic methanotrophic communities in hydrothermal sediments in the Guaymas Basin (AF419646) [67], in sediments from deep-sea hydrothermal vent (AB019723) [65], in the waters from deep South African gold mines (AB050240) [66], in the Lake Michigan sediments (U87519) [41], in forest soils in the range of Colorado (AY016470; GenBank description), and in metal-rich particles from a freshwater reservoir (AF418935) [63]. Five sequences formed the second cluster, and they were related to archaeal communities associated with the geological horizons in the coastal subseafloor sediments from the Sea of Okhotsk (97% similarity, AB094513) [29] and those retrieved from the deep subsurface sediments from the Nankai Trough and East Sea (Sea of Japan; 93% similarity, AY345168; GenBank description). Seventeen sequences formed the third cluster, and they were closely related to (90–99% similarity) those archaeal sequences recovered from the geological horizons in the coastal subseafloor sediments from the Sea of Okhotsk (AB095518) [29].

Neighbor-joining tree (partial sequences, ∼600 bp) showing phylogenetic relationships of archaeal 16S rRNA gene sequences cloned from the Qinghai Lake sediments to closely related sequences from GenBank. The same algorithms as those used for the bacterial tree were employed. A. pyrophilus is used as an outer group.
Discussion
Microbial Biomass
The overall decrease in biomass with increased depth was consistent with the general trend reported for subsurface terrestrial sediments [49] and suggests that geochemical conditions may be controlling cell biomass. At the water–sediment interface (the redox boundary), the fraction of biomass that represented the Proteobacteria (monoenoic PLFA) was most abundant among all the samples (approximately 34% of all biomass), and this fraction decreased deeper into the anoxic sediments. Thus, the data suggest that the initial decrease of biomass from the redox boundary to anoxic environment (depth of a few centimeters) may be caused by the change in the redox state. The further decrease in biomass with depth within the anoxic zone suggests that other variables, such as nutrients and stress, may become important factors. The PLFA data revealed that biomass was inversely correlated with indicators of starvation, suggesting that the system may be limited by certain nutrients. High concentrations of DOC and acetate and low concentrations of iron and sulfate suggest that the anoxic portion of the Qinghai Lake sediments may be electron acceptor limited. This observation was consistent with the PLFA, enrichment, and SSU rRNA gene sequence data, indicating the absence of Fe reducers and a negligible fraction of sulfate reducers. High levels of acetate in pore water may be driving heterotrophic methanogenesis, as verified by our enrichment cultures.
Microbial Diversity
In our study, most identified clone sequences had <95% sequence similarity to previously cultivated microorganisms. The phylogenetic analysis of clone sequences resulted in overlaps with sequences from other lakes with similar habitats and from many methane-rich sediments. The Proteobacteria and CFB bacteria are commonly observed in waters and sediments from other saline and alkaline lakes [12, 22, 28, 33, 40, 54, 62]. These two groups do not appear to be specific to saline environments because of observations from freshwater lake environments as well [21, 27]. The gram-positive, Acidobacteria/Holophaga, and green nonsulfur bacterial groups appear to be unique in saline and alkaline lakes. Whereas the dominance of the low G + C gram-positive group has been observed recently in another saline and alkaline lake [28], this study reports the presence of Acidobacteria/Holophaga and green nonsulfur bacteria in an alkaline, saline environment.
The major groups of bacteria detected in this study have also been previously detected in methane-rich sediments [37, 53, 67], indicating the presence of methanogens in the Qinghai Lake sediments. Our successful enrichments of methanogens support this conclusion. Indeed, the presence of methane gas in the sediments of Qinghai Lake was reported in a previous study [58]. In addition, the relatedness of the majority of our detected archaeal sequences to those previously recovered from methanogenic sediments further indicated that methanogenesis may be an important metabolic process in Qinghai Lake. These data were also consistent with the observation of low terminal electron acceptors such as sulfate and iron.
Archaeal diversity in saline and alkaline lake sediments has not been studied extensively in the past. Relative to the high bacterial diversity, the archaeal diversity was low. There were only five clusters within the Euryarchaeota and Crenarchaeota. Our detected sequences were not related to any known cultivated archaea but were most closely related to sequences previously recovered from methane-rich sediments. The archaeal diversity in methane-rich sediments has always been described as quite low [2, 26, 37, 42, 50, 53, 67, 69].
Depth Distribution of Microbial Diversity
Results from the analysis of phospholipids and nucleic acids indicated a shift of microbial community with depth. The PLFA data showed a decrease in the proportion of the Proteobacteria and an increase in the Firmicutes with increased depth. Characterization of SSU rRNA genes amplified from the sediments indicated a shift in the bacterial community with depth. Whereas the α-, β-, and γ-Proteobacteria and the Cytophaga/Flavobacterium/Bacteroides (CFB) were dominant at the water–sediment interface, low G + C gram-positive bacteria, a subgroup with the Firmicutes, became the predominant group in the deeper anoxic sediments (Table 2). The PLFA and the sequence data were consistent and showed changes of microbial community with depth.
The depth-related shift in the microbial composition may reflect the major changes in geochemical conditions. Among these, the most dramatic change was the redox condition and electron acceptor/donor availability. The entire water column in Qinghai Lake was oxic, including the bottom (23 m), but within a few centimeters, the sediments become anoxic as determined from pore water chemistry. Across the redox boundary, other conditions changed as well, including sulfate, DOC, and acetate concentrations. In the water column, sulfate was abundant, but organic matter levels were low. Qinghai Lake is considered mesotrophic to oligotrophic in productivity [58], and the water column lacks significant loads of organic matter with a biological oxygen demand of 1.4 mg/L. In contrast, in the sediments, sulfate concentrations are generally low, but organic materials are abundant [58] with a biological oxygen demand of 44.3 mg/L. Thus, the water column and the sediments represent two contrasting geochemical environments. These contrasting differences may have accounted for the observed differences in the microbial community between the oxic water–sediment interface and the anoxic sediments.
In the water–sediment sample (QLS0), the clone sequences belonged to the α-, β-, and γ-Proteobacteria and CFB (Table 2), and they were similar to those previously observed in saline lake sediments, hydrothermal vents, hot springs, active sludge, phosphorous removal ecosystems, benzene-contaminated groundwater, and forest wetland. This wide range of environments could reflect the diverse inputs of material to the lake from multiple sources (rivers). There are 40 rivers of different sizes that discharge into the lake and account for about 95% of the lake's total water input with the rest coming from groundwater [58]. Although it is difficult to estimate the size of drainage basins of the 40 rivers, it is clear that the basins consist of many different types of soils, vegetations, rocks, and possible weather patterns because of the unique location of Qinghai Lake. The relatedness of QLS0 sequences to selenate and selenite reducers was consistent with the recent findings of selenate reducers in arid and saline Mono Lake [4] and other hypersaline environment [5]. The two isolates, both belonging to low G + C gram-positives, obtained from Mono Lake were capable of coupling selenate reduction, Se(VI) to Se(IV) and Se(IV) to Se(0), with oxidation of organic matter. The QLS0 clone sequences belonged to the γ-Proteobacteria.
The samples from the lower depths (QLS10 through QLS40) differed in the dominant clone types among the samples. The low G + C gram-positive sequences were common to all samples from the anoxic zone (QLS10–QLS40; Table 2) and were related to those retrieved from Mono Lake, CA. Low G + C gram-positive bacteria are characteristic of sediments from lake [28] and deep-sea environment [38]. Previous studies [8, 38] suggest that low G + C gram-positive bacteria may be common in sediments containing methane-rich gas hydrates, but definitive functions of low G + C gram-positives remain to be determined. In addition, further work is needed to increase sequence coverage and to identify the predominant, active members from the respective depths.
The majority of clone sequences in subsample QLS20 were gram-positive bacteria, and they were closely related to previously retrieved sequences from gas hydrates or hydrocarbon seeps in the Gulf of Mexico, Cascade margin, and Nakai Trench, Japan [32, 37, 43, 53]. The relatedness of the QLS20 sequences to the sulfate-reducing and methane-oxidizing sequences detected in the gas hydrates from the Gulf of Mexico [43] suggests that sulfate reducers and methane oxidizers may be present in subsample QLS20. Indeed, high concentration of sulfate was detected near this depth (Fig. 2A), but the enrichment attempts failed to cultivate any sulfate reducers.
The majority of clone sequences from QLS30 dominated the δ-Proteobacteria and may be related to SRB (i.e., Desulfosarcina). However, sulfate-reducing activity was not detected in our enrichment cultures. Sequences within the Desulfosarcina branch of SRB have been previously reported to be abundant in the clone library in hydrate ridge sediments from Cascadia Margin, Oregon [32]. The presence of SRB and methanogens at the same depth suggests possible interactions of potential SRB and methanogens. The clone sequences from QLS40 clustered in two groups: Acidobacteria and low G + C gram-positive. They were related to uncultured Holophaga sp. recovered from sulfate-reducing and methane-oxidizing sediments above gas hydrate in Cascadia Margin, Oregon [32].
Biogeochemical Processes
The absence of sulfate and ferric iron and the presence of abundant acetate suggest that methanogenesis is possibly the dominant metabolic process in the Qinghai Lake sediments. The high concentration of acetate and alkaline pH suggests that the pathway may be via acetate decarboxylation: CH3COO− + H2O → CH4 + HCO3 − + (−31 kJ/mol CH4). Indeed, our enrichment cultures have verified the presence of acetate-utilizing methanogens. Zinder [82] reported that acetate decarboxylation is carried out only by two methanogenic genera (i.e., Methanosarcina and Methanosaeta) but has been shown to be responsible for approximately two thirds of the methane produced in most freshwater sediments and in anoxic bioreactors. In our study, however, the majority of methanogenic sequences in Cluster 1 of the Euryarchaeota were not particularly related to these two genera. More work is necessary to confirm these observations.
It is unclear what role(s) bacteria play in association with methanogenic archaea. Because most of the bacterial clone sequences were related to uncultivated gram-positives, the possible functions cannot be determined. However, as mentioned above, some of the gram-positive sequences were most closely related to known sulfate reducers, and the sequences might represent organisms that could hydrogen-transfer with the methanogens.
Acetogens are generally considered to be obligate anaerobes but can colonize habitats that are not completely anoxic. Recently, Boga and Brune [6] hypothesized that homoacetogens (e.g., such as those observed from termite guts) could reestablish anoxic conditions because of tolerance to temporarily low pO2 and the capacity to reduce O2. The deeper sediments might contain fermentative acetogens able to tolerate the increased pH values. The low iron and sulfate levels then might allow for acetoclastic methanogenesis to predominate. Our enrichment experiments appear to indicate the presence of fermentative bacteria. Methane was produced when starch and acetate were used as carbon sources. Because there is no known methanogen that utilizes starch directly, our current hypothesis is that fermentative acetogens may be responsible for the production of acetate from starch, which was utilized by methanogens.
This study is one of the few microbial diversity studies in saline and alkaline lakes at high altitude, and the data indicated the presence of different microbial populations with respect to depth of saline and alkaline sediments. In addition, because of the unique position of the lake and the diversity of water sources, the clonal libraries contained sequences previously observed from many different environments. The clonal sequences suggested the presence of unique and novel microorganisms associated with phosphate and electron-acceptor limitation that has allowed the development of acetoclastic methanogens. Current work is underway to further characterize and to quantify the unique populations as well as to obtain microbial isolates.
References
ZS An (2003) Scientific Drilling at Qinghai Lake on the Northwestern Tibetan Plateau: High-Resolution Paleoenvironmental Records of Eastern Asia and Their Significance for Global Change Lake Qinghai Workshop, Institute of Earth Environment, Chinese Academy of Sciences Xining, China 1–2
KA Bidle M Kastner DH Bartlett (1999) ArticleTitleA phylogenetic analysis of microbial communities associated with methane hydrate containing marine fluids and sediments in the Cascadia Margin (ODP8 site 892B) FEMS Microbiol Lett 177 101–108 Occurrence Handle1:CAS:528:DyaK1MXls1SmsbY%3D Occurrence Handle10436927 Occurrence Handle10.1111/j.1574-6968.1999.tb13719.x
MM Blanco A Gibello AI Vela MA Moreno L Dominguez JF Fernandez-Garayzabal (2002) ArticleTitlePCR detection and PFGE DNA macrorestriction analyses of clinical isolates of Pseudomonas anguilliseptica from winter disease outbreaks in sea bream Sparus aurata Dis Aquat Org 50 19–27 Occurrence Handle1:CAS:528:DC%2BD38XmvVWit7w%3D Occurrence Handle12152901 Occurrence Handle10.3354/dao050019
JS Blum AB Bindi J Buzzelli JF Stolz RS Oremland (1998) ArticleTitle Bacillus arsenicoselenatis, sp nov, and Bacillus selenitireducens, sp nov: two haloalkaliphiles from Mono Lake, California that respire oxyanions of selenium and arsenic Arch Microbiol 171 19–30 Occurrence Handle1:CAS:528:DyaK1MXltlWgsQ%3D%3D Occurrence Handle10.1007/s002030050673
JS Blum JF Stolz A Oren RS Oremland (2001) ArticleTitle Selenihalanaerobacter shriftii gen. nov., sp nov., a halophilic anaerobe from Dead Sea sediments that respires selenate Arch Microbiol 175 208–219 Occurrence Handle1:CAS:528:DC%2BD3MXjtlGjurs%3D Occurrence Handle11357513 Occurrence Handle10.1007/s002030100257
HI Boga A Brune (2003) ArticleTitleHydrogen-dependent oxygen reduction by homoacetogenic bacteria isolated from termite guts Appl Environ Microbiol 69 779–786 Occurrence Handle10.1128/AEM.69.2.779-786.2003 Occurrence Handle1:CAS:528:DC%2BD3sXhtF2isrc%3D Occurrence Handle12570995
PJ Bottomley (1994) Light microscopic methods for studying soil microorganisms RW Weaver (Eds) Methods of Soil Analysis, Part 2. Microbiological and Biochemical Properties-SSSA Book Series, No. 5 Soil Science Society of American Publisher Madison, WI 81–105
JP Bowman SA McCammon JAE Gibson L Robertson PD Nichols (2003) ArticleTitleProkaryotic metabolic activity and community structure in Antarctic continental shelf sediments Appl Environ Microbiol 69 2448–2462 Occurrence Handle1:CAS:528:DC%2BD3sXjslOlsr8%3D Occurrence Handle12732510 Occurrence Handle10.1128/AEM.69.5.2448-2462.2003
I Brettar R Christen MG Hofle (2002) ArticleTitle Rheinheimera baltica gen. nov., sp nov., a blue-coloured bacterium isolated from the central Baltic Sea Int J Syst Evol Microbiol 52 1851–1857 Occurrence Handle1:CAS:528:DC%2BD38XnvFyhsLs%3D Occurrence Handle12361297 Occurrence Handle10.1099/ijs.0.02151-0
H Castro A Ogram KR Reddy (2004) ArticleTitlePhylogenetic characterization of methanogenic assemblages in eutrophic and oligotrophic areas of the Florida Everglades Appl Environ Microbiol 70 6559–6568 Occurrence Handle10.1128/AEM.70.11.6559-6568.2004 Occurrence Handle1:CAS:528:DC%2BD2cXhtVSju7%2FM Occurrence Handle15528519
P Dabert B Sialve JP Delgenes R Moletta JJ Godon (2001) ArticleTitleCharacterisation of the microbial 16S rDNA diversity of an aerobic phosphorus-removal ecosystem and monitoring of its transition to nitrate respiration Appl Microbiol Biotechnol 55 500–509 Occurrence Handle10.1007/s002530000529 Occurrence Handle1:CAS:528:DC%2BD3MXktlOgsro%3D Occurrence Handle11398934
C Demergasso EO Casamayor G Chong P Galleguillos L Escudero C Pedros-Alio (2004) ArticleTitleDistribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile FEMS Microbiol Ecol 48 57–69 Occurrence Handle1:CAS:528:DC%2BD2cXisF2gt74%3D Occurrence Handle10.1016/j.femsec.2003.12.013
NJE Dowling F Widdel DC White (1986) ArticleTitlePhospholipid ester-linked fatty acid biomarkers of acetate-oxidizing sulfate reducersand other sulfide forming bacteria J Gen Microbiol 132 1815–1825 Occurrence Handle1:CAS:528:DyaL28XkslGmt70%3D
AW Duckworth WD Grant BE Jones R vanSteenbergen (1996) ArticleTitlePhylogenetic diversity of soda lake alkaliphiles FEMS Microbiol Ecol 19 181–191 Occurrence Handle1:CAS:528:DyaK28XjtFejtrc%3D Occurrence Handle10.1111/j.1574-6941.1996.tb00211.x
RS Dungan SR Yates WT Frankenberger SuffixJr (2003) ArticleTitleTransformations of selenate and selenite by Stenotrophomonas maltophilia isolated from a seleniferous agricultural drainage pond sediment Environ Microbiol 5 287–295 Occurrence Handle10.1046/j.1462-2920.2003.00410.x Occurrence Handle1:CAS:528:DC%2BD3sXjt1yktbw%3D Occurrence Handle12662176
A Edlund PD Nichols R Roffey DC White (1985) ArticleTitleExtractable and lipopolysaccharide fatty acid and hydroxy acid profiles from Desulfovibrio species J Lipid Res 26 982–988 Occurrence Handle1:CAS:528:DyaL2MXlsFKqtLc%3D Occurrence Handle4045322
MS Elshahed FZ Najar BA Roe A Oren TA Dewers LR Krumholz (2004) ArticleTitleSurvey of archaeal diversity reveals an abundance of halophilic Archaea in a low-salt, sulfide- and sulfur-rich spring Appl Environ Microbiol 70 2230–2239 Occurrence Handle10.1128/AEM.70.4.2230-2239.2004 Occurrence Handle1:CAS:528:DC%2BD2cXjtlOiu7w%3D Occurrence Handle15066817
TE Freitag JI Prosser (2003) ArticleTitleCommunity structure of ammonia-oxidizing bacteria within anoxic marine sediments Appl Environ Microbiol 69 1359–1371 Occurrence Handle10.1128/AEM.69.3.1359-1371.2003 Occurrence Handle1:CAS:528:DC%2BD3sXitlCmu7w%3D Occurrence Handle12620817
MR Fries J Zhou J Chee-Sanford JM Tiedje (1994) ArticleTitleIsolation, characterization, and distribution of denitrifying toluene degraders from a variety of habitats Appl Environ Microbiol 60 2802–2810 Occurrence Handle1:CAS:528:DyaK2cXltVeqtbk%3D Occurrence Handle8085824
WC Ghiorse DL Balkwill (1983) ArticleTitleEnumeration and morphological characterization of bacteria indigenous to subsurface sediments Dev Ind Microbiol 24 213–224
FO Glockner E Zaichikov N Belkova L Denissova J Pernthaler A Pernthaler R Amann (2000) ArticleTitleComparative 16S rRNA analysis oflake bacterioplankton reveals globally distributed phylogenetic clusters including an abundant group of actinobacteria Appl Environ Microbiol 66 5053–5065 Occurrence Handle10.1128/AEM.66.11.5053-5065.2000 Occurrence Handle1:CAS:528:DC%2BD3cXnvFyqsLY%3D Occurrence Handle11055963
S Grant DY Sorokin WD Grant BE Jones S Heaphy (2004) ArticleTitleA phylogenetic analysis of Wadi el Natrun soda lake cellulase enrichment cultures and identification of cellulase genes from these cultures Extremephiles 8 421–429 Occurrence Handle1:CAS:528:DC%2BD2cXot1yltLg%3D Occurrence Handle10.1007/s00792-004-0402-7
JB Guckert CP Antworth PD Nichols DC White (1985) ArticleTitlePhospholipid ester-linked fatty acid profiles as reproducible assays for changes in prokaryotic community structure of estuarine sediments FEMS Microbiol Ecol 31 147–158 Occurrence Handle10.1016/0378-1097(85)90016-3 Occurrence Handle1:CAS:528:DyaL2MXlsFOjs7Y%3D
JB Guckert MA Hood DC White (1986) ArticleTitlePhospholipid ester-linked fatty acid profile changes during nutrient deprivation of Vibrio cholerae: increases in the trans/cis ratio and proportions of cyclopropyl fatty acids Appl Environ Microbiol 52 794–801 Occurrence Handle1:CAS:528:DyaL28XlvVOgtbY%3D Occurrence Handle3777927
ACG Henderson JA Holmes J Zhang MJ Leng LR Carvalho (2003) ArticleTitleA carbon- and oxygen-isotope record of recent environmental change from Qinghai Lake, NE Tibetan Plateau Chin Sci Bull 48 1463–1467 Occurrence Handle1:CAS:528:DC%2BD3sXmslOrsrY%3D Occurrence Handle10.1360/02wd0272
KU Hinrichs JM Hayes SP Sylva PG Brewer EF DeLong (1999) ArticleTitleMethane-consuming archaebacteria in marine sediments Nature 398 802–805 Occurrence Handle1:CAS:528:DyaK1MXjtVSrsLc%3D Occurrence Handle10235261 Occurrence Handle10.1038/19751
WD Hirons BA Methe SA Nierzwicki-Bauer JP Zehr (1997) ArticleTitleBacterial diversity in Adirondack Mountain lakes as revealed by 16S rRNA gene sequences Appl Environ Microbiol 63 2957–2960
SB Humayoun N Bano JT Hollibaugh (2003) ArticleTitleDepth distribution of microbial diversity in Mono Lake, a meromictic soda lake in California Appl Environ Microbiol 69 1030–1042 Occurrence Handle10.1128/AEM.69.2.1030-1042.2003 Occurrence Handle1:CAS:528:DC%2BD3sXhtF2itr4%3D Occurrence Handle12571026
F Inagaki M Suzuki K Takai H Oida T Sakamoto K Aoki KH Nealson K Horikoshi (2003) ArticleTitleMicrobial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk Appl Environ Microbiol 69 7224–7235 Occurrence Handle10.1128/AEM.69.12.7224-7235.2003 Occurrence Handle1:CAS:528:DC%2BD3sXpvFCls74%3D Occurrence Handle14660370
BE Jones WD Grant AW Duckworth GG Owenson (1998) ArticleTitleMicrobial diversity of soda lakes Extremephiles 2 191–200 Occurrence Handle1:CAS:528:DyaK1cXmtVOqt70%3D Occurrence Handle10.1007/s007920050060
G Jurgens FO Glockner R Amann A Saano L Montonen M Likolammi U Munster (2000) ArticleTitleIdentification of novel Archaea in bacterioplankton of a boreal forest lake by phylogenetic analysis and fluorescent in situ hybridization FEMS Microbiol Ecol 34 45–56 Occurrence Handle1:CAS:528:DC%2BD3cXosVyrtL8%3D Occurrence Handle11053735
K Knittel A Boetius A Lemke H Eilers K Lochte O Pfannkuche P Linke (2003) ArticleTitleActivity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia Margin, Oregon) Geomicrobiol J 20 1362–3087 Occurrence Handle10.1080/01490450303896
Y Koizumi H Kojima M Fukui (2004) ArticleTitleDominant microbial composition and its vertical distribution in saline meromictic Lake Kaiike (Japan) as revealed by quantitative oligonucleotide probe membrane hybridization Appl Environ Microbiol 70 4930–4940 Occurrence Handle10.1128/AEM.70.8.4930-4940.2004 Occurrence Handle1:CAS:528:DC%2BD2cXms1eks7w%3D Occurrence Handle15294833
Y Koizumi H Kojima K Oguri H Kitazato M Fukui (2004) ArticleTitleVertical and temporal shifts in microbial communities in the water column and sediment of saline meromictic Lake Kaiike (Japan), as determined by a 16S rDNA-based analysis, and related to physicochemical gradients Environ Microbiol 6 622–637 Occurrence Handle10.1111/j.1462-2920.2004.00620.x Occurrence Handle1:CAS:528:DC%2BD2cXlsVSmtr0%3D Occurrence Handle15142251
JE Kostka KH Nealson (1998) Isolation, cultivation, and characterization of iron- and manganese-reducing bacteria RS Burlage (Eds) Techniques in Microbial Ecology Oxford University Press Oxford 58–78
S Kuo (1996) Phosphorus, Methods of Soil Analysis. Part 3: Chemical Methods Soil Science Society of America Madison, WI, USA 894–895
BD Lanoil R Sassen MT Duc ParticleLa ST Sweet KH Nealson (2001) ArticleTitleBacteria and Archaea physically associated with Gulf of Mexico Gas Hydrate Appl Environ Microbiol 67 5143–5153 Occurrence Handle10.1128/AEM.67.11.5143-5153.2001 Occurrence Handle1:CAS:528:DC%2BD3MXotlSnsrg%3D Occurrence Handle11679338
L Li C Kato K Horikoshi (1999) ArticleTitleBacterial diversity in deep-sea sediments from different depths Biodivers Conserv 8 659–677 Occurrence Handle10.1023/A:1008848203739
C Lizama M Monteoliva-Sanchez B Prado A Ramos-Cormenzana J Weckesser V Campos (2001) ArticleTitleTaxonomic study of extreme halophilic archaea isolated from the “Salar de Atacama”, Chile Syst Appl Microbiol 24 464–474 Occurrence Handle10.1078/0723-2020-00053 Occurrence Handle1:STN:280:DC%2BD38%2FptlOhuw%3D%3D Occurrence Handle11822685
YH Ma WZ Zhang YF Xue PJ Zhou A Ventosa WD Grant (2004) ArticleTitleBacterial diversity of the Inner Mongolian Baer Soda Lake as revealed by 16S rRNA gene sequence analyses Extremephiles 8 45–51 Occurrence Handle1:CAS:528:DC%2BD2cXos1ersw%3D%3D Occurrence Handle10.1007/s00792-003-0358-z
BJ MacGregor DP Moser EW Alm KH Nealson DA Stahl (1997) ArticleTitleCrenarchaeota in Lake Michigan Sediment Appl Environ Microbiol 63 1178–1181 Occurrence Handle1:CAS:528:DyaK2sXhsFSms7o%3D Occurrence Handle9055434
JR Marchesi AJ Weightman BA Cragg RJ Parkes JC Fry (2001) ArticleTitleMethanogen and bacterial diversity and distribution indeep gas hydrate sediments from the Cascadia Margin as revealed by 16S rRNA molecular analysis FEMS Microbiol Ecol 34 221–228 Occurrence Handle1:CAS:528:DC%2BD3MXht1Ghur8%3D Occurrence Handle11137602 Occurrence Handle10.1111/j.1574-6941.2001.tb00773.x
HJ Mills C Hodges K Wilson IR MacDonald PA Sobecky (2003) ArticleTitleMicrobial diversity in sediments associated with surface-breaching gas hydrate mounds in the Gulf of Mexico FEMS Microbiol Ecol 46 39–52 Occurrence Handle1:CAS:528:DC%2BD3sXns12lsbc%3D Occurrence Handle10.1016/S0168-6496(03)00191-0
DP Moser TC Onstott JK Fredrickson FJ Brockman DL Balkwill GR Drake SM Pfiffner DC White K Takai LM Pratt J Fong BS Lollar G Slater TJ Phelps N Spoelstra M Deflaun G Southam AT Welty BJ Baker J Hoek (2003) ArticleTitleTemporal shifts in the geochemistry and microbial community structure of an ultradeep mine borehole following isolation Geomicrobiol J 20 517–548 Occurrence Handle1:CAS:528:DC%2BD3sXpvV2ltLs%3D Occurrence Handle10.1080/713851170
K Nelson CR Fisher (2000) ArticleTitleAbsence of cospeciation in deep-sea vestimentiferan tube worms and their bacterial endosymbionts Symbiosis 28 1–15
SC Nold ED Kopczynski DM Ward (1996) ArticleTitleCultivation of aerobic chemoorganotrophic proteobacteria and gram-positive bacteria from a hot spring microbial mat Appl Environ Microbiol 62 3917–3921 Occurrence Handle1:CAS:528:DyaK28Xms1KitLw%3D Occurrence Handle8899976
B Nusslein KJ Chin W Eckert R Conrad (2001) ArticleTitleEvidence for anaerobic syntrophic acetate oxidation during methane production in the profundal sediment of subtropical Lake Kinneret (Israel) Environ Microbiol 3 460–470 Occurrence Handle10.1046/j.1462-2920.2001.00215.x Occurrence Handle1:CAS:528:DC%2BD3MXntVOjs7s%3D Occurrence Handle11553236
B Ollivier CE Hatchikian G Prensier J Guezennec JL Garcia (1991) ArticleTitle Desulfohalobium retbaense gen. nov., sp. nov., a halophilic sulfate-reducing bacterium from sediments of a hypersaline lake in Senegal Int J Sys Bacteriol 41 74–81 Occurrence Handle1:CAS:528:DyaK3MXktVCrsLk%3D
Onstott, TC, Phelps, TJ, Kieft, T, Colwell, FS, Balkwill, DL, Fredrickson, JK, Brockman, FJ 1999. A global perspective on the microbial abundance and activity in the deep subsurface. In: Seckbach, J (Ed.) Enigmatic Microorganisms and Life in Extreme Environments. Kluwer Academic Publishers, pp 489–499
VJ Orphan CH House KU Hinrichs KD McKeegan EF DeLong (2001) ArticleTitleMethane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis Science 293 484– 487 Occurrence Handle10.1126/science.1061338 Occurrence Handle1:CAS:528:DC%2BD3MXlsFGjs7k%3D Occurrence Handle11463914
CD Phelps LJ Kerkhof LY Young (1998) ArticleTitleMolecular characterization of a sulfate-reducing consortium which mineralizes benzene FEMS Microbiol Ecol 27 269–279 Occurrence Handle1:CAS:528:DyaK1cXnt1aquro%3D Occurrence Handle10.1111/j.1574-6941.1998.tb00543.x
SG Prowe G Antranikian (2001) ArticleTitle Anaerobranca gottschalkii sp nov., a novel thermoalkaliphilic bacterium that grows anaerobically at high pH and temperature Int J Syst Evol Microbiol 51 457–465 Occurrence Handle1:CAS:528:DC%2BD3MXivVOrtbk%3D Occurrence Handle11321091
DW Reed Y Fujita ME Delwiche DB Blackwelder PP Sheridan T Uchida FS Colwell (2002) ArticleTitleMicrobial communities from methane hydrate-bearing deep marine sediments in a forearc basin Appl Environ Microbiol 68 3759–3770 Occurrence Handle1:CAS:528:DC%2BD38XmtVaqtbc%3D Occurrence Handle12147470 Occurrence Handle10.1128/AEM.68.8.3759-3770.2002
HC Rees WD Grant BE Jones S Heaphy (2004) ArticleTitleDiversity of Kenyan soda lake alkaliphiles assessed by molecular methods Extremephiles 8 63–71 Occurrence Handle1:CAS:528:DC%2BD2cXos1ertw%3D%3D Occurrence Handle10.1007/s00792-003-0361-4
DB Ringelberg GT Townsend KA DeWeerd JM Sulita DC White (1994) ArticleTitleDetection of the anaerobic dechlorinating microorganism Desulfomonile tiedjei in environmental matrices by its signature lipopolysaccharide branch-long-chain hydroxy fatty acids FEMS Microbiol Ecol 14 9–18 Occurrence Handle1:CAS:528:DyaK2cXivFeks78%3D Occurrence Handle10.1111/j.1574-6941.1994.tb00085.x
L Romanenko M Uchino E Falsen NV Zhukova VV Mikhailov T Uchimura (2003) ArticleTitle Rheinheimera pacifica sp. nov., a novel halotolerant bacterium isolated from deep sea water of the Pacific Int J Syst Evol Microbiol 53 1973–1977 Occurrence Handle1:CAS:528:DC%2BD2cXhtF2gug%3D%3D Occurrence Handle14657132 Occurrence Handle10.1099/ijs.0.02252-0
S Schnell B Schink (1991) ArticleTitleAnaerobic aniline degradation via reductive deamination of 4-aminobenzoyl-coa in Desulfobacterium anilini Arch Microbiol 152 183–190 Occurrence Handle10.1007/BF00248615
InstitutionalAuthorNameSciences (1979) Comprehensive Survey Report of Qinghai Lake Scientific Publishing House Beijing
S Selenska-Pobell G Kampf K Flemming G Radeva G Satchanska (2001) ArticleTitleBacterial diversity in soil samples from uranium waste piles as determined by rep-APD, RISA, and 16S rDNA retrieval Antonie van Leeuwenhoek 79 149–161 Occurrence Handle10.1023/A:1010237711077 Occurrence Handle1:CAS:528:DC%2BD3MXmslWkt7s%3D Occurrence Handle11520001
DY Sorokin VM Gorlenko BB Namsaraev ZB Namsaraev AM Lysenko BT Eshinimaev VN Khmelenina YA Trotsenko JG Kuenen (2004) ArticleTitleProkaryotic communities of the north-eastern Mongolian soda lakes Hydrobiologia 522 235–248 Occurrence Handle10.1023/B:HYDR.0000029989.73279.e4
S Spring R Schulze J Overmann K-H Schleifer (2000) ArticleTitleIdentification and characterization of ecologically significant prokaryotes in the sediment of freshwater lakes: molecular and cultivation studies FEMS Microbiol Rev 24 573–590 Occurrence Handle1:CAS:528:DC%2BD3cXotVCksLo%3D Occurrence Handle11077151 Occurrence Handle10.1111/j.1574-6976.2000.tb00559.x
E Stackbrandt E Brambilla (2002) Life in cold lakes G Horneck C Baumstark-Khan (Eds) Astrobiology: The Quest for the Conditions of Life Springer Koln, Germany 161–168
LY Stein G Jones B Alexander K Elmund C Wright-Jones KH Nealson (2002) ArticleTitleIntriguing microbial diversity associated with metal-rich particles from a freshwater reservoir FEMS Microbiol Ecol 42 431–440 Occurrence Handle1:CAS:528:DC%2BD38XoslGiurw%3D Occurrence Handle10.1111/j.1574-6941.2002.tb01032.x
P Stougaard F Jorgensen MG Johnsen OC Hansen (2002) ArticleTitleMicrobial diversity in ikaite tufa columns: an alkaline, cold ecological niche in Greenland Environ Microbiol 4 487–493 Occurrence Handle10.1046/j.1462-2920.2002.00327.x Occurrence Handle1:CAS:528:DC%2BD38XntFSltb8%3D Occurrence Handle12153590
K Takai K Horikoshi (1999) ArticleTitleGenetic diversity of archaea in deep-sea hydrothermal vent environments Genetics 152 1285–1297 Occurrence Handle1:CAS:528:DyaK1MXlslSrtr0%3D Occurrence Handle10430559
K Takai DP Moser DF DeFlaun TC Onstott JK Fredrickson (2001) ArticleTitleArchaeal diversity in waters from deep South African gold mines Appl Environ Microbiol 67 5750–5760 Occurrence Handle1:CAS:528:DC%2BD3MXovFehtLg%3D Occurrence Handle11722932 Occurrence Handle10.1128/AEM.67.21.5750-5760.2001
AP Teske K-U Hinrichs VP Edgcomb A Gomez ParticledeVera DT Kysela ML Sogin HW Jannasch (2002) ArticleTitleMicrobial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities Appl Environ Microbiol 68 1994–2007 Occurrence Handle10.1128/AEM.68.4.1994-2007.2002 Occurrence Handle1:CAS:528:DC%2BD38XivFGlu7Y%3D Occurrence Handle11916723
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleClustal-w— improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acid Res 22 4637–4680
TR Thomsen K Finster NB Ramsing (2001) ArticleTitleBiogeochemical and molecular signatures of anaerobic methane oxidation in a marine sediment Appl Environ Microbiol 67 1646–1656 Occurrence Handle10.1128/AEM.67.4.1646-1656.2001 Occurrence Handle1:CAS:528:DC%2BD3MXis1egtrc%3D Occurrence Handle11282617
BJ Tindall (2004) ArticleTitleProkaryotic diversity in the Antarctic: the tip of the iceberg Microb Ecol 47 271–283 Occurrence Handle10.1007/s00248-003-1050-7 Occurrence Handle1:STN:280:DC%2BD2cvotFOktw%3D%3D Occurrence Handle15054676
IV Tsitko GM Zaitsev AG Lobanok MS Salkinoja-Salonen (1999) ArticleTitleEffect of aromatic compounds on cellular fatty acid composition of Rhodococcus opacus Appl Environ Microbiol 65 853–855 Occurrence Handle1:STN:280:DC%2BD2M3is1ClsA%3D%3D Occurrence Handle9925629
DC White WM Davis JS Nickels JD King RJ Bobbie (1979) ArticleTitleDetermination of the sedimentary microbial biomass by extractable lipid phosphate Oecologia 40 51–62 Occurrence Handle10.1007/BF00388810
DC White HC Pinkart DB Ringelberg (1997) Biomass measurements: biochemical approaches CJ Hurst GR Knudsen MJ McInerney LD Stetzenbach MV Walter (Eds) Manual of Environmental Microbio ASM Press Washington 91–101
DC White DB Ringelberg (1995) Utility of signature lipid biomarker analysis in determining in situ viable biomass, community structure, and nutritional/physiological status of the deep subsurface microbiota PS Amy DL Halderman (Eds) The Microbiology of the Terrestrial Subsurface CRC Press Boca Raton 119–136
DC White JO Stair DB Ringelberg (1996) ArticleTitleQuantitative comparisons of in situ microbial biodiversity by signature biomarker analysis J Ind Microbiol 17 185–196 Occurrence Handle10.1007/BF01570054 Occurrence Handle1:CAS:528:DyaK2sXltFOkug%3D%3D
SC Xie XL Lai Y Yi YS Gu YY Liu XY Wang G Liu B Liang (2003) ArticleTitleMolecular fossils in a Pleistocene river terrace in southern China related to paleoclimate variation Org Geochem 34 789–797 Occurrence Handle1:CAS:528:DC%2BD3sXjsFOkurs%3D Occurrence Handle10.1016/S0146-6380(03)00026-3
Q Ye Y Roh BB Blair C Zhang J Zhou MW Fields (2004) ArticleTitleAlkaline anaerobic respiration: isolation and characterization of a novel, alkaliphilic and metal-reducing bacterium Appl Environ Microbiol 70 5595–5602 Occurrence Handle15345448 Occurrence Handle10.1128/AEM.70.9.5595-5602.2004
K Zengler G Toledo M Rappe J Elkins EJ Mathur JM Short M Keller (2002) ArticleTitleCultivating the uncultured Proc Natl Acad Sci 99 15681–15686 Occurrence Handle10.1073/pnas.252630999 Occurrence Handle1:CAS:528:DC%2BD3sXjvFSh Occurrence Handle12438682
CL Zhang BW Fouke G Bonheyo A Peacock DC White Y Huang CS Romanek (2004) ArticleTitleLipid biomarkers and carbon-isotopes of modern travertine deposits (Yellowstone National Park, USA): implications for biogeochemical dynamics in hot-spring systems Geochim Cosmochim Acta 68 3157–3169 Occurrence Handle10.1016/j.gca.2004.03.005 Occurrence Handle1:CAS:528:DC%2BD2cXlvFamtLs%3D
CL Zhang Y Li JD Wall L Larsen R Sassen Y Huang Y Wang A Peacock DC White J Horita DR Cole (2002) ArticleTitleLipid and carbon isotopic evidence of methane-oxidizing and sulfate-reducing bacteria in association with gas hydrates from the Gulf of Mexico Geology 30 239–242 Occurrence Handle1:CAS:528:DC%2BD38XivFOlt70%3D Occurrence Handle10.1130/0091-7613(2002)030<0239:LACIEO>2.0.CO;2
CLL Zhang YL Li Q Ye J Fong AD Peacock E Blunt JS Fang DR Lovley DC White (2003) ArticleTitleCarbon isotope signatures of fatty acids in Geobacter metallireducens and Shewanella algae Chem Geol 195 17–28 Occurrence Handle10.1016/S0009-2541(02)00386-8 Occurrence Handle1:CAS:528:DC%2BD3sXisVGlsL0%3D
SH Zinder (1998) Methanogens RS Burlage (Eds) Techniques in Microbial Ecology Oxford University Press Oxford 113–136
Acknowledgments
This work was supported by National Science Foundation grant EAR-0345307 and National Science Foundation of China grant 40228004 to HD. C.R. Lucas was supported by an undergraduate research grant from Miami University. We are grateful to W. Green for his help in the field sampling equipments, Dong Chen for doing DOC analysis, and John Morton for his help in cation and anion analyses. We are grateful to three anonymous reviewers for substantially improving the quality of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dong, H., Zhang, G., Jiang, H. et al. Microbial Diversity in Sediments of Saline Qinghai Lake, China: Linking Geochemical Controls to Microbial Ecology. Microb Ecol 51, 65–82 (2006). https://doi.org/10.1007/s00248-005-0228-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-005-0228-6