
Abstract
The use of corticosteroids in the treatment of steroid-sensitive nephrotic (SSNS) syndrome in children has evolved surprisingly slowly since the ISKDC consensus over 50 years ago. From a move towards longer courses of corticosteroid to treat the first episode in the 1990s and 2000s, more recent large, well-designed randomized controlled trials (RCTs) have unequivocally shown no benefit from an extended course, although doubt remains whether this applies across all age groups. With regard to prevention of relapses, daily ultra-low-dose prednisolone has recently been shown to be more effective than low-dose alternate-day prednisolone. Daily low-dose prednisolone for a week at the time of acute viral infection seems to be effective in the prevention of relapses but the results of a larger RCT are awaited. Recently, corticosteroid dosing to treat relapses has been questioned, with data suggesting lower doses may be as effective. The need for large RCTs to address the question of whether corticosteroid doses can be reduced was the conclusion of the authors of the recent corticosteroid therapy for nephrotic syndrome in children Cochrane update. This review summarizes development in thinking on corticosteroid use in SSNS and makes suggestions for areas that merit further scrutiny.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
What’s in a name? That which we call a rose
By any other name would smell as sweet. [1]
Names carry meaning, deliberately so in the case of medical diagnoses, and thereby help us as clinicians to orientate ourselves. There would be few medical school prizes for guessing what first-line treatment for steroid-sensitive nephrotic syndrome (SSNS) might be. However, definition names can encourage tunnel vision and limit out-of-the-box thinking. Such a mindset is all too common with this most prevalent of childhood kidney diseases. It is now five decades since the International Study for Kidney Disease in Children (ISKDC) as a consensus of international experts first recommended a schedule of corticosteroid treatment for the treatment of idiopathic nephrotic syndrome in children [2]. Though the duration of treatment for induction during the first episode (or flare) has been questioned over the last 20 years, it is perhaps surprising that, overall, corticosteroid schedules for initial episode, treatment of relapses, and prevention of relapses have not been more thoroughly challenged.
Small research studies have tried to question the prevailing consensus, but their size has limited impact; multiplicity of study design, often without sufficient care to remove potential bias, has limited the collective evidence to be drawn out through repeated Cochrane reviews [3,4,5].
The use of corticosteroids themselves can vary greatly: the type of corticosteroid used, what dose to give to treat a relapse, when to commence treatment, when to individualize dosing, how to wean the dose, and when to give prophylactically are just some examples. This review, following the recent publication of an updated Cochrane meta-analysis [6] and anticipating publication of the revised KDIGO guidelines for glomerular disease, has been written to cause us to reflect on how the name steroid-sensitive nephrotic syndrome shapes our understanding of the condition. It will discuss the pertinent aspects of current corticosteroid use in SSNS, highlighting areas which call for new creative uses through a balance between efficacy and avoidance of adverse effects, and explore the future role for corticosteroids in this condition which bears its name.
What is the optimum way to treat the first episode?
Over 50 years ago, the International Study of Kidney Disease in Children (ISKDC) suggested a regimen of 60 mg/m2 (maximum dose 80 mg) of daily steroids for 4 weeks (the induction dose), followed by a consolidation regimen of 40 mg/m2 (maximum dose 60 mg) given on three consecutive days out of seven, based on a consensus of international experts [2]. KDIGO amended maximal doses for induction and consolidation phases to 60 mg and 40 mg respectively based on dosing used in adult minimal change disease [7].
This consolidation phase was subsequently modified to alternate-day prednisolone as it was associated with fewer relapses [8]. A shorter consolidation phase was shown to be associated with a shorter cumulative time in remission by the German Arbetsgemeinschaft für Pädiatrische Nephrologie study group in 1988 [9], the same year that a Japanese study first demonstrated benefits in longer term tapering consolidation [10]. Subsequent trials appeared to confirm the finding that a prolonged tapering consolidation course of up to 6 months to treat the initial episode, had a disease-modifying effect resulting in a more sustained remission and this was the conclusion of the first Cochrane review in 2002 [3]. It recommended that patients should be treated at first presentation for at least 3 months, with some benefit gained from up to 7 months of treatment. A subsequent meta-analysis and Cochrane review affirmed that longer initial courses seemed to be more effective [4, 11]. Based upon this, KDIGO published guidance in 2012 recommending 60 mg/m2 daily induction treatment for 4–6 weeks, followed by a tapering consolidation phase from 40 mg/m2 on alternate days over the next 2–5 months (Table 1) [7].
However, many pediatric nephrologists continued to treat patients with an initial ISKDC 8-week course, as highlighted in a recent European survey of practice [12] and their view has now been vindicated.
Prior to the penultimate Cochrane update in 2015 [5], three well-designed randomized controlled trials in 552 children showed no difference in the incidence of future relapses through extending the duration [13] and the dose [14, 15] of the initial corticosteroid course.
The 2015 Cochrane update [5] commented that meta-analysis continued to favor the initial extended courses in reducing risk of relapse by 12–24 months; however, when only studies at low risk of bias (for allocation concealment, attrition, and performance/detection) were included, there was no significant difference between initial corticosteroid courses of 2–3 months compared to 3–7 months.
The PREDNOS study [16], published in 2019, confirmed these newer findings in a multi-center, double-blind, placebo-controlled trial that randomized 237 children with newly presenting nephrotic syndrome to receive either a standard 8-week course of prednisolone (cumulative dose 2240 mg/m2) or an extended course of 16 weeks (cumulative dose 3150 mg/m2). Although there was no difference in the primary outcome of time to first relapse, there was a small difference in cost-effectiveness and quality of life favoring the extended regimen, something that was interpreted by the authors as small, clinically insignificant differences combined with the low cost of prednisolone [17].
The 2020 Cochrane update [6] now includes the PREDNOS data [16] and confirms the authors’ previous view [5] that a difference in results between earlier and more recent trials is explained by study design that better addresses bias risk. With 823 children included in these recent well-designed trials, the Cochrane conclusion to the question of initial corticosteroid dose is that the case is now made against extended initial corticosteroid duration and future resources would be better spent addressing other unanswered treatment questions.
PREDNOS did suggest a possible advantage of an extended course in children under 6 years, acknowledging that this finding replicated that of Sinha’s study [15], which supported retrospective [18] and prospective studies [19] that have shown a greater risk of steroid dependency with young age at onset that is ameliorated by early increased corticosteroid dosing. A meta-analysis is currently underway and two clinical trials of 3 versus 6 months are on-going: a Chinese trial in children under 6 years (NCT04536181) and an Indian trial in children under 4 years (CTRI/2015/06/005939).
Should we dose by body weight or body surface area?
The original ISKDC guidance suggested dosing of prednisone standardized for body surface area (BSA) and not for weight [2]. The majority of guidance and studies following on from this have worked on a BSA- rather than weight-based dosing regimen (as with, for example, the use of prednisolone in the treatment of asthma) [20], with the theory being it more accurately reflects total blood volume (and therefore drug distribution), particularly in children and when dosing with chemotherapy [21]. However, some North American guidelines only use dosing by weight [22] and KDIGO [7] allows for both options (2 mg/kg instead of 60 mg/m2 initially followed by 1.5 mg/kg instead of 40 mg/m2) though without evidence to support. Does it matter which is used?
A commonly used equation to calculate BSA is that of Mosteller [23]:
This evolved in turn from the DuBois formula of 1916 [24] and Haycock in 1978 [25] and is the most commonly accepted formula used in clinical practice. It does require an accurate height, and hence the desire to have a simplified weight-based dosing regimen is attractive. Dosing according to BSA has been used for many years; Feber et al. [26] showed that children receiving a weight-based dose of 2 mg/kg received 0.85 of the 60 mg/m2 BSA dose, resulting in potential under-dosing when one considers original studies showed effectiveness based upon a BSA dosing regimen. This difference is more apparent at lower weights (as much as 8 mg at 15 kg), supported by data from a Japanese retrospective study [27], but is less relevant in children over 30 kg (the average weight of a 9-year-old boy). The fact this dosing difference is more apparent at a weight consistent with most children at their first presentation of nephrotic syndrome makes this more important. Conversely, it is worth noting that as BSA uses both weight and height, the difference with weight only calculations will be reduced in obese children (or those where non-dry weight is used at initial presentation) [28]. Saadeh et al. [29] confirmed the findings in a small study of 43 children and showed that lower dosing at onset had an impact on longer term outcomes. This finding was not supported by two more recent comparisons of weight- and BSA-based corticosteroid dosing although follow-up was relatively short [30, 31].
During the current COVID-19 pandemic, remote care and tele-healthcare have become quickly adapted by patients and healthcare workers. While seeing patients face to face is likely to return, there seems to be a desire from many to utilize some of the technological advances seen going forward. It would be reasonable for parents to obtain weights (and serially, to reduce differences in measuring scales), but an accurate height could be harder to achieve. Emma et al.’s [28] weight-only formula may help with this and is more practical than nomograms [32]. Their equations were:
Using the formula above there was over-estimation of 3.4% for 60 mg/m2 and 2.2% for 40 mg/m2; rarely was there dose under-estimation. When one considers physicians would usually round to the nearest 5 mg this is unlikely to infer clinically significant doses and using the above weight-adjusted formula during the initial episode would avoid under-dosing.
Which is the ideal corticosteroid to use?
Glucocorticoids have been the mainstay of therapy in idiopathic nephrotic syndrome for over 50 years. The exact mechanism of action of glucocorticoids is not fully understood in nephrotic syndrome, but is believed to be both genomic (related to the regulation of nuclear gene expression and the suppression of pro-inflammatory genes) and non-genomic [33, 34].
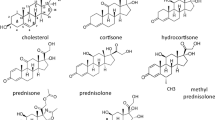
There are a number of different corticosteroids, varying in their glucocorticoid and mineralocorticoid effects (Table 2).
For treatment options, both prednisone and the active metabolite prednisolone are currently frequently the first-choice glucocorticoid steroid [22]. Studies have shown that prednisone has a high first-pass conversion to prednisolone. Both drugs reach peak plasma concentrations at 0.5–3 h and the nephrotic state does not affect the conversion time; therefore, the same dosing regimen has been used interchangeably [7, 35,36,37].
Studies have looked at the use of other corticosteroids. Oral methylprednisolone may be as effective as prednisolone but data are limited [38]. Intravenous (IV) methylprednisolone followed by oral prednisolone was shown to be only marginally less effective than high dose oral prednisolone [39]. With proven efficacy of oral prednisolone in multiple subsequent randomized controlled trials (RCTs), early IV methylprednisolone does not seem justified, but remains useful in attempting to induce complete remission after 4 weeks of oral prednisolone has failed [40,41,42,43]. There is evidence of similar efficacy of intravenous dexamethasone and its cost makes it a more attractive choice in this context in resource-poor countries [44, 45], but there is little evidence to advocate for its use in the treatment of SSNS otherwise.
Questions are frequently raised about the need for intravenous corticosteroid during the nephrotic state when gut edema might theoretically reduce absorption, but historic studies show no difference between children in relapse or remission [46, 47], and in children with active inflammatory bowel disease, there were no differences in pharmacokinetics between oral prednisone and intravenous methylprednisolone [48].
Deflazacort is derived from prednisolone and has similar glucocorticoid and mineralocorticoid properties. Small studies have shown it may be more effective in maintaining remission and may be associated with less corticosteroid toxicity [49, 50]. However, the latest Cochrane meta-analysis shows no increased chance of remission with deflazacort, but concludes that it may reduce the number of relapses by 9–12 months.
In clinical practice, crushed prednisone/prednisolone tablets are generally well-tolerated but some have found better tolerability from liquid preparations [51, 52]. Whether the issue of disease course in young children is related to corticosteroid delivery remains another issue for further study. Certainly, literature on formulations of oral corticosteroids in children is sparse and better data on tolerability and bioequivalence of different preparations is needed [53].
Corticosteroid treatment to prevent relapses
Despite long-term alternate-day prednisolone being the first choice of therapy to prevent relapses in SSNS, there is little quality evidence to support its use [54, 55]. In a recent open-label RCT, Yadav et al. [56] showed greater efficacy when the same 48-hour averaged dose of prednisolone (0.26 mg/kg) was given on a daily rather than alternate-day basis. Not only were relapse rates significantly lower in the daily prednisolone arm over the 12-month study duration, but there was also a tendency for lower obesity and other corticosteroid adverse effects but no difference in growth. The findings of this study also question the long-established practice of giving alternate-day corticosteroid once remission has been achieved.
It has long been recognized that most relapses of SSNS are precipitated by viral upper respiratory tract infection (URTI) and that in children with SSNS, intercurrent infections frequently precipitate a relapse [57,58,59,60]. Most of these observations come from the Indian sub-continent where the pattern of intercurrent illness includes respiratory infection (both upper and lower respiratory tract) and gastroenteritis [57, 58, 60, 61].
In the last two decades, four trials (232 patients) have demonstrated effectiveness of daily low-dose prednisolone for 5–7 days in preventing SSNS relapses associated with intercurrent infections [61,62,63,64]. The applicability of these for children taking a range of background treatment and the generalizability to children in countries where patterns of intercurrent illness differ led to the setting up of the PREDNOS 2 study in the UK, a double-blinded, placebo-controlled trial [65]. The study aimed to address, in a representative sample of children with relapsing SSNS on a range of background treatments, whether a short daily course of low-dose prednisolone given at the time of an URTI prevented a subsequent URTI-related relapse. Over a 7-year period from early 2013, 365 children with frequently relapsing SSNS were randomized to receive either prednisolone or a matched placebo for 6 days at the start of an URTI (www.birmingham.ac.uk/research/bctu/trials/renal/prednos2/investigators/recruitment.aspx, accessed on 7 January 2021). Assessment of corticosteroid adverse effects was from parental reporting and the more objective Achenbach child behavior checklist [66]. Quality of life was evaluated with validated questionnaires and a health-economic evaluation built into the analysis in the same way as was done for the group’s previous PREDNOS trial [17]. The trial completed in early 2020 but the global lockdown for COVID-19 meant a delay in data analysis and results release, which are now anticipated in early 2021. On the theme of prevention of URTI-related relapses with daily corticosteroid dosing, Cochrane’s 2020 update [6] concludes that the practice may be of benefit but anticipates results of the larger PREDNOS 2 trial.
What is the optimum way to treat relapses?
The recommended dose of prednisolone to treat INS relapses is an induction dose of 60 mg/m2 daily until urinary remission (defined as trace or negative urine protein on urine dipstick testing for three consecutive days) followed by a consolidation phase of 40 mg/m2 on alternate days for at least 4 weeks [7]. This is based on the consensus dosing for the initial episode and unlike the initial dosing, treatment of infrequent relapses has not been the subject of large RCTs [12].
The issue has not been completely neglected, but one has to look back 32 years to 1989, when a single-arm, uncontrolled study demonstrated efficacy of half conventional dosing in 17 children with INS [67] and a retrospective review [68] showed benefits of less corticosteroid toxicity through treating relapses with lower doses.
Despite the clear call for an RCT to follow Choonara et al.’s [67] uncontrolled study, only in recent years has this area attracted interest again. In 2017, Raja et al. [69] published a retrospective case series of a selected group of 50 children with frequently-relapsing SSNS without previous complex relapses who were treated with 1 mg/kg of prednisolone daily until remission and then gradually tapered over 4 weeks. They showed 77% of relapses entered remission within 10 days and showed some evidence of better quality of life for those treated with lower doses of prednisolone. These findings were supported by a retrospective Japanese study of 49 patients [70].
In the last 2 years, the effectiveness of half standard doses of corticosteroid to induce and maintain remission has been demonstrated by two small RCTs: Borovitz et al. [71] in a study of 30 children with SSNS showed that this smaller dose resulted in the requirement for a significantly smaller cumulative dose to induce remission, and Sheikh et al. [72] showed no difference between time to remission and a significantly reduced cumulative prednisone dose in a study of 60 children.
These RCTs have been included in the latest Cochrane update [6] and the authors conclude that it is now imperative that large trials address the efficacy of lower dosing, initially in the treatment of relapsing disease, but ultimately in addressing the initial presentation dose.
Other recent studies of dosing for INS relapses have focused on duration of therapy rather than total dose, specifically on the consolidation rather than the induction phase. The Italian PROPINE trial found no difference in time to next relapse when comparing corticosteroid courses that delivered the same cumulative dose over different durations [73], translating findings of prolonged corticosteroids for the initial episode into treatment for relapses, and thus questioning the practice of treating relapses with tapering consolidation corticosteroids. The on-going Dutch RESTERN trial will evaluate both cumulative dose (280 mg/m2 versus 840 mg/m2) and duration (2 versus 6 weeks) of the consolidation phase [74].
These recent studies rightly make the case for a trial to investigate lower dosing as the attractiveness of lower corticosteroid-dosing is self-evident. However, we know that some individual children will respond less well to a reduction in standard corticosteroid dosing. Mehls and Hoyer, in their 2011 editorial commentary [33], suggest the initial ISKDC dosing was based on an “intention to successfully treat as many patients as possible as ‘steroid sensitive’ and to define ‘steroid resistance’ (non-responders),” highlighting the relativity of the term ‘steroid resistance’ in this case. Moves to lower dosing would push more children towards an unhelpful label and this direction of treatment regimen makes the case for strategies to identify those requiring higher doses to achieve remission and the inclusion of biomarker evaluation within future clinical trials [35].
Between the use of lower -dose corticosteroid to treat relapses and the use of prophylactic corticosteroid to prevent URTI-related relapses lies an important hinterland, where long-established management has remained unchallenged for many years. The generally accepted definition of a relapse is 3 days of +++ or more proteinuria on home testing [12], but the consensus recommendation to commence high-dose corticosteroid at that particular point has never been subjected to a clinical trial. In a “muddying” practice of using lower-doses to treat relapses and prophylactic dosing to prevent URTI-related relapses, we have observed clinicians commencing low-dose prednisolone within 1 or 2 days of an URTI first triggering any degree of proteinuria and often with success. In contrast, it is common clinical practice to wait up to 5 days of +++ proteinuria before commencing a relapse corticosteroid regimen out of the experience of spontaneous remission in some children. These divergent practices highlight a need for mechanistic studies to evaluate pathophysiology in the early stages of a relapse, alongside clinical trials that address the relationship between corticosteroid dose and timing of initiation.
What are the adverse effects of corticosteroids in nephrotic syndrome?
There are few other chronic diseases of childhood which require the repeated courses of high-dose and cumulative doses of corticosteroid that are given for many children with SSNS. Due to the genomic and non-genomic properties of corticosteroids, their use comes with a wide range of side-effects, both in the short and long term [75, 76]. In children with SSNS, the spectrum of adverse effects of medication, assumed to be related predominantly to corticosteroids, was detailed in a 10-year trial follow-up cohort study of 46 patients [77]. Adverse effects listed included obesity, short stature, effects on bone mineral density, cataracts, and hypertension. These adverse effects mirror those published elsewhere [78,79,80,81]. In adult studies of childhood SSNS survivors, the adverse effects of corticosteroids remain apparent with higher than expected rates of hypertension, osteoporosis, and short stature [82, 83].
The effect of corticosteroids on behavior is also well-described [84,85,86,87,88] and parents describe behavioral difficulties as the adverse effect most commonly encountered and of most significance to them [17]. These behavioral changes have been documented objectively using the Achenbach Child Behavior Score [65, 86, 89]. It remains unclear why there is such a wide range in severity of adverse effects [33] but individual variability may also be explained by individual variations in corticosteroid pharmacogenomics as discussed above.
Historical and more recent publications noted that adrenal suppression occurs in children with INS [90,91,92,93,94], but this has been another neglected area in nephrotic syndrome management over many years. In clinical practice, few children with SSNS appear to have Addisonian crises; however, the prevalence, timing, duration, and symptoms of adrenal suppression following the treatment of INS using prednisolone remain largely undocumented. Adrenal suppression and adrenal crisis are well documented in a number of other conditions treated with prednisolone or other synthetic glucocorticoids [91,92,93,94,95]. It seems unlikely that children with nephrotic syndrome are not equally susceptible to adrenal insufficiency on completion of treatment.
Clinical trials involving corticosteroids in SSNS lack standardization of corticosteroid adverse effects, limiting the possible comparisons. Such standardization is essential in the design of future trials and might be achieved with tools such as the Glucocorticoid Toxicity Index [96]. Initial work in adults has been adapted for children and the launching of an app is eagerly awaited [97], but it remains to be seen whether the dependence on repeated blood sampling and ionizing radiation will impact on its adoption into pediatric trials.
We know that patients are very rightly concerned about corticosteroid adverse effects [98] and the patient voice must be included in any design of a corticosteroid adverse effect standardization score. Standardized Outcomes in Nephrology (SONG) in children and adolescents [99] has already published key patient-led outcomes for chronic kidney disease trials [100] and this group would be an appropriate forum to determine what patients and their caregivers rank as the most important adverse effects of corticosteroids.
Should all patients be treated the same way?
Application of the current interest in biomarkers to predict steroid sensitivity in childhood nephrotic syndrome has attracted attention in the last year [101], but this mostly relates to binary categorization of steroid sensitivity versus steroid resistance. The quest for an early clinical marker that will accurately predict degree of responsiveness to corticosteroids, and hence individualize dosing, has not yet improved on the clinical observations of time to initial remission [102,103,104], age [18, 42, 105] or time to first relapse [104]. The identification of “personal profiles” of patients and their impact on disease response and adverse effects, that can define individually-tailored approaches to treatment remains a goal still out of reach [106] but one for which it is imperative we strive.
As well as variations in the disease process itself and variations in the individual immune response, there are individual variations in the pharmacokinetics and pharmacodynamics of corticosteroids, which may contribute towards explanation for the effect of age at first presentation [35, 107]. While relatively little is known about the impact of genetic polymorphisms on glucocorticoid response and steroid-related toxicities in children with SSNS, pharmacogenomics offers an important tool in precision medicine applied to nephrotic syndrome and the inclusion of pharmacogenomic testing in large trials of corticosteroid RCTs is recommended to gain greater understanding of their predictive value and eventual application of personalized corticosteroid regimens.
Adrenal suppression may also predict risk of relapse [108,109,110]. As highlighted in the conclusions of the recent Cochrane update [6], there is a need for further study in this area as the prevalence of adrenal insufficiency and its effect on future course of INS would add important information about future treatment stratification. Little has been published since Leisti et al. [111] found in 1978 that cortisol replacement in children with SSNS and adrenal insufficiency reduces the risk of relapse. Particularly in the light of Yadav’s [56] findings of the increased efficacy of maintenance corticosteroids given daily rather than on alternate days, there is a need to revisit this area of study. Screening for adrenal suppression may be now carried out non-invasively through salivary cortisol sampling [112], thus making this an easier bolt-on to large future nephrotic syndrome trials.
Is there a future for corticosteroids in the treatment of steroid-sensitive nephrotic syndrome?
It is perhaps somewhat surprising that, for a disease that recent clinical trials [16] have confirmed progresses with a chronic course in the majority of cases [113], until now, so few clinical trials have considered non-corticosteroid, disease-modifying options for the initial treatment. Hoyer and Brodehl’s 2006 [114] finding that ciclosporin in comparison to standard consolidation corticosteroid dosing made no difference to disease course stands alone as a published trial of a non-corticosteroid treatment used for the initial episode.
Current trials are evaluating early use of drugs traditionally used once relapsing disease is confirmed. NEPHROVIR3 (NCT02818738) and LEARNS [115] started recruiting in 2016 and are multi-center, double-blind, placebo-controlled randomized control trials comparing levamisole with placebo at the first manifestation of INS on the incidence of early relapses. As a low adverse effect [116] immunomodulatory agent that may have disease-modifying properties [117,118,119], levamisole is an attractive adjunct to initial treatment.
INTENT [120] is a large multi-center, open-label RCT designed to compare the efficacy of early-use mycophenolate mofetil with standard corticosteroid treatment in influencing future disease course.
The results of RITURNS [121] support use of rituximab as the initial choice of steroid-sparing agent in SSNS and now results from a Japanese placebo-controlled study of rituximab as first-line steroid-sparing agent are eagerly anticipated [122].
All these trials only consider children who have entered remission on conventional high-dose corticosteroids and, as yet, there is only one published trial where non-corticosteroids are used as part of the induction regimen, showing some benefit of 3 days of azithromycin with the initial corticosteroid course [123]. In adult minimal change disease, a French study of considerable interest to pediatric nephrologists has recently opened, which will evaluate rituximab used at initial presentation (NCT03970577). As yet, there are no published or current trials of non-corticosteroid treatments without corticosteroids and it looks as though the presence of corticosteroids in the treatment of nephrotic syndrome will remain for many years yet.
Conclusions
For now, the name is safe and children with SSNS will continue to be dependent on corticosteroids to induce remission in the first episode and future relapses! There would appear to be a current tension in research direction at present between, on one hand, those seeking to trial alternative treatments ever sooner in the disease course and those seeking to reduce the corticosteroid burden for those who may not need such high or prolonged doses. More than ever, our understanding of individual response is key.
Recent large RCTs can now begin to inform guidelines, and plans for IPNA clinical practice guidelines on SSNS following publication of SRNS guidelines last year [124] are particularly welcome as a tool to harmonize treatment strategy. Though they may be challenging in terms of sufficient sample size and high administration costs, the importance of placebo-controlled double-blind trial designs cannot be underplayed. The future of SSNS depends on large, high-quality RCTs with standardization of corticosteroid adverse effects and embedded mechanistic studies. Placebo-controlled design is important to generate robust evidence, not so much for objective assessment of behavioral effects of corticosteroids, but as much for removing the risk of non-adherence with trial protocols in a condition where expert parents will be tempted to give the treatment they think best for their child. It is possible that something of this same mentality among clinicians has been a factor in the study design bias that has been repeatedly seen in clinical trials of SSNS. As a collaborative international pediatric nephrology community, particularly at a time of accelerated use of video meeting technology, we have the opportunity to form the clinical trials network infrastructure needed to ensure the research recommendations are implemented and that the robustness of study designs continues to improve.
Change history
24 February 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00467-022-05471-y
References
Shakespeare, Romeo and Juliet, Act 2, Scene 2.
Arneil GC (1971) The nephrotic syndrome. Pediatr Clin N Am 18:547–559. https://doi.org/10.1016/S0031-3955(16)32565-2
Hodson E, Knight J, Willis N, Craig J (2002) Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev 2:CD001533. https://doi.org/10.1002/14651858.cd001533
Hodson EM, Knight JF, Willis NS, Craig JC (2005) Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev. Jan 25;(1):CD001533. https://doi.org/10.1002/14651858.CD001533.pub3
Hahn D, Hodson EM, Willis NS, Craig JC (2015) Corticosteroid therapy for nephrotic syndrome in children (Review). Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD001533.pub5.www.cochranelibrary.com
Hahn D, Samuel SM, Willis NS, Narelle S, Craig JC, Hodson EM (2020) Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD001533.pub6
KDIGO (2012) Clinical practice guideline for glomerulonephritis. Chapter 3: steroid-sensitive nephrotic syndrome in children. Kidney Int Supp 2, 143:163–171. https://doi.org/10.1038/kisup.2012.12
(1979) Alternate-day versus intermittent prednisone in frequently relapsing nephrotic syndrome. A report of “Arbetsgemeinschaft für Pädiatrische Nephrologie.” Lancet 24:401–403
(1988) Short versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Lancet 331:380–383.
Ueda N, Chihara M, Kawaguchi S, Niinomi Y, Nonoda T, Matsumoto J, Ohnishi M, Yasaki T (1988) Intermittent versus long-term tapering prednisolone for initial therapy in children with idiopathic nephrotic syndrome. J Pediatr 112:122–126
Hodson EM, Craig JC, Willis NS (2005) Evidence-based management of steroid-sensitive nephrotic syndrome. Pediatr Nephrol 20:1523–1530
Deschênes G, Vivarelli M, Peruzzi L (2017) Variability of diagnostic criteria and treatment of idiopathic nephrotic syndrome across European countries. Eur J Pediatr 176:647–654
Teeninga N, Kist-van Holthe JE, Van Rijswijk N, de Mos NI, Hop WCJ, Wetzels JFM, van der Heijden AJ, Nauta J (2013) Extending prednisolone treatment does not reduce relapses in childhood nephrotic syndrome. J Am Soc Nephrol 24:149–159
Yoshikawa N, Nakanishi K, Sako M, Oba MS, Mori R, Ota E, Ishikura K, Hataya H, Honda M, Ito S, Shima Y, Kaito H, Nozu K, Nakamura H, Igarashi T, Ohashi Y, Iijima K (2015) A multicenter randomized trial indicates initial prednisolone treatment for childhood nephrotic syndrome for two months is not inferior to six-month treatment. Kidney Int 87:227–232
Sinha A, Saha A, Kumar M, Sharma S, Afzal K, Mehta A, Kalaivani M, Hari P, Bagga A (2015) Extending initial prednisolone treatment in a randomized control trial from 3 to 6 months did not significantly influence The course of illness in children with steroid-sensitive nephrotic syndrome. Kidney Int 87:217–224
Webb NJA, Woolley RL, Lambe T, Frew E, Brettell EA, Barsoum EN, Trompeter RS, Cummins C, Deeks JJ, Wheatley K, Ives NJ (2019) Long term tapering versus standard prednisolone treatment for first episode of childhood nephrotic syndrome: phase III randomised controlled trial and economic evaluation. BMJ 365:l1800. https://doi.org/10.1136/bmj.l1800
Webb NJA, Woolley RL, Lambe T, Frew E, Brettell EA, Barsoum EN, Trompeter RS, Cummins C, Wheatley K, Ives NJ (2019) Sixteen-week versus standard eight-week prednisolone therapy for childhood nephrotic syndrome: the PREDNOS RCT. Health Technol Assess (Rockv) 23:1–108. https://doi.org/10.3310/hta23260
Andersen RF, Thrane N, Noergaard K, Rytter L, Jespersen B, Rittig S (2010) Early age at debut is a predictor of steroid-dependent and frequent relapsing nephrotic syndrome. Pediatr Nephrol 25:1299–1304
Hiraoka M, Tsukahara H, Matsubara K, Tsurusawa M, Takeda N, Haruki S, Hayashi S, Ohta K, Momoi T, Ohshima Y, Suganuma N, Mayumi M (2003) A randomized study of two long-course prednisolone regimens for nephrotic syndrome in children. Am J Kidney Dis 41:1155–1162
Alangari AA (2014) Corticosteroids in the treatment of acute asthma. Ann Thorac Med 9:187–192
(2010) Body surface area for adjustment of drug dose. Drug Ther Bull 48:33–36
Gipson DS, Massengill SF, Yao L, Nagaraj S, Smoyer WE, Mahan JD, Wigfall D, Miles P, Powell L, Lin JJ, Trachtman H, Greenbaum LA (2009) Management of childhood onset nephrotic syndrome. Pediatrics 124:747–757
Mosteller RD (1987) Simplified calculation of body-surface area. N Engl J Med 317:1098
Du Bois D, Du Bois E (1916) A formula to estimate the approximate surface area if height and weight be known. Arch Int Med 17:863–871
Haycock GB, Schwartz GJ, Wisotsky DH (1978) Geometric method for measuring body surface area: A height-weight formula validated in infants, children, and adults. J Pediatr 93:62–66
Feber J, Al-Matrafi J, Farhadi E, Vaillancourt R, Wolfish N (2009) Prednisone dosing per body weight or body surface area in children with nephrotic syndrome - is it equivalent? Pediatr Nephrol 24:1027–1031
Hirano D, Fujinaga S (2014) Two dosing regimens for steroid therapy in nephrotic syndrome. Pediatr Nephrol 29:325
Emma F, Montini G, Gargiulo A (2019) Equations to estimate prednisone dose using body weight. Pediatr Nephrol 34:685–688
Saadeh SA, Baracco R, Jain A, Kapur G, Mattoo TK, Valentini RP (2011) Weight or body surface area dosing of steroids in nephrotic syndrome: is there an outcome difference? Pediatr Nephrol 26:2167–2171
Basu B, Bhattacharyya S, Barua S, Naskar A, Roy B (2020) Efficacy of body weight vs body surface area-based prednisolone regimen in nephrotic syndrome. Clin Exp Nephrol 24:622–629
Raman V, Krishnamurthy S, Harichandrakumar KT (2016) Body weight-based prednisolone versus body surface area-based prednisolone regimen for induction of remission in children with nephrotic syndrome: a randomized, open-label, equivalence clinical trial. Pediatr Nephrol 31:595–604
Sharkey I, Boddy AV, Wallace H, Mycroft J, Hollis R, Picton S (2001) Body surface area estimation in children using weight alone: Application in paediatric oncology. Br J Cancer 85:23–28
Mehls O, Hoyer PF (2011) Dosing of glucocorticosteroids in nephrotic syndrome. Pediatr Nephrol 26:2095–2098
Groeneweg FL, Karst H, de Kloet ER, Joëls M (2011) Rapid non-genomic effects of corticosteroids and their role in the central stress response. J Endocrinol 209:153–167
Schijvens AM, ter Heine R, de Wildt SN, Schreuder MF (2019) Pharmacology and pharmacogenetics of prednisone and prednisolone in patients with nephrotic syndrome. Pediatr Nephrol 34:389–403
Jusko WJ, Rose JQ (1980) Monitoring prednisone and prednisolone. Ther Drug Monit 2:169–176
Noone DG, Iijima K, Parekh R (2018) Seminar: Idiopathic nephrotic syndrome in children. Lancet 392:61–74
Mocan H, Erduran E, Karagüzel G (1999) High dose methylprednisolone therapy in nephrotic syndrome. Indian J Pediatr 66:171–174
Imbasciati E, Gusmano R, Edefonti A, Zucchelli P, Pozzi C, Grassi C, Della Volpe M, Perfumo F, Petrone P, Picca M, Appiani AC, Pasquali S, Ponticelli C (1985) Controlled trial of methylprednisolone pulses and low dose oral prednisone for the minimal change nephrotic syndrome. Br Med J (Clin Res Ed) 291:1305–1308
Deschênes G, Dossier C, Hogan J (2019) Treating the idiopathic nephrotic syndrome: are steroids the answer? Pediatr Nephrol 34:777–785
Shenoy M, Plant ND, Lewis MA, Bradbury MG, Lennon R, Webb NJA (2010) Intravenous methylprednisolone in idiopathic childhood nephrotic syndrome. Pediatr Nephrol 25:899–903
Dossier C, Delbet JD, Boyer O, Dossier C, Delbet JD, Boyer O, Daoud P, Mesples B, Pellegrino B, See H, Benoist G, Chace A, Larakeb A, Hogan J, Deschênes G (2019) Five-year outcome of children with idiopathic nephrotic syndrome: the NEPHROVIR population-based cohort study. Pediatr Nephrol 34:671–678
Pasini A, Benetti E, Conti G, Ghio L, Lepore M, Massella L, Molino D, Peruzzi L, Emma F, Fede C, Trivelli A, Maringhini S, Materassi M, Messina G, Montini G, Murer L, Pecoraro C, Pennesi M (2017) The Italian Society for Pediatric Nephrology (SINePe) consensus document on the management of nephrotic syndrome in children: Part i - Diagnosis and treatment of the first episode and the first relapse. Ital J Pediatr 43:41. https://doi.org/10.1186/s13052-017-0356-x
Hari P, Bagga A, Mantan M (2004) Short term efficacy of intravenous dexamethasone and methylprednisolone therapy in steroid resistant nephrotic syndrome. Indian Pediatr 41:993–1000
Colquitt JL, Kirby J, Green C, Cooper K, Trompeter RS (2007) The clinical effectiveness and cost-effectiveness of treatments for children with idiopathic steroid-resistant nephrotic syndrome: a systematic review. Health Technol Assess (Rockv) 11:iii-iv, ix-xi, 1-93. https://doi.org/10.3310/hta11210
Rocci M, Assael B, Appiani AC, Edefonti A, Jusko WJ (1982) Effect on nephrotic syndrome on absorption and disposition of prednisolone in children. Int J Pediatr Nephrol 3:159–166
Gatti G, Perucca E, Frigo G, Notarangelo LD, Barberis L, Martini A (1984) Pharmacokinetics of prednisone and its metabolite prednisolone in children with nephrotic syndrome during the active phase and in remission. Br J Clin Pharmacol 17:423–431
Faure C, André J, Pelatan C, Munck A, Giraud M, Cèzard JP, Jacqz-Aigrain E (1998) Pharmacokinetics of intravenous methylprednisolone and oral prednisone in paediatric patients with inflammatory bowel disease during the acute phase and in remission. Eur J Clin Pharmacol 54:555–560
Broyer M, Terzi F, Lehnert A, Gagnadoux MF, Guest G, Niaudet P (1997) A controlled study of deflazacort in the treatment of idiopathic nephrotic syndrome. Pediatr Nephrol 11:418–422
Singhal R, Pandit S, Dhawan N (2015) Deflazacort versus prednisolone: Randomized controlled trial in treatment of children with idiopathic nephrotic syndrome. Iran J Pediatr 25:e510. https://doi.org/10.5812/ijp.510
Aljebab F, Alanazi M, Choonara I, Conroy S (2018) Observational study on the palatability and tolerability of oral prednisolone and oral dexamethasone in children in Saudi Arabia and the UK. Arch Dis Child 103:83–88
Lucas-Bouwman ME, Roorda RJ, Jansman FGA, Brand PLP (2001) Crushed prednisolone tablets or oral solution for acute asthma? Arch Dis Child 84:347–348
Haslund-Krog SS, Schmidt M, Mathot R, Jensen AK, Jørgensen IM, Holst H (2019) Pharmacokinetics of prednisolone in children: An open-label, randomised, two-treatment cross-over trial investigating the bioequivalence of different prednisolone formulations in children with airway disease. BMJ Paediatr Open 3:1–7. https://doi.org/10.1136/bmjpo-2019-000520
Elzouki AY, Jaiswal OP (1988) Long-term, small dose prednisonetherapy in frequently relapsing nephrotic syndrome of childhood: effect on remission, statural growth, obesity, and infection rate. Clin Pediatr (Phila) 27:387–392
Srivastava RN, Vasudev AS, Bagga A, Sunderam KR (1992) Long-term, low-dose prednisolone therapy in frequently relapsing nephrotic syndrome. Pediatr Nephrol 6:247–250
Yadav M, Sinha A, Khandelwal P, Hari P, Bagga A (2019) Efficacy of low-dose daily versus alternate-day prednisolone in frequently relapsing nephrotic syndrome: an open-label randomized controlled trial. Pediatr Nephrol 34:829–835
Alwadhi RK, Mathew JL, Rath B (2004) Clinical profile of children with nephrotic syndrome not on glucorticoid therapy, but presenting with infection. J Paediatr Child Health 40:28–32
Arun S, Bhatnagar S, Menon S, Savita S, Hari P, Bagga A (2009) Efficacy of zinc supplements in reducing relapses in steroid-sensitive nephrotic syndrome. Pediatr Nephrol 24:1583–1586
MacDonald NE, Wolfish N, McLaine P, Phipps P, Rossier E (1986) Role of respiratory viruses in exacerbations of primary nephrotic syndrome. J Pediatr 108:378–382
Moorani KN, Khan KMA, Ramzan A (2003) Infections in children with nephrotic syndrome. J Coll Physicians Surg Pak 13:337–339
Gulati A, Sinha A, Sreenivas V, Math A, Hari P, Bagga A (2011) Daily corticosteroids reduce infection-associated relapses in frequently relapsing nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol 6:63–69
Mattoo TK, Mahmoud MA (2000) Increased maintenance corticosteroids during upper respiratory infection decrease the risk of relapse in nephrotic syndrome. Nephron 85:343–345
Abeyagunawardena AS, Trompeter RS (2008) Increasing the dose of prednisolone during viral infections reduces the risk of relapse in nephrotic syndrome: a randomised controlled trial. Arch Dis Child 93:226–228
Abeyagunawardena AS, Thalgahagoda RS, Dissanayake PV, Abeyagunawardena S, Illangasekera YA, Karunadasa UI, Trompeter RS (2017) Short courses of daily prednisolone during upper respiratory tract infections reduce relapse frequency in childhood nephrotic syndrome. Pediatr Nephrol 32:1377–1382
Webb NJA, Frew E, Brettell EA, Milford DV, Bockenhauer D, Saleem MA, Christian M, Hall AS, Koziell A, Maxwell H, Hegde S, Finlay ER, Gilbert RD, Booth J, Jones C, McKeever K, Cook W, Ives NJ (2014) Short course daily prednisolone therapy during an upper respiratory tract infection in children with relapsing steroid-sensitive nephrotic syndrome (PREDNOS 2): Protocol for a randomised controlled trial. Trials 15:147. https://doi.org/10.1186/1745-6215-15-147
Achenbach T (2001) Achenbach system of empirically based assessment (ASEBA) Manual for the Child Behaviour Checklist pre-school (1.5–5 year), 2000 and school (6–18 year). Research Centre for Children Youth and Families, Burlington, VT
Choonara IA, Heney D, Meadow SR (1989) Low dose prednisolone in nephrotic syndrome. Arch Dis Child 64:610–611
Warshaw BL, Hymes LC (1989) Daily single-dose and daily reduced-dose prednisone therapy for children with the nephrotic syndrome. Pediatrics 83:694–699
Raja K, Parikh A, Webb H, Hothi D (2017) Use of a low-dose prednisolone regimen to treat a relapse of steroid-sensitive nephrotic syndrome in children. Pediatr Nephrol 32:99–105
Fujinaga S, Sakuraya K (2018) Initial prednisolone dosing for the first relapse of steroid-sensitive nephrotic syndrome in Japanese children. Pediatr Nephrol 33:2205–2206
Borovitz Y, Alfandary H, Haskin O, Levi S, Shulamit K, Davidovits M, Dagan A (2019) Lower prednisone dosing for steroid-sensitive nephrotic syndrome relapse: a prospective randomized pilot study. Eur J Pediatr 179:279–283. https://doi.org/10.1007/s00431-019-03506-5
Sheikh S, Mishra K, Kumar M (2019) Low versus conventional dose prednisolone for nephrotic syndrome relapses: randomised-controlled, non-inferiority trial. Pediatr Nephrol 34:1980
Gargiulo A, Massella L, Ruggiero B, Ravà L, Ciofi degli Atti M, Materassi M, Lugani F, Benetti E, Morello W, Molino D, Mattozzi F, Pennesi M, Maringhini S, Pasini A, Gianoglio B, Pecoraro D, Montini G, Murer L, Ghiggeri GM, Romagnani P, Vivarelli M, Emma F (2021) Results of the PROPINE randomized controlled study suggest tapering of prednisone treatment for relapses of steroid sensitive nephrotic syndrome is not necessary in children. Kidney Int 99:475–483. https://doi.org/10.1016/j.kint.2020.09.024
Schijvens AM, Dorresteijn EM, Roeleveld N, ter Heine R, van Wijk JAE, Bouts AHM, Keijzer-Veen MG, van de Kar NCAJ, van den Heuvel LPWJ, Schreuder MF (2017) REducing STEroids in Relapsing Nephrotic syndrome: The RESTERN study-protocol of a national, double-blind, randomised, placebo-controlled, non-inferiority intervention study. BMJ Open 7:e018148. https://doi.org/10.1136/bmjopen-2017-018148
Aljebab F, Choonara I, Conroy S (2016) Systematic review of the toxicity of short-course oral corticosteroids in children. Arch Dis Child 101:365–370. https://doi.org/10.1136/archdischild-2015-309522
Aljebab F, Choonara I, Conroy S (2016) Systematic review of the toxicity of long-course oral corticosteroids in children. Arch Dis Child 101:365–370
Ishikura K, Yoshikawa N, Nakazato H, Sasaki S, Nakanishi K, Matsuyama T, Ito S, Hamasaki Y, Yata N, Ando T, Iijima K, Honda M (2014) Morbidity in children with frequently relapsing nephrosis: 10-year follow-up of a randomized controlled trial. Pediatr Nephrol 30:459–468
Leonard MB, Feldman HI, Shults J, Zemel BS, Foster BJ, Stallings VA (2004) Long-term, high-dose glucocorticoids and bone mineral content in childhood glucocorticoid-sensitive nephrotic syndrome. N Engl J Med 351:868–875
Wetzsteon RJ, Shults J, Zemel BS, Gupta PU, Burnham JM, Herskovitz RM, Howard KM, Leonard MB (2009) Divergent effects of glucocorticoids on cortical and trabecular compartment BMD in childhood nephrotic syndrome. J Bone Miner Res 24:503–513
Tsampalieros A, Gupta P, Denburg MR, Shults J, Zemel BS, Mostoufi-Moab S, Wetzsteon RJ, Whitehead KM, Leonard MB (2013) Glucocorticoid effects on changes in bone mineral density and cortical structure in childhood nephrotic syndrome. J Bone Miner Res 28:480–488
Ribeiro D, Zawadynski S, Pittet LF, Chevalley T, Giradin E, Parvex P (2015) Effect of glucocorticoids on growth and bone mineral density in children with nephrotic syndrome. Eur J Pediatr 174:911–917
Kyrieleis HAC, Löwik MM, Pronk I, Cruysberg HRM, Kremer JAM, Oyen WJG, van den Heuvel BLP, Wetzels JFM, Levtchenko EN (2009) Long-term outcome of biopsy-proven, frequently relapsing minimal-change nephrotic syndrome in children. Clin J Am Soc Nephrol 4:1593–1600
Skrzypczyk P, Pańczyk-Tomaszewska M, Roszkowska-Blaim M, Wawer Z, Bienias B, Zajgzkowska M, Kilis-Pstrusinska K, Jakubowska A, Szczepaniak M, Pawlak-Bratkowska M, Tkaczyk M (2014) Long-term outcomes in idiopathic nephrotic syndrome: From childhood to adulthood. Clin Nephrol 81:166–173
Hall AS, Thorley G, Houtman PN (2003) The effects of corticosteroids on behavior in children with nephrotic syndrome. Pediatr Nephrol 18:1220–1223
Mishra OP, Basu B, Upadhyay SK, Prasad R, Schaefer F (2010) Behavioural abnormalities in children with nephrotic syndrome. Nephrol Dial Transplant 25:2537–2541
Upadhyay A, Mishra OP, Prasad R, Upadhyay SK, Schaefer F (2016) Behavioural abnormalities in children with new-onset nephrotic syndrome receiving corticosteroid therapy: results of a prospective longitudinal study. Pediatr Nephrol 31:233–238
Soliday E, Grey S, Lande MB (1999) Behavioral effects of corticosteroids in steroid-sensitive nephrotic syndrome. Pediatrics 104:e51–e51. https://doi.org/10.1542/peds.104.4.e51
Mehta M, Bagga A, Pande P, Bajaj G, Srivastava RN (1995) Behavior problems in nephrotic syndrome. Indian Pediatr 32:1281–1286
Achenbach T, Edelbrock C (1983) Manual for the child behavior checklist and revised behavior profile. University of Vermont Department of Psychiatry
Leisti S, Koskimies O (1983) Risk of relapse in steroid-sensitive nephrotic syndrome: Effect of stage of post-prednisone adrenocortical suppression. J Pediatr 103:553–557
Bowden SA, Connolly AM, Kinnett K, Zeitler PS (2019) Management of adrenal insufficiency risk after long-term systemic glucocorticoid therapy in Duchenne muscular dystrophy : clinical practice recommendations. J Neuromusc Dis 6:31–41
Rensen N, Gemke RJBJ, van Dalen EC, Rotteveel J, Kaspers GJL (2017) Hypothalamic-pituitary-adrenal ( HPA ) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia (Review). https://doi.org/10.1002/14651858.CD008727.pub4.www.cochranelibrary.com
Ahmet A, Benchimol EI, Goldbloom EB, Barkey JL (2016) Adrenal suppression in children treated with swallowed fluticasone and oral viscous budesonide for eosinophilic esophagitis. Allergy Asthma Clin Immunol 12:49. https://doi.org/10.1186/s13223-016-0154-9
Hawcutt DB, Jorgensen AL, Wallin N, Thompson B, Peak M, Lacy D, Newland P, Didi M, Couriel J, Blair J, Pirmohamed M, Smyth RL (2015) Adrenal responses to a low-dose short synacthen test in children with asthma. Clin Endocrinol (Oxf) 82:648–656
Hawcutt DB, Jorgensen AL, Wallin N, Thompson B, Peak M, Lacy D, Newland P, Didi M, Couriel J, Blair J, Pirmohamed M, Smyth RL (2016) Erratum: Adrenal responses to a low-dose short synacthen test in children with asthma. Clin Endocrinol (Oxf) 82:648–656
Miloslavsky EM, Naden RP, Bijlsma JWJ, Brogan PA, Brown ES, Brunetta P, Buttgereit F, Choi HK, DiCaire JF, Gelfand JM, Heaney LG, Lightstone E, Lu N, Murrell DF, Petri M, Rosenbaum JT, Saag KS, Urowitz MB, Winthrop KL, Stone JH (2016) Development of a Glucocorticoid Toxicity Index (GTI) using multicriteria decision analysis. Ann Rheum Dis 76:543–546
Brogan P, Naden R, Ardoin S, Cooper JC, De Benedetti F, DiCaire JF, Eleftheriou D, Feldman BM, Goldin J, Karol SE, Miloslavsky EM, Price-Kuehne F, Skuse D, Stratakis CA, Webb N, Stone JH (2018) Development of a Pediatric Glucocorticoid Toxicity Index. Arthritis. Rheumatol 70(suppl 10) https://acrabstracts.org/abstract/development-of-a-pediatric-glucocorticoid-toxicity-index/
Nephrotic girl. https://nephroticgirl.home.blog/. Accessed 27 Sep 2020
Tong A, Samuel S, Zappitelli M, Dart A, Furth S, Eddy A, Groothoff J, Webb NJA, Yap H-K, Bockenhauer D, Sinha A, Alexander SI, Goldstein SL, Gipson DS, Hanson CS, Evangelidis N, CroweS HT, Hemmelgarn BR, Manns B, Gill J, Tugwell P, Van Biesen W, Wheeler DC, Winkelmayer WC, Craig JC (2016) Standardised Outcomes in Nephrology-Children and Adolescents (SONG-Kids): a protocol for establishing a core outcome set for children with chronic kidney disease. Trials 17:401. https://doi.org/10.1186/s13063-016-1528-5
Hanson CS, Gutman T, Craig JC, Bernays S, Raman G, Zhang Y, James LJ, Ralph AF, Ju A, Manera KE, Teixeira-Pinto A, Viecelli AK, Alexander SI, Blydt-Hansen TD, Dionne J, McTaggart S, Michael M, Walker A, Carter S, Wenderfer SE, Winkelmayer WC, Bockenhauer D, Dart A, Eddy AA, Furth SL, Gipson DS, Goldstein SL, Groothoff J, Samuel S, Sinha A, Webb NJA, Yap H-K, Zappitelli M, Currier H, Tong A (2019) Identifying important outcomes for young people with CKD and their caregivers: a nominal group technique study. Am J Kidney Dis 74:82–94. https://doi.org/10.1053/j.ajkd.2018.12.040
Uwaezuoke SN (2017) The role of novel biomarkers in childhood idiopathic nephrotic syndrome: a narrative review of published evidence. Int J Nephrol Renovasc Dis 10:123–128
Vivarelli M, Moscaritolo E, Tsalkidis A, Massella L, Emma F (2010) Time for initial response to steroids is a major prognostic factor in idiopathic nephrotic syndrome. J Pediatr 156:965–971. https://doi.org/10.1016/j.jpeds.2009.12.020
Harambat J, Godron A, Ernould S, Rigothier C, Llanas B, Leroy S (2013) Prediction of steroid-sparing agent use in childhood idiopathic nephrotic syndrome. Pediatr Nephrol 28:631–638
Nakanishi K, Iijima K, Ishikura K, Hataya H, Nakazato H, Sasaki S, Honda M, Yoshikawa N (2013) Two-year outcome of the ISKDC regimen and frequent-relapsing risk in children with idiopathic nephrotic syndrome. Clin J Am Soc Nephrol 8:756–762
Mishra OP, Abhinay A, Mishra RN, Prasad R, Pohl M (2013) Can we predict relapses in children with idiopathic steroid-sensitive nephrotic syndrome? J Trop Pediatr 59:358–364
Colucci M, Cascioli S, Vivarelli M, Serafinelli J, Emma F, Vivarelli M (2018) B cell phenotype in pediatric idiopathic nephrotic syndrome. Pediatr Nephrol 34:177–181
Cuzzoni E, De Iudicibus S, Franca R, Stocco G, Lucafò M, Pelin M, Favretto D, Pasini A, Montini G, Decorti G (2015) Glucocorticoid pharmacogenetics in pediatric idiopathic nephrotic syndrome. Pharmacogenomics 16:1631–1648
Leisti S, Koskimies O, Rapola J, Hallman N, Perheentupa J, Vilska J (1977) Association of post-medication hypocortisolism with early first relapse of idiopathic nephrotic syndrome. Lancet 2:795–796
Leisti S, Vilska J, Hallman N (1977) Adrenocortical insufficiency and relapsing in the idiopathic nephrotic syndrome of childhood. Pediatrics 60:334–342
Abeyagunawardena AS, Hindmarsh P, Trompeter RS (2007) Adrenocortical suppression increases the risk of relapse in nephrotic syndrome. Arch Dis Child 92:585–588
Leisti S, Koskimies O, Perheentupa J, Vilska J, Hallman N (1978) Idiopathic nephrotic syndrome : prevention of early relapse. Br Med J (Clin Res Ed) 1:892
Blair J, Lancaster G, Titman A, Peak M, Newlands P, Collingwood C, Chesters C, Moorcroft T, Wallin N, Hawcutt D, Gardner C, Didi M, Lacy D, Couriel J (2014) Early morning salivary cortisol and cortisone, and adrenal responses to a simplified low-dose short Synacthen test in children with asthma. Clin Endocrinol (Oxf) 80:376–383
Tarshish P, Tobin JN, Bernstein J, Edelmann CM (1997) Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol 8:769–776
Hoyer PF, Brodehl J (2006) Initial treatment of idiopathic nephrotic syndrome in children: Prednisone versus prednisone plus cyclosporine A: A prospective, randomized trial. J Am Soc Nephrol 17:1151–1157
Veltkamp F, Khan DH, Reefman C, Veissi S, van Oers HA, Levtchenko E, Mathôt RAA, Florquin S, van Wijk JAE, Schreuder MF, Haverman L, Bouts AHM (2019) Prevention of relapses with levamisole as adjuvant therapy in children with a first episode of idiopathic nephrotic syndrome: Study protocol for a double blind, randomised placebo-controlled trial (the LEARNS study). BMJ Open 9. https://doi.org/10.1136/bmjopen-2018-027011
Mühlig AK, Lee JY, Kemper MJ, Kronbichler A, Yang JW, Lee JM, Shin JI, Oh J (2019) Levamisole in children with idiopathic nephrotic syndrome: clinical efficacy and pathophysiological aspects. J Clin Med 8:860. https://doi.org/10.3390/jcm8060860
Boyer O, Moulder JK, Grandin L, Somers MJG (2008) Short- and long-term efficacy of levamisole as adjunctive therapy in childhood nephrotic syndrome. Pediatr Nephrol 23:575–580
Madani A, Isfahani ST, Rahimzadeh N, Fereshtehnejad SM, Hoseini R, Moghtaderi M, Mohseni P, Ataiee N (2010) Effect of levamisole in steroid-dependent nephrotic syndrome. Iran J Kidney Dis 4:292–296
Elmas AT, Tabel Y, Elmas ÖN (2013) Short- and long-term efficacy of levamisole in children with steroid-sensitive nephrotic syndrome. Int Urol Nephrol 45:1047–1055
Ehren R, Benz MR, Doetsch J, Fichtner A, Gellermann J, Haffner D, Höcker B, Hoyer PF, Kästner B, Kemper MJ, Konrad M, Luntz S, Querfeld U, Sander A, Toenshoff B, Weber LT (2018) Initial treatment of steroid-sensitive idiopathic nephrotic syndrome in children with mycophenolate mofetil versus prednisone: protocol for a randomised, controlled, multicentre trial (INTENT study). BMJ Open. https://doi.org/10.1136/bmjopen-2018-024882
Basu B, Sander A, Roy B, Preussler S, Barua S, Mahapatra TKS, Schaefer F (2018) Efficacy of rituximab vs tacrolimus in pediatric corticosteroid-dependent nephrotic syndrome a randomized clinical trial. JAMA Pediatr 172:757–764
Nagano C, Sako M, Kamei K, Ishikura K, Nakamura H, Nakanishi K, Omori T, Nozu K, Iijima K (2019) Study protocol: Multicenter double-blind, randomized, placebo-controlled trial of rituximab for the treatment of childhood-onset early-stage uncomplicated frequently relapsing or steroid-dependent nephrotic syndrome (JSKDC10 trial). BMC Nephrol 20:293. https://doi.org/10.1186/s12882-019-1470-3
Zhang B, Liu T, Wang W, Zhang X, Fan S, Liu Z, Liu Z, Wu X (2014) A prospective randomly controlled clinical trial on azithromycin therapy for induction treatment of children with nephrotic syndrome. Eur J Pediatr 173:509–515
Trautmann A, Vivarelli M, Samuel S, Gipson D, Sinha A, Schaefer F, Hui NK, Boyer O, Saleem MA, Feltran L, Müller-Deile J, Becker JU, Cano F, Xu H, Lim YN, Smoyer W, Anochie I, Nakanishi K, Hodson E, Haffner D (2020) IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 35:1529–1561
Acknowledgements
The authors would like to thank Professor Nicholas Webb for his helpful comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Dr. Christian and Dr. Maxted contributed equally to writing and reviewing the manuscript.
Corresponding author
Ethics declarations
Ethics approval
No ethical approval required.
Consent for publication
Martin Christian and Andrew Maxted give consent for publication of this review. There are no copyright issues with any part of it.
Competing interests
Martin Christian is the chief investigator for the PREDNOS 2 study and is currently a member of the IPNA clinical practice recommendations on steroid-sensitive nephrotic syndrome (SSNS).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In the fifth paragraph of the section entitled “Is there a future for corticosteroids in the treatment of steroid-sensitive nephrotic syndrome?”, the authors refer to a study that used a non-corticosteroid drug as part of initial regimen. That drug should be azithromycin not azathioprine as they noted. This has been corrected.
Rights and permissions
About this article

Cite this article
Christian, M.T., Maxted, A.P. Optimizing the corticosteroid dose in steroid-sensitive nephrotic syndrome. Pediatr Nephrol 37, 37–47 (2022). https://doi.org/10.1007/s00467-021-04985-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-04985-1