
Abstract
Objectives
To determine the clinical, anatomical, genetic and pathological features of dual frontotemporal lobar degeneration (FTLD) pathology: FTLD-tau and FTLD-TDP-43 in a large clinicopathological cohort.
Methods
We selected subjects with mixed FTLD-TDP and FTLD-tau from 247 FTLD cases from the University of California, San Francisco, Neurodegenerative Disease Brain Bank collected between 2000 and 2016 and compared their clinical, anatomical, genetic, imaging and pathological signatures with those of subjects with pure FTLD.
Results
We found nine cases (3.6%) with prominent FTLD-TDP and FTLD-tau. Six cases were sporadic, whereas one case had a C9ORF72 expansion, another had a TARDBP A90V variant, and the other had an MAPT p.A152T variant. The subtypes of FTLD-TDP and FTLD-tau varied. Mixed FTLD cases were older and tended to show a higher burden of Alzheimer disease pathology (3/9, 33%). The neuroimaging signature of mixed cases, in general, included more widespread atrophy than that of pure groups. Specifically, cases of mixed corticobasal degeneration (CBD) with FTLD-TDP showed more prominent asymmetric left-sided atrophy than did those of pure CBD. However, the clinical phenotype of mixed cases was similar to that seen in pure FTLD.
Conclusions
Although patients with mixed FTLD-TDP and FTLD-tau are rare, in-depth clinical, pathological and genetic investigations may shed light on the genetic and biochemical pathways that cause the accumulation of multiple proteinaceous inclusions and inform therapeutic targets that may be beneficial to each one of these abnormal protein misfoldings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration (FTLD) is a neuropathological umbrella term applied to cases featuring superficial neuronal loss, vacuolation, and astrogliosis that in most cases manifest clinically as one of frontotemporal dementia (FTD) syndromes [1]. The clinically, genetically, and pathologically heterogeneous neuropathological entities grouped under the term “FTLD” can be classified into three major categories based on the biochemical signature of their proteinaceous neuronal and glial inclusions: FTLD-tau, FTLD-TAR-DNA binding protein-43 (TDP), and FTLD-fused sarcoma (FUS) [2]. Of these, approximately 90% of FTLD cases are either FTLD-TDP, which is slightly more common, or FTLD-tau [3].
Even within each category, the neuropathological entities are quite heterogeneous. FTLD-tau is sub-classified in a predominantly 3-repeat tau inclusion (i.e., Pick’s disease), 4-repeat tau [i.e., corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and globular glial tauopathy], or both 3 and 4-repeat tau forms. MAPT mutations can produce either a 3-repeat, 4-repeat, or 3 and 4-repeat FTLD-tau, depending on the mutation [4]. Although all FTLD-tau are tauopathies, not all tauopathies are FTLD-tau. Alzheimer’s disease (AD), argyrophilic grain disease (AGD) and chronic traumatic encephalopathy are usually not considered forms of FTLD-tau. In comparison, FTLD-TDP is less heterogeneous but still has four major histological subtypes (from A to D) based on the morphology, anatomical distribution, and cellular location of inclusions [5]. Moreover, not all TDP-43 proteinopathies are FTLD-TDP, and TDP-43 inclusions with a predominant limbic distribution are a relatively common finding in normal aging and hippocampal sclerosis of aging and overlap with AD pathology [6].
It is generally accepted that FTLD cases feature either TDP-43 or FUS or tau deposits without substantial co-occurrence within patients [5, 7]. Scattered literature suggests that rare cases may present both FTLD-tau and FTLD-TDP, but systematic reviews are lacking [8,9,10,11].
To investigate how frequently FTLD-TDP and FTLD-tau co-occur (mixed FTLD), we reviewed all 247 FTLD cases from the University of California, San Francisco (UCSF), Neurodegenerative Disease Brain Bank (NDBB) collected during a period of 16 years. Subsequently, to investigate whether mixed FTLD pathology differs from pure FTLD, we compared the clinical, genetic, imaging and neuropathological features of the nine cases with prominent FTLD-TDP and FTLD-tau pathology (3.6% of the total FTLD cases) with those of pure FTLD cases and normal controls.
Materials and methods
Case selection
The UCSF/NDBB serves all research projects of the UCSF/Memory and Aging Center (MAC), a center of reference for FTD research. The brains were procured by the UCSF/NDBB between 2000 and 2016 from subjects who participated in the UCSF/MAC research projects. Inclusion criteria for mixed FTLD were as follows: (1) a neuropathological diagnosis of primary or contributing (severe and spread enough to have significantly contributed to the clinical outcome) FTLD-tau, and (2) a neuropathological diagnosis of primary or contributing FTLD-TDP. Because of the low number of mixed FTLD cases, we maintained cases with other neuropathological diagnosis, too. One of the cases (Case 7), showed contributing Lewy body disease and, another case (Case 8) showed contributing AD. Clinical and genetic features of Case 2 and 7 were previously reported elsewhere [12, 13]. Table 1 summarizes the neuropathological diagnosis assigned to each case. We also created four disease control groups with pure pathology for clinical, genetic and anatomical (neuroimaging) comparisons: (1) pure FTLD-tau (CBD, n = 17) cases, (2–4) pure FTLD-TDP (type A, n = 10, type B, n = 16 and type C, n = 14) cases. Cases in the pure FTLD group lacked additional primary or contributing neuropathological diagnoses but showed a variety of incidental neuropathological changes including low levels of AD-type pathology [none or low AD neuropathological changes (ADNC)] [14], small and isolated cerebrovascular lesions, AGD [15], primary age-related tauopathy (PART) [16], or age-related tau astrogliopathy [17]. Finally, we included a clinical group of neurologically healthy controls (n = 288, mean age 66.3 ± 10.8, male:female = 116:172) for neuroimaging comparisons. The UCSF institutional review boards for Human Research approved the study. All participants or their surrogates consented to study protocols.
Clinical, neuropathological, genetic and neuroimaging assessment
All patients had undergone neurological evaluation, including extensive neuropsychological assessment and neuroimaging, at least once, at the UCSF, MAC and an extensive dementia-oriented postmortem assessment at UCSF/NDBB (n = 7) or UCSF-Department of Pathology (n = 2). Seven out of nine patients performed genetic assessment. Eight out of nine patients underwent structural magnetic resonance imaging (MRI) on a 1.5- or 3-T scanner (Fig. 1). Case 6 was scanned in another hospital and the images could not be analyzed by our group. If a subject had more than one MRI, then the MRI obtained closest to death was selected for the study. As we could not conduct voxel-based morphometry group-level analyses due to the small number of patients in each mixed pathology groups, we generated a W-score map which shows the relative involvement of each brain region for each patient with mixed pathology compared to 288 clinically normal controls. The detailed methods are provided as online supplementary 1.

T1-weighted axial and coronal images of brain MRIs for each case
Results
The demographic, clinical, genetic, and neuropathological characteristics of all nine patients with mixed FTLD-TDP and FTLD-tau are summarized in Table 1. Detailed case descriptions are provided as online supplementary 1.
Briefly, we found five subjects with primary FTLD-TDP and contributing FTLD-tau (Cases 1–5) and four subjects with primary FTLD-tau and contributing FTLD-TDP (Cases 6–9). Among the five subjects with primary FTLD-TDP and contributing FTLD-tau, Case 1 presented as a bvFTD due to a C9ORF72 hexanucleotide repeat expansion, Case 2 presented as bvFTD-motor neuron disease (MND) with a TARDBP A90V variant [12] and the other three cases (Cases 3–5) presented as semantic variant primary progressive aphasia (svPPA). In four subjects with primary FTLD-tau and contributing FTLD-TDP, two subjects presented with nonfluent/agrammatic variant PPA (nfvPPA) (Cases 7 and 8), one with corticobasal syndrome (CBS) (Case 6), and the other with bvFTD (Case 9). Case 7 harbored a MAPT p.A152T variant [18]. As expected, the brain MRI of subjects with bvFTD (Case 1, Case 2 and Case 9) revealed severe dorsolateral frontal, insular, and temporal atrophy, and that of subjects with svPPA revealed asymmetrical left (Case 3 and 5) or right anterior temporal atrophy (Case 4) (Fig. 1).
Neuropathological comparison
The density and distribution of tau immunoreactivity in the nine patients are summarized in Table 2.
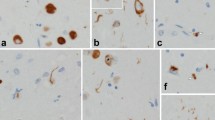
In brief, the pattern of concurrent FTLD-TDP and FTLD-tau was not specific. Primary FTLD-TDP showed overlapping unclassifiable FTLD-tau with 3R or 4R inclusions (Cases 1–3) (Fig. 2a–d) or PSP (Cases 4 and 5) (Fig. 2e–h). The primary FTLD-tau, CBD cases showed overlapping FTLD-TDP, type B (Case 6), unclassifiable FTLD-TDP pathology (Cases 7 and 9) or FTLD-TDP, type A (Case 8). Interestingly, three cases (Cases 6, 7, and 9) with FTLD-tau, CBD and FTLD-TDP pathology showed hippocampal sclerosis (HS) and two of them had atypical forms; neuronal loss in all CA subfields with subiculum (Case 7, Fig. 2i), neuronal loss in selective CA2 and CA3 (Case 9, Fig. 2j).

Neuropathological features of each case. Astrocytic plaque (a), granulo/fuzzy astrocyte (b), or tufted astrocyte (c) were seen in the angular gyrus in Case 1 (CP-13 antibody, scale bar 25 µm). Pretangle-like diffuse granular tau-positive NCIs, sometimes with perinuclear halos, and astrocytic plaques were found in middle frontal gyrus in Case 2 (d CP-13 antibody, scale bar 25 µm). Globose tangles were observed in substantia nigra in Case 4 (e CP-13 antibody, scale bar 25 µm). Thorny astrocyte (f) and tufted astrocyte (g) in Putamen and globose tangles (h) in the substantia nigra were seen in Case 5 (CP-13 antibody, Scale bar 25 µm for f and g, 250 µm for h). Atypical hippocampal sclerosis was identified in Case 7 (i neuronal loss in all CA fields and subiculum, Scale bar 500 µm) and Case 9 (j neuronal loss only in the CA2 and CA3, scale bar 500 µm). Scattered TDP-immunoreactive processes appeared to lace astrocytic plaques (j) in the inferior temporal gyrus in Case 7 (TDP-43, Scale bar 25 µm)
Comparison of demographics, genetics, and MRI atrophy patterns between mixed FTLD-TDP and FTLD-tau versus pure FTLD-TDP or FTLD-tau
Due to the small number of mixed cases with primary FTLD-TDP [type A group (n = 1), type B group (n = 2), and type C group (n = 2)], statistical comparisons between mixed and pure groups were conducted only in the primary FTLD-tau group.
We failed to detect any difference in demographic characteristics between mixed FTLD-tau, CBD with FTLD-TDP and pure FTLD-tau, CBD groups. On the other hand, both mixed FTLD-TDP, type B with FTLD-tau and FTLD-TDP, type C with FTLD-tau groups were older at death than pure FTLD-TDP, type B or C. The mixed FTLD-TDP, type C with FTLD-tau had an older age of onset and a shorter disease duration than pure FTLD-TDP, type C (Table 3).
Regarding genetics, the frequency of the apolipoprotein E (APOE) ε4 allele was higher in the mixed FTLD-tau, CBD with FTLD-TDP (2/3, 67%) than that in the pure FTLD-tau, CBD group (2/15, 13%), however, it is noteworthy that two of the mixed cases also had AD pathology (intermediate ADNC) (Table 3).
Figure 3 demonstrates the mean W-score maps for comparisons of MRI findings between the mixed and pure groups. Compared with controls, the mixed FTLD-TDP, type A with FTLD-tau patients had widespread atrophy (W-score > 2.5) in regions including the bilateral frontal, temporal, insula, basal ganglia, thalamus, and cerebellum, whereas the pure FTLD-TDP, type A group showed atrophy only in the paralimbic fronto–insular–striatal circuit. Both mixed and pure FTLD-TDP, type B groups also showed atrophy in the paralimbic fronto-insular-striatal region, but the mixed FTLD-TDP, type B with FTLD-tau group had a much greater degree of atrophy involving temporal lobes than did the pure FTLD-TDP, type B group. The differences in atrophy pattern and severity between pure and mixed FTLD-TDP, type C groups were mild. The mixed FTLD-tau, CBD with FTLD-TDP group demonstrated gray matter loss predominantly in the left frontal lobe, insula, and striatum, extending to the temporal lobe and amygdala. In contrast, the pure FTLD-tau, CBD group showed decreased gray matter only in the bilateral frontal lobes, insula, and striatum.

Mean gray matter atrophy pattern (W-map) in the mixed and pure pathology groups
Discussion
This study investigated the clinical, anatomical, and genetic characteristics of nine subjects with mixed FTLD-TDP and FTLD-tau pathology and revealed the following: (1) the subtyping of FTLD-tau and FTLD-TDP varies, (2) although both pathologies were considered severe enough to contribute to clinical outcomes, the clinical phenotypes met the criteria for a known clinical phenotype associated with FTLD and showed better correlations with the most severe pathology. For instance, the two cases with primary FTLD-TDP, type C manifested as svPPA, whereas the clinical phenotypes of the four cases of primary FTLD-tau, CBD were CBS, nfvPPA and bvFTD. Since svPPA is usually associated with underlying FTLD-TDP, type C, Case 3 with svPPA who had FTLD-TDP, type B seems to be an exception; such exceptions may occur in up to 10% of svPPA cases [19]. Finally, we found that (3) three out of five patients (Cases 1, 2, and 3) with mixed FTLD-TDP with FTLD-tau showed unclassifiable FTLD-tauopathies which are partially comparable with the “complex tauopathy” described by Kovacs et al. that has characteristics including diffuse granular immunopositivity of astrocytic processes and patchy accumulation of thin threads variably combined with AD-related neurofibrillary tangle (NFT) [20].
Primary FTLD-TDP with contributing FTLD-tau pathology
Most previously published research on overlapping tau and TDP-43 pathology has focused on 3R- or 4R-tauopathies (i.e., PSP, CBD) bearing co-occurring TDP-43 pathology, rather than TDP-43 pathology with a concomitant tauopathy. Recently, Robinson et al. investigated tau pathology in 45 patients with FTLD-TDP and 23 patients with MND. They failed to find cases with mixed FTLD-TDP and FTLD-tau, but reported tau pathology consistent with low levels of AD pathology, in most of their subjects [21]. In fact, patients showing tau pathology were older and tended to have an APOE ε4 allele. Our two mixed cases with a primary FTLD-TDP, type B and the one mixed case with a primary FTLD-TDP, type C had an older age at onset and age at death than the respective pure groups. However, we found no differences in the frequency of the APOE ε4 allele between mixed and pure groups and our mixed FTLD-TDP (primary) with FTLD-tau cases showed widespread neuronal and glial tau pathologies not consistent with AD or PART. Out of five cases with mixed FTLD-TDP and tau pathology, three (one with FTLD-TDP, type A and two with FTLD-TDP, type B) had unclassifiable 3R/4R or 4R tauopathy, and the other two with FTLD-TDP, type C were accompanied by FTLD-tau, PSP. There have been a few studies demonstrating contributing TDP-43 pathology in FTLD-tau, PSP. Conversely, primary FTLD-TDP, type C with overlapping FTLD-tau, PSP type has not been reported yet [10, 22, 23]. This concurrent PSP pathology in our FTLD-TDP, type C cases (Cases 4 and 5) is similar to what is expected in PSP cases. Even though Case 4 had no clinical features of PSP, the clinical history of late-emerging gait imbalance, a prominent stare, and swallowing difficulties in Case 5 (Supplementary material) suggested that the PSP co-pathology had clinical impact.
One of our mixed FTLD-TDP and tau cases (Case 1) harbored a C9ORF72 hexanucleotide repeat expansion. Although mixed FTLD-TDP, mostly type A and type B, is usually known to be associated with the C9ORF72 mutation, there have been only a few reports of FTLD-tau in subjects with a C9ORF72 abnormal expansion [21, 24,25,26]. Robinson et al. found that patients with the C9ORF72 expansion had significantly more tau pathology than those with a GRN mutation. This is consistent with the report by Bieniek et al. that suggested that the C9ORF72 mutation may enhance tau pathology [21, 24]. King et al. also observed a patient with mixed Pick body-like tau inclusions and TDP-43 pathology who had both a C9ORF72 mutation and a rare MAPT A239T variant [26]. However, in contrast to Case 1 that showed varied concurrent tau pathology, including both neuronal and glial inclusions, other studies demonstrated predominant Alzheimer-type NFT pathology in the background of TDP-43 pathology in patients with the C9ORF72 mutation, which is different from the atypical tauopathy we found in our case [21, 24, 25].
The other mixed FTLD-TDP and tau case (Case 2) carried a TARDBP A90V variant which was previously reported as a potential genetic risk factor for FTLD/amyotrophic lateral sclerosis [12]. However, the pathogenicity of this variant is uncertain and the pathologic characteristics of cases with a TARDBP A90V and associations between TARDBP A90V and mixed FTLD-TDP with tau pathology have not been described yet.
There has been no report comparing neuroimaging features between mixed and pure FTLD groups. In the cases described in the present study, the primary FTLD-TDP with FTLD-tau group had more widely distributed atrophy than pure FTLD-TDP group, further demonstrating the negative effect of double pathology in these subjects.
Primary FTLD-tau with FTLD-TDP pathology
We found concomitant TDP-43 pathology in four FTLD-tau, CBD cases. No clinical and demographic differences were found between mixed FTLD-tau, CBD with FTLD-TDP and pure FTLD-tau, CBD groups.
After TDP-43 inclusions were recognized as the most common changes in FTLD, several groups reported TDP-43 proteinopathy in AD [27] and controls [6]. Thirty to seventy percent of AD cases show TDP-43 proteinopathy, with a predilection for limbic areas in a distribution that differs from that observed in classical FTLD-TDP [28]. Although several studies investigating the implication of TDP-43 pathology in AD have provided inconsistent results, TDP-43 pathology in AD was more frequent in cases with HS than in those without HS [11, 29,30,31,32]. Three cases (Cases 6, 7, and 9) with FTLD-tau, CBD and FTLD-TDP pathology showed HS. Intriguingly, the HS in two cases (Cases 7 and 9) was somewhat different from the typical HS characterized by selective neuronal loss in the subiculum and CA1 regions of the hippocampus with sparing of CA2–CA4 regions [33], in that there was neuronal loss in all CA subfields, including the subiculum (Case 7), and in selective CA2 and CA3 subregions (Case 9). Little is known about the pathophysiological differences between atypical and typical HS. It remains unclear whether there are any differences in the clinical and pathological effects of typical or atypical HS on concomitant TDP-43 pathology detected in mixed FTLD-tau, CBD.
FTLD-CBD is the most common FTLD-tau with concomitant TDP-43. About 16% of CBD cases show TPD-43 pathology, mostly limited to TDP-43-positive annular clusters around astrocytic tau-positive plaques [11, 23]. In addition to the overlapping FTLD-TDP, we also observed these peri-plaque TDP-43 deposition in our mixed FTLD-CBD cases (Fig. 2k). Along with this, the distribution of TDP-43 pathology in our mixed FTLD-tau, CBD cases was widespread, showing the extension of TDP-43 pathology to regions beyond the limbic areas, such as the middle frontal gyrus, inferior frontal gyrus, and inferior temporal gyrus, which were severely affected; the impairment in these areas corresponds to the observed clinical features, such as frontotemporal abnormal behaviors or nonfluent aphasia. These findings may support the suggestion by Kouri et al. that concomitant TDP-43 pathology in primary tauopathies is more prominent in brain areas vulnerable to the primary tauopathy, and such individuals likely share genetic risk factors predisposing them to poly-proteinopathies [34].
Intriguingly, Case 7 carried the rare MAPT variant p.A152T, which has been suggested to be a risk factor for both FTD spectrum disorders and AD [13, 18, 35]. Neuropathological features of the p.A152T variation have so far been reported in only six cases [35,36,37]. Our Case 7 showing nfvPPA with CBD mixed with FTLD-TDP pathology was most consistent with one of the cases exhibiting asymmetrical parkinsonism with mixed CBD and TDP-43 pathology described by Kara et al. [37]. Recently, the association between p.A152T and α-synucleinopathy has been proposed [38]. Case 7, along with a few previously reported cases, indeed exhibited α-synucleinopathy as either the primary or contributing pathology during autopsy, and the patient had a family history of Parkinson’s disease. Thus, apart from clinical variability, p.A152T may be related to proteostasis changes common to several proteinopathies.
To our knowledge, this is the first study specifically exploring the neuroanatomical differences between FTLD-tau, CBD with and without TDP-43 pathology. Compared with healthy controls, patients with mixed FTLD-tau, CBD and FTLD-TDP pathology showed prominent left asymmetric frontotemporoparietal, hippocampal, amygdala and striatal atrophy, whereas those with pure FTLD-tau, CBD had atrophy in the bilateral frontoparietal and basal ganglia, sparing the medial temporal lobe. Severe medial temporal atrophy was also identified in AD with TDP-43 pathology [29]. Given the strong age-related association of TDP-43 pathology with HS [39], the medial temporal atrophy in AD with TDP-43 pathology might be a consequence of the accompanying HS in AD. However, Josephs et al. showed that within an AD with TDP-43 pathology group, no difference was observed in medial temporal volume loss between subjects with and without HS [29]. This suggests that TDP-43 might be independently related with medial temporal atrophy regardless of the presence of HS. In our series, three out of four FTLD-tau, CBD with FTLD-TDP cases had typical or atypical HS, but the one case without HS had intermediate ADNC, which can also be an underlying cause of the medial temporal atrophy. Another interesting neuroimaging finding was the asymmetric and symmetric atrophic pattern in the mixed and pure FTLD-tau, CBD groups, respectively. The prevalence of clinical PPA syndrome in the mixed group (2/4, 50%) was higher than that in the pure group (5/17, 33%), although the difference was not significant. Hence, it is possible that the left asymmetric involvement in the CBD with FTLD-TDP group could be attributed to the clinical nfvPPA syndrome. Considering the relatively small number of cases analyzed in this study, a larger data set should be used to clarify the significance of the asymmetric vs. symmetric neurodegeneration between the CBD with and without FTLD-TDP groups.
In summary, the overlap between prominent FTLD-tau and FTLD-TDP is rare (3.6% in our series). It may be present in familial and in sporadic cases and can comprise different combinations of FTLD-tau and FTLD-TDP. Although all primary FTLD-tau cases were of the CBD type, that other less common forms of FTLD-tau may overlap with FTLD-TDP cannot be ruled out. In our series, mixed cases had an older age at onset and a low or intermediate burden of AD pathology, which may suggest that these mixed cases have a higher propensity of developing polyproteinopathies. Investigating the molecular differences between the mixed and pure pathology groups may help us understand the general mechanisms of proteostasis failure in neurodegenerative diseases. We failed to identify striking clinical and radiological differences between pure and mixed cases, however, in general, mixed cases showed more severe atrophy than pure cases, and specifically, the mixed CBD with FTLD-TDP group showed prominent asymmetric left-sided atrophy compared to the pure CBD group. This corroborates the negative contribution of the second pathology.
Lastly, it is important to note that this study is limited to nine cases with mixed FTLD pathology. Therefore, the results should not be generalized until replicated in a larger sample.
References
McKhann GM, Albert MS, Grossman M et al (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58(11):1803–1809
Mackenzie I, Neumann M, Bigio E et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119(1):1–4
Baborie A, Griffiths TD, Jaros E et al (2011) Pathological correlates of frontotemporal lobar degeneration in the elderly. Acta Neuropathol 121(3):365–371
Kovacs GG (2015) Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 41(1):3–23
Mackenzie IR, Neumann M, Baborie A et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122(1):111–113
Nascimento C, Di Lorenzo Alho AT, Bazan Conceição Amaral C et al (2017) Prevalence of transactive response DNA-binding protein 43 (TDP-43) proteinopathy in cognitively normal older adults: systematic review and meta-analysis. Neuropathol Appl Neurobiol 44(3):286–297
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114(1):5–22
Flanagan EP, Duffy JR, Whitwell JL et al (2016) Mixed tau and TDP-43 pathology in a patient with unclassifiable primary progressive aphasia. Neurocase 22(1):55–59
Freeman SH, Spires-Jones T, Hyman BT, Growdon JH, Frosch MP (2008) TAR-DNA binding protein 43 in Pick disease. J Neuropathol Exp Neurol 67(1):62–67
Koga S, Sanchez-Contreras M, Josephs KA et al (2016) Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord 32(2):246–255
Uryu K, Nakashima-Yasuda H, Forman MS et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67(6):555–564
Winton MJ, Van Deerlin VM, Kwong LK et al (2008) A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBS Lett 582(15):2252–2256
Lee SE, Tartaglia MC, Yener G et al (2013) Neurodegenerative disease phenotypes in carriers of MAPT p.A152T, a risk factor for frontotemporal dementia spectrum disorders and Alzheimer disease. Alzheimer Dis Assoc Disord 27(4):302–309
Montine TJ, Phelps CH, Beach TG et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11
Rodriguez RD, Suemoto CK, Molina M et al (2016) Argyrophilic Grain Disease: Demographics, Clinical, and Neuropathological Features From a Large Autopsy Study. J Neuropathol Exp Neurol 75(7):628–635
Crary JF, Trojanowski JQ, Schneider JA et al (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128(6):755–766
Kovacs GG, Ferrer I, Grinberg LT et al (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131(1):87–102
Coppola G, Chinnathambi S, Lee JJ et al (2012) Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet 21(15):3500–3512
Josephs KA, Hodges JR, Snowden JS et al (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122(2):137–153
Kovacs GG, Molnár K, László L et al (2011) A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 122(2):205–222
Robinson AC, Thompson JC, Weedon L et al (2014) No interaction between tau and TDP-43 pathologies in either frontotemporal lobar degeneration or motor neurone disease. Neuropathol Appl Neurobiol 40(7):844–854
Storey K, Johanidesová S, Matěj R et al (2016) FTLD-TDP and progressive supranuclear palsy in comorbidity-a report of two cases with different clinical presentations. Neurocase 1–7
Yokota O, Davidson Y, Bigio EH et al (2010) Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 120(1):55–66
Bieniek KF, Murray ME, Rutherford NJ et al (2013) Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol 125(2):289–302
Hsiung GY, DeJesus-Hernandez M, Feldman HH et al (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135(Pt 3):709–722
King A, Al-Sarraj S, Troakes C et al (2013) Mixed tau, TDP-43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and p.Ala239Thr MAPT (tau) variant. Acta Neuropathol 125(2):303–310
Amador-Ortiz C, Lin WL, Ahmed Z et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61(5):435–445
Josephs KA, Murray ME, Whitwell JL et al (2016) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131(4):571–585
Josephs KA, Whitwell JL, Knopman DS et al (2008) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70(19 Pt 2):1850–1857
Bigio EH, Mishra M, Hatanpaa KJ et al (2010) TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 120(1):43–54
Davidson YS, Raby S, Foulds PG et al (2011) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122(6):703–713
Josephs KA, Whitwell JL, Weigand SD et al (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127(6):811–824
Dickson DW, Davies P, Bevona C et al (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol 88(3):212–221
Kouri N, Oshima K, Takahashi M et al (2013) Corticobasal degeneration with olivopontocerebellar atrophy and TDP-43 pathology: an unusual clinicopathologic variant of CBD. Acta Neuropathol 125(5):741–752
Kovacs GG, Wöhrer A, Ströbel T et al (2011) Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol 30(1):3–10
Graff-Radford J, Whitwell JL, Dickson DW, Josephs KA (2013) Pallidonigroluysian atrophy associated with p.A152T variant in MAPT. Parkinsonism Relat Disord 19(9):838–841
Kara E, Ling H, Pittman AM et al (2012) The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol Aging 33(9):2231.e7-.e14
Labbé C, Ogaki K, Lorenzo-Betancor O et al (2015) Role for the microtubule-associated protein tau variant p.A152T in risk of α-synucleinopathies. Neurology 85(19):1680–1686
Nag S, Yu L, Capuano AW et al (2015) Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol 77(6):942–952
Acknowledgments
This study was supported by the Financial Supporting Project of Long-term Overseas Dispatch of PNU's Tenure-track Faculty, 2015 to Eun-Joo Kim.
Funding
Funding was provided by the NIH Grant #K24AG053435, P50AG023501 and P01AG019724, Rainwater Foundation to Lea T. Grinberg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have nothing to disclose.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Kim, EJ., Brown, J.A., Deng, J. et al. Mixed TDP-43 proteinopathy and tauopathy in frontotemporal lobar degeneration: nine case series. J Neurol 265, 2960–2971 (2018). https://doi.org/10.1007/s00415-018-9086-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9086-2