
Abstract
FAD-dependent glucose dehydrogenase (FAD-GDH), which contains FAD as a cofactor, catalyzes the oxidation of d-glucose to d-glucono-1,5-lactone, and plays an important role in biosensors measuring blood glucose levels. In order to obtain a novel FAD-GDH gene homolog, we performed degenerate PCR screening of genomic DNAs from 17 species of thermophilic filamentous fungi. Two FAD-GDH gene homologs were identified and cloned from Talaromyces emersonii NBRC 31232 and Thermoascus crustaceus NBRC 9129. We then prepared the recombinant enzymes produced by Escherichia coli and Pichia pastoris. Absorption spectra and enzymatic assays revealed that the resulting enzymes contained oxidized FAD as a cofactor and exhibited glucose dehydrogenase activity. The transition midpoint temperatures (T m) were 66.4 and 62.5 °C for glycosylated FAD-GDHs of T. emersonii and T. crustaceus prepared by using P. pastoris as a host, respectively. Therefore, both FAD-GDHs exhibited high thermostability. In conclusion, we propose that these thermostable FAD-GDHs could be ideal enzymes for use as thermotolerant glucose sensors with high accuracy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Self-monitoring of blood glucose (SMBG) is a key clinical test for diabetes patients who need to know their blood glucose levels. Traditional glucose sensors used for the clinical test are equipped with glucose oxidase (GOD), NAD(P)-dependent glucose dehydrogenase (NAD(P)-GDH), or pyrroloquinoline quinone (PQQ)-dependent GDH (PQQ-GDH).
GOD (EC 1.1.3.4), NAD(P)-GDH (EC 1.1.1.47), and PQQ-GDH (EC 1.1.5.2) are oxidoreductases that catalyze the oxidation of d-glucose to d-glucono-1,5-lactone. Use of these enzymes for glucose sensors has the following limitations: (1) GOD requires oxygen as an electron acceptor, and dissolved oxygen in the blood influences the accuracy of measurements for glucose concentration; (2) NAD(P)-GDH requires continuous coenzyme addition during measurements; and (3) PQQ-GDH exhibits broad substrate specificity (Ferri et al. 2011; Heller and Feldman 2008; Yoo and Lee 2010).
Recently, FAD-dependent GDH (FAD-GDH; EC 1.1.5.9), which contains FAD as a cofactor, has become a focus of research related to glucose sensing. FAD-GDH exhibits strict substrate specificity, and glucose sensors employing the enzyme are insensitive to dissolved oxygen and require no cofactor addition (Ferri et al. 2011; Tsujimura et al. 2006). Several studies on FAD-GDHs from filamentous fungi, including Aspergillus oryzae (Bak 1967a, b; Ogura 1951), Aspergillus terreus (Tsujimura et al. 2006; Yang et al. 2014, 2015), Aspergillus flavus (Mori et al. 2011; Yoshida et al. 2015), Aspergillus niger (Mori et al. 2011), Glomerella cingulata (Sygmund et al. 2011a, 2011b), Mucor prainii (Satake et al. 2015), and Pycnoporus cinnabarinus (Piumi et al. 2014) have been published. These FAD-GDHs have suitable enzymatic properties for the biosensors of SMBG. Preferably, more thermostable FAD-GDH is required for industrial application. Thermophilic filamentous fungi are capable of growing at temperatures between 20 and 50 °C, or above 50 °C (Maheshwari et al. 2000), and many thermostable enzymes from these fungi were characterized (Berka et al. 2011; Maheshwari et al. 2000). All previous FAD-GDHs have originated from filamentous fungi that favor and grow at ordinary temperatures, while thermophilic filamentous fungal FAD-GDHs remain unidentified.
In order to obtain a novel FAD-GDH gene homolog, we performed degenerate PCR screening of genomic DNAs from thermophilic filamentous fungi. As a result, novel FAD-GDH gene homologs were identified from the genomic DNAs of Talaromyces emersonii NBRC 31232, Thermoascus crustaceus NBRC 9129, and T. crustaceus NBRC 9816. Furthermore, we successfully cloned and expressed the FAD-GDH gene homologs from T. emersonii NBRC 31232 and T. crustaceus NBRC 9129 to prepare recombinant enzymes by using Escherichia coli and Pichia pastoris as hosts, and then characterized the properties of the resulting enzymes. The results indicated that the novel FAD-GDHs from the thermophilic filamentous fungi exhibited high thermostability.
Materials and methods
Strains and culture media
Thermophilic filamentous fungi used in this study are listed in Table S1. The fungal strains were purchased from the Biological Resource Center at the National Institute of Technology and Evaluation (NBRC; Chiba, Japan) and cultured in malt extract medium or potato dextrose medium as directed by NBRC. E. coli strains DH5α (Takara Bio, Shiga, Japan) and BLR(DE3) (Merck Millipore, Darmstadt, Germany) were cultured in Luria-Bertani (LB) medium (Sambrook and Russell 2001). Methylotrophic yeast P. pastoris strain GS115 (Life Technologies, Carlsbad, CA, USA) was cultured in media (yeast extract peptone dextrose medium, minimal dextrose medium, buffered minimal glycerol medium, and buffered minimal methanol medium) with a Pichia expression kit (Life Technologies) according to manufacturer instructions.
DNA manipulation
All DNA experiments followed standard protocols (Sambrook and Russell 2001). Genomic DNA was extracted with a Wizard genomic DNA purification kit (Promega, Madison, WI, USA). Thermostable DNA polymerases and oligonucleotide primers (Table S2) were purchased from Toyobo (Osaka, Japan) and Life Technologies, respectively. GeneAmp PCR System 9700 (Life Technologies) was used for PCR. Each DNA fragment amplified by PCR was subcloned into a pCR-Blunt II-TOPO vector (Life Technologies), and its nucleotide sequence was determined using an Applied Biosystems 3130xl genetic analyzer (Life Technologies). Restriction and modification enzymes were purchased from New England BioLabs (Ipswich, MA, USA) or Takara Bio. Southern hybridization analysis was performed according to a standard protocol (Sambrook and Russell 2001) with a DIG DNA labeling and detection kit (Roche Diagnostics, Basel, Switzerland).
Degenerate PCR screening for FAD-GDH gene homologs
Seventeen species of thermophilic filamentous fungi were screened for FAD-GDH gene homologs by degenerate PCR using the individual fungal genomic DNA as a template. Three sense degenerate primers (HCA, HCB, and HCC) and two antisense degenerate primers (HCD and HCE) were designed based on the primary structures of A. oryzae and A. terreus FAD-GDHs (28-YDYIVVGGG-36, 100-QVLRAGKALG-109, 120-TRAEDVQI-127, 525-NFHPVGTAAMM-535, and 565-FQVCGHLVST-574, where the numbers correspond to the amino acid sequence of A. oryzae FAD-GDH). The PCR mixture (50 μl) contained 1 U KOD FX (Toyobo), PCR buffer for KOD FX, 0.3 μM sense primer, 0.3 μM antisense primer, dNTP mixture (0.4 mM each), and the individual fungal genomic DNA as the template. Each reaction consisted of 30 cycles of the following steps: denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 68 °C for 2 min. The amplified DNA fragments in the PCR products were separated by agarose gel electrophoresis and then extracted from preparative agarose gels. Each extracted DNA fragment was subcloned and sequenced. The predicted amino acid sequences were compared to those of A. oryzae and A. terreus FAD-GDHs.
Cloning of FAD-GDH gene homologs
Prior to gene cloning, the genomic DNA from T. emersonii or T. crustaceus was digested by various restriction enzymes, and analyzed by Southern hybridization. The DNA fragment amplified by degenerate PCR, representing a portion of the FAD-GDH gene homolog, was used as a probe. As a result, a single band hybridized with the probe was detected at ~4.0 kb in the lane of EcoRI-digested T. emersonii genomic DNA, or at ~5.0 kb in the lane of HindIII-digested Th. crustaceus genomic DNA.
Inverse PCR was performed to clone the upstream region of the start codon and the downstream region of the stop codon of the FAD-GDH gene homolog. The EcoRI-digested T. emersonii genomic DNA or the HindIII-digested T. crustaceus genomic DNA was subjected to preparative agarose gel electrophoresis, and the DNA fragments around 4.0 or 5.0 kb were extracted from the gels. These DNA fragments were self-ligated with T4 DNA ligase and used as the template for inverse PCR. The primers for inverse PCR, GSP3, and GSP5, were designed based on the nucleotide sequence of the internal FAD-GDH gene homolog from T. emersonii or T. crustaceus. The PCR mixture (50 μl) contained 1 U KOD-Plus-Neo (Toyobo), PCR buffer for KOD-Plus-Neo, 0.2 μM sense primer, 0.2 μM antisense primer, dNTP mixture (0.2 mM each), 1.5 mM magnesium sulfate, and the circularized DNA as the template. Each reaction consisted of 30 cycles of the following steps: denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 68 °C for 3 min.
The genomic DNA fragment containing the FAD-GDH gene homolog was amplified by PCR using KOD-Plus- Neo, sense primer GSP9, antisense primer GSP10, and either T. emersonii or T. crustaceus genomic DNA. Primers GSP9 and GSP10 were designed based on the nucleotide sequence of the inverse PCR-amplified DNA fragment.
Genetic analysis
Exon-intron splicing sites were predicted by the GT-AG rule (Breathnach et al. 1978; Catterall et al. 1978). Secretion signal sequence was predicted by SignalP 4.0 (Petersen et al. 2011), and N-glycosylation sites were predicted by NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/). The amino acid sequences of the FAD-GDHs were aligned using ClustalW (http://clustalw.ddbj.nig.ac.jp/) (Thompson et al. 1994).
Synthesis of the coding sequence
The coding sequence (CDS) of the FAD-GDH gene homolog from T. emersonii or T. crustaceus was artificially synthesized by overlap extension PCR (Horton et al. 1989). Four exons were individually amplified and connected to synthesize the complete structural gene. The primers used were as follows: GSP11 and E1A for exon 1, E2S and E2A for exon 2, E3S and E3A for exon 3, and E4S and GSP12 for exon 4.
Preparation of recombinant enzymes produced by E. coli
The gene encoding the mature FAD-GDH was generated by PCR using primers Esc2 and Esc4 to construct an E. coli expression plasmid. Primers Esc2 and Esc4 contained NdeI site, and HindIII site and six CAC codons encoding a histidine tag located at the C-terminus, respectively. The amplified DNA fragment was inserted between the NdeI and HindIII sites in the E. coli pET-21b(+) expression vector (Merck Millipore).
E. coli BLR(DE3) cells harboring the expression plasmid were cultured in LB medium containing 100 μg/ml ampicillin at 37 °C for approximately 4 h, and isopropyl-β-d-thiogalactopyranoside was added into the culture medium (1 mM final concentration) to induce the gene expression. The cells were further cultured at 25 °C overnight.
Cells were collected and disrupted by sonication in PBS [137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM disodium hydrogen phosphate, and 2 mM potassium dihydrogen phosphate (pH 7.4)]. The extract was centrifuged at 8000g for 10 min at 4 °C to remove unbroken cells, and further centrifuged at 12,000g for 20 min at 4 °C to obtain the soluble fraction. The soluble fraction was applied to a Ni-NTA agarose column (QIAGEN, Hilden, Germany) equilibrated with PBS. The adsorbed proteins were eluted using 20 mM HEPES buffer (pH 7.5) containing 100 mM imidazole. The eluate was applied to a Q Sepharose Fast Flow column (GE Healthcare, Little Chalfont, UK) equilibrated with 20 mM HEPES buffer (pH 7.5). The adsorbed proteins were eluted using 20 mM HEPES buffer (pH 7.5) containing 100 mM sodium chloride. The eluate was desalted with an Amicon Ultra-15 10 K centrifugal filter device (Merck Millipore) and applied to a RESOURCE Q (1 ml) column (GE Healthcare) equilibrated with 20 mM HEPES buffer (pH 7.5). The adsorbed proteins were eluted by a linear gradient of 0–1.0 M sodium chloride in 20 mM HEPES buffer (pH 7.5). The fractions containing recombinant FAD-GDH were desalted and stored at 4 °C until use.
Preparation of recombinant enzymes produced by P. pastoris
The gene encoding mature FAD-GDH was generated by PCR using primers Pic1 and Pic2 to construct a P. pastoris expression plasmid. Primers Pic1 and Pic2 contained EcoRI site, and NotI site and six CAC codons encoding a histidine tag located at the C-terminus, respectively. Before PCR amplification, the structural gene was modified by silent mutagenesis to synthesize the artificial gene lacking SalI sites. The amplified DNA fragment was inserted between the EcoRI and NotI sites in the P. pastoris pPIC9 expression vector (Life Technologies). By inserting the DNA fragment into the vector, the yeast secretion signal peptide was fused to the mature FAD-GDH. The resulting plasmid was cleaved with SalI, and the linearized DNA was introduced into P. pastoris GS115 genomic DNA at the his4 locus by electroporation.
The recombinant strain was cultured in buffered minimal methanol medium at 30 °C for 5 days. Methanol was added into the culture medium every 24 h (0.5 % final concentration).
Culture supernatant was dialyzed against PBS, and applied to a Ni-NTA agarose column equilibrated with PBS. The adsorbed proteins were eluted using 20 mM HEPES buffer (pH 7.5) containing 200 mM imidazole. The eluate was desalted and stored at 4 °C until use.
Electrophoresis
SDS-PAGE was performed by the method of Laemmli (Laemmli 1970). Proteins were stained with Coomassie brilliant blue (CBB) G-250 (CBB Stain One; Nacalai Tesque, Kyoto, Japan), and glycosylated proteins were detected by periodic acid-Schiff (PAS) staining with a Pierce glycoprotein staining kit (Thermo Fisher Scientific, Waltham, MA, USA). Protein concentration was estimated with a Pierce BCA protein assay kit (Thermo Fisher Scientific) (Smith et al. 1985). Bovine serum albumin was used as a standard. Purity of the purified proteins was evaluated with ImageJ (http://imagej.nih.gov/ij/) (Abramoff et al. 2004).
Absorption spectrum
The absorption spectrum of purified recombinant enzyme (2 mg/ml) was measured with a DU-800 UV/visible spectrophotometer (Beckman Coulter, Brea, CA, USA) using a micro cell (1-cm path length). The molar absorption coefficient of FAD at 450 nm (11.3 mM−1 cm−1) (Macheroux 1999) was used to calculate the concentration of FAD in the enzyme.
GDH assay
GDH activity was measured by the 2,6-dichloroindophenol (DCIP) method. The reaction mixture (3 ml) contained 50 mM potassium phosphate buffer (pH 7.0), 0.14 mM 1-methoxy-5-methylphenazinium methylsulfate (1-mPMS; Dojindo, Kumamoto, Japan), 0.07 mM DCIP (2,6-dichlorophenol-indophenol sodium salt dihydrate; Merck Millipore), 0.2 % Triton X-100, and 300 mM glucose. The enzymatic reaction was initiated by the addition of purified recombinant enzyme (0.1 ml). The absorbance of oxidized DCIP at 600 nm was monitored with a UV-1700 Pharma Spec UV–Vis spectrophotometer (Shimadzu, Kyoto, Japan). One unit was defined as the amount of FAD-GDH producing 1 μmol of reduced DCIP per minute at pH 7.0 and 37 °C. The molar absorption coefficient of oxidized DCIP at 600 nm (16.3 mM−1 cm−1) was used to determine the enzyme activity. Specific activity was shown as units per 1 μmol of holo-enzyme (U/μmol).
Assay of oxygen-reducing activity
Oxygen-reducing activity was measured by the method of Swoboda and Massey (1965). A. niger GOD (Wako Pure Chemical Industries, Osaka, Japan) was used as a positive control.
Accession numbers
The nucleotide sequences of the FAD-GDH genes have been deposited in the DNA Data Bank of Japan (DDBJ) under the accession numbers LC069047 and HW503402 for T. emersonii NBRC 31232 and LC069048 and HW503403 for T. crustaceus NBRC 9129.
Results
Screening for FAD-GDH gene homologs
We performed degenerate PCR screening of genomic DNAs from 17 species of thermophilic filamentous fungi to identify novel FAD-GDH gene homologs. Genomic DNA extracted from each thermophilic filamentous fungus was used as the template for the degenerate PCR, and results of agarose gel electrophoresis of the amplified DNA fragments are shown in Fig. S1. DNA bands of ~1.3–2.0 kb were detected from the PCR products derived from the genomic DNAs of T. emersonii NBRC 31232, T. crustaceus NBRC 9129, and T. crustaceus NBRC 9816. These DNA fragments were successfully amplified by all primer pairs for T. emersonii NBRC 31232 (Fig. S1a), in contrast to only two primer pairs successfully amplified for both T. crustaceus strains (Fig. S1b, c). Nucleotide sequencing of these DNA fragments revealed that the predicted amino acid sequences were homologous to those of A. oryzae and A. terreus FAD-GDHs. These results indicated that the genomic DNA of T. emersonii NBRC 31232 and the two T. crustaceus strains contained FAD-GDH gene homologs. Furthermore, the nucleotide sequences of the two T. crustaceus FAD-GDH gene homologs were identical each other.
Cloning of FAD-GDH gene homologs
The genomic DNA fragments containing the FAD-GDH gene homologs were cloned from T. emersonii NBRC 31232 and T. crustaceus NBRC 9129. Nucleotide sequencing revealed that these fragments contained open reading frames of the FAD-GDH gene homologs, and that the structural genes were divided into four exons by three introns. The putative FAD-GDH gene homologs from T. emersonii and T. crustaceus contained 1782-bp encoding 593 amino acids and 1755-bp encoding 584 amino acids, respectively.
The secretion signal peptides were identified as 17 residues from Met1 to Ala17 in T. emersonii FAD-GDH, and 16 residues from Met1 to Ala16 in T. crustaceus FAD-GDH. The amino acid sequences implicated in FAD-binding (Gly-X-Gly-X-X-Gly, where X represents any amino acid residue) (Dym and Eisenberg 2001) were conserved in the FAD-GDHs of both strains. The 12 and 11 asparagine residues that undergo N-glycosylation (Asn-X-Ser/Thr, where X represents any residue, except for proline) (Tschopp et al. 1987) were predicted in T. emersonii FAD-GDH and T. crustaceus FAD-GDH, respectively. The alignment of the amino acid sequences of the FAD-GDHs from T. emersonii, T. crustaceus, A. oryzae, and A. terreus is shown in Fig. S2. T. emersonii FAD-GDH showed 63 % identity with both A. oryzae and A. terreus FAD-GDHs, whereas T. crustaceus FAD-GDH showed 54 and 56 % identity with A. oryzae and A. terreus FAD-GDHs, respectively. Additionally, T. emersonii FAD-GDH and T. crustaceus FAD-GDH showed 62 % identity with each other.
Preparation of recombinant FAD-GDHs
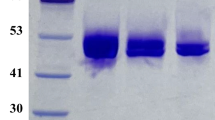
Talaromyces emersonii mature FAD-GDH (TeGDH) and T. crustaceus mature FAD-GDH (TcGDH) were successfully produced in the soluble fractions of E. coli, and then purified (>95 % purity). The yields per 1 l of E. coli culture media were 30 mg for TeGDH and 5 mg for TcGDH. Protein bands denoting the purified recombinant enzymes were detected by SDS-PAGE at ~60 kDa for TeGDH and ~58 kDa for TcGDH, which were consistent with the theoretical molecular masses of both enzymes (Fig. 1a). The theoretical molecular masses of these enzymes with their respective histidine tags were calculated at ~63 kDa for TeGDH and ~62 kDa for TcGDH. When produced by P. pastoris, both enzymes were effectively secreted into the culture media, and then purified (>95 % purity). The yields per 1 l of P. pastoris culture media were 46 mg for TeGDH and 39 mg for TcGDH. Protein bands denoting the purified recombinant enzymes were detected at 90–200 kDa for TeGDH, and 80–200 kDa for TcGDH by CBB staining (Fig. 1b, left). These enzymes were also stained with PAS, indicating that they had undergone glycosylation during post-translational modification in P. pastoris (Fig. 1b, right). Deglycosylation under the denaturing condition with PNGase F exhibited decreased molecular masses, which were ~65 kDa for TeGDH and ~63 kDa for TcGDH (Fig. S3). The results revealed that the glycosylated enzymes were modified by N-linked sugar chains as major components of glycosylation. The molecular masses of glycosylated enzymes would be overestimated by SDS-PAGE. Glycosylated proteins are known to have decreased charge-mass ratios when complexed to SDS, resulting in decreased migration rates and overestimated molecular masses (Werner et al. 1993).

SDS-PAGE of FAD-GDHs. Purified FAD-GDHs were subjected to SDS-PAGE (5 μg/lane). a FAD-GDHs produced by E. coli. The proteins in the gels were stained with CBB. b FAD-GDHs produced by P. pastoris. The proteins in the left gel were stained with CBB, and the glycosylated proteins in the right gel were stained with PAS. M molecular mass markers. In both panels, lane 1 TeGDH and lane 2 TcGDH
The solutions of the purified TeGDHs and TcGDHs were yellow. Absorption spectra of the enzymes produced by E. coli (unglycosylated FAD-GDH) (Fig. 2a) and P. pastoris (glycosylated FAD-GDH) (Fig. 2b) were similar to the typical spectrum observed for oxidized flavin with major peaks at 380 and 450 nm. The peaks corresponding to oxidized flavin disappeared when glucose was added, and also when sodium dithionite was added as a reductant. These results suggested that TeGDHs and TcGDHs produced by E. coli and P. pastoris were purified as holo forms containing oxidized FAD as a cofactor.

Absorption spectra of FAD-GDHs. a Unglycosylated FAD-GDHs produced by E. coli. b Glycosylated FAD-GDHs produced by P. pastoris. The spectra of TeGDHs and TcGDHs are on the left and right sides, respectively. The spectra of purified FAD-GDHs are drawn as solid lines, the spectra of FAD-GDHs in the presence of 100 mM glucose are drawn as dashed lines, and the spectra of FAD-GDHs in the presence of sodium dithionite are drawn as dotted and dashed lines
GDH activity
GDH activity at pH 7.0 and 37 °C was measured. The specific activities of unglycosylated and glycosylated TeGDHs were (9.21 ± 0.24) × 103 U/μmol and (8.79 ± 0.12) × 103 U/μmol, respectively, whereas those of unglycosylated and glycosylated TcGDHs were (16.0 ± 1.2) × 103 U/μmol and (13.5 ± 0.9) × 103 U/μmol, respectively. The specific activities of TcGDHs were slightly higher than those of TeGDHs.
The activities of TeGDHs and TcGDHs depended on glucose concentration (Fig. 3a, b). The maximum velocity (V max) and the Michaelis-Menten constant (K m) were calculated by the Michaelis-Menten curve-fitting with the least square method. The V max values of unglycosylated FAD-GDHs were higher than those observed for glycosylated FAD-GDHs. The K m values were calculated at (4.06 ± 0.40) × 102 mM for unglycosylated TeGDH and (3.37 ± 0.31) × 102 mM for glycosylated TeGDH. Unglycosylated and glycosylated TcGDHs exhibited higher substrate affinities with the K m values of 4.63 ± 0.19 and 2.82 ± 0.17 mM, respectively.

Glucose concentration dependence of FAD-GDH activity. a TeGDHs. b TcGDHs. In both panels, closed circles unglycosylated FAD-GDHs and open circles glycosylated FAD-GDHs
The oxygen-reducing activities of TeGDHs and TcGDHs were also investigated, using A. niger GOD as a positive control. However, none of the enzymes prepared in this study exhibited oxygen reducing activity. Therefore, our results revealed that these enzymes were FAD-dependent GDHs.
Effect of pH on FAD-GDH stability
The stability of the FAD-GDHs at various pH values was examined (Fig. 4). Unglycosylated TeGDH was stable at a range of pH values between 3.0 and 8.0 (Fig. 4a). Additionally, glycosylated TeGDH showed high stability at a wide range of pH values between 2.0 and 9.0. Also, the stability of unglycosylated and glycosylated TcGDHs appeared similar to that of unglycosylated and glycosylated TeGDHs (Fig. 4b). These results indicated that TeGDHs and TcGDHs exhibited stability over a broad pH range, and that glycosylated FAD-GDHs were more stable than unglycosylated FAD-GDHs.

Effect of pH on FAD-GDH stability. FAD-GDH (7 U/ml) was incubated in 50 mM Britton-Robinson universal buffer at indicated pH at 25 °C for 20 h, and then diluted with 50 mM potassium phosphate buffer (pH 7.0) containing 0.1 % Triton X-100. This diluted solution was used for assay. a TeGDHs. b TcGDHs. In both panels, closed circles unglycosylated FAD-GDHs and open circles glycosylated FAD-GDHs
Effect of reaction pH on FAD-GDH activity
The pH profile of FAD-GDH activity was examined (Fig. 5). The relative activities of TeGDHs were over 50 % at a wide range of pH values between 3.0 and 10.0. The optimum reaction pH values were 6.0 for unglycosylated TeGDH and 5.0 for glycosylated TeGDH (Fig. 5a). The optimum reaction pH values for unglycosylated and glycosylated TcGDHs were 5.0, although the activities of TcGDHs were relatively low at a range of pH values between 8.0 and 10.0 (Fig. 5b).

Effect of reaction pH on FAD-GDH activity. Reaction mixture containing FAD-GDH was incubated in 50 mM Britton-Robinson universal buffer at the indicated pH at 37 °C for 2 min. The oxidized DCIP was quantified after adding 0.2 M Tris–HCl buffer (pH 8.0) containing 8 M urea into the mixture to stop the reaction. The reaction mixture was finally diluted fivefold with the stopping buffer. a TeGDHs. b TcGDHs. In both panels, closed circles unglycosylated FAD-GDHs and open circles glycosylated FAD-GDHs
Effect of temperature on FAD-GDH stability
In order to investigate whether thermophilic filamentous fungal FAD-GDHs are highly thermostable, the enzymatic activities of TeGDHs and TcGDHs were measured after heat treatment at 40–70 °C for 15 min (Fig. 6). Unglycosylated TeGDH maintained nearly 100 % activity at temperatures up to 45 °C, but completely lost activity at 60 °C (Fig. 6a). Glycosylated TeGDH displayed high thermostability, maintaining 89 % activity even at 60 °C. Unglycosylated TcGDH was stable at temperatures up to 45 °C (Fig. 6b), while glycosylated TcGDH maintained 75 % activity even at 60 °C. The apparent T m values were 52.7 °C for unglycosylated TeGDH, 66.4 °C for glycosylated TeGDH, 51.6 °C for unglycosylated TcGDH, and 62.5 °C for glycosylated TcGDH. These results revealed that glycosylated TeGDH and glycosylated TcGDH exhibited high thermostability.

Effect of temperature on FAD-GDH stability. FAD-GDH (7 U/ml) was incubated at indicated temperatures at pH 5.0 for 15 min, and then diluted with 50 mM potassium phosphate buffer (pH 7.0) containing 0.1 % Triton X-100. This diluted solution was used for assay. a TeGDHs. b TcGDHs. In both panels, closed circles unglycosylated FAD-GDHs and open circles glycosylated FAD-GDHs
Effect of reaction temperature on FAD-GDH activity
The temperature profile of FAD-GDH activity was examined (Fig. 7). The optimum reaction temperatures of unglycosylated and glycosylated TeGDHs were observed at 55–60 and 65–70 °C, respectively (Fig. 7a). The optimum reaction temperatures of unglycosylated and glycosylated TcGDHs were observed at 45–60 and 60–65 °C, respectively (Fig. 7b). Our results revealed that the optimum reaction temperatures of unglycosylated TeGDH and unglycosylated TcGDH were ~60 °C and that glycosylated TeGDH and glycosylated TcGDH were ~65 °C. The activities of glycosylated TeGDH and glycosylated TcGDH at their optimum reaction temperatures were ~4.2 and ~3.3 times higher relative to those observed at 37 °C, respectively.

Effect of reaction temperature on FAD-GDH activity. Reaction mixture containing FAD-GDH was incubated at the indicated temperatures at pH 5.0 for 2 min. The oxidized DCIP was quantified after adding 0.2 M Tris–HCl buffer (pH 8.0) containing 8 M urea into the mixture to stop the reaction. The reaction mixture was finally diluted fivefold with the stopping buffer. a TeGDHs. b TcGDHs. In both panels, closed circles unglycosylated FAD-GDHs and open circles glycosylated FAD-GDHs
Substrate specificity of FAD-GDHs
The substrate specificity of TeGDHs and TcGDHs was examined (Table 1). TeGDHs and TcGDHs exhibited the highest specificity for d-glucose, while they exhibited no activity in the presence of maltose, to which PQQ-GDH exhibited cross-reactivity. TeGDHs and TcGDHs exhibited low reactivity to maltotriose and d-mannose, respectively, although TeGDHs and TcGDHs exhibited cross-reactivity toward 2-deoxy-d-glucose and d-xylose. These results revealed that TeGDHs and TcGDHs exhibited high substrate specificity for d-glucose.
Discussion
Here, we identified novel FAD-GDH gene homologs from T. emersonii NBRC 31232, T. crustaceus NBRC 9129, and T. crustaceus NBRC 9816. Since the partial nucleotide sequences of the T. crustaceus FAD-GDH gene homologs were identical with each other, we cloned two FAD-GDH gene homologs from T. emersonii NBRC 31232 and T. crustaceus NBRC 9129, and characterized the enzymatic properties of the gene products.
The culture media of A. oryzae and A. terreus exhibited GDH activity, and then FAD-GDHs were purified and their enzymatic properties were examined (Bak 1967a, b; Omura et al. 2010). In order to obtain thermostable FAD-GDHs from thermophilic filamentous fungi, we first attempted to detect GDH activity in the culture media of the fungi. However, no GDH activity was detected from the culture media, indicating that the production level of FAD-GDH depended on the culture conditions. Thus, the purification of native FAD-GDHs from any fungal culture media was resigned, and thermophilic filamentous fungi with FAD-GDH gene homologs on their genomic DNAs were screened by degenerate PCR. Successfully, novel FAD-GDH gene homologs were identified from the genomic DNAs of T. emersonii NBRC 31232, T. crustaceus NBRC 9129, and T. crustaceus NBRC 9816 by degenerate PCR using primers designed based on the primary structures of A. oryzae and A. terreus FAD-GDHs. The genomic DNA fragments containing the FAD-GDH gene homologs were subsequently cloned from T. emersonii NBRC 31232 and T. crustaceus NBRC 9129.
The enzymatic properties of the recombinant FAD-GDHs are summarized in Table 2. In the experiment of the effect of temperature on stability, the apparent T m values of glycosylated TeGDH and glycosylated TcGDH were 66.4 and 62.5 °C, respectively (Fig. 6). Studies on the thermostability of native A. oryzae and A. terreus FAD-GDHs purified from their culture media were previously reported (Bak 1967a; Omura et al. 2010). The apparent T m value of A. oryzae FAD-GDH was ~53 °C after heat treatment for 15 min. A. terreus FAD-GDH retained ~60 % of the activity after heat treatment at 55 °C for 15 min. These results revealed that glycosylated TeGDH and glycosylated TcGDH were highly thermostable. Additionally, glycosylated TeGDH and glycosylated TcGDH were stable at a broad pH range. The thermostability and pH stability of TeGDH and TcGDH were improved when the enzymes were produced by P. pastoris, likely a result of glycosylation. Several studies on glycosylated proteins produced by P. pastoris support our results (Daly and Hearn 2005). The numbers of predicted N-glycosylation sites were 12 for TeGDH and 11 for TcGDH. The evidence supporting the glycosylation of the FAD-GDHs produced by P. pastoris was confirmed by PAS staining (Fig. 1b) and PNGase F treatments (Fig. S3).
Generally, the structures of N-glycans produced by filamentous fungi and P. pastoris are high-mannose type. N-glycans produced by filamentous fungi are Man6–9GlcNAc2 or Man5–12GlcNAc2 sizes (Maras et al. 1999). The majority of N-glycans produced by P. pastoris are Man8–14GlcNAc2 sizes, with the remainder being much larger Man30–50GlcNAc2 sizes (Bretthauer and Castellino 1999). These suggest that the degree of N-glycosylation of recombinant protein produced by P. pastoris, which can be observed by SDS-PAGE, is similar to or higher than that of N-glycosylation of native protein produced by filamentous fungus. In the case of G. cingulata FAD-GDH, native enzyme and recombinant enzyme produced by P. pastoris exhibited similar degree of N-glycosylation, as well as similar optimum temperatures (Sygmund et al. 2011a; Sygmund et al. 2011b). In other examples, A. niger GOD, Penicillium variabile GOD and Agaricus meleagris FAD-dependent pyranose dehydrogenase (EC 1.1.99.29), which were produced by P. pastoris, exhibited higher degree of N-glycosylation than native enzymes (Crognale et al. 2006; Guo et al. 2010; Sygmund et al. 2012). In the matter of stability, while these recombinant GODs exhibited increased pH stability, decreased thermostability was unexpectedly exhibited (Crognale et al. 2006; Guo et al. 2010). Therefore, among native FAD-GDHs produced by filamentous fungi and recombinant FAD-GDHs produced by P. pastoris, the stability of the glycosylated FAD-GDHs would depend on the enzymes, rather than the degree of glycosylation.
The kinetic parameters, including V max and K m, were calculated. The V max values were 1.57 × 104–2.08 × 104 U/μmol for unglycosylated and glycosylated FAD-GDHs. The K m values of TcGDHs were ~100-fold lower relative to those of TeGDHs. The reported K m values of A. oryzae FAD-GDH and A. terreus FAD-GDH were 25 and 49.7 mM, respectively (Bak 1967b; Omura et al. 2010). These data indicated that TcGDHs exhibited higher affinities for glucose. The fasting blood glucose level in a healthy individual is almost 5 mM. On the other hand, in a diabetes patient, the fasting blood glucose level is over 7.0 mM, or the blood glucose level after an oral glucose tolerance test is over 11.1 mM (American Diabetes Association 2016). The blood glucose level of a severe diabetes patient sometimes reaches over 33.3 mM (American Diabetes Association 2004). Therefore, at the glucose concentrations, T. crustaceus FAD-GDH can exhibits high activity, but the activity of T. emersonii FAD-GDH becomes low. By a site-directed mutagenesis approach, improvement of T. emersonii FAD-GDH would be required to decrease the K m value.
TeGDHs and TcGDHs highly recognized glucose as a substrate (Table 1). Glucose sensors employing PQQ-GDH that react with non-glucose saccharides, including maltose, galactose, and xylose, would lead to potentially fatal errors for blood glucose monitoring (US Food and Drug Administration 2009). Protein engineering studies have succeeded in improving its substrate specificity (Hamamatsu et al. 2006; Igarashi et al. 2004; Sode et al. 2002). In our present experiments, TeGDHs and TcGDHs exhibited cross-reactivity toward 2-deoxy-d-glucose and d-xylose. A. oryzae FAD-GDH and A. terreus FAD-GDH also exhibited reactivity to these saccharides (Bak 1967b; Omura et al. 2010). However, glucose sensors employing these enzymes would provide appropriate glucose concentrations for SMBG, because these saccharides are generally absent in the blood of diabetes patients. Based on the findings, we suggested that T. emersonii FAD-GDH and T. crustaceus FAD-GDH could be used for biosensors to SMBG, although improvement of the substrate specificity would be required to minimalize cross-reactivity to non-glucose saccharides.
In the present study, we cloned novel FAD-GDH gene homologs from the thermophilic filamentous fungi T. emersonii NBRC 31232 and T. crustaceus NBRC 9129, and characterized the enzymatic properties of the gene products. The results revealed that these FAD-GDHs exhibited high thermostability and substrate specificity. Moreover, these thermostable FAD-GDHs have potential to be ideal enzymes for biosensors of SMBG, enabling long-term storage and high accuracy.
References
Abramoff MD, Magalhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42
American Diabetes Association (2016) Standards of medical care in diabetes—2016. Diabetes Care 39:S1–S112
American Diabetes Association (2004) Hospital admission guidelines for diabetes. Diabetes Care 27:S103. doi:10.2337/diacare.27.2007.S103
Bak TG (1967a) Studies on glucose dehydrogenase of Aspergillus oryzae: II. purification and physical and chemical properties. Biochim Biophys Acta 139:277–293. doi:10.1016/0005-2744(67)90032-0
Bak TG (1967b) Studies on glucose dehydrogenase of Aspergillus oryzae: III. General enzymatic properties. Biochim Biophys Acta 146:317–327. doi:10.1016/0005-2744(67)90218-5
Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, Ishmael N, John T, Darmond C, Moisan MC, Henrissat B, Coutinho PM, Lombard V, Natvig DO, Lindquist E, Schmutz J, Lucas S, Harris P, Powlowski J, Bellemare A, Taylor D, Butler G, de Vries RP, Allijn IE, van den Brink J, Ushinsky S, Storms R, Powell AJ, Paulsen IT, Elbourne LD, Baker SE, Magnuson J, Laboissiere S, Clutterbuck AJ, Martinez D, Wogulis M, de Leon AL, Rey MW, Tsang A (2011) Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat Biotechnol 29:922–927. doi:10.1038/nbt.1976
Breathnach R, Benoist C, O’Hare K, Gannon F, Chambon P (1978) Ovalbumin gene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc Natl Acad Sci U S A 75:4853–4857
Bretthauer RK, Castellino FJ (1999) Glycosylation of Pichia pastoris-derived proteins. Biotechnol Appl Biochem 30:193–200. doi:10.1111/j.1470-8744.1999.tb00770.x
Catterall JF, O’Malley BW, Robertson MA, Staden R, Tanaka Y, Brownlee GG (1978) Nucleotide sequence homology at 12 intron-exon junctions in the chick ovalbumin gene. Nature 275:510–513. doi:10.1038/275510a0
Crognale S, Pulci V, Brozzoli V, Petruccioli M, Federici F (2006) Expression of Penicillium variabile P16 glucose oxidase gene in Pichia pastoris and characterization of the recombinant enzyme. Enzyme Microb Technol 39:1230–1235. doi:10.1016/j.enzmictec.2006.03.005
Daly R, Hearn MTW (2005) Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit 18:119–138. doi:10.1002/jmr.687
Dym O, Eisenberg D (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci 10:1712–1728. doi:10.1110/ps.12801
Ferri S, Kojima K, Sode K (2011) Review of glucose oxidases and glucose dehydrogenases: a bird’s eye view of glucose sensing enzymes. J Diabetes Sci Technol 5:1068–1076. doi:10.1177/193229681100500507
Guo Y, Lu F, Zhao H, Tang Y, Lu Z (2010) Cloning and heterologous expression of glucose oxidase gene from Aspergillus niger Z-25 in Pichia pastoris. Appl Biochem Biotechnol 162:498–509. doi:10.1007/s12010-009-8778-6
Hamamatsu N, Suzumura A, Nomiya Y, Sato M, Aita T, Nakajima M, Husimi Y, Shibanaka Y (2006) Modified substrate specificity of pyrroloquinoline quinone glucose dehydrogenase by biased mutation assembling with optimized amino acid substitution. Appl Microbiol Biotechnol 73:607–617. doi:10.1007/s00253-006-0521-4
Heller A, Feldman B (2008) Electrochemical glucose sensors and their applications in diabetes management. Chem Rev 108:2482–2505. doi:10.1021/cr068069y
Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi:10.1016/0378-1119(89)90359-4
Igarashi S, Hirokawa T, Sode K (2004) Engineering PQQ glucose dehydrogenase with improved substrate specificity. Site-directed mutagenesis studies on the active center of PQQ glucose dehydrogenase. Biomol Eng 21:81–89. doi:10.1016/j.bioeng.2003.12.001
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Macheroux P (1999) UV-visible spectroscopy as a tool to study flavoproteins. In: Chapman SK, Reid GA (eds) Flavoprotein protocols. Humana Press, New York, pp 1–7
Maheshwari R, Bharadwaj G, Bhat MK (2000) Thermophilic fungi: their physiology and enzymes. Microbiol Mol Biol Rev 64:461–488. doi:10.1128/MMBR.64.3.461-488.2000
Maras M, van Die I, Contreras R, van den Hondel CA (1999) Filamentous fungi as production organisms for glycoproteins of bio-medical interest. Glycoconj J 16:99–107. doi:10.1023/A:1026436424881
Mori K, Nakajima M, Kojima K, Murakami K, Ferri S, Sode K (2011) Screening of Aspergillus-derived FAD-glucose dehydrogenases from fungal genome database. Biotechnol Lett 33:2255–2263. doi:10.1007/s10529-011-0694-5
Ogura Y (1951) Studies on the glucose dehydrogenase of Aspergillus oryzae. J Biochem 38:75–84
Omura H, Sanada H, Yada T, Morita T, Kuyama M, Ikeda T, Kano K, Tsujimura S (2010) Coenzyme-binding glucose dehydrogenase. EP Patent 1584675:B1
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi:10.1038/nmeth.1701
Piumi F, Levasseur A, Navarro D, Zhou S, Mathieu Y, Ropartz D, Ludwig R, Faulds CB, Record E (2014) A novel glucose dehydrogenase from the white-rot fungus Pycnoporus cinnabarinus: production in Aspergillus niger and physicochemical characterization of the recombinant enzyme. Appl Microbiol Biotechnol 98:10105–10118. doi:10.1007/s00253-014-5891-4
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Satake R, Ichiyanagi A, Ichikawa K, Hirokawa K, Araki Y, Yoshimura T, Gomi K (2015) Novel glucose dehydrogenase from Mucor prainii: purification, characterization, molecular cloning and gene expression in Aspergillus sojae. J Biosci Bioeng 120:498–503. doi:10.1016/j.jbiosc.2015.03.012
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. doi:10.1016/0003-2697(85)90442-7
Sode K, Igarashi S, Morimoto A, Yoshida H (2002) Construction of engineered water-soluble PQQ glucose dehydrogenase with improved substrate specificity. Biocatal Biotransformation 20:405–412. doi:10.1080/1024242021000058694
Swoboda BE, Massey V (1965) Purification and properties of the glucose oxidase from Aspergillus niger. J Biol Chem 240:2209–2215
Sygmund C, Klausberger M, Felice AK, Ludwig R (2011a) Reduction of quinones and phenoxy radicals by extracellular glucose dehydrogenase from Glomerella cingulata suggests a role in plant pathogenicity. Microbiology 157:3203–3212. doi:10.1099/mic.0.051904-0
Sygmund C, Staudigl P, Klausberger M, Pinotsis N, Djinović-Carugo K, Gorton L, Haltrich D, Ludwig R (2011b) Heterologous overexpression of Glomerella cingulata FAD-dependent glucose dehydrogenase in Escherichia coli and Pichia pastoris. Microb Cell Fact 10:106. doi:10.1186/1475-2859-10-106
Sygmund C, Gutmann A, Krondorfer I, Kujawa M, Glieder A, Pscheidt B, Haltrich D, Peterbauer C, Kittl R (2012) Simple and efficient expression of Agaricus meleagris pyranose dehydrogenase in Pichia pastoris. Appl Microbiol Biotechnol 94:695–704. doi:10.1007/s00253-011-3667-7
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi:10.1093/nar/22.22.4673
Tschopp JF, Sverlow G, Kosson R, Craig W, Grinna L (1987) High-level secretion of glycosylated invertase in the methylotrophic yeast, Pichia pastoris. Nat Biotechnol 5:1305–1308. doi:10.1038/nbt1287-1305
Tsujimura S, Kojima S, Kano K, Ikeda T, Sato M, Sanada H, Omura H (2006) Novel FAD-dependent glucose dehydrogenase for a dioxygen-insensitive glucose biosensor. Biosci Biotechnol Biochem 70:654–659. doi:10.1271/bbb.70.654
U.S. Food and Drug Administration (2009) FDA public health notification: potentially fatal errors with GDH-PQQ glucose monitoring technology. http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/PublicHealthNotifications/ucm176992.htm. Accessed 5 November 2015
Werner WE, Demorest DM, Stevens J, Wiktorowicz JE (1993) Size-dependent separation of proteins denatured in SDS by capillary electrophoresis using a replaceable sieving matrix. Anal Biochem 212:253–258. doi:10.1006/abio.1993.1319
Yang Y, Huang L, Wang J, Wang X, Xu Z (2014) Efficient expression, purification, and characterization of a novel FAD-dependent glucose dehydrogenase from Aspergillus terreus in Pichia pastoris. J Microbiol Biotechnol 24:1516–1524
Yang Y, Huang L, Wang J, Xu Z (2015) Expression, characterization and mutagenesis of an FAD-dependent glucose dehydrogenase from Aspergillus terreus. Enzyme Microb Technol 68:43–49. doi:10.1016/j.enzmictec.2014.10.00
Yoo EH, Lee SY (2010) Glucose biosensors: an overview of use in clinical practice. Sensors 10:4558–4576. doi:10.3390/s100504558
Yoshida H, Sakai G, Mori K, Kojima K, Kamitori S, Sode K (2015) Structural analysis of fungus-derived FAD glucose dehydrogenase. Sci Rep 5:13498. doi:10.1038/srep13498
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethical statement
This articles does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Kazumichi Ozawa and Hisanori Iwasa contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 846 kb)
Rights and permissions
About this article

Cite this article
Ozawa, K., Iwasa, H., Sasaki, N. et al. Identification and characterization of thermostable glucose dehydrogenases from thermophilic filamentous fungi. Appl Microbiol Biotechnol 101, 173–183 (2017). https://doi.org/10.1007/s00253-016-7754-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7754-7