
Abstract
d-Tagatose is a ketohexose, which presents unique properties as a low-calorie functional sweetener possessing a sweet flavor profile similar to d-sucrose and having no aftertaste. Considered a generally recognized as safe (GRAS) substance by FAO/WHO, d-tagatose can be used as an intermediate for the synthesis of other optically active compounds as well as an additive in detergent, cosmetic, and pharmaceutical formulations. This study reports important features for l-arabinose isomerase (EC 5.3.1.4) (L-AI) use in industry. We describe arabinose (araA) gene virulence analysis, gene isolation, sequencing, cloning, and heterologous overexpression of L-AI from the food-grade GRAS bacterium Enterococcus faecium DBFIQ E36 in Escherichia coli and assess biochemical properties of this recombinant enzyme. Recombinant L-AI (rL-AI) was one-step purified to homogeneity by Ni2+-agarose resin affinity chromatography and biochemical characterization revealed low identity with both thermophilic and mesophilic L-AIs but high degree of conservation in residues involved in substrate recognition. Optimal conditions for rL-AI activity were 50 °C, pH 5.5, and 0.3 mM Mn2+, exhibiting a low cofactor concentration requirement and an acidic optimum pH. Half-life at 45 °C and 50 °C were 1427 h and 11 h, respectively, and 21.5 h and 39.5 h at pH 4.5 and 5.6, respectively, showing the high stability of the enzyme in the presence of a metallic cofactor. Bioconversion yield for d-tagatose biosynthesis was 45% at 50 °C after 48 h. These properties highlight the technological potential of E. faecium rL-AI as biocatalyst for d-tagatose production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
d-Tagatose is a rare nutraceutic ketohexose, enantiomer of d-fructose in C-4, found in the gum produced by Sterculia setigera tree and present in trace amounts in powder and UHT cow milk as well as in other dairy products [1, 2]. It is employed as a low caloric value bulk sweetener (1.5 kcal/g) with a sweetness of 92% compared with d-sucrose [3, 4] and presenting no aftertaste. In addition, d-tagatose is a recognized prebiotic due to its partial absorption in the small intestine (20%), which allows most of it to be metabolized to lactate, butyrate, and other beneficial short fatty acid chains by probiotic bifido and lactic acid bacteria [5, 6]. Furthermore, it has been demonstrated that d-tagatose can be employed for the treatment of type II diabetes, obesity, and other related disorders [7] due to its low insulinemic and glycaemic index [8, 9]. It has also been described as a non-carcinogenic sugar [10], and to inhibit dental plate and biofilm formation, thereby preventing halitosis [11]. Therefore, in 2001 d-tagatose was announced as a generally recognized safe (GRAS) substance by the WHO and FAO with varying recommended daily doses depending on the food product it has been added to Bär [12] and Favara [13].
Due to its rareness and scarcity, several technological approaches for the synthesis of d-tagatose have been employed such as chemical, chemo-enzymatic, and biocatalytic [14,15,16]. Currently, enzymatic synthesis of d-tagatose is the most suitable method because it provides specificity, stereoselectivity, and high conversion yields under mild temperature and pH conditions [17,18,19].
Although several (bio)processes employing different substrates for d-tagatose production have been proposed [14, 20,21,22], none of them have proven to be cost-effective in large scale. Currently, l-arabinose isomerase (EC 5.3.1.4, L-AI) biosynthesis of d-tagatose from d-galactose (a common sugar found in cheese whey) is the most economical, environmental-friendly method for obtaining this valuable sugar. This is verified by the ever-increasing number of patents related to d-tagatose synthesis from this sugar [23].
In vivo, L-AI catalyzes the reversible aldose-ketose isomerization from l-arabinose to l-ribulose [24]. However, in vitro, it can convert d-galactose (to d-tagatose) and other substrates, depending on the selectivity of the target enzyme [25, 26]. For industrial production of the ketohexose, reaction temperatures must be as high as 50–70 °C and pH moderately acid (5.0–7.0). Such conditions reduce both browning reaction and by-product formation and simultaneously encourage aldose-ketose equilibrium and reaction rate [18]. Thus for d-tagatose biosynthesis, the use of thermophilic, thermotolerant, and occasionally mesophilic bacteria would be most suitable. In addition, L-AIs preferentially use Co2+ as cofactor, which is forbidden in food and additives [27, 28], whereas Mn2+ and Mg2+ are non-toxic cations employed preferentially by mesophyll bacteria and therefore more feasible for d-tagatose industrial production [29].
In a previous report, Enterococcus faecium DBFIQ E36 L-AI was purified to homogeneity and primary characterization (monomer molecular weight, native mass, isoelectric point) was performed [30]. Furthermore, L-AI was immobilized onto several chitosan and agarose supports and then tested for the stability and activity of these derivatives [31, 32]. Herein, we report gene virulence analysis of the thermotolerant L-AI producer Enterococcus faecium DBFIQ E36 and sequencing, primer design, cloning, and heterologous expression of L-AI in E. coli BL21 cells as well as further biochemical characterization of rL-AI such as ion metal requirement and thermal stability at different pHs and temperatures.
Materials and Methods
Chemicals and Enzymes
Restriction endonucleases, T4 DNA ligase, buffers, isopropyl-β-d-thiogalactopyranoside (IPTG), Factor Xa, and Taq DNA polymerase were obtained from NEB Inc. (Ipswich, MA, USA). The source of strains and plasmids are shown in Table 1. Primers were synthesized at Macrogen Inc. (Seoul, Korea) and are displayed in Table 2. Affinity chromatography resins were acquired from GE Healthcare (Uppsala, Sweden). All other enzymes, antibiotics (Carbenicillin and Ampicillin), chemicals, and reagents were of analytical grade and were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated.
Bacterial Strains and Culture Conditions
Enterococcus faecium DBFIQ E36 isolated from raw cow milk was chosen as the l-arabinose isomerase producer bacterium [33]. E. faecium strain was kept at − 80 °C in MRS (Difco Laboratories, Detroit, MI, USA) supplemented with 15% (v/v) glycerol [32].
Genomic DNA Extraction from E. faecium DBFIQ E36
For genomic DNA extraction, cells were harvested by centrifugation at 12,000×g for 2 min at room temperature from 5 ml of an overnight MRS liquid culture grown in a shaker at 37 °C. Pellets were resuspended in 200 µl 10 mM TRIS (hydroxymethyl aminomethane)-1 mM EDTA buffer (TE, pH 8.0) with the addition of 10 µl of 50 mg/ml lysozyme solution prepared in sterile Milli-Q water. The suspension was incubated in a water bath at 37 °C for 30 min. Next, 30 µl of 10% (w/v) SDS and 20 µl of 20 mg/ml proteinase K solution were added to lysozyme-treated suspension and incubated for 1 h at 65 °C with periodical mechanical stirring. Next, DNA extraction protocol was continued according to Warner’s report [34]. Quality of DNA was assessed both through 0.6% (w/v) agarose gel electrophoresis (Bio-Rad Laboratories, Hercules, USA) and Nanodrop™ UV–Vis spectrophotometer model ND-1000 (Thermo Fisher Scientific Inc., Waltham, USA).
Virulence Analysis of Enterococcus faecium DBFIQ E36 Strain
To confirm GRAS state of the enterococcus strain, several tests were done. Firstly, hemolysis reaction, gelatinase assay, and vancomycin sensitivity test were performed using plate culture methods following the methodology proposed by Foulquié-Moreno et al. [35, 36]. Virulence was further assessed by PCR amplification of the strain’s pathogenicity-related genes agg, gelE, cylB, cpd, esp, ccf, and efaAfm followed by 2% agarose gel electrophoresis as described by Abouelnaga et al. [37].
Amplification and Sub-cloning of the araA Gene
The araA gene was amplified from E. faecium DBFIQ E36 genomic DNA by PCR. Primers were designed based on the araA sequence from the genomic DNA belonging to E. faecium Aus0004 and DO strains which are available in the NCBI databank under the accession numbers CP003351.1 and NC_017960.1, respectively [38, 39]. In order to clone the araA gene into pET302/NT-His (+) vector, EcoRI and AvrII (BlnI) restriction sites were included in the forward and reverse primers, respectively, and two PCRs were performed. In the first PCR, LaisoFOR and LaisoREV primers were employed for the amplification of the araA sequence using 1 µl of a 1120 ng/µl solution E. faecium DBFIQ E36 genomic DNA. Then, 1 µl of the PCR product was employed as template using the LaisoXaFOR and LaisoREV primers for obtaining the final chimeric gene sequence (Table 2). Both PCRs (50 µl) contained a final concentration of 1 µM forward and reverse primers, 0.2 mM dNTP, 1.5 mM MgCl2, 1 U of Taq DNA polymerase (NEB), 1 × PCR Taq buffer, and nuclease-free sterile water. PCR reactions were performed using a GeneAmp PCR system 2400 (Perkin Elmer, Wellesley, USA) according to the following cycling conditions: in the first PCR, an initial step was performed at 95 °C for 5 min followed by 35 cycles of 1 min at 95 °C, 2 min at 41 °C, 2 min at 68 °C and, a final step at 68 °C for 10 min. The second PCR was performed with an initial step at 95 °C for 5 min followed by 35 cycles of 1 min at 95 °C, 2 min at 53 °C, 2 min at 68 °C, and a final step at 68 °C for 10 min. PCR products were purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, USA) and sequenced at Macrogen (Seoul, Korea) using the primers LaisoXaFOR and LaisoREV. High-quality (Phred > 20) partial sequences were used to generate consensus sequences using the Codon Code Aligner 5.1 package [40,41,42]. The final L-AI sequence was deposited in the GenBank under the accession number KU221400 (ANS10198.1).
Construction of the Expression Vector and Overexpression of Recombinant L-AI in E. coli BL21 Cells
PCR products were digested with EcoRI and AvrII and cloned into pET302/NT-His (+) plasmid vector for expression and characterization of the araA gene product. Transformation was performed by electroporation of 500 ng DNA (10 µl) ligation into a 60 µl suspension of electrocompetent E. coli BL21 or TOP10F’ cells [43]. Carbenicillin-resistant colonies were selected after overnight incubation in LB agar plates supplemented with carbenicillin (100 µg/ml) at 37 °C. After DNA extraction and purification of the selected colonies using QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany), plasmids were sequenced to confirm the presence of the araA gene.
For enzyme expression, BL21 cells transformed with the vector harboring the recombinant araA gene were grown overnight in 10 ml of LB broth supplemented with 100 µg/ml ampicillin (or 50 µg/ml carbenicillin) at 37 °C. Then, 2 ml of cell culture was transferred to 250 ml of LB broth with 100 µg/ml ampicillin and incubated at 37 °C with an orbital agitation of 200 rpm until an optical density (OD) between 0.8 and 1.0 was reached. Induction of araA expression was achieved by the addition of IPTG to the final concentrations of 0.1, 0.5, or 1.0 mM and incubation at 20 °C, 200 rpm for 16 h and 37 °C, 200 rpm for 4 h. Next, cells were centrifuged at 7000×g for 15 min at 4 °C and the pellet was washed with chilled 0.1 M NaCl, after which another centrifugation was done at 7000×g for 1 h at 4 °C and the pellet suspended in 150 mM NaCl buffer (pH 8.0). Cells were disrupted through sonication (M.S.E.-Mullard Ultrasonic Disintegrator, Mullard Ltd., London, UK) for 10 min (100 W, pulse on, 4 s; pulse off, 3 s) at 4 °C and cell debris was removed by centrifugation at 12,000×g at 4 °C for 20 min. Immediately after, cell-free supernatant was sterilized by filtration using a 0.22-µm pore diameter membrane (Sartorius, Göttingen, Germany), fractionated and frozen at − 20 °C until further use. In addition, cell debris was also stored for inclusion bodies analysis.
Purification of the Recombinant L-AI
rL-AI was purified by Nickel (Ni2+) immobilized metal affinity chromatography (IMAC) employing imidazole as eluting agent. First, 5 ml of 50 mg/ml sterile crude lysate was applied onto the affinity column with a column volume of 7.5 ml and recirculated for 2 h for appropriate attachment of the tagged L-AI enzyme. The column was washed with three column volumes of 10 mM washing buffer [1 M NaCl, 10 mM imidazole, and 10% (v/v) glycerol prepared in potassium phosphate buffer (KPB)] for removal of non-bound protein and five column volumes of 20 mM washing buffer [1 M NaCl, 20 mM imidazole, and 10% (v/v) glycerol prepared in KPB] in order to eliminate weakly bound protein. rL-AI was eluted with three column volumes of elution buffer [1 M NaCl, 250 mM imidazole, and 10% (v/v) glycerol prepared in KPB]. Absorbance at 230 nm and 280 nm of 5 ml fractions were measured throughout the process. Fractions with the highest OD at 280 nm were pooled together and desalted employing PD-10 prepacked columns containing Sephadex G-25 medium (GE Healthcare). Imidazole concentration was optimized for elution of the target enzyme through SDS-PAGE analysis of all eluted fractions.
Protein concentration was determined following the Coomassie Blue G-250 protocol [44] or the bicinchoninic acid methodology [45] using Bovine Serum Albumin (Sigma-Aldrich, St. Louis, MI, USA) as standard.
Determination of purity and protein profiles were performed by SDS-PAGE at constant voltage in a Bio-Rad (model Mini-PROTEAN 3 Cell, USA) equipment and using a 12.5%T polyacrylamide gel [46]. Broad-range molecular weight standards (6.5–200 kDa, GE Healthcare) were used for molecular weight estimation.
Sequence Alignment and Homology Modeling
Multiple alignments of the L-AI amino acid sequences from various reported mesophilic bacteria were performed using Clustal X and Mega6 softwares [47, 48]. Strains reported included Enterococcus faecium DBFIQ E36, Lactobacillus fermentum IFO3956, Lactobacillus sakei subsp. sakei 23K, Lactobacillus plantarum ssp. plantarum NC8, Pediococcus pentosaceus ATCC 25745, Bacillus subtilis, Parageobacillus thermoglucosidans, Bifidobacterium longum NRRLB-41409, Shewanella sp. ANA-3, and Escherichia coli str. K-12 substr. W3110. The L-AI sequence was submitted to the SWISS-MODEL server (automated comparative protein modeling sever) (http://swissmodel.expasy.org/) for comparative structural modeling [49]. The crystal structure of the mesophilic L-AI E. coli K-12 (UniProtKB P08202) presented 49.4% identity and was therefore used as a template for homology modeling.
Enzymatic Activity Assays
The enzyme assay reaction was prepared by mixing 500 µl of 1000 mM d-galactose (500 mM final concentration) and 30 µl of 10 mM MnCl2 (0.3 mM final concentration) and allowed to reach 50 °C. Next, 470 µl of a 31.9 µM rL-AI enzyme solution was added to the reaction mix to bring rL-AI to a final concentration of 15 µM. Enzyme addition immediately started the reaction, which was allowed to run for 20 min. The reaction was stopped by cooling on wet ice. All solutions used in the assay were prepared with 50 mM (pH 5.6) acetate buffer [50]. Enzymatic activity was quantified from the amount of d-tagatose produced according to Dische and Borenfreund [51]. One unit of enzymatic activity was defined as the enzyme quantity required to generate 1 µmol of ketosugar per minute under the assay conditions.
Effect of Metal Ions on Enzymatic Activity
The effect of the addition of divalent cations on L-AI enzymatic activity was evaluated at 1 mM concentration for each metal ion. For the assay, 15 ml of enzyme solution (90 µM) was dialyzed 2-times for 24 h each against 4 L of 50 mM (pH 7.0) phosphate buffer containing 10 mM EDTA at 4 °C for 48 h employing a 10 kDa MWCO CelluSep™ regenerated cellulose membrane (Membrane Filtration Products Inc., Austin, USA). Remaining metal content in dialyzed rL-AI solutions was assessed through inductively coupled plasma (ICP) mass spectrometry following the conditions proposed by Salonen et al. [52]. Next, free-cation enzyme samples were dialyzed for 24 h in the same conditions as described above against the same volume of 50 mM (pH 5.6) acetate buffer (enzyme/solvent relation: 1/250) in order to eliminate the EDTA. Next, the enzyme preparation was pre-incubated for 30 min in the presence of 1 mM MgCl2, NH4Cl, MnCl2, CoCl2, ZnCl2, CuCl2, BaCl2, CaCl2, FeCl2, FeCl3, PdCl2, SrCl2, SnCl2, and mixtures (1:1, 1:3, and 3:1 Mn2+:Co2+ 1 mM final concentration). Enzymatic assay and the measurement of ketoses produced were done under standard conditions as mentioned above. Finally, cation-added enzyme activity was compared with non-treated enzyme activity [53, 54]. On the other hand, the evaluation of the optimal manganese concentration experiment was performed at a 15 µM final concentration of free-cation enzyme preparation with different MnCl2 solutions ranging from 0 to 30 mM and performing enzymatic activity as described previously.
Optimum Temperature and pH
Analysis of temperature over enzyme activity was evaluated through the incubation of 15 µM final concentration purified enzyme at different temperatures ranging from 25 to 70 °C in 50 mM (pH 5.5) acetate buffer for 20 min. For the evaluation of enzyme optimum pH, the same enzyme preparation was incubated at different pH values employing: 50 mM sodium citrate buffer (pH 3.0 to 4.0), 50 mM sodium acetate buffer (pH 4.5 to 5.5), 50 mM sodium phosphate buffer (pH 6.0 to 7.5), and 50 mM TRIS–HCl (pH 8.0 to 9.0) and incubated at 50 °C for 20 min. Enzymatic activity was expressed as relative activity in relation with maximum activity achieved in the assays [55].
Temperature and pH Stability Profiles
Purified enzyme preparation (15 µM final concentration) was assessed for thermal stability at 45, 50, and 55 °C in the absence and presence of 0.3 mM Mn2+ cation and at four different pHs (4.5, 5.6, 7.0, and 8.0). For each temperature, cation presence, and pH condition tested, samples were collected at different time points during an 8-h time-frame and placed on cold ice bath (0 °C) until determination of enzyme activity. Soluble enzyme inactivation kinetics was evaluated through measuring residual activity at 50 °C during 20 min and proceeding as described above. Relative activity curves as a function of time were plotted employing Origin v7.5 software (OriginLab Corporation, Northampton, USA) and Arrhenius thermal inactivation constants and half-life were estimated as a first-order kinetics [56, 57].
Kinetic Parameters
The kinetic parameters of L-AI (15 µM final concentration) were assessed in 50 mM sodium acetate buffer (pH 5.6) with 0.3 mM MnCl2 and 10–800 mM d-galactose in a final volume of 1 ml. Reaction mixtures were incubated for 20 min at 50 °C. Initial velocities achieved were fitted following a non-linear curve fit model using Origin v7.5 software and kinetic parameters (Vmax, Km, kcat, and Kcat/Km) were estimated.
Bioconversion Assays
The evaluation of bioconversion yield of d-galactose to d-tagatose was performed in a volume of 250 ml containing 50 mM (pH 5.6) acetate buffer, 0.3 mM Mn2+, and 50 or 500 mM of d-galactose with the addition of rL-AI at a 15 µM final concentration. Conversion levels were measured for 48 h with samples being removed at defined intervals and stopping the reaction on ice. Quantification of d-tagatose synthesis was performed colorimetrically at 560 nm using the method proposed by Dische and Borenfreund [51]. Enzymatic activity was expressed as percentage of converted substrate and results were expressed as the mean and standard deviation. Experiments were performed in triplicate.
Results and Discussion
Cloning and Sequence Analysis of E. faecium L-AI
The 1.4 kb araA gene was amplified from E. faecium genomic DNA and cloned into pET302/NT-His (+) to yield a rL-AI sequence fused with an N-terminal His-tag and the Factor Xa recognition sequence. Sequencing analysis confirmed in frame cloning of the araA gene and revealed an open reading frame of 1425 bp, which codes for a protein of 474 amino acids with a predicted MW of 54.2 kDa and a theoretical isoelectric point (pI) of 4.88. Nucleotide sequence of E. faecium DBFIQ E36 was submitted to GenBank and accession number ANS10198.1 was assigned to it.
The amino acid sequence of E. faecium DBFIQ E36 shared 59.5%, 58.9%, 58.9%, and 53.6% identity with lactic acid bacteria such as L. sakei 23K, L. plantarum NC8, P. pentosaceus ATCC 25745, and L. fermentum IFO 3956, respectively (Fig. 1). In addition, non-lactic acid bacteria revealed identity percentages of 55.2%, 55.2%, 48.9%, 43.9%, and 43.6% for B. subtilis, P. thermoglucosidans, E. coli str. K-12, B. longum, and Shewanella sp. ANA-3, respectively. From these results, it can be seen that Bacillus strains show higher identity percentages than some lactic acid bacteria, such as L. fermentum. Multiple sequence alignment of L-AI amino acid sequences from different microorganisms showed low identity with the L-AI of E. faecium DBFIQ E36, confirming that the gene isolated here codes for a unique member of the l-arabinose isomerase superfamily [58].

Analysis for the presence of virulence genes in E. faecium DBFIQ E36 strain complete PCR. MWM molecular weight marker, efaAfm cell-wall adhesin EfaA for E. faecium, cylB β-hemolysin or cytolysin, agg aggregation protein, ccf sex pheromone for E. faecium, gelE enzyme gelatinase, esp Enterococcus surface protein
As expected, identity of the L-AI isolated here was highest with other Enterococcus faecium strains (99.4%). The amino acid sequence of E. faecium DBFIQ E36 differed only at positions R91, D227, and V446 relative to the most phylogenetically related strains E. faecium Aus0004, E. faecium UW7606 × 64/3 , and E. faecium DO (Fig. S3).
Additionally, we performed homology modeling by comparing the primary amino acid sequence of E. faecium L-AI with the crystal structure of E. coli L-AI [59] and identified a high degree of structural conservation between both enzymes. Residues E306, E331, H348, and H447, which are essential for enzyme activity in E. faecium L-AI, corresponded to residues E306, E333, H350, and H450 in E. coli L-AI, showing an evolutionary conserved catalytic site. A high degree of conservation was also identified for residues involved in substrate recognition and isomerization reaction: Q16, L18, Y19, Q125, H128, M185, F279, and Y335 were present in the same position in both E. faecium and E. coli L-AI.
A high identity between the L-AI isolated here and L-AI sequences from thermophilic and hyperthermophilic bacteria was observed. In fact, the highest amino acid identity was shared with Geobacillus stearothermophilus L-AI (57.6%, Figs. S1, S3), which may explain the thermotolerant nature of the enzyme used in the current work.
Evaluation of Virulence Activity and Genes
Enterococcus faecium DBFIQ E36 strain virulence and potential pathogenicity were evaluated through culture-dependent and culture-independent methods. Plate culture methods revealed that both α-, β-, and γ-hemolysis reaction and gelatin hydrolysis were not evidenced. Vancomycin antibiotic was employed in order to detect resistance of the enterococci strain to this glycopeptide antibiotic considered as a drug of last resort for the treatment of enterococcal infections [35]. Minimal inhibitory concentration was below 2 µg/ml which indicates this strain was sensitive to this antibiotic in agreement with Sabia et al. [60] report. Furthermore, the presence of pathogenicity-related genes was assessed (Fig. 2). With the exception of efaAfm gene, which expresses a cell-wall adhesin, a commonly found virulence trait related to E. faecium strains isolated from unfermented foods (it appeared in more than 95% of E. faecium isolates), such as cow raw milk where this strain was isolated, no other gene was detected.

Multiple sequence alignment of the amino acid sequences of L-AI from different mesophilic bacterial strains. Shewanella sp. ANA-3 (Sc_ANA-3; GenBank accession No. ABK48296.1); Escherichia coli str. K-12 substr. W3110 (Ec_AAA23463; BAB96631.2); Bifidobacterium longum NRRL B-41409 (Bl_NRRLB-41409; AFL93470.1); Lactobacillus sakei subsp. sakei 23K (Ls_23K; CAI56163.1); Pediococcus pentosaceus ATCC 25745 (Pp_ATCC25745; Q03HQ0.1); Lactobacillus plantarum subsp. plantarum NC8 (Lp_NC8; EHS84470.1), Lactobacillus fermentum IFO3956 (Lf_IFO3956; BAG27890.1); Enterococcus faecium DBFIQ E36 (Ef_DBFIQ_E36; ANS10198.1); Bacillus subtilis (Bs_ACT82395; ACT82395.1); Parageobacillus thermoglucosidans KCTC 1828 (Gt_AAQ23079; AAQ23079.1). Alignment was carried out using Clustal W. Amino acids residues shaded in red (or with an asterisk) indicate single and strongly conserved residues, while residues with a colon show conservation between groups of amino acids residues with strongly similar properties (scoring > 0.5 in the Gonnet PAM 250 matrix). A period sign displays conservation between groups of weakly similar properties (those who scored ≤ 0.5 in the Gonnet PAM 250 matrix). (Color figure online)
This virulence factor is mainly related to their high adaptability to different environments. Thus, the presence of this particular gene alone among all analyzed virulence factor genes drastically reduces their potential pathogenicity [61]. In addition, expression of these genes is regulated in a complex manner and influenced by environmental factors; therefore, pathogenicity is not solely related to the presence of these genes [62]. Hence, employment of this food-grade strain should be adequate in several biotechnological processes if continuous assessment for the presence of these virulence characters through time and different environmental conditions is ensured [37]. In order to minimize production costs and virulence risks, E. faeciuml-arabinose isomerase overexpression in E. coli was performed.
Expression and Purification of Recombinant E. faecium L-AI
The expressed rL-AI possesses 14 amino acid residues beyond those coded by the araA gene. These include a methionine, the 6 × His-tag, a valine, an asparagine, a serine , and the four amino acids corresponding to the cleaving site of Factor Xa protease. Thus, the L-AI reported herein is a 488 amino acid protein with a MW of 55.87 kDa and a pI of 5.07. rL-AI expression in E. coli BL21 was tested with IPTG concentrations of 0.1, 0.5, and 1 mM. Optimal IPTG concentration was shown to be 0.5 mM as 1 mM led to the formation of inclusion bodies and 0.1 mM was not enough to achieve robust expression. Two induction conditions were tested (20 °C for 16 h and 37 °C for 4 h). Induction at the lower temperature yielded 40% more recombinant protein in soluble form. Thus, 20 °C for 16 h was selected as the induction condition for L-AI overexpression.
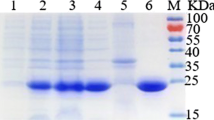
Electrophoresis analysis revealed a strong band around 55–56 kDa corresponding to rL-AI. In agreement with [58], this was the major protein present in the cell crude extract, reaching 40% of total protein. It was observed that elution with 250 mM imidazole resulted in the recovery of rL-AI with a minimal loss in the enzyme’s activity (18% loss of activity). The resulting purified product had a specific activity of 0.75 U/mg (Fig. 3 and Table S1).

SDS-PAGE protein profiles produced after E. faecium L-AI overexpression in E. coli BL21. Lane 1: broad-range SDS-PAGE molecular weight marker standards from Sigma (11–245 kDa); Lane 2: crude extract; Lane 3: Ni-Sepharose flow-through fraction; Lane 4: Ni-Sepharose washing solution fraction; Lane 5: 250 mM imidazole elution fraction
Analysis of Optimum Temperature and pH
The rL-AI isolated here presented the same optimum pH and temperature as shown by de Sousa et al. [31]. The observed temperature curve (Fig. S5A) is typical of enzymes from thermotolerant mesophilic organisms [50, 63,64,65]. In addition, analysis of activity as function of pH revealed the optimal range for E. faecium DBFIQ E36 rL-AI to be 4.5-6.0 (Fig. S5B), indicating the enzyme’s acid-tolerant trait as previously reported for other L-AI enzymes [63, 29]. This is a desired characteristic for L-AI industrial application because activity at low pH avoids non-enzymatic browning and by-product formation while promoting simultaneous lactose hydrolysis followed by d-galactose and d-glucose isomerization [66]. Thus, the isolation of acid-tolerant organisms, such as lactic acid bacteria, arises as a potential source for novel L-AI with industrial applications.
Effect of Different Metal Ions in Enzyme Activity
The effect of different cations at 1 mM was tested in order to evaluate their enhancing or inhibitory effect on enzymatic activity. At 1 mM, EDTA significantly decreased enzymatic activity: L-AI presented only 20% activity relative to non-treated enzyme at optimum temperature (Fig. 4a). These findings are in line with reports for G. stearothermophilus [67] and L. sakei [29] L-AIs as well as A. acidocaldarius [68], for which total inactivation was observed. These results also confirm the high dependency of thermophilic L-AIs on divalent metal ions for both stability and activity in comparison with mesophilic L-AIs [69]. Furthermore, the treatment of purified E. faecium L-AI with chelating agents followed by dialysis was seen to be an effective technique for metal removal as demonstrated by ICP-MS analysis where a concentration of 0.12 ± 0.04 µM of Mn2+ cation was detected in the dialyzed enzyme solution. This value was approximately 2500 times lower than optimum metal ion concentration.

Effect of divalent metal ions at 1 mM concentration at 50 °C and pH 5.6. a. Non-supplemented (NS) indicates enzyme activity for the purified EDTA-treated and dialyzed enzyme with no addition of any divalent cation. NS activity was defined as 100% of relative activity. b Effect of various concentrations of Mn2+ cation on EDTA-treated and purified enzyme. Results were stated as relative activity and 100% was defined as the cation concentration where maximum activity was reached. Error bars represent standard deviation from three independent experiments
The best cation for E. faecium DBFIQ E36 L-AI activity was Mn2+ followed by Co2+, which led to a 17-fold and 13-fold increase in enzyme activity, respectively, relative to non-supplemented L-AI. Mn2+ has been described as the best L-AI cofactor in mesophilic bacteria, whereas Co2+ is favored by thermophilic and hyperthermophilic bacteria [27, 70, 71]. We also observed a synergistic effect of both cations, with a Mn2+/Co2+ ratio of 3/1 leading to a 27-fold increase in activity (Fig. 4a). As a matter of fact, both cations have been previously described as the most common cofactors for L-AI [18]. However, due to its toxicity, Co2+ is not recommended for human consumption. Therefore, the isolation of isomerases with low metal requirement is highly desired in order to reduce costs associated with the elimination of metal ions during industrial d-tagatose enzymatic synthesis [72, 73].
Addition of Ba2+ completely inhibited L-AI enzymatic activity. Our findings are in line with the results from Cheng et al. [74] but in contrast to Xu et al. [58] that reported Ba2+ to slightly increase L-AI enzymatic activity [58].
Mg2+ and Ca2+ also had an inhibitory effect on enzyme activity, whereas Sr2+, Sn2+, Zn2+, and Cu2+ did not affect L-AI activity. Despite Mg2+ having been reported as an activating cofactor in different studies [64, 29], our results are in line with Staudigl et al. [73], who reported a slightly inhibitory effect for this cation. Salonen et al. [52] also reported an enhancing effect for Mg2+ at concentrations lower than 1 mM in B. longum L-AI expressed in L. lactis subsp. lactis. However, enzymatic activity decreased steadily with increasing Mg2+ concentrations to up to 10 mM. It is noteworthy that, in contrast to most L-AI previously characterized, Salonen et al. [52] observed an inhibitory effect of Mn2+ and Co2+. They also reported B. longum L-AI to be strongly inhibited by Fe2+ and Fe3+. In our hands, Fe2+ did not affect enzymatic activity, whereas Fe3+ slightly increased it (1.1-fold) (data not shown). These results partially agree with the report from Patel et al. [75], which show a slight activation of L-AI in the presence of both Fe2+ and Fe3+.
Enzymatic activity remained virtually unaltered in the presence of Cu2+, a cation generally deemed as inhibitory. Addition of Zn2+ modestly increased enzymatic activity. This latter observation is in line with the report from Xu et al. [58] but contrasts with those from Yamanaka [76], Yoon et al. [77], and Seo [78], which demonstrated an inhibitor effect of Zn2+.
We also assessed the effect of NH4+ on L-AI activity and observed no alteration. In general, it is observed that the bigger the atomic weight of a cation, the stronger its inhibitory effect on the activity of L-AI. As a matter of fact, all cations with an atomic weight above that of Co2+ are inhibitory. Those with a molecular weight around Co2+, such as iron, can be either inhibitory or enhancing, whereas cations with a lower molecular weight, such as Mn2+ and Mg2+, are likely to be activators of L-AI activity. Our results show that the rL-AI described here has a low metal concentration requirement to be fully active and a broad range of several divalent metal ions acceptability, where activity is not altered, which may be useful for the different industrial applications reported for d-tagatose production.
Optimum Mn2+ Concentration
L-AI is a metalloprotein and requires millimolar concentrations of a divalent cation for its stability and optimal activity. L-AIs from most mesophilic bacteria require Mn2+ to be fully active, and therefore the optimum manganese concentration for enzyme activity of rL-AI from E. faecium was assessed. Although Co2+ and Mn2+ presented a synergistic effect relative to Mn2+ alone, the addition of Co2+ cation to any food product is strictly forbidden due to its toxicity at high concentrations and its occurrence in food products consumed by humans, such as meat and vegetables [79, 63]. Thus, only Mn2+ was assessed for its optimum concentration for maximum L-AI activity.
Our results revealed that 0.3 mM Mn2+ is the optimal concentration for L-AI activity (Fig. 4b). Mn2+ had an enhancing effect on enzymatic activity at concentrations below 0.3 mM, which stabilized at 0.3–5 mM. At higher concentrations, a decrease in enzyme activity was observed, probably due to competitive inhibition with the substrate (Fig. 4b). A concentration of 0.3 mM Mn2+ for optimal enzymatic activity is one of the lowest reported to date for L-AIs, especially those from food-grade and other mesophilic bacteria (Table 3). This requirement is only comparable to that seen in L-AIs isolated from (A) cellulolyticus ATCC 43068, T. saccharolyticum NTOU1, and (B) stearothermophilus US100, which require a mix of Mn2+ plus Co2+ at concentrations of 1 mM plus 0.5 mM, 0.1 mM plus 0.05 mM, and 1 mM plus 0.2 mM, respectively [53, 74, 79]. To date, only the L-AI of B. stearothermophilus US100, B. halodurans and Arthrobacter sp. 22c are known not to have any metal requirement for enzymatic activity [28, 54, 72].
Effect of Temperature, pH, and Cofactor Presence on L-AI Stability
We performed a thermostability assay on rL-AI and determined that in the absence of Mn2+ L-AI becomes rapidly inactive at temperatures of 50 °C and 55 °C (Fig. 5a). Thermal stability was significantly increased in the presence of Mn2+, with residual activity at 45 °C remaining above 50% for at least 60 days (Table S2).

Thermal and pH stability profiles of EDTA-treated E. faecium L-AI with no addition of cofactor (a, c) and in the presence of 0.3 mM of Mn2+ (b, d). Initial activity was set as 100% and residual activity was measured as time progressed. Error bars represent standard deviation from three independent experiments
rL-AI exhibited activity in the absence of Mn2+ at temperatures below 30 °C (data not shown), confirming that the cofactor is not required for the enzymatic activity per se [29]. However, given that the velocity of the isomerization reaction increases at higher temperatures, the cofactor becomes essential for the maintenance of L-AI thermal stability.
A similar effect, albeit less pronounced, was also observed when L-AI was exposed to different pHs (Fig. 5c). In the presence of cofactor, L-AI was extremely stable in an acid milieu (Fig. 5d). Thus, E. faecium L-AI could be easily coupled to d-lactose hydrolysis reaction with β-galactosidase as demonstrated by Torres and Batista-Viera [66].
rL-AI half-life (t1/2) at different temperatures, pHs, and cofactor conditions was calculated as first-order inactivation kinetics (Arrhenius model of denaturation) [57] (Table S2). E. faecium L-AI half-life was of 11 h and 1427 h at 50 °C and 45 °C, respectively (Fig. 5b). These findings are of great technological relevance due to the fact that a very long t1/2 was observed at L-AI optimum temperature. Such t1/2 are only comparable to those of the mesophilic bacteria L. sakei 23K, S. flexneri, and B. longum (Table 3). The highest enzyme stability was observed at pH 5.6, 50 °C: t1/2 of 39.47 h and 10.78 h in the presence and absence of cofactor, respectively. These data show that the presence of Mn2+ promotes greater stability of L-AI at different pHs but the effect is not as relevant as it is for different temperatures.
Bioconversion Yield
Conversion assay showed maximum isomerization is achieved after 48-h incubation (Fig. 6). The evolution at two different d-galactose concentrations allowed reaching different conversion yields where a tenfold increase in substrate concentration caused rise in conversion from 30 to 45%. This result demonstrates that the isomerization reaction is substrate-dependent given that the equilibrium was favored towards d-tagatose synthesis when substrate concentration was increased, as described by Le Chatellier’s principle.

Bioconversion yield of d-tagatose from d-galactose by recombinant L-AI from E. faecium DBFIQ E36. d-tagatose synthesis was measured employing the cysteine-carbazole-sulfuric acid methodology [51]. Enzyme reactions were carried out in 50 mM sodium acetate buffer (pH 5.6) with the addition of 0.3 mM Mn2+ at a temperature of 50 °C
The bioconversion yield reported here is one of the highest described so far for mesophilic bacteria (Table 3), being surpassed only by L. fermentum [58] and P. pentosaceus PC-5 [63] L-AIs. Thus, the bioconversion yield of E. faecium DBFIQ E36 rL-AI is outstanding among L-AIs, including those from thermophilic bacteria. Nevertheless, the highest conversion yields are still found among thermophile and hyperthermophile organisms due to the fact that isomerization reaction equilibrium is favored by higher temperatures.
With respect to the biochemical properties of non-recombinant enzyme from E. faecium DBFIQ E36 L-AI characterized by Torres and Batista-Viera [66], slight differences were observed as shown in Table 3 (optimum pH of 7.0, optimum cofactor concentration of 10 mM, and half-life time of 360 min at 50 °C). Furthermore, Km and Vmax values were 34 mM and 0.081 U/mg, respectively, and bioconversion yield of d-galactose to d-tagatose was 22% after 6 h at 50 °C.
In addition, de Sousa et al. [31] heterologously expressed two different tagged forms of recombinant L-AI from E. faecium DBFIQ E36 in E. coli. C-terminal and N-terminal His-tag L-AI forms presented bioconversion yields of 11% and 26%, respectively. In contrast to Torres and Batista-Viera [66], N-terminal His-tag L-AI showed Km and Vmax values of 252 mM and 0.092 U/mg, optimum temperature, and pH of 50 °C and pH 5.6, respectively.
In a similar fashion, we expressed a N-terminal His-tag L-AI from E. faecium DBFIQ E36 in E. coli obtaining Km and Vmax values of 225 mM and 0.41 U/mg, respectively. Furthermore, we determined kcat of 151 min−1 and catalytic efficiency of 0.68 min−1 mM−1. These values are in line with L-AIs from other mesophilic bacteria [73, 82, 83]. Our enzyme presented increased bioconversion to d-tagatose (45% at 50 °C for 48 h), a lower cofactor concentration requirement (0.3 mM Mn2+) and better thermal stability (660 min at 50 °C and pH 5.6) relative to the same enzyme expressed by de Sousa et al. [31]. Nevertheless, optimum pH, temperature, and kinetic parameters did not show any modifications. We believe the 14-residue N-terminal fragment (approximately 1.7 kDa), which includes a 6 × His-tag, confers a structural advantage to our enzyme relative to the rL-AI with a 7-residue fragment, which also has the 6 × His-tag, as reported by Sousa et al. [31].
Currently, several studies are being performed to shed light into the structure and improvement of the properties of recombinant E. faecium L-AI using different plasmids, tags, and mutagenesis strategies in order to make it industrially feasible.
Conclusions
In this study, we report isolation of the araA gene from E. faecium DBFIQ E36, its cloning, and overexpression in E. coli BL21. Characterization performed on our rL-AI revealed biochemical properties of great technological interest such as high enzymatic activity at low pH, low cofactor requirement for maximum activity, excellent bioconversion yield (45% at 50 °C), and a long half-life at temperatures of 45 °C, 50 °C, and pH 4.5, 5.6, and 6.0. Although a metallic cofactor was not essential for L-AI activity, it was proven to be relevant for its thermal stability, tolerance at low pH, and maximal activity.
These results demonstrate stronger technological potential of L-AI from the mesophilic thermotolerant bacterium E. faecium DBFIQ E36 for d-galactose to d-tagatose bioconversion relative to the previously reported L-AIs. In addition, analysis for virulence traits in E. faecium revealed that this strain should be considered as a food-grade GRAS microorganism with a potential technological applicability and use in the food industry.
References
Troyano, E., Villamiel, M., Olano, A., Sanz, J., & Martínez-Castro, I. (1996). Monosaccharides and Myo-inositol in commercial milks. Journal of Agricultural and Food Chemistry, 44, 815–817.
Mendoza, M. R., Olano, A., & Villamiel, M. (2005). Chemical indicators of heat treatment in fortified and special milks. Journal of Agricultural and Food Chemistry, 53, 2995–2999.
Bertelsen, H., Jensen, B. B., Buemann, B.. d-Tagatose (1999). A novel low-caloric bulk sweetener with prebiotic properties. World Review of Nutrition and Dietetics, 85, 98–109.
Patra, F., Tomar, S. K., & Arora, S. (2009). Technological and functional applications of low-calorie sweeteners from lactic acid bacteria. Journal of Food Science, 74, 16–23.
Bertelsen, H., Andersen, H., & Tvede, M. (2001). Fermentation of d-tagatose by human intestinal bacteria and dairy lactic acid bacteria. Microbial Ecology in Health and Disease, 3, 87–95.
Mortensen, P. B. 1999. In vitro fermentation of tagatose to short-chain fatty acids (SCFA) and D + l-lactate in human faecal homogenates, internal report.
Moore, M. C. (2006). Drug evaluation: Tagatose in the treatment of Type 2 diabetes and obesity. Current Opinion in Investigational Drugs, 7, 924–935.
Livesey, G. (2003). Health potential of polyols as sugar replacers, with emphasis on low glycaemic properties. Nutrition Research Reviews, 16, 163–191.
Lu, Y., Levin, G. V., & Donner, T. W. (2008). Tagatose, a new antidiabetic and obesity control drug. Diabetes, Obesity and Metabolism, 10, 109–134.
Levin, G. V. (2002). Tagatose, the new GRAS sweetener and health product. Journal of Medicinal Food, 5, 23–36.
Wong, D. (2000). Sweetener determined safe in drugs, mouthwashes, and toothpastes. Dentistry Today, 19, 34–35.
Bär, A. (2004). d-tagatose, Dossier prepared and submitted on behalf of Arla food ingredients, Amba, Viby, Denmark, for evaluation pursuant to EU novel foods regulation (EC) 258/97 by the UK advisory committee on novel foods and processes, bioresco, food scientific and regulatory services.
Favara, A. F. & Federal Drug Administration (2003). Food labelling: Health claims; d-tagatose and dental caries. Final rule. Federal Register, 68, 39831–39833.
Beadle, J. R., Saunder, J. P., & Wajada, T. J. (1992). Process for manufacturing tagatose. US Patent 5.078.796.
Freimund, S., Huwig, A., Giffhorn, F., & Köpper, S. (1996). Convenient chemo-enzymatic synthesis of d-tagatose. Journal of Carbohydrate Chemistry, 15, 115–120.
Yun, M., Moon, H. R., Kim, H. O., Choi, W. J., Kim, Y. C., Park, C. S., & Jeong, L. S. (2005). A highly efficient synthesis of unnatural l-sugars from d-ribose. Tetrahedron Letters, 46, 5903–5905.
Kim, H. J., Kang, S. Y., Park, J. J., & Kim, P. (2011). Novel activity of UDP-galactose-4-epimerase for free monosaccharide and activity improvement by active site-saturation mutagenesis. Applied Biochemistry and Biotechnology, 163, 444–451.
Oh, D. K. (2007). Tagatose: Properties, applications, and biotechnological processes. Applied Microbiology and Biotechnology, 76, 1–8.
Yeom, S. J., Kim, N. H., Park, C. S., & Oh, D. K. (2009). l-ribose production from l-arabinose by using purified l-arabinose Isomerase and mannose-6-phosphate isomerase from Geobacillus thermodenitrificans. Applied and Environmental Microbiology, 75, 6941–6943.
Hugenholtz, J., & Smid, E. J. (2002). Nutraceutical production with food-grade microorganisms. Current Opinion in Biotechnology, 13, 497–507.
Kim, H. J., Kim, J. H., Oh, H. J., & Oh, D. K. (2006). Characterization of a mutated Geobacillus stearothermophilus l-arabinose isomerase that increases the production rate of d-tagatose. Journal of Applied Microbiology, 101, 213–221.
Yoshida, H., Yamada, M., Nishitani, T., Takada, G., Izumori, K., & Kamitori, S. (2007). Purification, crystallization and preliminary X-ray diffraction studies of d-tagatose 3-epimerase from Pseudomonas cichorii. Acta Crystallographica Section F, 63, 123–125.
Boudebbouze, S., Maguin, E., & Rhimi, M. (2011). Bacterial l-arabinose isomerases: Industrial application for d-tagatose production. Recent Patents on DNA and Gene Sequences, 5, 194–201.
Kim, P. (2004). Current studies on biological tagatose production using l-arabinose isomerase: A review and future perspective. Applied Microbiology and Biotechnology, 65, 243–249.
Cheetham, P. S. J., & Wootton, A. N. (1993). Bioconversion of d-galactose into d-tagatose. Enzyme and Microbial Technology, 15, 105–108.
Xu, Z., Li, S., Feng, X., Liang, J., & Xu, H. (2014). l-arabinose isomerase and its use for biotechnological production of rare sugars. Applied Microbiology and Biotechnology, 98, 8869–8878.
Kim, B. C., Lee, Y. H., Lee, H. S., Lee, D. W., Choe, E. A., & Pyun, Y. R. (2002). Cloning, expression and characterization of l-arabinose isomerase from Thermotoga neapolitana: Bioconversion of d-galactose to d-tagatose using the enzyme. FEMS Microbiology Letters, 212, 121–126.
Lee, D. W., Choe, E. A., Kim, S. B., Eom, S. H., Hong, Y. H., Lee, S. J., Lee, H. S., Lee, D. Y., & Pyun, Y. R. (2005). Distinct metal dependence for catalytic and structural functions in the l-arabinose isomerase from the mesophilic Bacillus halodurans and the thermophilic Geobacillus stearothermophilus. Archives of Biochemistry and Biophysics, 434, 333–343.
Rhimi, M., Ilhammami, R., Bajic, G., Boudebbouze, S., Maguin, E., Haser, R., & Aghajari, N. (2010). The acid tolerant l-arabinose isomerase from the food grade Lactobacillus sakei 23K is an attractive d-tagatose producer. Bioresource Technology, 101, 9171–9177.
Torres, P. R., Manzo, R. M., Rubiolo, A. C., Batista-Viera, F. D., & Mammarella, E. J. (2014). Purification of an l-arabinose isomerase from Enterococcus faecium DBFIQ E36 employing a biospecific affinity strategy. Journal of Molecular Catalysis B: Enzymatic, 102, 99–105.
de Sousa, M., Manzo, R. M., García, J. L., Mammarella, E. J., Gonçalves, L. R. B., & Pessela, B. C. J. (2017). Engineering the l-arabinose isomerase from Enterococcus faecium for d-tagatose synthesis. Molecules, 22, 2164.
Manzo, R. M., Sousa, M., Fenoglio, C. L., Gonçalves, L. R. B., & Mammarella, E. J. (2015). Chemical improvement of chitosan-modified beads for the immobilization of Enterococcus faecium DBFIQ E36 l-arabinose isomerase through multipoint covalent attachment approach. Journal of Industrial Microbiology and Biotechnology, 42, 1325–1340.
Manzo, R. M., Simonetta, A. C., Rubiolo, A. C., & Mammarella, E. J. (2013). Screening and selection of wild strains for l-arabinose isomerase production. Brazilian Journal of Chemical Engineering, 30, 711–720.
Warner, S. A. J. (1996). Genomic DNA isolation and lambda library construction. In: G.D. Foster and D. Twell (ed) Plant gene isolation (pp. 56–58) Hoboken: Wiley.
Foulquié Moreno, M. R., Callewaert, R., Devreese, B., Van Beeumen, J., & De Vuyst, L. (2003). Isolation and biochemical characterisation of enterocins produced by enterococci from different sources. Journal of Applied Microbiology, 94, 214–229.
Manzo, R. M., Cardoso, M. D. L. M., Tonarelli, G. G., & Simonetta, A. C. (2016). Purification of two bacteriocins produced by Enterococcus faecalis DBFIQ E24 strain isolated from raw bovine milk. International Journal of Dairy Technology, 69, 282–293.
Abouelnaga, M., Lamas, A., Quintela-Baluja, M., Osman, M., Miranda, J. M., Cepeda, A., & Franco, C. M. (2016). Evaluation of the extent of spreading of virulence factors and antibiotic resistance in Enterococci isolated from fermented and unfermented foods. Annals of Microbiology, 66, 577–585.
Lam, M. M. C., Seemann, T., Bulach, D. M., Gladman, S. L., Chen, H., Haring, V., Moore, R. J., Ballard, S., Grayson, M. L., Johnson, P. D. R., Howden, B. P., & Stinear, T. P. (2012). Comparative analysis of the first complete Enterococcus faecium genome. Journal of Bacteriology, 194, 2334–2341.
Qin, X., Galloway-Peña, J. R., Sillanpaa, J., Roh, J. H., Nallapareddy, S. R., Chowdhury, S., Bourgogne, A., Choudhury, T., Muzny, D. M., Buhay, C. J., Ding, Y., Dugan-Rocha, S., Liu, W., Kovar, C., Sodergren, E., Highlander, S., Petrosino, J. F., Worley, K. C., Gibbs, R. A., Weinstock, G. M., & Murray, B. E. (2012). Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiology, 12, 135–155.
Ewing, B., & Green, P. (1998). Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Research, 8, 186–194.
Ewing, B., Miller, L., Wendl, M. C., & Green, P. (1998). Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Research, 8, 175–185.
Gordon, D., Abaijan, C., & Green, P. (1998). Consed: A graphical tool for sequence finishing. Genome Research, 8, 195–202.
Lessard, J. C. (2013). Transformation of E. coli via electroporation. Methods in Enzymology, 529, 321–327.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Walker, J. M. (2002). The bicinchoninic acid (BCA) assay for protein quantitation. In J. M. Walker (Ed.), The protein protocols handbook (2nd edn., pp. 11–14). Totowa: Humana Press Inc.
Laemmli, U. K. (1970). Cleavage of structural protein during the assembly of the heat of bacteriophage T4. Nature, 227, 680–685.
Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., & Higgins, D. G. (2007). Clustal W and clustal X version 2.0. Bioinformatics, 23, 2947–2948.
Schwede, T., Kopp, J., Guex, N., & Peitsch, M. C. (2003). SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Research, 31, 3381–3385.
Chouayekh, H., Bejar, W., Rhimi, M., Jelleli, K., Mseddi, M., & Bejar, S. (2007). Characterization of an l-arabinose isomerase from the Lactobacillus plantarum NC8 strain showing pronounced stability at acidic pH. FEMS Microbiology Letters, 277, 260–267.
Dische, Z., & Borenfreund, E. (1951). A New spectrophotometric method for the detection and determination of keto sugars and trioses. Journal of Biological Chemistry, 192, 583–587.
Salonen, N., Nyyssölä, A., Salonen, K., & Turunen, O. (2012). Bifidobacterium longum l-arabinose isomerase overexpression in Lactococcus lactis, purification, and characterization. Applied Biochemistry and Biotechnology, 168, 392–405.
Rhimi, M., Aghajari, N., Juy, M., Chouayekh, H., Maguin, E., Haser, R., & Bejar, S. (2009). Rational design of Bacillus stearothermophilus US100 l-arabinose Isomerase: Potential applications for d-tagatose production. Biochimie, 91, 650–653.
Wanarska, M., & Kur, J. (2012). A method for the production of d-tagatose using a recombinant Pichia pastoris strain secreting ß-d-GALACTOSIDASE from Arthrobacter chlorophenolicus and a recombinant l-arabinose isomerase from Arthrobacter sp. 22c. Microbial Cell Factories, 11, 113–128.
Kim, H. J., & Oh, D. K. (2005). Purification and characterization of an l-arabinose isomerase from an isolated strain of Geobacillus thermodenitrificans producing d-tagatose. Journal of Biotechnology, 120, 162–173.
Torres, P., & Batista-Viera, F. (2012). Immobilization of β-galactosidase from Bacillus circulans onto Epoxy-Activated Acrylic Supports. Journal of Molecular Catalysis B: Enzymatic, 83, 57–64.
Torres, P., & Batista-Viera, F. (2012). Improved biocatalysts based on Bacillus circulans β-galactosidase immobilized onto epoxy-activated acrylic supports: Applications in whey processing. Journal of Molecular Catalysis B: Enzymatic, 74, 230–235.
Xu, Z., Qing, Y., Li, S., Feng, X., Xu, H., & Ouyang, P. (2011). A Novel l-arabinose isomerase from Lactobacillus fermentum CGMCC2921 for d-tagatose production: Gene cloning, purification and characterization. Journal of Molecular Catalysis B: Enzymatic, 70, 1–7.
Manjasetty, B. A., & Chance, M. R. (2006). Crystal structure of Escherichia coli l-arabinose isomerase (ECAI), the putative target of biological tagatose production. Journal of Molecular Biology, 360, 297–309.
Sabia, C., De Niederhäusern, S., Guerrieri, E., Messi, P., Anacarso, I., Manicardi, G., & Bondi, M. (2008). Detection of bacteriocin production and virulence traits in vancomycin-resistant enterococci of different sources. Journal of Applied Microbiology, 104, 970–979.
Semedo, T., Santos, M. A., Lopes, M. F., Marques, J. J. F., Crespo, M. T., & Tenreiro, R. (2003). Virulence factors in food, clinical and reference enterococci: A common trait in the genus? Systematic and Applied Microbiology, 26, 13–22.
Martin, B., Garriga, M., Hugas, M., & Aymerich, T. (2005). Genetic diversity and safety aspects of enterococci from slightly fermented sausages. Journal of Applied Microbiology, 98, 1177–1190.
Men, Y., Zhu, Y., Zhang, L., Kang, Z., Izumori, K., Sun, Y., & Ma, Y. (2014). Enzymatic conversion of d-galactose to d-tagatose: Cloning, overexpression and characterization of l-arabinose isomerase from Pediococcus pentosaceus PC-5. Microbiological Research, 169, 171–178.
Fan, C., Liu, K., Zhang, T., Zhou, L., Xue, D., Jiang, B., & Mu, W. (2014). Biochemical characterization of a thermostable l-arabinose isomerase from a thermoacidophilic bacterium, Alicyclobacillus hesperidum URH17-3-68. Journal of Molecular Catalysis B: Enzymatic, 102, 120–125.
Zhang, Y., Fan, Y., Hu, H., Yang, H., Luo, X., Li, Z., Zhou, H., Ma, W., Song, Y., & Zhang, T. (2017). d-Tagatose production by Lactococcus lactis NZ9000 Cells Harboring Lactobacillus plantarum l-arabinose isomerase. Indian Journal of Pharmaceutical Education and Research, 51, 288–294.
Torres, P., & Batista-Viera, F. (2017). Immobilized trienzymatic system with enhanced stabilization for the biotransformation of lactose. Molecules, 22, 284–298.
Cheng, L., Mu, W., Zhang, T., & Jiang, B. (2010). An l-arabinose isomerase from Acidothermus cellulolyticus ATCC 43068: Cloning, expression, purification, and characterization. Applied Microbiology and Biotechnology, 86, 1089–1097.
Lee, S. J., Lee, D. W., Choe, E. A., Hong, Y. H., Kim, S. B., Kim, B. C., & Pyun, Y. R. (2005). Characterization of a thermoacidophilic l-arabinose isomerase from Alicyclobacillus acidocaldarius: role of Lys-269 in pH optimum. Applied and Environmental Microbiology, 71, 7888–7896.
Choi, J. M., Lee, Y. J., Cao, T. P., Shin, S. M., Park, M. K., Lee, H. S., di Luccio, E., Kim, S. B., Lee, S. J., Lee, S. J., Lee, S. H., & Lee, D. W. (2016). Structure of the thermophilic l-arabinose isomerase from Geobacillus kaustophilus reveals metal-mediated intersubunit interactions for activity and thermostability. Archives of Biochemistry and Biophysics, 596, 51–62.
Lee, D. W., Jang, H. J., Choe, E. A., Kim, B. C., Lee, S. J., Kim, S. B., Hong, Y. H., & Pyun, Y. R. (2004). Characterization of a thermostable l-arabinose (d-galactose) isomerase from the hyperthermophilic eubacterium Thermotoga maritima. Applied and Environmental Microbiology, 70, 1397–1404.
Patrick, J. W., & Lee, N. (1968). Purification and properties of an l-arabinose isomerase from Escherichia coli. Journal of Biological Chemistry, 243, 4312–4318.
Rhimi, M., & Bejar, S. (2006). Cloning, purification and biochemical characterization of metallic-ions independent and thermoactive l-arabinose isomerase from the Bacillus stearothermophilus US100 strain. Biochimica et Biophysica Acta, 1760, 191–199.
Staudigl, P., Haltrich, D., & Peterbauer, K. (2014). l-arabinose isomerase and d-xylose isomerase from Lactobacillus reuteri: Characterization, coexpression in the food grade host Lactobacillus plantarum, and application in the conversion od d-galactose and d-glucose. Journal of Agricultural and Food Chemistry, 62, 1617–1624.
Cheng, L., Mu, W., & Jiang, B. (2010). Thermostable l-arabinose isomerase from Bacillus stearothermophilus IAM 11001 for d-tagatose production: Gene cloning, purification and characterization. Journal of the Science of Food and Agriculture, 90, 1327–1333.
Patel, M. J., Akhani, R. C., Patel, A. T., Dedania, S. R., & Patel, D. H. (2017). A single and two step isomerization process for d-tagatose and l-ribose bioproduction using l-arabinose isomerase and d-lyxose isomerase. Enzyme and Microbial Technology, 97, 27–33.
Yamanaka, K. (1975). l-Arabinose isomerase from Lactobacillus gayonii. Methods in Enzymology, 41, 458–461.
Yoon, S. H., Kim, P., & Oh, D. K. (2003). Properties of l-arabinose isomerase from Escherichia coli as biocatalyst for tagatose production. World Journal of Microbiology and Biotechnology, 19, 47–51.
Seo, M. J. (2013). Characterization of an l-arabinose isomerase from Bacillus thermoglucosidasius for d-tagatose production. Bioscience, Biotechnology, and Biochemistry, 77, 385–388.
Hung, X. G., Tseng, W. C., Liu, S. M., & Tzou, W. S. (2014). Characterization of a thermophilic l-arabinose isomerase from Thermoanaerobacterium saccharolyticum NTOU1. Biochemical Engineering Journal, 83, 121–128.
Kim, J. H., Prabhu, P., Jeya, M., Tiwari, M. K., Moon, H. J., Singh, R. K., & Lee, J. K. (2010). Characterization of an l-arabinose isomerase from Bacillus subtilis. Applied Microbiology and Biotechnology, 85, 1839–1847.
Zhan, Y., Xu, Z., Li, S., Liu, X., Xu, L., Feng, X., & Xu, H. (2014). Coexpression of β-d-galactosidase and l-arabinose isomerase in the production of d-tagatose: A functional sweetener. Journal of Agricultural and Food Chemistry, 62, 2412–2417.
Rhimi, M., Bajic, G., Ilhammami, R., Boudebbouze, S., Maguin, E., Haser, R., & Aghajari, N. (2011). The acid-tolerant l-arabinose isomerase from the mesophilic Shewanella sp. ANA-3 is highly active at low temperatures. Microbial Cell Factories, 10, 96–107.
Zhang, H., Jiang, B., & Pan, B. (2007). Purification and characterization of l-arabinose isomerase from Lactobacillus plantarum producing d-tagatose. World Journal of Microbiology and Biotechnology, 23, 641–646.
Acknowledgements
This study was partially sponsored with funds of the Projects CAI + D 2016 50420150100051 LI (Universidad Nacional del Litoral, Santa Fe, Argentina), PIP 2015–2017 No. 11220150100606CO (CONICET, Buenos Aires, Argentina), PICT-201-0249 (Agencia Nacional de Promoción Científica y Tecnológica, Buenos Aires, Argentina). Also, the authors are grateful for the financial support provided by the Brazilian research agencies CAPES, CNPq (407363/2013-0) and FUNCAP (Project Number PRONEX PR2-0101-00012.01.00/15).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manzo, R.M., Antunes, A.S.L.M., de Sousa Mendes, J. et al. Biochemical Characterization of Heat-Tolerant Recombinant l-Arabinose Isomerase from Enterococcus faecium DBFIQ E36 Strain with Feasible Applications in d-Tagatose Production. Mol Biotechnol 61, 385–399 (2019). https://doi.org/10.1007/s12033-019-00161-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00161-x