
Abstract
Inorganic phosphate is a vital constituent of cells and cell membranes, body fluids, and hard tissues. It is a major intracellular divalent anion, participates in many genetic, energy and intermediary metabolic pathways, and is important for bone health. Although we usually think of phosphate mostly in terms of its level in the serum, it is needed for many biological and structural functions of the body. Availability of adequate calcium and inorganic phosphate in the right proportions at the right place is essential for proper acquisition, biomineralization, and maintenance of mass and strength of the skeleton. The three specialized mineralized tissues, bones, teeth, and ossicles, differ from all other tissues in the human body because of their unique ability to mineralize, and the degree and process of mineralization in these tissues also differ to suit the specific functions: locomotion, chewing, and hearing, respectively. Biomineralization is a dynamic, complex, and lifelong process by which precipitations of inorganic calcium and inorganic phosphate divalent ions form biological hard tissues. Understanding the biomineralization process is important for the management of diseases caused by both defective and abnormal mineralization. Hypophosphatemia results in mineralization defects and osteomalacia, and hyperphosphatemia is implicated in abnormal excess calcification and/or ossification, but the exact mechanisms underlying these processes are not fully understood. In this review, we summarize available evidence on the role of phosphate in biomineralization. Other manuscripts in this issue of the journal deal with other relevant aspects of phosphate homeostasis, phosphate signaling and sensing, and disorders resulting from hypo- and hyperphosphatemic states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphate (Pi) is a vital constituent of cells and cell membranes as well as of body fluids and tissues, it is a major intracellular divalent anion, participates in many genetic, energy and intermediary metabolic pathways, and is important for bone health [1,2,3]. Availability of calcium (Ca) and inorganic phosphate (Pi) in the right proportions at the right place is crucial for proper acquisition, biomineralization of collagen fibrils, and maintenance of mass and strength of bones and teeth [4], the two hardest tissues in human body. Among the various micronutrients in bone, Ca and Pi are the two major components of hydroxyapatite, the crystalline mineral component of the extracellular organic matrix of bone. Phosphate metabolism, distribution, intracellular signaling, physiological and pathological perturbations causing hypo- and hyperphosphatemia, and clinical aspects of phosphate excess and depletion are dealt with in other sections in this issue of the journal. Accordingly, in this section, we will focus exclusively on the role of phosphate in biomineralization, and more specifically, in bone and other tissue mineralization. A knowledge and understanding the role of phosphate in biomineralization processes are essential to manage patients with disorders of phosphate metabolism and the associated abnormal biomineralization of bone and other tissues.
Distribution and Role of Phosphate in the Body
Although we think of phosphate mostly in terms of its level in the serum, which is maintained within a narrow range for many biological and structural functions of the body, phosphate is also an integral part of bones, tissues, cells and cell membranes. In a typical Western diet contains 1000–1600 mg of Pi/day, of which 3 mg/kg bodyweight/day enters the extracellular fluid with consequent exchange with bone as required [5, 6]. Bone contains about 99 and 80% of the total body content of Ca and Pi with a mass ratio of 2:2. Both these divalent ions exist in soluble and semi-soluble form in body fluids, cells and cell membranes, and phosphate circulates in the blood in free form, bound to protein, and as complex with Ca. A 70 kg individual has approximately 500–800 g of total body phosphate, 80% of which is in the form of hydroxyapatite crystal in bone and 20% as intracellular component. Intracellularly, phosphate is present predominantly in the organic form in nucleic acids and nucleoproteins. In contrast, both organic and inorganic forms of phosphate are present in serum, the latter is measured routinely by standard methods in clinical laboratories. At physiological pH of 7.40, phosphate exists as a mixture of ions (orthophosphates): HPO2− and H2PO4− in a ratio of 4:1. Normal physiological functions of phosphate are manifold: it is the major source of high energy phosphate bonds (ATP) required ubiquitously for cellular homeostasis-muscle contraction, electrolyte transport, etc.; it is an integral part of intracellular messenger system including cyclic adenosine and guanosine monophosphates (c-AMP; c-GMP); it is required, for the synthesis phospholipid bilayer of all cell membranes; it is involved in the formation of 2,3-diphosphoglycerol that regulates oxygen delivery to tissues, and acts as a buffer to maintain normal blood pH and plays a significant role in immune functions and coagulation cascade (also see other chapters in this issue).
The Process of Biomineralization
The three most mineralized tissues (bones, teeth, and ossicles) are specialized organs that differ from all other tissues in the human body because of their unique ability to mineralize [7, 8]. Interestingly, the degree and process of mineralization in bone, teeth, and ossicles also differ to suit the specific functions of these hard tissues: locomotion, chewing, and hearing, respectively. Biomineralization is a dynamic, complex, and lifelong process by which precipitations of inorganic Ca and Pi to form biological hard tissues such as bone, cementum, dentin, and enamel [8, 9]. Understanding the biomineralization process is important for the management of diseases caused by both defective and abnormal mineralization [10,11,12,13,14,15]. Throughout life bone and teeth, but not ossicles, are subject to processes of mineralization and demineralization on a constant and continual basis required for renovation of these hard tissues [9]. Notwithstanding the significant progress made in our understanding of bone biology several questions remain: What are the initial steps in biomineralization? How is the temporal and spatial regulation of matrix production and biomineralization integrated and accomplished? Why does biologic mineralization normally occur only in certain types of tissues but not in others? Why, and under what circumstances, does abnormal biomineralization occur? Is non-skeletal biomineralization such as that occurs in muscles, tendons, cartilage, and blood vessels dependent on ambient Pi or due to underlying tissue characteristics? Are any other cells involved in biomineralization besides the osteogenic cells? Nevertheless, considering that mineralized hard tissue formation in vivo is governed by a combination of cellularly driven processes and thermodynamics, biomineralization should be considered both biological and chemical in nature [16].
Phosphate, Ca and type-1 collagen fibrils are the major building blocks of bone tissue aided by key enzymes [17,18,19,20]. In humans, free phosphates also have control over the formation of new mineral by influencing a wide variety of cells (chondrocytes, osteoblasts, and osteocytes), signaling molecules, and enzymes [16] (see other chapters in this issue). Not surprisingly serum phosphate concentrations vary considerably with age, higher in infants and children (1.5–2.65 mM) and decline during adulthood (0.8–1.5 mM) [21]. This is most likely because of higher requirements for phosphate needed for bone growth, optimal bone mineralization, and to achieve peak bone mass in infants and growing children.
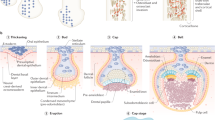
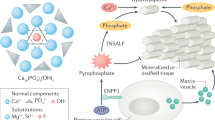
The first step in biomineralization process, at least in bone, appears to be the nucleation of Ca–Pi crystals within the matrix vesicles (Fig. 1) [4, 22, 23], followed by formation of amorphous Ca–Pi (ACP) phase with gradual transition to Ca–Pi crystal nucleation resulting in hydroxyapatite crystal [4, 24]. The Ca–Pi crystal nucleation takes place within the matrix vesicles that bud from the plasma membrane of osteogenic cells [25]. The matrix vesicles are endowed with two key enzymes, tissues non-specific alkaline phosphatase (TNSALP) and PHOSPHO-1, and Na/Pi cotransporter to generate and accumulate Pi from organic phosphate compounds (Fig. 1). TNSALP hydrolyzes inorganic pyrophosphate (PPi), adenosine triphosphate (ATP) and protein-P, whereas PHOSPHO-1 generates Pi from phosphatidyl choline (PC) and phospho-ethanol amine (PEA). In addition, Pi is actively transported into the matrix vesicles from the plasma by Na/Pi cotransporters (Fig. 1). In humans, biologic mineralization occurs by precipitation Ca and Pi in the presence of collagenous and non-collagenous proteins, polysaccharides at a pH of 7.4, and an ambient temperature of 37 °C [16]. Both H2PO4− and HPO42−, the two most important orthophosphate ions, react avidly with aqueous Ca ions resulting in the nucleation and subsequent growth of biologically relevant mineral complexes, providing a chemical basis for biomineralization [16]. Whether such a sequence occurs in tissues other than bone is less clear.

Schematic depiction of initial step in bone mineralization. ATP adenosine triphosphate; Ca calcium; HA hydroxyapatite; Na sodium; Pi inorganic phosphate; PPi inorganic pyrophosphate; PEA phosphoethanolamine; TNSLAP tissue non-specific alkaline phosphatase
Additional supporting evidence for biomineralization process comes from the classical in vivo electron microprobe studies that demonstrated a rapid initial deposition of mineral when the Ca–Pi ratio of 1.35 with a further slow increase in the deposition of minerals up to a ratio of 1.6 over a few days [26]. One possible sequence in the formation of bone mineral (or hydroxyapatite crystals) is that brushite, tricalcium phosphate, octocalcium phosphate, and hydroxyapatite are formed in succession (Fig. 2a, b). An alternate pathway is that a trimer of amorphous tricalcium brushite or three dimers of amorphous brushite are intermediate products before transitioning to hydroxyapatite crystal in bone. In either case, there is sequential addition of calcium and phosphate from the extracellular fluid to bone, but these steps are phase transformations [27], not exactly true chemical reactions. In bone, the mineral ultrastructure organization, morphology, and composition are crucial for its mechanical and biological functions. Osteocalcin and osteopontin play specific roles in the biomolecular regulation of mineral content in bone, the quality of bone mineral, and regulate bone mineral crystal size, shape, and organization. While osteocalcin predominantly regulates the physical properties of bone mineral, osteopontin plays a major role in the regulation of mineral composition [28].

a A simplified schematic depiction of sequential phase transformation of amorphous calcium phosphate to hydroxyapatite crystal formation. b Schematic depiction of different phases in bone mineralization. A Ca (red color filled circles) and Pi (lavender color filled circles) ions are co-localized in the physiological milieu. B The two divalent ions associate in a ratio of 1.5 to form Posner’s clusters. C Posner’s clusters agglomerate to form amorphous calcium phosphate (ACP) particulates. D ACP transforms into crystalline hydroxyapatite (HA) phase with a Ca:Pi ratio of 1.67. E The HA nanocrystals are then incorporated into collagen fibrils, mineralizing the organic scaffold (modified from [16]
Bone Mineralization
As discussed broadly with respect to the process of biomineralization, the basic template for bone formation is osteoid deposited by osteoblasts [29]. This unmineralized matrix, or osteoid, forms a scaffold for subsequent mineral deposition and bone formation. Osteoid is composed of organic materials, the major component of which is type-1 collagen [30]. The exact role of this phase in the infiltration of mineral precursors and the subsequent evolution of highly oriented hydroxyapatite crystals remains unknown. Several non-collagenous proteins, pH, and enzymes influence hydroxyapatite crystal formation in addition to the availability of appropriate ratio of Ca–Pi ions. There is growing evidence that orthophosphate mineral precursors are formed separately before integrating with collagen [16, 18]. The orthophosphates required for biomineralization is provided in the form of inorganic phosphate (Pi) and the optimal Ca × Pi product for proper mineralization is ~ 40. At a Ca × P product of 60 represents the saturation product above which spontaneous precipitation of ca-phosphate salt may occur in non-skeletal tissues [31].
As best as we currently understand, for proper and optimal mineralization of bone, at a minimum, requires two principal processes: synthesis of mature lamellar bone matrix by osteoblasts and exposure of the newly synthesized lamellar bone matrix to optimal calcium × phosphate product insured by the mineral homeostatic system regulated by parathyroid hormone (PTH), vitamin D, and fibroblast growth factor-23 (FGF-23; and see other chapters) [32, 33]. Any abnormality in either component will result in defective mineralization (Tables 1, 2).
Role of Phosphate in Chondrocytes and Bone Cells
Attaining the full potential of adult height and achieving maximal peak bone mass require longitudinal bone growth and maximal consolidation of mineral into bones during growth period. Therefore, it is not surprising that these two important biological processes require participation of cells, hormones and minerals, each of which are interconnected and interdependent [2, 7, 34,35,36,37,38,39]. Both Ca and Pi influence bone cells, and their differentiation and function as well as mineralization process. Since hypophosphatemia is common in all types of rickets, much of the research is focussed on the study of the effect of hypophosphatemia on chondrocytes. An orderly process of proliferation of resting chondrocytes and their differentiation into pre-hypertrophic, hypertrophic and terminally differentiated mature chondrocytes is necessary for longitudinal bone growth [40]. Adequate amounts of Pi are critical for the induction of apoptosis of mature chondrocytes in the growth plate [36, 38], without which the normal physiological chain of events fail resulting in expansion of growth plate, which is manifested as rickets and delayed growth [41, 42]. Under normal conditions, hypertrophic chondrocytes secrete angiogenic factors that promote vascular invasion [37], undergo apoptosis [36,37,38], and are replaced by mineralized bone. The chondrocyte apoptotic pathway is facilitated by phosphate-regulated activation of the caspase-9-mediated mitochondrial pathway [38]. Since rickets caused either by vitamin D deficiency or ablation of vitamin D receptor (VDR) can be rescued by adequate dietary Ca and Pi suggest that rickets is not a direct consequence of impaired VDR action, but rather, is due to the resultant hypocalcemia, hypophosphatemia, or hyperparathyroidism [38, 39].
Other osteogenic cells are also involved in bone mineralization. Crucial to the activity of osteoblasts and osteocytes in the process of matrix mineralization is the maintenance of adequate ambient Pi levels [2, 3]. Matrix vesicles arise from the cell membranes of osteoblasts and osteoblast lineage [7, 23, 25], and osteocytes produce FGF-23 to regulate phosphate homeostasis to protect osteogenic cells from hyperphosphatemia, which negatively impacts osteoblasts and practically result in cell death [2, 32].
Abnormal Biomineralization
In contrast to some understanding of the physiological normal mineralization of bone, teeth and ossicles, the pathogenesis of abnormal biomineralization is poorly understood. Nevertheless, research in the last two decades has shed light on our understanding of the non-skeletal calcifications and mineralization processes. The most extensively studied is the vascular calcifications in the context of chronic kidney disease (CKD) in which hyperphosphatemia plays a dominant role [43,44,45,46,47,48]. Several complex pathological mechanisms are implicated in the development of vascular, valvular and soft tissue calcifications, including trans-differentiation of vascular smooth muscle cells to osteo/chondrogenic phenotype [49], apoptosis of vascular smooth muscle cells [50], instability and release of extracellular vesicles loaded calcium and phosphate [51], and elastin degradation [52]. Also, deficiency of inhibitors of mineralization such as pyrophosphate contributes to abnormal calcification process [47, 49]. However, it is unclear why abnormal biomineralization outside the skeleton necessarily does not become bone.
In classical osteomalacia, deficiency of key divalent ions (calcium and phosphate), however produced, results in the accumulation of unmineralized bone matrix (or osteoid) [10, 11, 41]. In contrast, in bone disorders that resemble osteomalacia or “osteomalacia like” or sometimes referred to interchangeably as “hyperosteoidosis”, the osteoid accumulation is a consequence of disturbances outside of these two principal components. In hypophosphatasia [53], it is the enzyme deficiency, whereas in Paget disease of bone [54], fibrous dysplasia [55], fibrogenesis imperfecta ossium [56], and osteogenesis imperfecta [57], it is the abnormal bone matrix due either to abnormal collagen fibrillar arrangement or to mutations in type-1 collagen genes. In drug-induced bone disorders that are associated with prolonged treatment with etidronate [58], fluoride [59, 60], aluminum [61, 62], or iron excess [63], the mineralization defect is the result of the toxic effects of these drugs inhibiting matrix mineralization [64]. Understanding the fundamental differences in the pathogenesis of defective mineralization of bone in different disease states and conditions is critical for clinical management as many disorders that mimic osteomalacia do not respond to vitamin D therapy as the word osteomalacia might imply [41]. With a very few exceptions (adefovir, adefovir, and tenofovir-induced osteomalacia [65,66,67], and that associated with renal failure [62], the serum phosphate levels are generally normal in these various bone disorders.
Role of Phosphate in Abnormal Mineralization
Several types of calcifications, enthesopathy, and ossification occur in a variety of conditions and disorders; some are associated with hyperphosphatemia, others with hypophosphatemia, and still others without any abnormalities in calcium and phosphate homeostasis (Table 3). Calcification of tendons (calcific tendinitis) [68], cartilage (chondrocalcinosis) [69], and soft tissues (metastatic and dystrophic calcifications [70, 71], can occur in various conditions and in aging, but their pathogenesis is poorly understood. In most such instances the serum phosphate levels are normal except in patients with associated CKD [48, 49]. Chondrocalcinosis and corneal calcifications (band keratopathy) have been described both in patients with primary hyperparathyroidism with hypophosphatemia, but with significant hypercalcemia [72, 73], and in patients with uremic secondary hyperparathyroidism with hyperphosphatemia with variable concentrations of serum Ca levels [74, 75]. Calciphylaxis, an uncommon serious complication seen in patients with CKD is often, but not always, associated with hyperphosphatemia, and responds sometimes to parathyroidectomy [76]. Enthesopathy, a common complication of X-linked hypophosphatemic disorders (XLH), is associated with hypophosphatemia rather than hyperphosphatemia, and its pathogenesis remains largely elusive, but FGF-23-Klotho axis has been implicated [77, 78]. In contrast, basal ganglion calcifications, a characteristic feature of patients with all varieties of hypoparathyroidism, is associated with hyperphosphatemia and hypocalcemia [79, 80], but such intracranial calcifications have also been described in patients without the abnormalities in divalent ion mineral homeostasis [81]. More recently, therapeutic use of FGFR inhibitors to treat certain cancers is associated with hyperphosphatemia and calcinosis cutis [82]. Tumoral calcinosis is another interesting entity, first described in 1943, and occurs with or without hyperphosphatemia [83]. Even the least understood abnormal biomineralization is heterotopic ossification, the pathogenesis of which remains unknown [84].
Summary
Biomineralization is a complex and dynamic lifelong process necessary to maintain both the structural and the functional integrity of the skeleton. Inorganic phosphate is an essential nutrient needed for many genetic, energy, and intermediary metabolic pathways as well as function of the osteogenic cells. Availability of adequate calcium and inorganic phosphate in the right proportions, at the right place, and at the right time is critical for proper acquisition, biomineralization, and maintenance of mass and strength of the skeleton. Hypophosphatemia results in mineralization defects and osteomalacia, and hyperphosphatemia is implicated in abnormal excess calcification and/or ossification, but the exact mechanisms underlying these processes are not fully understood. In this review we summarize available evidence on the role of phosphate in biomineralization. Other papers in this issue of the journal deal with other relevant aspects of phosphate homeostasis, phosphate signaling and sensing, and disorders resulting from hypo- and hyperphosphatemic states.
References
Chande S, Bergwitz C (2018) Role of phosphate sensing in bone and mineral metabolism. Nat Rev Endocrinol 14(11):637–655. https://doi.org/10.1038/s41574-018-0076-3
Goretti Penido M, Alon US (2012) Phosphate homeostasis and its role in bone health. Pediatr Nephrol (Berlin, Germany) 27(11):2039–2048. https://doi.org/10.1007/s00467-012-2175-z
Michigami T, Kawai M, Yamazaki M, Ozono K (2018) Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev 98(4):2317–2348. https://doi.org/10.1152/physrev.00022.2017
Bonjour JP (2011) Calcium and phosphate: a duet of ions playing for bone health. J Am Coll Nutr 30(5 Suppl 1):438s–448s. https://doi.org/10.1080/07315724.2011.10719988
Tenenhouse HS (2007) Phosphate transport: molecular basis, regulation and pathophysiology. J Steroid Biochem Mol Biol 103(3–5):572–577. https://doi.org/10.1016/j.jsbmb.2006.12.090
Sapio L, Naviglio S (2015) Inorganic phosphate in the development and treatment of cancer: a Janus Bifrons? World J Clin Oncol 6(6):198–201. https://doi.org/10.5306/wjco.v6.i6.198
Michigami T (2019) Skeletal mineralization: mechanisms and diseases. Ann Pediatr Endocrinol Metab 24(4):213–219. https://doi.org/10.6065/apem.2019.24.4.213
Kawasaki K, Buchanan AV, Weiss KM (2009) Biomineralization in humans: making the hard choices in life. Annu Rev Genet 43:119–142. https://doi.org/10.1146/annurev-genet-102108-134242
Abou Neel EA, Aljabo A, Strange A, Ibrahim S, Coathup M, Young AM, Bozec L, Mudera V (2016) Demineralization–remineralization dynamics in teeth and bone. Int J Nanomed 11:4743–4763. https://doi.org/10.2147/ijn.S107624
Bhan A, Qiu SJ, Rao SD (2018) Bone histomorphometry in the evaluation of osteomalacia. Bone Rep 8:124–135
Basha B, Rao DS, Han ZH, Parfitt AM (2000) Osteomalacia due to vitamin D depletion: a neglected consequence of intestinal malabsorption. Am J Med 108:296–300
Minisola S, Peacock M, Fukumoto S, Cipriani C, Pepe J, Tella SH, Collins MT (2017) Tumour-induced osteomalacia. Nat Rev Dis Primers 3:17044. https://doi.org/10.1038/nrdp.2017.44
Claes KJ, Viaene L, Heye S, Meijers B, d'Haese P, Evenepoel P (2013) Sclerostin: another vascular calcification inhibitor? J Clin Endocrinol Metab 98(8):3221–3228. https://doi.org/10.1210/jc.2013-1521
Brandenburg VM, Kramann R, Koos R, Kruger T, Schurgers L, Muhlenbruch G, Hubner S, Gladziwa U, Drechsler C, Ketteler M (2013) Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: a cross-sectional study. BMC Nephrol 14:219. https://doi.org/10.1186/1471-2369-14-219
Komarova SV, Safranek L, Gopalakrishnan J, Ou MJ, McKee MD, Murshed M, Rauch F, Zuhr E (2015) Mathematical model for bone mineralization. Front Cell Dev Biol 3:51. https://doi.org/10.3389/fcell.2015.00051
Hughes EAB, Robinson TE, Bassett DB, Cox SC, Grover LM (2019) Critical and diverse roles of phosphates in human bone formation. J Mater Chem B 7(47):7460–7470. https://doi.org/10.1039/C9TB02011J
Reznikov N, Shahar R, Weiner S (2014) Bone hierarchical structure in three dimensions. Acta Biomater 10(9):3815–3826. https://doi.org/10.1016/j.actbio.2014.05.024
Dillon S, Staines KA, Millan JL, Farquharson C (2019) How to build a bone: PHOSPHO1, biomineralization, and beyond. JBMR Plus 3(7):e10202. https://doi.org/10.1002/jbm4.10202
Orimo H, Shimada T (2008) The role of tissue-nonspecific alkaline phosphatase in the phosphate-induced activation of alkaline phosphatase and mineralization in SaOS-2 human osteoblast-like cells. Mol Cell Biochem 315(1–2):51–60. https://doi.org/10.1007/s11010-008-9788-3
Halling Linder C, Ek-Rylander B, Krumpel M, Norgard M, Narisawa S, Millan JL, Andersson G, Magnusson P (2017) Bone alkaline phosphatase and tartrate-resistant acid phosphatase: potential co-regulators of bone mineralization. Calcif Tissue Int 101(1):92–101. https://doi.org/10.1007/s00223-017-0259-2
Florenzano P, Cipriani C, Roszko KL, Fukumoto S, Collins MT, Minisola S, Pepe J (2020) Approach to patients with hypophosphataemia. Lancet Diabetes Endocrinol 8(2):163–174. https://doi.org/10.1016/S2213-8587(19)30426-7
Hasegawa T (2018) Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem Cell Biol 149(4):289–304. https://doi.org/10.1007/s00418-018-1646-0
Chaudhary SC, Kuzynski M, Bottini M, Beniash E, Dokland T, Mobley CG, Yadav MC, Poliard A, Kellermann O, Millan JL, Napierala D (2016) Phosphate induces formation of matrix vesicles during odontoblast-initiated mineralization in vitro. Matrix Biol J Int Soc Matrix Biol 52–54:284–300. https://doi.org/10.1016/j.matbio.2016.02.003
Zhang J, Wang L, Putnis CV (2019) Underlying role of brushite in pathological mineralization of hydroxyapatite. J Phys Chem B 123(13):2874–2881. https://doi.org/10.1021/acs.jpcb.9b00728
Cui L, Houston DA, Farquharson C, MacRae VE (2016) Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 87:147–158. https://doi.org/10.1016/j.bone.2016.04.007
Wergedal JE, Baylink DJ (1974) Electron microprobe measurements of bone mineralization rate in vivo. Am J Physiol 226(2):345–352. https://doi.org/10.1152/ajplegacy.1974.226.2.345
Zhang X, Huang X, Ma M (2017) Role of phosphorylation of phosvitin in the phase transformation of mineralization. Int J Biol Macromol 101:712–718. https://doi.org/10.1016/j.ijbiomac.2017.03.158
Poundarik AA, Boskey A, Gundberg C, Vashishth D (2018) Biomolecular regulation, composition and nanoarchitecture of bone mineral. Sci Rep 8(1):1191. https://doi.org/10.1038/s41598-018-19253-w
Parfitt AM (1992) Human bone mineralization studied by in vivo tetracycline labeling: application to the pathophysiology of osteomalacia. In: Excerpta medica international congress, 4th international symposium on chemistry and biology of mineralized tissues, Coronado Peninsula, California, pp 465–474
Ben Shoham A, Rot C, Stern T, Krief S, Akiva A, Dadosh T, Sabany H, Lu Y, Kadler KE, Zelzer E (2016) Deposition of collagen type I onto skeletal endothelium reveals a new role for blood vessels in regulating bone morphology. Development 143(21):3933–3943. https://doi.org/10.1242/dev.139253
Velentzas C, Meindok H, Oreopoulos DG, Meema HE, Rabinovich S, Jones M, Sutton D, Rapoport A, deVeber GA (1978) Visceral calcification and the CaXP product. Adv Exp Med Biol 103:195–201. https://doi.org/10.1007/978-1-4684-7758-0_21
Erben RG, Andrukhova O (2017) FGF23-Klotho signaling axis in the kidney. Bone 100:62–68. https://doi.org/10.1016/j.bone.2016.09.010
Tatsumi S, Miyagawa A, Kaneko I, Shiozaki Y, Segawa H, Miyamoto K (2016) Regulation of renal phosphate handling: inter-organ communication in health and disease. J Bone Miner Metab 34(1):1–10. https://doi.org/10.1007/s00774-015-0705-z
Michigami T, Ozono K (2019) Roles of phosphate in skeleton. Front Endocrinol 10:180. https://doi.org/10.3389/fendo.2019.00180
Liu ES, Zalutskaya A, Chae BT, Zhu ED, Gori F, Demay MB (2014) Phosphate interacts with PTHrP to regulate endochondral bone formation. Endocrinology 155(10):3750–3756. https://doi.org/10.1210/en.2014-1315
Miedlich SU, Zalutskaya A, Zhu ED, Demay MB (2010) Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J Biol Chem 285(24):18270–18275. https://doi.org/10.1074/jbc.M109.098616
Carlevaro MF, Cermelli S, Cancedda R, Descalzi Cancedda F (2000) Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci 113(Pt 1):59–69
Sabbagh Y, Carpenter TO, Demay MB (2005) Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA 102(27):9637–9642. https://doi.org/10.1073/pnas.0502249102
Donohue MM, Demay MB (2002) Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology 143(9):3691–3694. https://doi.org/10.1210/en.2002-220454
Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423(6937):332–336. https://doi.org/10.1038/nature01657
Bhan A, Rao AD, Bhadada SK, Rao DS (2020) Rickets and osteomalacia. In: Melmed S, Auchus RJ, Goldfine AB, Koenig RJ, Rosen CJ (eds) Williams textbook of endocrinology, 14th edn. Elsevier, Philadelphia, pp 1298–1317
Tiosano D, Hochberg Z (2009) Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab 27(4):392–401. https://doi.org/10.1007/s00774-009-0079-1
Shroff R (2013) Phosphate is a vascular toxin. Pediatr Nephrol (Berlin, Germany) 28(4):583–593. https://doi.org/10.1007/s00467-012-2347-x
Yamada S, Giachelli CM (2017) Vascular calcification in CKD-MBD: roles for phosphate, FGF23, and Klotho. Bone 100:87–93. https://doi.org/10.1016/j.bone.2016.11.012
Floege J (2004) When man turns to stone: extraosseous calcification in uremic patients. Kidney Int 65(6):2447–2462. https://doi.org/10.1111/j.1523-1755.2004.00664.x
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM, Towler DA (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109(6):697–711. https://doi.org/10.1161/CIRCRESAHA.110.234914
Alfrey AC, Solomons CC, Ciricillo J, Miller NL (1976) Extraosseous calcification. Evidence for abnormal pyrophosphate metabolism in uremia. J Clin Investig 57(3):692–699. https://doi.org/10.1172/JCI108326
Giachelli CM (2009) The emerging role of phosphate in vascular calcification. Kidney Int 75(9):890–897. https://doi.org/10.1038/ki.2008.644
Paloian NJ, Giachelli CM (2014) A current understanding of vascular calcification in CKD. Am J Physiol Ren Physiol 307(8):F891–F900. https://doi.org/10.1152/ajprenal.00163.2014
Pai AS, Giachelli CM (2010) Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol JASN 21(10):1637–1640. https://doi.org/10.1681/asn.2010040349
Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM (2004) Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol JASN 15(11):2857–2867. https://doi.org/10.1097/01.Asn.0000141960.01035.28
Hosaka N, Mizobuchi M, Ogata H, Kumata C, Kondo F, Koiwa F, Kinugasa E, Akizawa T (2009) Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif Tissue Int 85(6):523–529. https://doi.org/10.1007/s00223-009-9297-8
Barvencik F, Beil FT, Gebauer M, Busse B, Koehne T, Seitz S, Zustin J, Pogoda P, Schinke T, Amling M (2011) Skeletal mineralization defects in adult hypophosphatasia—a clinical and histological analysis. Osteoporos Int 22(10):2667–2675. https://doi.org/10.1007/s00198-011-1528-y
Singer FR (2016) Bone quality in Paget’s disease of bone. Curr Osteoporos Rep 14(2):39–42. https://doi.org/10.1007/s11914-016-0303-6
Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, Bianco P (2003) Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: core biopsy studies and clinical correlations. J Bone Miner Res 18(7):1235–1246. https://doi.org/10.1359/jbmr.2003.18.7.1235
Bhadada SK, Dhiman V, Mukherjee S, Aggarwal S, Bal A, Sukumar SP, Sood A, Sharma DC, Khandelwal N, Bhansali A, Van Hul W, Rao SD (2017) Fibrogenesis imperfecta ossium and response to human growth hormone: a potential therapy. J Clin Endocrinol Metab 102(5):1750–1756. https://doi.org/10.1210/jc.2016-3055
Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, Montpetit K, Semler O (2017) Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052. https://doi.org/10.1038/nrdp.2017.52
Hoppe E, Masson C, Laffitte A, Chappard D, Audran M (2012) Osteomalacia in a patient with Paget’s bone disease treated with long-term etidronate. Morphologie: bulletin de l'Association des anatomistes 96(313):40–43. https://doi.org/10.1016/j.morpho.2012.08.001
Balena R, Kleerekoper M, Foldes JA, Shih MS, Rao DS, Schober HC, Parfitt AM (1998) Effects of different regimens of sodium fluoride treatment for osteoporosis on the structure, remodeling and mineralization of bone. Osteoporos Int 8:428–435
Teotia M, Teotia SP, Singh KP (1998) Endemic chronic fluoride toxicity and dietary calcium deficiency interaction syndromes of metabolic bone disease and deformities in India. Indian J Pediatr 65:371–381
Quarles LD, Dennis VW, Gitelman HJ, Harrelson JM, Drezner MK (1985) Aluminum deposition at the osteoid–bone interface. An epiphenomenon of the osteomalacic state in vitamin D-deficient dogs. J Clin Invest 75(5):1441–1447. https://doi.org/10.1172/jci111846
Parfitt AM, Rao DS, Stanciu J, Villanueva AR (1986) Comparison of aluminum related with vitamin D related osteomalacia by tetracycline based bone histomorphometry. Adv Exp Med Biol 208:283–287
Matsushima S, Torii M, Ozaki K, Narama I (2003) Iron lactate-induced osteomalacia in association with osteoblast dynamics. Toxicol Pathol 31(6):646–654. https://doi.org/10.1080/01926230390241990
Parfitt AM, Qiu S, Rao DS (2004) The mineralization index—a new approach to the histomorphometric appraisal of osteomalacia. Bone 35:320–325
Chines A, Pacifici R (1990) Antacid and sucralfate-induced hypophosphatemic osteomalacia: a case report and review of the literature. Calcif Tissue Int 47(5):291–295
Fabbriciani G, de Socio GV, Massarotti M, Ceriani R, Marasini B (2011) Adefovir induced hypophosphatemic osteomalacia. Scand J Infect Dis 43(11–12):990–992. https://doi.org/10.3109/00365548.2011.581307
Mateo L, Holgado S, Marinoso ML, Perez-Andres R, Bonjoch A, Romeu J, Olive A (2016) Hypophosphatemic osteomalacia induced by tenofovir in HIV-infected patients. Clin Rheumatol 35(5):1271–1279. https://doi.org/10.1007/s10067-10014-12627-x
Sansone V, Maiorano E, Galluzzo A, Pascale V (2018) Calcific tendinopathy of the shoulder: clinical perspectives into the mechanisms, pathogenesis, and treatment. Orthop Res Rev 10:63–72. https://doi.org/10.2147/orr.S138225
Rosenthal AK, Ryan LM (2016) Calcium pyrophosphate deposition disease. N Engl J Med 374(26):2575–2584. https://doi.org/10.1056/NEJMra1511117
Taniwaki M, Kawamoto K, Yamasaki M, Funaishi K, Matsumoto Y, Matsumoto N, Ohashi N, Hattori N (2019) Severe metastatic pulmonary calcification. Am J Med 132(10):e733–e734. https://doi.org/10.1016/j.amjmed.2019.04.031
Mignemi NA, Yuasa M, Baker CE, Moore SN, Ihejirika RC, Oelsner WK, Wallace CS, Yoshii T, Okawa A, Revenko AS, MacLeod AR, Bhattacharjee G, Barnett JV, Schwartz HS, Degen JL, Flick MJ, Cates JM, Schoenecker JG (2017) Plasmin prevents dystrophic calcification after muscle injury. J Bone Miner Res 32(2):294–308. https://doi.org/10.1002/jbmr.2973
Lee DK, Eiferman RA (2006) Ocular calcifications in primary hyperparathyroidism. Arch Ophthalmol 124(1):136–137. https://doi.org/10.1001/archopht.124.1.136
Yashiro T, Okamoto T, Tanaka R, Ito K, Hara H, Yamashita T, Kanaji Y, Kodama T, Ito Y, Obara T et al (1991) Prevalence of chondrocalcinosis in patients with primary hyperparathyroidism in Japan. Endocrinol Jpn 38(5):457–464. https://doi.org/10.1507/endocrj1954.38.457
Klaassen-Broekema N, van Bijsterveld OP (1993) Limbal and corneal calcification in patients with chronic renal failure. Br J Ophthalmol 77(9):569–571. https://doi.org/10.1136/bjo.77.9.569
Braunstein EM, Menerey K, Martel W, Swartz R, Fox IH (1987) Radiologic features of a pyrophosphate-like arthropathy associated with long-term dialysis. Skeletal Radiol 16(6):437–441. https://doi.org/10.1007/bf00350536
Gaisne R, Pere M, Menoyo V, Hourmant M, Larmet-Burgeot D (2020) Calciphylaxis epidemiology, risk factors, treatment and survival among French chronic kidney disease patients: a case–control study. BMC Ephrology 21(1):63. https://doi.org/10.1186/s12882-020-01722-y
Lecoq AL, Brandi ML, Linglart A, Kamenicky P (2020) Management of X-linked hypophosphatemia in adults. Metabolism 103s:154049. https://doi.org/10.1016/j.metabol.2019.154049
Whyte MP, Amalnath SD, McAlister WH, McKee MD, Veis DJ, Huskey M, Duan S, Bijanki VN, Alur S, Mumm S (2020) Hypophosphatemic osteosclerosis, hyperostosis, and enthesopathy associated with novel homozygous mutations of DMP1 encoding dentin matrix protein 1 and SPP1 encoding osteopontin: the first digenic SIBLING protein osteopathy? Bone 132:115190. https://doi.org/10.1016/j.bone.2019.115190
Marcucci G, Cianferotti L, Brandi ML (2018) Clinical presentation and management of hypoparathyroidism. Best Pract Res Clin Endocrinol Metab 32(6):927–939. https://doi.org/10.1016/j.beem.2018.09.007
Goswami R, Sharma R, Sreenivas V, Gupta N, Ganapathy A, Das S (2012) Prevalence and progression of basal ganglia calcification and its pathogenic mechanism in patients with idiopathic hypoparathyroidism. Clin Endocrinol (Oxf) 77(2):200–206. https://doi.org/10.1111/j.1365-2265.2012.04353.x
Mufaddel AA, Al-Hassani GA (2014) Familial idiopathic basal ganglia calcification (Fahr’s disease). Neurosciences (Riyadh, Saudi Arabia) 19(3):171–177
Carr DR, Pootrakul L, Chen HZ, Chung CG (2019) Metastatic calcinosis cutis associated with a selective FGFR inhibitor. JAMA Dermatol 155(1):122–123. https://doi.org/10.1001/jamadermatol.2018.4070
Fathi I, Sakr M (2014) Review of tumoral calcinosis: a rare clinico-pathological entity. World J Clin Cases 2(9):409–414. https://doi.org/10.12998/wjcc.v2.i9.409
Yolcu YU, Wahood W, Goyal A, Alvi MA, Reeves RK, Qu W, Gerberi DJ, Bydon M (2020) Pharmacologic prophylaxis for heterotopic ossification following spinal cord injury: a systematic review and meta-analysis. Clin Neurol Neurosurg 193:105737. https://doi.org/10.1016/j.clineuro.2020.105737
Rao DS, Parfitt AM, Kleerekoper M, Pumo BS, Frame B (1985) Dissociation between the effects of endogenous parathyroid hormone on adenosine 3′,5′-monophosphate generation and phosphate reabsorption in hypocalcemia due to vitamin D depletion: an acquired disorder resembling pseudohypoparathyroidism type II. J Clin Endocrinol Metab 61(2):285–290. https://doi.org/10.1210/jcem-61-2-285
Bhadada SK, Palnitkar S, Qiu S, Parikh N, Talpos GB, Rao SD (2013) Deliberate total parathyroidectomy: a potentially novel therapy for tumor-induced hypophosphatemic osteomalacia. J Clin Endocrinol Metab 98(11):4273–4278. https://doi.org/10.1210/jc.2013-2705
Parfitt AM (1990) Osteomalacia and related disorders. In: Avioli LV, Krane SM (eds) Metabolic bone disease and clinically related disorders, vol 2. W.B. Saunders, Philadelphia, pp 329–396
Funding
The study was partly supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (AR062103).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Sudhaker D. Rao and Sanjay Kumar Bhadada declare that they have no conflict of interest.
Ethical Approval
The study was conducted in accordance with the ethical standards of the institutional research committee of Henry Ford Hospital and the 1964 Helsinki Declaration and its later amendments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bhadada, S.K., Rao, S.D. Role of Phosphate in Biomineralization. Calcif Tissue Int 108, 32–40 (2021). https://doi.org/10.1007/s00223-020-00729-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00729-9