
Abstract
Phosphate has extensive physiological roles including energy metabolism, genetic function, signal transduction and membrane integrity. Regarding the skeleton, not only do phosphate and calcium form the mineral component of the skeleton, but phosphate is also essential in regulating function of skeletal cells. Although our understanding of phosphate homeostasis has lagged behind and remains less than that for calcium, considerable advances have been made since the recognition of fibroblast growth factor-23 (FGF23) as a bone-derived phosphaturic hormone that is a major regulator of phosphate homeostasis. In this two-part review of disorders of phosphate homeostasis in children, part 1 covers the basics of mineral ion homeostasis and the roles of phosphate in skeletal biology. Part 1 includes phosphate-related disorders of mineralization for which overall circulating mineral ion homeostasis remains normal. Part 2 covers hypophosphatemic and hyperphosphatemic disorders, emphasizing, but not limited to, those related to increased and decreased FGF23 signaling, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction and overview
Body phosphorus exists predominantly as inorganic phosphate, which is PO4−3, whether in the form of free HPO4−2 or H2PO4−1 ions or incorporated into other inorganic molecules such as hydroxyapatite. Organic phosphates, such as adenosine triphosphate (ATP) or nucleotides, have an organic residue added via an ester bond. In most circumstances, “phosphate physiology” or “phosphate homeostasis” refers to that for inorganic phosphate, and that is the focus of this review.
Eighty-five percent of total body phosphate is contained in the skeleton and dentition in the form of hydroxyapatite; 14% is intracellular, mostly organic phosphate within molecules involved in membrane integrity, nucleotides essential for genetic function, and multiple molecules involved in energy metabolism and signal transduction; and 1% is in the extracellular fluid as HPO4−2 or H2PO4−1, functioning as a pH buffer [1,2,3]. Regarding the skeleton, not only do phosphate and calcium form its mineral component, but phosphate is also essential in regulating function of skeletal cells.
Despite these global physiological roles of phosphate, our understanding of phosphate homeostasis has lagged behind that of calcium. However, considerable advances have occurred in our understanding of phosphate homeostasis over the last few decades, particularly with respect to recognition of the role of fibroblast growth factor-23 (FGF23) as a bone-derived phosphaturic hormone and major regulator of phosphate homeostasis. Many hypophosphatemic disorders are now understood as being caused by excessive FGF23 signaling, with some hyperphosphatemic disorders caused by insufficient FGF23 signaling.
In this two-part review of phosphate-related disorders in children, part 1 covers the basic principles of mineral ion homeostasis and the roles of phosphate in biomineralization and other aspects of skeletal biology. A brief discussion of phosphate nutrition and the role of phosphate in metabolic bone disease of prematurity is included. Part 1 also includes phosphate-related disorders of biomineralization that have normal systemic mineral ion homeostasis. Part 2 covers hypophosphatemic and hyperphosphatemic disorders, emphasizing, but not limited to, those caused by abnormalities of FGF23 signaling.
Primer on mineral ion homeostasis
Background
Calcium and phosphate are the two ions that together form hydroxyapatite to mineralize the skeleton, and their homeostases are intertwined. Although parathyroid hormone (PTH) and 1,25-dihydroxy-vitamin D (the active vitamin D metabolite, henceforth called calcitriol) are the major hormones regulating calcium homeostasis (discussed later), it became evident that these factors alone could not explain alterations in phosphate in many pathological conditions. Rather, several lines of evidence showed that renal phosphate excretion is strongly influenced by circulating substances referred to generically as “phosphatonins.” For example, in the mouse model of X-linked hypophosphatemia (XLH, also known as X-linked hypophosphatemic vitamin-D-resistant rickets) it had been shown that a substance circulating in an affected mouse could induce phosphate-wasting in a normal mouse [4, 5]. A major clue to the identity of this phosphatonin was uncovered in 2000, when a less common variety of hereditary hypophosphatemic rickets, autosomal-dominant hypophosphatemic rickets (ADHR), was shown to be caused by a gain-of-function mutation for the gene encoding FGF23, leading to elevated levels of intact FGF23, suggesting that it might be the causative phosphatonin [5, 6]. Shortly thereafter, FGF23 was demonstrated to be the causative phosphatonin in most cases of tumor-induced rickets or osteomalacia [7,8,9]. Other forms of hereditary hypophosphatemic rickets, including XLH, were also shown to have high FGF23 levels, and FGF23 was demonstrated to have multiple effects on lowering the circulating phosphate concentration, primarily by inducing renal phosphate loss [5].
Calcium homeostasis
Calcium homeostasis is not the explicit topic of this manuscript and therefore is described briefly. The circulating ionized calcium concentration is monitored by the calcium-sensing receptor in the parathyroid gland. In response to hypocalcemia, there is increased synthesis and release of PTH, which has multiple effects to raise the serum and extracellular calcium concentrations. For bone, PTH promotes osteoclastic bone resorption, mobilizing calcium and phosphate. In the kidney, PTH enhances calcium reabsorption in the distal tubule and decreases phosphate reabsorption in the proximal tubule. PTH also acts to increase renal synthesis of calcitriol from 25-hydroxyvitamin D in the proximal tubule by up-regulating the enzyme 1-α-hydroxylase and decreasing its degradation by down-regulating the enzyme 24-hydroxylase. The major function of calcitriol in turn is to increase calcium absorption in the intestine. As the calcium concentration rises in response to these actions, the calcium-sensing receptor then turns off further release of PTH, having achieved homeostasis.
Phosphate homeostasis
Regulation of the circulating phosphate concentration is accomplished by control of intestinal absorption, renal excretion and exchange of phosphate between extracellular fluid and other compartments including bone and intracellular phosphate. Of these, renal excretion is the most important, with a constant rate of filtration and control accomplished by regulation of reabsorption in the proximal tubule. Reabsorption in the proximal tubule depends mostly on the actions of types 2a and 2c sodium-phosphate cotransporters, which are located along the brush border membrane lining the lumen of the proximal tubule. Regulation of the expression of these cotransporters is the major method of modulating phosphate reabsorption.
The major hormones regulating phosphate homeostasis include PTH, calcitriol and FGF23. PTH and calcitriol have been discussed regarding their roles in calcium homeostasis and additional effects on phosphate is described later. The role of FGF23 in phosphate homeostasis has been the subject of many reviews [1, 10,11,12,13] and is later discussed in greater detail.
Parathyroid hormone (PTH)
For bone, PTH-induced osteoclastic resorption mobilizes phosphate in addition to calcium. In the kidney, PTH has a phosphaturic effect by reducing the expression of sodium-phosphate cotransporters in the proximal tubule and thereby reducing tubular reabsorption of phosphate.
Calcitriol
In addition to enhancing intestinal absorption of calcium, calcitriol enhances intestinal absorption of phosphate. Calcitriol also promotes the production and release of FGF23.
Fibroblast growth factor-23 (FGF23)
Fibroblast growth factor-23 is produced in bone, mostly by osteocytes and to a lesser extent by osteoblasts. Along with FGF19 and FGF21, FGF23 is an “endocrine FGF” that is capable of being released into the circulation to act remotely, whereas the other FGFs are all paracrine factors acting locally. FGF23 synthesis and secretion from bone are stimulated by high dietary phosphate and calcitriol. Unlike PTH, which is controlled by the calcium-sensing receptor, no well-defined “phosphate-sensing receptor” has been identified to account for how dietary phosphate controls FGF23. However, there is increasing evidence for the role of fibroblast growth factor receptor 1 (FGFR1) on the surface of osteocytes and osteoblasts for phosphate-sensing in the regulation of FGF23 [14], with potential additional phosphate-sensing roles of the calcium-sensing receptor in the parathyroid and a phosphate transporter in bone and kidney [15]. In order for FGF23 to bind to FGFRs with high affinity, the co-factor Klotho is required, with FGF23 binding to the FGFR–Klotho complex. The distribution of Klotho indicates the sites of action for FGF23, with the kidney showing the highest expression of Klotho. In the kidney, FGF23 has two major effects: (1) it is a phosphaturic hormone that decreases phosphate reabsorption in the proximal tubule by down-regulating the sodium-phosphate cotransporters, an effect similar to PTH; and (2) it decreases calcitriol by down-regulating its production by 1-α-hydroxylase and up-regulating its degradation by 24-hydroxylase, effects opposite those of PTH.
Fibroblast growth factor-23 regulation is accomplished not only by its transcription but by its post-transcriptional processing. In particular, FGF23 is susceptible to enzymatic cleavage and inactivation at a specific site by proprotein convertases, with only intact FGF23 being active. Normally, O-glycosylation of FGF23 adjacent to this site protects it from cleavage. Several disorders are related to genes that control this process, and these are discussed individually.
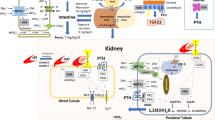
The effects of and factors regulating PTH, calcitriol and FGF23 along with their interactions with calcium and phosphate are summarized in Table 1 and Fig. 1 [13].

Mineral ion homeostasis. Net effects of fibroblast growth factor-23 (FGF23), parathyroid hormone (PTH) and calcitriol on phosphate and calcium homeostasis as well as on one another. Green arrows indicate positive regulatory effects and red arrows indicate negative regulatory effects. Diagram re-labeled from [13] and used with permission
Phosphate and skeletal biology
A sufficient circulating phosphate concentration is essential for multiple aspects of skeletal biology, most notably the processes of apoptosis of hypertrophic chondrocytes and mineralization of the extracellular skeletal matrix, with deficiencies in these processes leading to rickets and osteomalacia, respectively.
In rickets, mineral ion insufficiency leads to defects in both endochondral ossification and mineralization of cartilage and osteoid. The various causes of rickets can be divided pathophysiologically into those caused by insufficient calcium (“calcipenic rickets,” most often from vitamin D deficiency) versus those caused by insufficient phosphate (“phosphopenic rickets,” most often from phosphate-wasting as in XLH). With calcipenic forms, hypocalcemia then induces PTH synthesis and secretion to raise the calcium concentration. It is this secondary hyperparathyroidism of calcipenic rickets that then leads to hypophosphatemia by reducing renal tubular phosphate reabsorption. With phosphopenic rickets, renal tubular phosphate-wasting is the primary event. Hence, in both forms of rickets, hypophosphatemia develops and is believed to be the pathophysiological factor that leads to failure of normal apoptosis of hypertrophic chondrocytes. Apoptosis of hypertrophic chondrocytes is an essential step in the process of endochondral ossification, which removes those cells and promotes the ingrowth of vessels, osteoblasts and osteoclasts. For these reasons, hypophosphatemia is considered to be the “common denominator of all rickets” [16].
The other major skeletal process for which maintenance of adequate mineral ion concentrations is required, but not alone sufficient, is biomineralization of the extracellular matrix. Physiological concentrations of calcium and phosphate are high enough that extracellular fluid is supersaturated and hence precipitates the presence of nucleating agents such as collagen fibrils, leading to both normal and pathological biomineralization, unless prevented by mineralization inhibitors. Pyrophosphate, consisting of two phosphates connected by an ester bond, is a major mineralization inhibitor and accordingly has been nominated as the “body’s water softener” by Orriss [17]. A major determinant of biomineralization is the phosphate-to-pyrophosphate ratio in the extracellular matrix [18].
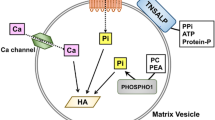
Although the details of biomineralization of the extracellular matrix are not completely understood, this process appears to begin in small (100–300-mm diameter) membrane-bound subcellular structures called matrix vesicles [19,20,21,22]. Matrix vesicles arise by budding from the plasma membranes of chondrocytes and osteoblasts and are the initial sites of hydroxyapatite crystal formation. To accomplish this, matrix vesicles possess an array of enzymes internally and on their surface as well as membrane-bound transporters in order to both generate sufficient concentrations of phosphate and calcium within the vesicles to initiate hydroxyapatite crystal formation and also prevent excessive accumulation of pyrophosphate in the perivesicular extracellular matrix. Transport of phosphate into matrix vesicles is accomplished by type 3 sodium–phosphate cotransporters, with transport of calcium mediated by annexins. The phosphate transported into matrix vesicles is largely derived from extracellular ATP and pyrophosphate by the action of tissue-nonspecific alkaline phosphatase (TNAP, also abbreviated as TNSALP), a membrane-bound phosphohydrolase. Matrix vesicle mediated mineralization depends on a sufficient supply of phosphate, and evidence suggests that TNAP on the surface of matrix vesicles is regulated to meet mineralization needs [23, 24]. Phosphate is also generated within matrix vesicles from phosphoethanolamine and phosphocholine by the action of phosphocholine phosphatase-1. Ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) is an enzyme located on the surface of matrix vesicles that also catabolizes ATP. However, in doing so, ENPP1 generates pyrophosphate; this is unlike TNAP, which generates phosphate from ATP and converts pyrophosphate to phosphate. TNAP and ENPP1 thus have opposing functions in regulating the phosphate-to-pyrophosphate ratio in the perivesicular extracellular matrix, which is a key determinant of mineralization. Within matrix vesicles, sufficient calcium and phosphate concentrations allow for precipitation of these ions forming amorphous calcium phosphate with subsequent staged transitions to hydroxyapatite crystals (Ca10(PO4)6(OH)2). These crystals grow within matrix vesicles and eventually break though the vesicle membrane to enter the extracellular matrix, where they are deposited on collagen fibrils and continue to grow. However, the extravesicular growth of hydroxyapatite crystals is inhibited by excessive pyrophosphate, with pyrophosphate normally degraded by TNAP. Osteopontin also inhibits propagation of hydroxyapatite crystals as well as initial precipitation of calcium and phosphate ions [19]. Many of these processes involved in matrix vesicle mediated biomineralization of the extracellular matrix are summarized in Fig. 2 [25].

Matrix vesicle mediated mineralization. Initial formation of hydroxyapatite crystals (HA) in matrix vesicles involves precipitation of phosphate (Pi) and calcium ions, first forming amorphous calcium phosphate (not shown), with subsequent transformation to hydroxyapatite. The calcium ions (not shown) are transported into matrix vesicles via annexins. Phosphate needed for hydroxyapatite is supplied by phosphohydrolysis of phosphocholine (PC) and phosphoethanolamine (not shown) by phosphocholine phosphatase (PHOSPHO1) as well as phosphate imported from the extracellular matrix (ECM) via type 3 sodium–phosphate cotransporters (PiT). Within the extracellular matrix, this phosphate is mostly generated by phosphohydrolysis of adenosine triphosphate (ATP) and pyrophosphate by tissue-nonspecific alkaline phosphatase (TNAP), thereby also lowering extracellular matrix pyrophosphate. Ectoucleotide pyrophosphatase/phosphodiesterase 1 (abbreviated here as NPP1) most often has the opposite effect of generating pyrophosphate from ATP (dashed arrow). Following formation of sufficient hydroxyapatite crystals within matrix vesicles, they are released into the extracellular matrix and deposited onto collagen fibrils where they continue to grow. The extravesicular growth of hydroxyapatite crystals can be inhibited by excessive pyrophosphate as well as other mineralization inhibitors such as osteopontin (OPN). Diagram used with permission [25]
To accomplish these processes of endochondral ossification and mineralization, skeletal cells are dependent on sufficient extracellular phosphate [26]. For chondrocytes, phosphate is needed for uptake into matrix vesicles; for gene expression of type X collagen, which is needed for hypertrophic differentiation; and for expression of caspase 3, which is required for apoptosis of hypertrophic chondrocytes. Sufficient phosphate is also needed by osteoblasts for matrix vesicles as well as for gene expression of osteopontin, which is necessary for formation of bone matrix and insulin-like growth factor 1, which is needed for osteoblast proliferation. When osteoblasts become embedded within bone matrix, they become osteocytes, branching into canaliculi and serving as mechano-sensors and carrying out most of the endocrine functions of bone. Elevated phosphate causes osteocytes to secrete FGF23, which acts to lower phosphate. Additionally, phosphate is involved in the regulation of many matrix proteins produced by osteocytes, including those encoded by the PHEX, DMP1 and ENPP1 genes, all of which are involved in the regulation of FGF23, with deficiencies of these resulting in excessive FGF23 and causing phosphate-wasting in XLH and autosomal-recessive hypophosphatemic rickets (ARHR) types 1 and 2, respectively. Regarding osteoclasts, normal osteoclast differentiation is stimulated by the binding of the receptor activator of nuclear factor κB ligand (RANKL) to RANK on the surface of osteoclast progenitors, and phosphate inhibits this process by decreasing RANKL production by osteocytes and osteoblasts. Similar signaling also limits osteoclast survival. These effects of phosphate on chondrocytes, osteoblasts, osteocytes and osteoclasts are summarized in Fig. 3 [26].

Effects of phosphate on skeletal cells. Phosphate (Pi) is essential for the major functions of chondrocytes, osteoblasts, osteocytes and osteoclasts, as illustrated in this diagram. Diagram is reformatted from [26] and used with permission. DMP1 dentin matrix acidic protein 1, ENPP1 Ectonucleotide pyrophosphatase/phosphodiesterase 1, FGF23 fibroblast growth factor-23, IGF1 insulin-like growth factor 1, NF-kB nuclear factor kappa B or nuclear factor κB, OPN osteopontin, PHEX phosphate-regulating endopeptidase homolog X-linked, Pi inorganic phosphate, RANKL receptor activator of nuclear factor kB ligand, ROS reactive oxygen species
Nutritional considerations
Phosphates, in both organic and inorganic forms, are important components of both plant and animal cells and are therefore present throughout diverse diets. Hence dietary phosphate deficiency is exceedingly rare other than in the event of generalized insufficient supply or absorption of all foods. While in most circumstances dietary intake of phosphate can be considered adequate, a notable exception concerns infants born significantly prematurely. Normally, 80% of in utero bone mineral accretion occurs during the 3rd trimester, with active transplacental transport of minerals against a concentration gradient [27, 28]. Hence, infants born prior to this time are challenged with being able to maintain normal bone mineral accretion and are at high risk for metabolic bone disease of prematurity. Nutrition is a major factor, with the need to supply sufficient amounts of phosphate, calcium and vitamin D. Of these, phosphate deficiency is considered to be the leading cause of metabolic bone disease of prematurity because the need to meet the intracellular phosphate requirements of the soft tissues places skeletal deposition at relatively greater risk [29, 30]. Indeed, the recognition of these nutritional needs and attention to mineral supplementation of feedings has led to a significant reduction in the incidence of metabolic bone disease of prematurity [31]. However, metabolic bone disease of prematurity is a complex disorder with multiple other risk factors including immobilization, several types of drugs, acidosis, cholestasis and others. Further discussion of the clinical and radiologic features of metabolic bone disease of prematurity is not within the scope of this review and can be found in selected references [27,28,29,30,31,32,33,34].
Disorders of biomineralization with normal circulating mineral ion concentrations
Although most disorders that alter biomineralization involve abnormality of mineral ion homeostasis, there are some for which circulating mineral ion concentrations are normal but abnormal biomineralization results from other conditions. Because the phosphate-to-pyrophosphate ratio in the perivesicular matrix is a major determinant of biomineralization, processes that alter this ratio form the major component of this group, although there are also conditions where abnormal biomineralization is related to other mineralization inhibitors or promoters.
Hypophosphatasia
Hypophosphatasia is the major disorder for which there is failure of skeletal mineralization despite normal mineral ion homeostasis. Hypophosphatasia is caused by loss of function mutations for the ALPL gene (1p36.12), which encodes TNAP. As of a 2018 review, more than 340 mutations for ALPL were known to be causative for hypophosphatasia, accounting for the large range in manifestations with varying degrees of severity [35]. The severe forms of hypophosphatasia are caused by biallelic recessive mutations, whereas the more benign forms might be from recessive mutations or dominant mutations with dominant-negative effects [25] whereby the mutant gene product inhibits the activity of the normal gene product or interferes with its localization [36,37,38]. In hypophosphatasia, lack of sufficient TNAP activity prevents normal phosphohydrolysis of pyrophosphate on the surface of matrix vesicles, which inhibits biomineralization by limiting the supply of phosphate that can be transported into the vesicle and by the inhibitory effect of pyrophosphate on hydroxyapatite crystal propagation. Additionally, in normal circumstances TNAP dephosphorylates and thereby inactivates osteopontin and thus deficient TNAP activity leads to increased osteopontin, another mineralization inhibitor [25, 35]. Although ossification and mineralization are distinct processes and TNAP deficiency in hypophosphatasia directly affects mineralization, endochondral ossification is also impaired, such that histopathological studies showed excessive amounts of proliferating and hypertrophic cartilage, often nonmineralized, at the growth plates [39, 40]. In addition to its well-established role in mineralization, recent evidence suggests that TNAP affects mitochondrial function and ATP production in bone and muscle progenitor cells, which might have a role in the weakness and fatigue associated with severe forms of hypophosphatasia [41].
Nosologically, several categories of hypophosphatasia range from the most severe perinatal lethal form to the least severe adult form and odontohypophosphatasia, which has only dental manifestations. The clinical manifestations of these categories are delineated in several reviews of hypophosphatasia [35, 42,43,44,45], and the present discussion concerns mostly their radiographic findings [46]. For radiographic description, the non-visualization or very poor visualization of bones is usually referred to as “deficiency of ossification,” and we use this here, recognizing that in hypophosphatasia the primary defect is mineralization.
In perinatal lethal hypophosphatasia, which is recognizable in utero, ossification is so profoundly impaired that substantial portions of the skeleton appear to be absent on radiographic evaluation. Although there is considerable variability among patients regarding the extent and pattern of involvement, such findings in 19 patients have been summarized by Shohat et al. [40]. Ossification of the calvarium might be completely absent or when present is usually limited to very poor ossification of the frontal or occipital bones. While spinal ossification is quite variable, the lower spine is often more severely involved than the upper spine. Individual vertebrae are often not visualized and vertebral morphology might be abnormal. Ossification of the posterior elements and ribs is diminished or absent. The scapulae and pubic bones are usually involved to varying degrees, and the clavicles tend to be the least involved region of the skeleton. In the extremities, variable findings include diminished ossification, shortening, bowing and metaphyseal irregularity. Additionally, spur-like projections from the midshafts of the fibula and occasionally the ulna (Bowdler spurs) with overlying skin dimpling are quite characteristic for hypophosphatasia. Many of the findings of perinatal lethal hypophosphatasia are illustrated in Fig. 4 [47]. Although most cases of hypophosphatasia that present in utero are the severe perinatal lethal form just described, a benign form has also been recognized with short bowed bones on fetal US that gradually regress during the 3rd trimester [48]. This prenatal benign form might result from dominant or recessive TNAP mutations, with most dominant cases related to maternal transmission, suggesting a maternal in utero effect. Postnatally, there is usually further improvement, although the overall range of severity is quite variable, extending from the infantile form to very mild.

Perinatal lethal hypophosphatasia in 1-day-old boy. a Lateral radiograph of the skull shows that ossification of the calvarium is limited to part of the occipital bone and minimal frontal ossification. The skull base is slightly better ossified. The upper cervical spine is very poorly ossified and there is no ossification of the lower cervical spine. b, c Anteroposterior (AP) (b) and lateral (c) radiographs of the trunk show no ossification of T1, L4, L5 or any posterior elements, with preservation of the other vertebral bodies, ribs and clavicles. d AP radiograph of the pelvis and lower extremities shows that the iliac bones are ossified but the pubic and ischial bones are not. The femurs are severely shortened and bowed and have metaphyseal defects extending deep into the shafts. The tibias are much less involved. The fibulas are very poorly ossified and no Bowdler spurs are evident in this example. Images used with permission [47]
Infantile hypophosphatasia presents between birth and 6 months, often with pulmonary complications secondary to severe rachitic-like involvement of the thorax. The skeleton is diffusely poorly mineralized, the long bones are often bowed, and there are prominent focal and often asymmetrical metaphyseal defects. Although some cases resemble rickets, these metaphyseal defects are usually more focal and extend from the growth plates deeper into the metaphyses or even into the diaphyses, appearing distinctively different from rickets, which has uniform involvement across the physis. In the skull, there is often craniosynostosis, although the sutures often initially appear wide rather than closed because the bones are so poorly ossified. Childhood hypophosphatasia presents after 6 months of age and can be subdivided into severe and mild types [43]. The radiographic findings become progressively less severe across this spectrum compared to the infantile form. In the mild form, the metaphyseal defects often have a “punched out” appearance. Figure 5 shows a relatively severe form of childhood hypophosphatasia. Adult hypophosphatasia resembles osteomalacia both clinically and radiographically. Metatarsal stress fractures and enthesopathy are common. However, femoral pseudofractures are distinct from those of osteomalacia in that pseudofractures are located at the lateral subtrochanteric region rather than the medial femoral necks, and thus resemble the fractures associated with anti-resorption therapeutic agents.

Severe childhood hypophosphatasia. a–d Radiographs in an 11-month-old boy include anteroposterior (AP) right shoulder view with cephalic angulation and abduction (a), posteroanterior view of right wrist (b), AP view of right knee (c) and AP view of right ankle (d). They show some findings suggestive of rickets, particularly at the wrist, as well as more focal defects extending into the metaphyses, especially prominent in the proximal humerus. e, f At 2.1 years, AP radiograph of the right upper extremity (e) shows the proximal humeral defect extending through the metaphysis into the proximal diaphysis. The prior rachitic findings in the distal radius and ulna are no longer present and there is a focal defect in the distal ulnar metaphysis. The lower extremities (AP radiograph, f) show prominent defects in the distal femoral, proximal fibular and distal fibular metaphyses as well as defect in the distal femoral epiphyses. Associated regions of sclerosis and abnormalities of tubulation, as well as sclerotic regions in the tibial metaphyses, are also evident
A relatively recent major advancement in the treatment of hypophosphatasia has been the development of enzyme replacement therapy with asfotase alfa, a recombinant TNAP dimer containing two identical moieties with enzymatic activity linked by an IgG1 immunoglobulin targeting the molecule to bone. Enzyme replacement therapy for perinatal and infantile hypophosphatasia has led to marked improvement in overall survival and bone mineralization as well as reduction in rachitic findings radiographically [49]. Other clinical finding such as weakness have also improved. For this group, there is agreement that therapy is indicated and needs to be lifelong. Although some data suggest that adolescents and adults with much less severe manifestations of hypophosphatasia have less pain and improved functional performance with enzyme replacement therapy, treatment for these patients is not as well established. Out of concern that such therapy could lead to pathological vascular calcifications, screening with radiography, renal US and ophthalmologic retinal examination has been performed with no significant problems found [49, 50]. Guidelines for which patients should receive enzyme replacement therapy are beyond the scope of this review but are discussed elsewhere [42, 51].
Generalized arterial calcification of infancy
Generalized arterial calcification of infancy is a disorder for which there is excessive biomineralization without elevated circulating mineral ion concentrations. Most cases (approximately 75%) result from autosomal-recessive loss-of-function mutations for ENPP1, the gene encoding ENPP1. Normally, ENPP1 hydrolyzes extracellular ATP to form adenosine monophosphate (AMP) and pyrophosphate, a strong inhibitor of biomineralization. Hence, loss of sufficient ENPP1 activity results in a reduction in pyrophosphate, thereby promoting both normal and pathological biomineralization. Vascular disease is caused not only by mineralization, but also by stenosis from the smooth muscle proliferation that can result from the diminished generation of AMP [52, 53]. Approximately half of all cases present in utero or very shortly after birth with signs of vascular compromise including hydrops fetalis, fetal distress and polyhydramnios. For this group, the major arteries involved are the hepatic artery, aorta and pulmonary arteries (Fig. 6) [52]. Extravascular ectopic calcifications might also be present in the neonatal period (Fig. 7 [54] and Fig. 4 of part 2). Those presenting later are often asymptomatic at birth and then develop cardiovascular symptoms including cyanosis and respiratory distress, and in this group the coronary arteries, renal arteries and pulmonary arteries predominate [52, 55]. In the neonatal period, mineral ion homeostasis is normal in generalized arterial calcification of infancy. However, survivors later develop FGF23-mediated hypophosphatemia, demonstrated to be present by 14 years of age in a prospective study of those who survived the neonatal period [54]. Indeed, autosomal-recessive hypophosphatemic rickets type 2 is also caused by loss-of-function mutations of ENPP1 and these are now considered to be within the spectrum of ENPP1 deficiency [56]. How this leads to increased FGF23 production is not clearly understood but is possibly mediated by increased sclerostin [52, 57]. Additional findings in survivors of generalized arterial calcification of infancy include a 25% lifetime risk of cervical spine fusions, mostly involving the posterior elements and posterior aspects of the vertebral bodies; a 75% risk of hearing loss, possibly caused by abnormal calcification within middle ear; and calcification of entheses in all adults [54]. Less often, 9% of cases are caused by autosomal-recessive mutations for ABCC6, the gene also associated with pseudoxanthoma elasticum. Although the role of ABCC6 in generalized arterial calcification of infancy is not well understood, decreased pyrophosphate is involved [56]. Despite similar prevalence of arterial and organ calcification, mortality is lower in those with ABCC6 mutations than ENPP1, and elevated FGF23 with phosphate-wasting is a manifestation only of ENPP1 mutations, not ABCC6 [56].

Generalized arterial calcification of infancy, severe early onset type. A prenatal US (not shown) revealed narrowing and calcification of abdominal aorta. The boy died shortly after delivery. This postmortem CT coronal reformation shows aortic calcification and focal narrowing with a reduction in diameter just below the diaphragm. There is also extensive arterial calcification including involvement of many major visceral arteries in the abdomen, the iliac arteries, and the pulmonary and coronary arteries in the chest. Image courtesy of Carlos R. Ferreira, MD, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD. Different image from same examination was previously published [52]

Periarticular calcification in generalized arterial calcification of infancy. CT axial bone window shows extensive right shoulder periarticular calcifications in 4-day-old girl with generalized arterial calcification of infancy. The proximal humeral shaft is indicated by the letter H and the body of the scapula is indicated by the letter S. All of the other calcifications between the scapula and humerus and anterior to the scapula comprise the abnormal periarticular calcifications. Image courtesy of Carlos R. Ferreira, MD, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD. Different image from same patient was previously published [54]
Summary
This first part of a two-part review of disorders of phosphate homeostasis in children has discussed the basic principles of mineral ion homeostasis with emphasis on the importance of FGF23 as a bone-derived phosphaturic hormone for regulating phosphate homeostasis as well as the roles of phosphate in skeletal biology and the process of mineralization. Phosphate is present throughout foods of plant and animal origin and hence dietary phosphate insufficiency is exceedingly uncommon. A notable exception is infants born significantly prematurely, for whom the difficulty in supplying sufficient minerals to match in utero mineral accretion rates is a major cause of metabolic bone disease of prematurity. Part 1 has also considered disorders of biomineralization for which circulating mineral ion concentrations are normal but where abnormal mineralization results from imbalance of the phosphate-to-pyrophosphate ratio in the perivesicular extracellular space. In the case of hypophosphatasia, deficient mineralization results from excessive pyrophosphate caused by deficient TNAP activity from loss-of-function mutations of the ALPL gene. In the case of generalized arterial calcification of infancy, pathological mineralization results from insufficient pyrophosphate, in most cases caused by deficient ENPP1 activity from loss-of-function mutations of ENPP1. Part 2 examines hypophosphatemic and hyperphosphatemic disorders, emphasizing those caused by increased and decreased FGF23 signaling, respectively.
References
Christov M, Jüppner H (2018) Phosphate homeostasis disorders. Best Pract Res Clin Endocrinol Metab 32:685–706
Kinoshita Y, Fukumoto S (2018) X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases: prospect for new treatment. Endocr Rev 39:274–291
Bitzan M, Goodyer PR (2019) Hypophosphatemic rickets. Pediatr Clin N Am 66:179–207
Meyer RA, Meyer MH, Gray RW (1989) Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res 4:493–500
Levine BS, Kleeman CR, Felsenfeld AJ (2009) The journey from vitamin D-resistant rickets to the regulation of renal phosphate transport. Clin J Am Soc Nephrol 4:1866–1877
ADHR Consortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Drezner MK (2001) Tumor-induced osteomalacia. Rev Endocr Metab Disord 2:175–186
Shimada T, Mizutani S, Muto T et al (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 98:6500–6505
Jan de Beur SM, Levine MA (2002) Molecular pathogenesis of hypophosphatemic rickets. J Clin Endocrinol Metab 87:2467–2473
Balani S, Perwad F (2019) Fibroblast growth factor 23 and phosphate homeostasis. Curr Opin Nephrol Hypertens 28:465–473
Bar L, Stournaras C, Lang F et al (2019) Regulation of fibroblast growth factor 23 (FGF23) in health and disease. FEBS Lett 593:1879–1900
Vervloet M (2019) Renal and extrarenal effects of fibroblast growth factor 23. Nat Rev Nephrol 15:109–120
Bacchetta J, Bardet C, Prié D (2020) Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism 103s:153865
Takashi Y, Fukumoto S (2020) Phosphate-sensing and regulatory mechanism of FGF23 production. J Endocrinol Investig 43:877–883
Figueres L, Beck-Cormier S, Beck L, Marks J (2021) The complexities of organ crosstalk in phosphate homeostasis: time to put phosphate sensing back in the limelight. Int J Mol Sci 22:5701
Tiosano D, Hochberg Z (2009) Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab 27:392–401
Orriss IR (2020) Extracellular pyrophosphate: the body's "water softener". Bone 134:115243
Orriss IR, Arnett TR, Russell RG (2016) Pyrophosphate: a key inhibitor of mineralisation. Curr Opin Pharmacol 28:57–68
Cui L, Houston DA, Farquharson C, MacRae VE (2016) Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 87:147–158
Anderson HC, Garimella R, Tague SE (2005) The role of matrix vesicles in growth plate development and biomineralization. Front Biosci 10:822–837
Millán JL (2013) The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 93:299–306
Bottini M, Mebarek S, Anderson KL et al (2018) Matrix vesicles from chondrocytes and osteoblasts: their biogenesis, properties, functions and biomimetic models. Biochim Biophys Acta Gen Subj 1862:532–546
Kimata M, Michigami T, Tachikawa K et al (2010) Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/pi cotransporter pit-1 and Raf/MEK/ERK pathway. Bone 47:938–947
Michigami T, Ozono K (2019) Roles of phosphate in skeleton. Front Endocrinol 10:180
Millán JL, Whyte MP (2016) Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int 98:398–416
Chande S, Bergwitz C (2018) Role of phosphate sensing in bone and mineral metabolism. Nat Rev Endocrinol 14:637–655
Montaner Ramón A (2020) Risk factors of bone mineral metabolic disorders. Semin Fetal Neonatal Med 25:101068
Chinoy A, Mughal MZ, Padidela R (2019) Metabolic bone disease of prematurity: causes, recognition, prevention, treatment and long-term consequences. Arch Dis Child Fetal Neonatal Ed 104:F560–f566
Schulz EV, Wagner CL (2020) History, epidemiology and prevalence of neonatal bone mineral metabolic disorders. Semin Fetal Neonatal Med 25:101069
Ryan S (1996) Nutritional aspects of metabolic bone disease in the newborn. Arch Dis Child Fetal Neonatal Ed 74:F145–F148
Greer FR (1994) Osteopenia of prematurity. Annu Rev Nutr 14:169–185
Done SL (2012) Fetal and neonatal bone health: update on bone growth and manifestations in health and disease. Pediatr Radiol 42:S158–S176
Viswanathan S, Khasawneh W, McNelis K et al (2014) Metabolic bone disease: a continued challenge in extremely low birth weight infants. JPEN J Parenter Enteral Nutr 38:982–990
Harrison CM, Gibson AT (2013) Osteopenia in preterm infants. Arch Dis Child Fetal Neonatal Ed 98:F272–F275
Mornet E (2018) Hypophosphatasia. Metabolism 82:142–155
Fauvert D, Brun-Heath I, Lia-Baldini AS et al (2009) Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet 10:51
Lia-Baldini AS, Brun-Heath I, Carrion C et al (2008) A new mechanism of dominance in hypophosphatasia: the mutated protein can disturb the cell localization of the wild-type protein. Hum Genet 123:429–432
Lia-Baldini AS, Muller F, Taillandier A et al (2001) A molecular approach to dominance in hypophosphatasia. Hum Genet 109:99–108
Ornoy A, Adomian GE, Rimoin DL (1985) Histologic and ultrastructural studies on the mineralization process in hypophosphatasia. Am J Med Genet 22:743–758
Shohat M, Rimoin DL, Gruber HE, Lachman RS (1991) Perinatal lethal hypophosphatasia; [sic] clinical, radiologic and morphologic findings. Pediatr Radiol 21:421–427
Zhang Z, Nam HK, Crouch S, Hatch NE (2021) Tissue nonspecific alkaline phosphatase function in bone and muscle progenitor cells: control of mitochondrial respiration and ATP production. Int J Mol Sci 22:1140
Khan AA, Josse R, Kannu P et al (2019) Hypophosphatasia: Canadian update on diagnosis and management. Osteoporos Int 30:1713–1722
Whyte MP, Zhang F, Wenkert D et al (2015) Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone 75:229–239
Linglart A, Biosse-Duplan M (2016) Hypophosphatasia. Curr Osteoporos Rep 14:95–105
Villa-Suárez JM, García-Fontana C, Andújar-Vera F et al (2021) Hypophosphatasia: a unique disorder of bone mineralization. Int J Mol Sci 22:4303
Kozlowski K, Sutcliffe J, Barylak A et al (1976) Hypophosphatasia. Review of 24 cases. Pediatr Radiol 5:103–117
Shore RM (2008) Metabolic bone disease. In: Slovis TL (ed) Caffey's pediatric diagnostic imaging, 11th edn. Elsevier, Philadelphia, pp 2726–2752
Wenkert D, McAlister WH, Coburn SP et al (2011) Hypophosphatasia: nonlethal disease despite skeletal presentation in utero (17 new cases and literature review). J Bone Miner Res 26:2389–2398
Whyte MP, Greenberg CR, Salman NJ et al (2012) Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 366:904–913
Whyte MP (2017) Hypophosphatasia: enzyme replacement therapy brings new opportunities and new challenges. J Bone Miner Res 32:667–675
Rush ET (2018) Childhood hypophosphatasia: to treat or not to treat. Orphanet J Rare Dis 13:116
Boyce AM, Gafni RI, Ferreira CR (2020) Generalized arterial calcification of infancy: new insights, controversies, and approach to management. Curr Osteoporos Rep 18:232–241
Nitschke Y, Yan Y, Buers I et al (2018) ENPP1-fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med 50:1–12
Ferreira CR, Hackbarth ME, Ziegler SG et al (2021) Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet Med 23:396–407
Chong CR, Hutchins GM (2008) Idiopathic infantile arterial calcification: the spectrum of clinical presentations. Pediatr Dev Pathol 11:405–415
Ferreira CR, Kintzinger K, Hackbarth ME et al (2021) Ectopic calcification and hypophosphatemic rickets: natural history of ENPP1 and ABCC6 deficiencies. J Bone Miner Res 36:2193–2202
Hajjawi MO, MacRae VE, Huesa C et al (2014) Mineralisation of collagen rich soft tissues and osteocyte lacunae in Enpp1(−/−) mice. Bone 69:139–147
Acknowledgments
I would like to thank Aaron L. Friedman, MD, for his helpful advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shore, R.M. Disorders of phosphate homeostasis in children, part 1: primer on mineral ion homeostasis and the roles of phosphate in skeletal biology. Pediatr Radiol 52, 2278–2289 (2022). https://doi.org/10.1007/s00247-022-05374-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-022-05374-y