
Abstract
Tissue-nonspecific alkaline phosphatase (TNAP) plays a key role in mineralization by degrading inorganic pyrophosphate and providing free inorganic phosphate. We have previously reported that TNAP is induced by β-glycerophosphate and NaH2PO4 in short-term cultures of SaOS-2 human osteoblast-like cells and that PHEX (phosphate-regulating gene with homologies to endopeptidase on the X chromosome) mRNA is also induced after TNAP induction. In the present study, we have investigated the effects of levamisole, a TNAP inhibitor, and phosphonoformic acid (PFA), a type III sodium-phosphate cotransporter inhibitor, on the phosphate-induced expression of TNAP and mineralization. Levamisole inhibited β-glycerophosphate-induced mineralization, TNAP and PHEX expression, and the increase in enzymatic activity of NPP1 (5′-nucleotide pyrophosphatase phosphodiesterase 1), but did not inhibit NaH2PO4-induced mineralization. PFA completely inhibited NaH2PO4-induced mineralization and NPP1 enzymatic activation, and partly inhibited β-glycerophosphate-induced mineralization, but did not affect the increase in TNAP activity. These results suggest that phosphate derived from TNAP-induced hydrolysis of β-glycerophosphate yields signals that induce TNAP expression and mineralization, and that PHEX expression may be linked to TNAP expression. However, luciferase assays failed to detect any transcriptional activation of the promoter region of the human TNAP gene by β-glycerophosphate or NaH2PO4, suggesting that the effects of these phosphates may be indirect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alkaline phosphatase (ALP, EC.3.1.3.1) is a biomarker of bone formation. Human enzymes are classified into four types: tissue-nonspecific, intestinal, placental, and germ cell, of which the tissue-nonspecific type is involved in bone formation [1, 2]. Tissue-nonspecific alkaline phosphatase (TNAP) is encoded by a gene located on chromosome 1p36.1-34 and is more than 50 kb in size [3]. TNAP plays a key role in bone mineralization by initiating and/or promoting the formation of hydroxyapatite crystals in the matrix vesicles of osteoblasts and hypertrophic chondrocytes and then propagates them into the extracellular matrix [4, 5]. Matrix vesicles are extracellular membrane-invested particles that serve as the initial site of mineralization [6]. On the outer surface of the matrix vesicle membrane, TNAP degrades inorganic pyrophosphate (PPi) in the extracellular matrix that inhibits hydroxyapatite formation, and also yields free inorganic phosphate (Pi) which reacts with calcium to form hydroxyapatite [4, 6]. PPi in the extracellular matrix is formed by NPP1 (an isoenzyme of 5′-nucleotide pyrophosphatase phosphodiesterase, PC-1) on the matrix vesicle membrane [7] and is also delivered by ANKH (human homolog of mouse progressive ankylosis (Ank)), a transporter of PPi on the hypertrophic chondrocyte membrane [8]. TNAP, NPP1 and ANKH together regulate hydroxyapatite formation [9–11]. The role of TNAP in bone mineralization has been confirmed in studies of patients with hypophosphatasia, a genetic defect of the TNAP gene [12–17], and knockout mice [5, 18, 19]. Hypophosphatasia is characterized by hypomineralization of hard tissues, and knockout mice have a phenotype that is similar to that of humans with hypophosphatasia [20]. Patients with hypophosphatasia have elevated serum levels of PPi [20]. Primary osteoblasts obtained from TNAP knockout mice have impaired capacity to undergo mineralization due to an inability to hydrolyze PPi [21]. These findings indicate that TNAP promotes mineralization through hydrolysis of PPi. The TNAP gene is also associated with bone mineral density in elderly women [22]. We have recently demonstrated that TNAP gene expression in SaOS-2 osteoblast-like cells is stimulated by β-glycerophosphate and slightly stimulated by sodium phosphate, both of which have been found to stimulate mineralization in an in vitro cell culture system [23].
Recent studies of phosphate metabolism have identified novel regulators of plasma Pi. In particular, FGF23 (fibroblast growth factor 23) may be a central regulator [24], and PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) and MEPE (matrix extracellular phosphoglycoprotein) have also been shown to regulate phosphate metabolism and mineralization [25, 26]. The relationship between these factors and TNAP, however, remains unclear. We have previously showed that gene expression of PHEX and MEPE is increased by β-glycerophosphate after induction of the TNAP gene in SaOS-2 cells [23].
Since hydroxyapatite is formed initially within matrix vesicles, intravesicular phosphate ion accumulation may precede calcium phosphate deposition [6]. Extracellular Pi is incorporated into the cells by the type III sodium–phosphate (Na/Pi) cotransporter, which is expressed on hypertrophic chondrocyte membranes [27] and in isolated matrix vesicles from murine chondrogenic cells and plays a role in matrix mineralization [28]. In order to clarify the role of TNAP in mineralization, it is important to understand the relationships among TNAP, the type III Na/Pi cotransporter and mineralization processes. In the present study, we used inhibitors of TNAP and the type III Na/Pi cotransporter to examine the effects of these inhibitors on the expression of TNAP, NPP1, and PHEX in human osteoblast-like cells, SaOS-2. In addition, we evaluated the influence of the inhibitors on mineralization in the cell culture system. Furthermore, we examined the effect of phosphates on transcription activity of the TNAP promoter and TNAP mRNA stability, and examined whether glycerol, a product of β-glycerophosphate hydrolysis, affects the enzymatic activity.
Materials and methods
Cell culture
The human osteoblastic osteosarcoma cell line SaOS-2 was obtained from the RIKEN cell bank (Tsukuba, Japan). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 50 U/ml penicillin, and 50 μg/ml streptomycin in an atmosphere of 5% CO2. Stock solutions of 1 M β-glycerophosphate disodium and 100 mM sodium dihydrogenphosphate were prepared in DMEM/10% FCS and adjusted to pH 7.3–7.4. In order to examine the effect of the phosphates, β-glycerophosphate and NaH2PO4, the stock solutions were added to the culture medium to yield a final concentration of 10 mM. Levamisole, an inhibitor of TNAP, and phosphonoformic acid (PFA), an inhibitor of the type III Na/Pi cotransporter, were also prepared as 100 mM stock solutions and added to the medium to yield a final concentration of 1 mM. Similarly, media containing a series of concentrations of glycerol were prepared to examine the effect of glycerol on ALP and NPP1 activity. In order to assess ALP and NPP1 activity, cells were seeded in 12-well plates and after the designated culture times they were washed with Tris-buffered saline (TBS; 10 mM Tris–HCl and 0.15 M NaCl, pH 7.4). The cells were then harvested in TBS containing 1% (v/v) Triton X-100 and 1 mM phenylmethylsulfonyl fluoride, sonicated, and centrifuged at 10,000×g for 10 min. The supernatant was collected and analyzed for the enzyme activity and protein concentration.
Alkaline phosphatase activity
ALP activity was assessed at 37°C using 10 mM p-nitrophenylphosphate as a substrate in 0.1 M 2-amino-2-methyl-1,3-propanediol-HCl buffer (pH 10.0) containing 5 mM MgCl2. Protein concentration was determined using a Protein DC Assay kit (BioRad Laboratories, Hercules, CA, USA). ALP activity was expressed as μmol/min/mg protein.
5′-Nucleotide pyrophosphatase phosphodiesterase (NPP1) activity
5′-Nucleotide pyrophosphatase phosphodiesterase 1 (NPP1) activity was assessed at 37°C using 1 mM p-nitrophenyl thymidine-5′-monophosphate as a substrate in 50 mM Tris–HCl, pH 8.9. NPP1 activity was expressed as nmol/h/μg protein.
Mineralization assay
SaOS-2 cells were cultured with 10 mM β-glycerophosphate or 10 mM NaH2PO4 plus 1 mM levamisole or 1 mM PFA for 5 days, and then mineralization was evaluated using Alizarin Red S staining [29]. Briefly, confluent cells in a 96-well culture plate were rinsed with PBS twice and fixed with ice-cold 70% ethanol for 1 h. The cells were then stained with 0.5% Alizarin Red S for 10 min at room temperature with rotation. After being washed with PBS, photographs of the cells were obtained. Any mineralized materials were solubilized by incubation with 100 mM cetylpyridinium chloride in 10 mM sodium phosphate (pH 7.0) for 1 h at room temperature, and optical density at 570 nm was measured. Mineralization was expressed as U/mg protein, where 1 U was equal to 1.00 unit of OD at 570 nm. Protein concentration was determined in the cells cultured in parallel by using a Protein DC Assay kit. The optical density and the protein concentration were both assayed in triplicate.
Assessment of cell proliferation
After cells were cultured in a 96-well culture plate for 5 days, they were stained with MTT (3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide) at 37°C for 3 h, then incubated with 0.04 N HCl in isopropanol for 10 min at room temperature to dissolve the formazan. Cell proliferation was calculated as a percentage as follows: 100 × (OD at 595 nm/OD at 595 nm in control cells).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using an RNeasy Mini Kit (Qiagen, Hilden, Germany), after which RT-PCR was carried out. Bone-type TNAP, ENPP1 (ecto-nucleotide pyrophosphatase phosphodiesterase, which encodes NPP1), PHEX, MEPE, and ANKH mRNAs were amplified using primers described elsewhere [23]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was also amplified as an internal standard. Quantitative real-time RT-PCR was carried out to estimate mRNA levels. In order to estimate the levels of TNAP mRNA, 5 ng of cDNA was transcribed using random hexamers, then primers and a FAM-labeled TaqMan probe described elsewhere [30] were used for amplification. Fluorescence was then detected using an ABI PRISM 7700 Sequence Detector (Applied Biosystems, Foster City, CA, USA). Levels of ENPP1, PHEX and MEPE mRNAs were estimated using the TaqMan Gene Expression Assays (Inventoried) kit with FAM-labeled MGB (minor groove binder) probes. Levels of ENPP1 mRNA were estimated using 10 ng of cDNA with the Assay ID Hs00196445_m1, those of PHEX mRNA were estimated using 20 ng of cDNA with the Assay ID Hs00166525_m1, and those of MEPE mRNA were estimated using 10 ng of cDNA with Hs00220237_m1. These mRNA levels were normalized according to the levels of GAPDH mRNA, which were estimated using primers and the VIC-labeled TaqMan Probe described elsewhere [30]. For the mRNA stability assay, we used 18S ribosomal RNA (rRNA) as an internal standard.
Luciferase assays
Plasmids containing the TNAP gene promoter region and the pGL3 basic vector (pGL3BV; Promega, Madison, WC, USA) with the firefly luciferase reporter gene were constructed as described elsewhere [30]. SaOS-2 cells were seeded in 6-well plates at a density of 6 × 105 cells/well, and were transfected for 24 h with 4 μg of the luciferase reporter plasmids containing portion of the TNAP promoter region extending from −4,556 to −122 or from −1,313 to −122 using Lipofectamine 2000 (10 μl/well; Invitrogen, Carlsberg, CA, USA) according to the manufacturer’s instructions. Eight nanograms of the pRL-Tk Renilla luciferase reporter plasmid was cotransfected into the cells as a transfection efficiency control. Thereafter, the cells were cultured for 48 h with or without 10 mM β-glycerophosphate or NaH2PO4, harvested, and lysed using the Picca-gene Dual kit (Toyo Ink, Tokyo, Japan). Twenty-microliter aliquots of lysate were added to the Picca-gene substrate solution for dual luciferase assays. Luciferase activity was measured using a Lumat LB 9506 luminometer (EG & G Berthold, Bad Wildbad, Germany) and normalized according to the levels of Renilla luciferase activity.
mRNA stability
SaOS-2 cells were cultured overnight and then treated with 10 mM β-glycerophosphate or 10 mM NaH2PO4 for 48 h. In order to assess mRNA stability, a final concentration of 7.5 μg/ml of actinomycin D was added, cells were harvested at 0, 4, 8, and 24 h, and then total RNA was extracted. The remaining TNAP mRNA levels were determined using quantitative real-time RT-PCR with 18S rRNA as an internal standard. TNAP mRNA levels were normalized according to 18S rRNA levels as estimated using the TaqMan Ribosomal RNA Control Reagents kit (Applied Biosystems). The half-life of the mRNA was calculated from a linear regression of decay of the mRNA levels.
Statistical analysis
All data except for responses to glycerol are expressed as mean ± SEM, and glycerol response data are expressed as mean ± SD. The statistical significance of differences between the groups was estimated using unpaired t-test. Values obtained from cells cultured in control medium and those from cells cultured with β-glycerophosphate or NaH2PO4 were compared. Similarly, values obtained from cells cultured with β-glycerophosphate or NaH2PO4 and those from cells cultured with the inhibitors were compared. For the glycerol response experiment, differences among the groups were analyzed using one-way analysis of variance (ANOVA). A P value less than 0.05 denoted the presence of a statistically significant difference.
Results
Effects of inhibitors on mineralization
Figure 1 shows the levels of mineralization in cell cultures incubated with the various phosphates and inhibitors. Addition of β-glycerophosphate resulted in a marked increase in mineralization relative to the control, which was completely inhibited by addition of levamisole, a TNAP inhibitor, and partly inhibited by addition of PFA, a Pi transporter inhibitor. The increase in mineralization relative to the control after addition of inorganic NaH2PO4 was smaller than that produced by β-glycerophosphate. This increase was inhibited completely by PFA but not by levamisole. Although addition of levamisole to the medium containing NaH2PO4 appeared to increase the levels of mineralization, no statistical difference in the levels of mineralization between addition of NaH2PO4 and that of NaH2PO4 plus levamisole was exhibited. This seeming increase in the levels of mineralization may be due to low protein concentration that was caused by reduced cell viability as shown in Fig. 2.

Effects of the inhibitors levamisole and PFA on mineralization. Mineralization was assessed 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors by using Alizarin Red S staining. The upper panel shows representative stained cell cultures for each treatment. Values are expressed as a ratio relative to mineralization unit of the cells cultured without β-glycerophosphate or NaH2PO4. Data are expressed as mean ± SEM for three independent experiments, and each experiment was carried out in triplicate. Differences between the groups were evaluated using unpaired t-test. ** P < 0.01, * P < 0.05. Lev.: levamisole, β-GP: β-glycerophosphate

Assessment of cell proliferation. Cell proliferation was assessed 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors by using MTT assay. Values are expressed as a percentage described in the Materials and methods section. Data are expressed as mean ± SEM for four independent experiments, and each experiment was carried out in triplicate. Differences between the groups were evaluated using unpaired t-test. ** P < 0.01, * P < 0.05
Assessment of cell proliferation
Cell viability was evaluated using an MTT assay 5 days after addition of the phosphates and inhibitors. Addition of β-glycerophosphate or NaH2PO4 reduced number of viable cells. Furthermore, addition of PFA to the medium containing β-glycerophosphate as well as addition of levamisole to the medium containing NaH2PO4 further reduced cell viability (Fig. 2). In particular, addition of levamisole to the medium containing NaH2PO4 exhibited low protein concentration.
Effects of inhibitors on TNAP activity and mRNA levels
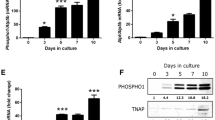
ALP enzymatic activity was determined at 5 days and TNAP mRNA levels were determined at 3 days after addition of the phosphates and inhibitors. β-Glycerophosphate-induced TNAP mRNA expression and ALP enzymatic activity were inhibited by levamisole but not PFA (Fig. 3). Although levamisole inhibited NaH2PO4-induced TNAP mRNA expression, the enzymatic activity did not change. Low protein concentration in levamisole plus NaH2PO4 condition may account for this discrepancy.

Effects of the inhibitors levamisole and PFA on expression of TNAP. (a) ALP enzymatic activity was assessed 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors. Values are expressed as a percentage relative to activity in cells cultured without β-glycerophosphate or NaH2PO4. Data are expressed as mean ± SEM for four independent experiments, and each experiment was carried out in triplicate. Differences between the groups were evaluated using unpaired t-test. *** P < 0.001, ** P < 0.01, * P < 0.05. (b) Levels of TNAP mRNA were assessed 3 days after addition of the phosphates and inhibitors by using a quantitative real-time RT-PCR analysis. Values are expressed as a percentage relative to the levels of TNAP mRNA in cells cultured without β-glycerophosphate or NaH2PO4. Data are expressed as mean ± SEM for five independent experiments, and each experiment was carried out in duplicate. Differences between the groups were evaluated using unpaired t-test. *** P < 0.001, * P < 0.05
Effects of inhibitors on NPP1 activity and mRNA levels
NPP1 enzymatic activity was determined at 5 days and ENPP1 mRNA levels were determined at 3 days after addition of the phosphates and inhibitors. Both β-glycerophosphate and inorganic NaH2PO4 induced NPP1 enzymatic activity. β-Glycerophosphate-induced NPP1 activity was inhibited by levamisole but not PFA, whereas NaH2PO4-induced NPP1 activity was inhibited by PFA. However, no statistically significant changes in ENPP1 mRNA levels were observed after addition of the phosphates, suggesting that the changes in NPP1 activity was posttranscriptional (Fig. 4).

Effects of the inhibitors levamisole and PFA on expression of NPP1. (a) NPP1 activity was assessed 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors. Values are expressed as a percentage relative to activity in cells cultured without β-glycerophosphate or NaH2PO4. Data are expressed as mean ± SEM for four independent experiments, and each experiment was carried out in triplicate. Differences between the groups were evaluated using unpaired t test. *** P < 0.001. (b) Levels of ENPP1 mRNA were assessed 3 days after addition of the phosphates and inhibitors by using quantitative RT-PCR analysis. Values are expressed as a percentage relative to the levels of ENPP1 mRNA in cells cultured without β-glycerophosphate or NaH2PO4. Data are expressed as mean ± SEM for three independent experiments, and each experiment was carried out in duplicate. No statistical differences between the groups were observed
Effects of inhibitors on PHEX and MEPE mRNA levels
RT-PCR analysis revealed an apparent increase in PHEX mRNA expression 5 days after the addition of β-glycerophosphate, which seemed to be inhibited by levamisole (Fig. 5). These findings were confirmed by quantitative RT-PCR analysis (Fig. 6). Levamisole completely inhibited and PFA partly inhibited β-glycerophosphate-induced PHEX expression. MEPE mRNA expression seemed to increase with addition of NaH2PO4, but the increase could not be confirmed by quantitative RT-PCR analysis (data not shown). Although quantitative RT-PCR analysis of TNAP mRNA carried out at 3 days after addition of various phosphates and inhibitors showed changes by phosphates and inhibitors (Fig. 3b), TNAP expression in Fig. 5 appeared not to change. In explanation of this data, intensity of the RT-PCR bands obtained from cells in control medium and that from cells treated with phosphates is almost similar at 5 days after addition of phosphates [23].

Effects of the inhibitors levamisole and PFA on expression of the phosphate regulating genes mRNA (RT-PCR). Total RNA was extracted 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors, and then RT-PCR was carried out using the primers described in the Materials and methods section. The PCR products were analyzed on a 1.5% agarose gel. C: control; β: β-glycerophosphate; βL: β-glycerophosphate + levamisole; βP: β-glycerophosphate + PFA; N: NaH2PO4; NL: NaH2PO4 + levamisole; NP: NaH2PO4 + PFA

Effects of the inhibitors levamisole and PFA on expression of PHEX mRNA (quantitative RT-PCR). Quantitative real-time RT-PCR was performed 5 days after addition of the phosphates (β-glycerophosphate and NaH2PO4) and inhibitors. PHEX mRNA levels are expressed as a percentage relative to the levels of PHEX mRNA in cells cultured without β-glycerophosphate or NaH2PO4. The PHEX mRNA level was normalized according to the level of GAPDH mRNA. Data are expressed as mean ± SEM for three independent experiments, and each experiment was carried out in duplicate. Differences between the groups were evaluated using unpaired t-test. ** P < 0.01, * P < 0.05
Luciferase assay
The dual luciferase assay showed that transcription activity over at least 4.5 kb of the region upstream of the human TNAP gene was not increased by addition of either β-glycerophosphate or NaH2PO4 (Fig. 7), suggesting that the phosphates do not directly transactivate the promoter region of the TNAP gene.

Effects of the phosphates β-glycerophosphate and NaH2PO4 on relative luciferase activity driven by the promoter region of the TNAP gene. Two reporter plasmids containing regions −4,556 to −122 and −1,313 to −122 of the TNAP promoter region were transfected for 24 h into SaOS-2 cells, then the cells were cultured for 48 h with β-glycerophosphate or NaH2PO4. Luciferase activity was measured 48 h after addition of the phosphates. Luciferase activity is expressed as a ratio relative to activity in the lysates of cells cultured without β-glycerophosphate or NaH2PO4
mRNA stability
Decay curves of the TNAP mRNA levels after addition of actinomycin D revealed that neither β-glycerophosphate nor NaH2PO4 had any effect on TNAP mRNA stability (data not shown).
Effects of glycerol on ALP and NPP1 activity
SaOS-2 cells were cultured for 5 days with 0.5–30 mM glycerol to evaluate the influence of glycerol on ALP and NPP1 activity. Glycerol did not induce ALP and NPP1 enzymatic activity (Fig. 8).

Effects of glycerol on ALP and NPP1 activity. SaOS-2 cells were cultured for 5 days with 0.5–30 mM glycerol to evaluate the influence of glycerol on ALP (a) and NPP1 (b) activity. Data are expressed as mean ± SD for an experiment carried out in triplicate. Differences among the groups were evaluated using one way ANOVA. No statistical differences among the groups were observed
Discussion
In the present study, we showed that a TNAP inhibitor, levamisole, inhibited β-glycerophosphate-induced mineralization and TNAP expression, whereas a Pi transporter inhibitor, PFA, inhibited Pi-induced mineralization but not TNAP expression. Induction of PHEX by β-glycerophosphate was inhibited by levamisole. These results suggest that the products of the extracellular TNAP-induced hydrolysis of β-glycerophosphate induced TNAP expression and mineralization and that PHEX expression may be linked to TNAP expression. Since one of the products of TNAP-induced hydrolysis of β-glycerophosphate, glycerol, was found to not affect TNAP expression, the factor that induced TNAP expression and mineralization is phosphate. However, phosphates did not directly transactivate the promoter region of the TNAP gene and did not affect TNAP mRNA stability.
We used human osteoblast-like cells, SaOS-2, derived from osteosarcoma in this study. SaOS-2 cells have several osteoblastic properties such as elevated ALP activity, release of matrix-vesicle-like structures with the capacity to mineralize, and production of mineralized matrix in chambers implanted in nude mice [31, 32], suggesting that they mimic primary osteoblast to a certain extent. SaOS-2 cells show extremely high ALP enzymatic activity without abnormal TNAP gene structure [33], and deposit around them an extensive collagenous matrix that is able to mineralize in the presence of an exogenous phosphate donor, β-glycerophosphate [34]. We previously demonstrated that SaOS-2 cells show either β-glycerophosphate-induced or retinoic acid-induced TNAP expression [23, 30]. β-Glycerophosphate was used as an artificial substrate for TNAP in the present study as well as in many of the in vitro studies, which increases the local phosphate concentration. When β-glycerophosphate supplements are kept at levels that produce Pi concentration in agreement with physiologic levels, its use may reflect mineralization per se rather than ALP expression [35]. β-Glycerophosphate and elevated levels of Pi are required for the initiation of mineralization, but once the process is initiated, mineralization will continue at non-elevated levels of phosphates [36]. A study using Fourier transform infrared imaging spectroscopy revealed that hydrolysis of AMP or β-glycerophosphate yields Pi which leads to the formation of mature crystalline, apatite mineral, while the hydrolysis of ATP inhibits the formation of mature hydroxyapatite [37]. As regards concentration of phosphates, we used equimolar concentration of β-glycerophosphate and inorganic NaH2PO4. In a study using mouse MC3T3-E1 cells, supplementation of 10 mM β-glycerophosphate yields 4.8 mM Pi after 48 h culture [38], suggesting that degraded products of β-glycerophosphate stimulate TNAP expression during the time lag and that actual concentration of β-glycerophosphate and Pi may not be equivalent in a cell culture system. Accordingly, further analysis of quantitative comparison of effects caused by β-glycerophosphate with those caused by NaH2PO4 may be necessary.
We have previously demonstrated that β-glycerophosphate and NaH2PO4 induced TNAP mRNA expression and enzymatic activity [23]. In the present study, we confirmed these results and exhibited that β-glycerophosphate and NaH2PO4 reduced cell viability (Fig. 2). Inhibition of cell proliferation occurs by apoptosis due to incorporation of Pi into the cells through Pi transporters in osteoblast-like cells [39]. In addition, PFA or levamisole further contributed to reduction of cell viability when they were added to the media containing β-glycerophosphate or NaH2PO4, respectively. Levamisole reduces thymidine incorporation into DNA of chick osteoblastic cells [40]. Inhibition of TNAP by levamisole resulted in complete inhibition of β-glycerophosphate-induced mineralization (Fig. 1), consistent with the previous report that levamisole and PPi inhibit β-glycerophosphate-induced mineralization [40]. Furthermore, β-glycerophosphate-induced ALP mRNA expression was inhibited by levamisole, in parallel with inhibition of mineralization and TNAP enzymatic activity (Fig. 3), which indicates that levamisole acts at the transcription step. Levamisole also inhibits expression of the type X collagen α1 chain mRNA in the chick chondrocytes, though the mechanism is not clearly defined [41]. Nevertheless, because levamisole is a low-molecular-weight uncompetitive inhibitor of TNAP enzymatic activity [42], the products of TNAP-induced hydrolysis of β-glycerophosphate may induce transcription of TNAP, given that inhibition of TNAP activity caused inhibition of TNAP mRNA expression. However, although levamisole inhibited NaH2PO4-induced TNAP mRNA, TNAP enzymatic activity of the cells cultured with NaH2PO4 and levamisole was not affected. This inconsistency may be caused by seeming high specific activity due to low protein concentration in NaH2PO4 plus levamisole that may exert slightly toxic effect on the cells.
PFA inhibited both β-glycerophosphate-induced and NaH2PO4-induced mineralization, but did not affect TNAP expression (Figs. 1 and 3). PFA is a competitive inhibitor of the type III Na/Pi cotransporter that does not inhibit ALP activity [43]. These results indicate that NaH2PO4-induced mineralization requires an influx of Pi through the type III Na/Pi cotransporter. The type III Na/Pi cotransporter is a widely expressed transmembrane protein and transports Pi into cells [27]. SaOS-2 cells express the type III Na/Pi cotransporter, and an osteotropic factor, insulin-like growth factor (IGF) I, stimulates expression of the Na/Pi cotransporter [44]. The type III Na/Pi cotransporter expressed in isolated matrix vesicles plays a role in matrix mineralization of ATDC5 chondrogenic cells [28], and a study using PFA showed that the type III Na/Pi cotransporter also plays an important role in bone mineralization in rat [45]. It is likely that β-glycerophosphate-induced mineralization requires both TNAP activity and an influx of Pi through the Pi transporter.
Levamisole inhibited β-glycerophosphate-induced NPP1 activity and PFA inhibited NaH2PO4-induced NPP1 activity (Fig. 4). NPP1 forms PPi from ATP, which inhibits hydroxyapatite formation [9, 40]. ENPP1 mRNA expression, however, was neither enhanced by addition of β-glycerophosphate or NaH2PO4 nor inhibited by levamisole or PFA, suggesting that the changes in NPP1 activity were not caused by changes in transcription, but were posttranscriptionally responded to the changes in TNAP activity and Pi influx. Addition of exogenous TNAP and transfection of TNAP cDNA into mouse osteoblastic cells increase ENPP1 expression and NPP1 activity accompanied by enhancement of mineralization [46, 47]. The increase in NPP1 activity when β-glycerophosphate was added (Fig. 4a) may be a response to the increase in TNAP activity, which may account for why mineralization proceeded under this condition. Although regulation of NPP1 by Pi has not been investigated, inhibition of Pi influx by PFA resulted in the decrease in NPP1 activity.
PHEX is a transmembrane endopeptidase that is predominantly expressed in bone and teeth [48]. It increases Pi reabsorption in the renal proximal tubules by clearance of a phosphaturic factor or activation of a phosphate-conserving factor, resulting in retention of Pi [48]. Mutations in the human PHEX gene cause X-linked hypophosphatemic rickets, which is characterized by hypophosphatemia and hypomineralization of bone [49]. β-Glycerophosphate upregulates the PHEX gene in primary mouse osteoblasts [50] and in SaOS-2 cells after induction of TNAP [23]. In the present study, levamisole completely inhibited and PFA partly inhibited β-glycerophosphate-induced PHEX expression (Fig. 6). This pattern of inhibition is similar to that observed for inhibition of mineralization, that is, levamisole completely inhibited and PFA partly inhibited mineralization. As regards the relationships between PHEX and mineralization, although the PHEX induction and the increased mineralization do not seem to be related directly each other in the in vitro experiment, patients with X-linked hypophosphatemic rickets show skeletal hypomineralization [49]. In addition, U2OS osteoblast-like cells transfected with active TNAP cDNA exhibit increased levels of PHEX mRNA accompanied by mineralization when cultured with β-glycerophosphate, whereas U2OS cells transfected with mutant TNAP cDNA do not respond to β-glycerophosphate [51]. Therefore, although the mechanism by which this occurs is unknown at present, TNAP may be involved in PHEX induction. In contrast, NaH2PO4 did not affect PHEX expression.
The mechanism by which phosphates induce TNAP is unknown. A luciferase assay using reporter plasmids containing the promoter region of the human TNAP gene showed no direct transactivation of the gene by the phosphates (Fig. 7). Thus, the induction mechanism may be indirect or a region responsive to the phosphates may be situated in a region far from the coding region. We identified a retinoic acid response element in the promoter region of the human TNAP gene [30]. In contrast, we found that there was no direct transactivation of the TNAP gene by the active form of vitamin D [52]. In fact, vitamin D modulated TNAP gene expression posttranscriptionally by increasing mRNA stability [52]. In the present study, therefore, we determined whether addition of the phosphates altered mRNA stability, but the phosphates had no effect on mRNA stability.
These results raise the question of what stimulated the expression of TNAP. One of the products of TNAP-induced hydrolysis of β-glycerophosphate is glycerol; however, we found that glycerol has no effect on ALP and NPP1 activity (Fig. 8). Glycerol induces osmotic stress [53] but has not been shown to be involved in bone metabolism. Thus, expression of TNAP is induced by signals obtaining from the phosphates derived from degraded β-glycerophosphate and transported by the Na/Pi cotransporter. Pi is a specific signal for induction of osteopontin gene expression in mouse MC3T3-E1 cells through a member of the MAPK (mitogen activated protein kinases) ERK1/2 (extracellular signal-regulated kinase), protein kinase C, and proteasome [54]. Pi also stimulates matrix Gla protein expression in mouse chondrocytes through ERK1/2 signaling pathway [55]. Further analysis whether MAPK and other kinase cascades involve in phosphate-stimulated TNAP expression is needed.
References
Harris H (1989) The human alkaline phosphatases: what we know and what we don’t know. Clin Chim Acta 186:133–150. doi:10.1016/0009-8981(90)90031-M
Millán JL (2006) Mammalian alkaline phosphatases. Wiley-VCH, Weinheim
Weiss MJ, Ray K, Henthorn PS et al (1988) Structure of the human liver/bone/kidney alkaline phosphatase gene. J Biol Chem 263:12002–12010
Hui M, Tenenbaum HC (1998) New face of an old enzyme: alkaline phosphatases may contribute to human tissue aging by inducing tissue hardening and calcification. Anat Rec 253:91–94. doi:10.1002/(SICI)1097-0185(199806)253:3<91::AID-AR5>3.0.CO;2-H
Anderson HC, Sipe JB, Hessle L et al (2004) Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol 164:841–847
Anderson HC (1995) Molecular biology of matrix vesicles. Clin Orthop Relat Res 314:266–280
Johnson K, Moffa A, Chen Y (1999) Matrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res 14:883–892. doi:10.1359/jbmr.1999.14.6.883
Ho AM, Johnson MD, Kingsley DM (2000) Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289:265–270. doi:10.1126/science.289.5477.265
Terkeltaub RA (2001) Inorganic pyrophosphate generation and disposition on pathophysiology. Am J Physiol 281:C1–C11
Hessle L, Johnson KA, Anderson HC et al (2002) Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA 99:9445–9449. doi:10.1073/pnas.142063399
Harmey D, Hessle L, Narisawa S et al (2004) Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank. Am J Pathol 164:1199–1209
Weiss MJ, Cole DEC, Ray K et al (1988) A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc Natl Acad Sci USA 85:7666–7669. doi:10.1073/pnas.85.20.7666
Henthorn PS, Raducha M, Fedde KN et al (1992) Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc Natl Acad Sci USA 89:9924–9928. doi:10.1073/pnas.89.20.9924
Orimo H, Hayashi Z, Watanabe A et al (1994) Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patients with hypophosphatasia. Hum Mol Genet 3:1683–1684. doi:10.1093/hmg/3.9.1683
Orimo H, Goseki-Sone M, Sato S et al (1997) Detection of deletion 1154-1156 hypophosphatasia mutation using TNSALP exon amplification. Genomics 42:364–366. doi:10.1006/geno.1997.4733
Mornet E (2000) Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat 15:309–315. doi:10.1002/(SICI)1098-1004(200004)15:4<309::AID-HUMU2>3.0.CO;2-C
Orimo H, Girschick HJ, Goseki-Sone M et al (2001) Mutational analysis and functional correlation with phenotype in German patients with childhood-type hypophosphatasia. J Bone Miner Res 16:2313–2319. doi:10.1359/jbmr.2001.16.12.2313
Waymire KG, Mahunen JD, Jaje JM et al (1995) Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet 11:45–50. doi:10.1038/ng0995-45
Fedde KN, Blair L, Silverstein J et al (1999) Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res 14:2015–2026. doi:10.1359/jbmr.1999.14.12.2015
Whyte MP (2001) Hypophosphatasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn., vol 4. McGraw-Hill, New York, pp 5313-5329
Wennberg C, Hessle L, Lundberg P et al (2000) Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res 15:1879–1888. doi:10.1359/jbmr.2000.15.10.1879
Goseki-Sone M, Sogabe N, Fukushi-Irie M et al (2005) Functional analysis of the single nucleotide polymorphism (787T>C) in the tissue-nonspecific alkaline phosphatase gene associated with BMD. J Bone Miner Res 20:773–782. doi:10.1359/JBMR.041229
Orimo H, Shimada T (2006) Effects of phosphates on the expression of tissue-nonspecific alkaline phosphatase gene and phosphate-regulating genes in short-term cultures of human osteosarcoma cell lines. Mol Cell Biochem 282:101–108. doi:10.1007/s11010-006-1520-6
Quarles LD (2003) FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol 285:E1–E9
Liu S, Zhou J, Tang W et al (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol 291:E38–E46
Rowe PSN, Kumagai Y, Gutierrez G et al (2004) MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 34:303–319
Collins JF, Bai L, Ghishan FK (2004) The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch-Eur J Physiol 447:647–652
Guicheux J, Palmer G, Shukunami C et al (2000) A novel in vitro culture system for analysis of functional role of phosphate transport in endochondral ossification. Bone 27:69–74
Johnson K, Hashimoto S, Lotz M et al (2001) Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum 44:1071–1081
Orimo H, Shimada T (2005) Regulation of the human tissue-nonspecific alkaline phosphatase gene expression by all-trans-retinoic acid in SaOS-2 osteosarcoma cell line. Bone 36:866–876
Rodan SB, Imai Y, Thiede MA et al (1987) Characterization of a human osteosarcoma cell line (Saos-2) with osteoblastic properties. Cancer Res 47:4961–4966
Fedde KN (1992) Human osteosarcoma cells spontaneously release matrix-vesicle-like structures with the capacity to mineralize. Bone Miner 17:145–151
Stinson RA, Thacker JD, Lin CC (1993) Expression and nature of the alkaline phosphatase gene in cultured osteosarcoma cells. Clin Chim Acta 221:105–114
McQuillan DJ, Richardson MD, Bateman JF (1995) Matrix deposition by a calcifying human osteogenic sarcoma cell line (SAOS-2). Bone 16:415–426
Boskey AL (1998) Biomineralization: conflicts, challenges, and opportunities. J Cell Biochem Suppl 30/31:83–91
Beck GR Jr (2003) Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem 90:234–243
Garimella R, Bi X, Anderson HC et al (2006) Nature of phosphate substrate as a major determinant of mineral type formed in matrix vesicle-mediated in vitro mineralization: an FTIR imaging study. Bone 38:811–817
Nakano Y, Addison WN, Kaartinen MT (2007) ATP-mediated mineralization of MC3T3-E1 osteoblast cultures. Bone 41:549–561
Adams CS, Mansfield K, Perlot RL et al (2001) Matrix regulation of skeletal cell apoptosis: role of calcium and phosphate ions. J Biol Chem 276:20316–20322
Tenenbaum HC (1987) Levamisole and inorganic pyrophosphate inhibit beta-glycerophosphate induced mineralization of bone formed in vitro. Bone Miner 3:13–26
Thomas JT, Boot-Handford RP, Grant ME (1990) Modulation of type X collagen gene expression by calcium β-glycerophosphate and levamisole: implications for a possible role for type X collagen in endochondral bone formation. J Cell Sci 95:639–648
Van Bell H (1976) Alkaline phosphatase. I. Kinetics and inhibition by levamisole of purified isoenzymes from humans. Clin Chem 22:972–976
Szczepanska-Konkel M, Yusufi ANK, Van Scoy M et al (1986) Phosphonocarboxylic acids as specific inhibitors of Na+-dependent transport of phosphate across renal brush border membrane. J Biol Chem 261:6375–6383
Palmer G, Bonjour J-P, Caverzasio J (1997) Expression of a newly identified phosphate transporter/retrovirus receptor in human SaOS-2 osteoblast-like cells and its regulation by insulin-like growth factor I. Endocrinology 138:5202–5209
Yoshiko Y, Candeliere GA, Maeda N et al (2007) Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol 27:4465–4474
Addison WN, Azari F, Sørensen ES et al (2007) Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem 282:15872–15883
Johnson KA, Hessle L, Vaingankar S et al (2000) Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol 279:R1365–R1377
Bielesz B, Klaushofer K, Oberbauer R (2004) Renal phosphate loss in hereditary and acquired disorders of bone mineralization. Bone 35:1229–1239
The HYP Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136
Ecarot B, Desbarats M (1999) 1,25-(OH)2D3 down-regulates expression of Phex, a marker of the mature osteoblast. Endocrinology 140:1192–1199
Orimo H, Goseki-Sone M, Hosoi T et al (2008) Functional assay of the mutant tissue-nonspecific alkaline phosphatase gene using U2OS osteoblast-like cells. Mol Genet Metab. doi:10.1016/j.ymgme.2008.03.015
Orimo H, Shimada T (2006) Posttranscriptional modulation of the human tissue-nonspecific alkaline phosphatase gene expression by 1,25-dihydroxyvitamin D3 in MG-63 osteoblastic osteosarcoma cells. Nutr Res 26:227–234
Brisson D, Vahl M-C, St-Pierre J et al (2001) Glycerol: a neglected variable in metabolic processes? BioEssays 23:534–542
Beck GR Jr, Knecht N (2003) Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem 278:41921–41929
Julien M, Magne D, Masson M et al (2007) Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology 148:530–537
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Orimo, H., Shimada, T. The role of tissue-nonspecific alkaline phosphatase in the phosphate-induced activation of alkaline phosphatase and mineralization in SaOS-2 human osteoblast-like cells. Mol Cell Biochem 315, 51–60 (2008). https://doi.org/10.1007/s11010-008-9788-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9788-3