
Abstract
The majority of people with autoimmune disorders, including those with rheumatoid arthritis, osteoarthritis, and tendonitis report pain, stiffness, and inflammation as major contributors to their worse quality of life in terms of overall health. Of all the available treatment options, COX inhibitors are the ones that are utilized most frequently to ease the symptoms. Various signaling cascades have been reported to be involved in the pathogenesis of rheumatoid arthritis which includes JAK/STAT, MAPK, and NF-kB signaling pathways, and several allopathic inhibitors (tofacitinib and baricitinib) have been reported to target the components of these cascades and have received approval for RA treatment. However, the prolonged use of these COX inhibitors and other allopathic drugs can pose serious health challenges due to their significant side effects. Therefore, searching for a more effective and side effect–free treatment for rheumatoid arthritis has unveiled phytochemicals as both productive and promising. Their therapeutic ability helps develop potent and safe drugs targeting immune-inflammatory diseases including RA. Various scientific databases were used for searching articles such as NCBI, SpringerLink, BioMed Central, ResearchGate, Google Scholar, Scopus, Nature, Wiley Online Library, and ScienceDirect. This review lists various phytochemicals and discusses their potential molecular targets in RA treatment, as demonstrated by various in vitro, in vivo (pre-clinical), and clinical studies. Several pre-clinical and clinical studies suggest that various phytochemicals can be an alternative promising intervention for attenuating and managing inflammation-associated pathogenesis of rheumatoid arthritis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
COX inhibitors are a class of drugs that are commonly used for the treatment of pain, fever, and inflammation. They have been used for decades for a variety of medical conditions and have become one of the most commonly used groups of drugs in the world (Toda 2021). The history of COX inhibitors dates back to the late nineteenth century when salicylates (such as aspirin) were first discovered to have pain-relieving and anti-inflammatory properties. Since then, many other COX inhibitors have been developed, including ibuprofen, naproxen, and celecoxib (Hacker and Satre 2021). The mechanism of action of COX inhibitors is related to their ability to inhibit the activity of enzymes called cyclooxygenases (COX), which are involved in the production of prostaglandins. Prostaglandins are chemical mediators that play a role in inflammation, pain, and fever. By inhibiting the activity of COX enzymes, COX inhibitors reduce the production of prostaglandins, thereby reducing pain, fever, and inflammation (Ngo and Addison 2018). Owing to their anti-inflammatory properties, COX inhibitors remain a popular and effective option for treating RA and tendinitis. They continue to be prescribed by healthcare providers and widely used by patients. There are currently at least 20 distinct allopathic COX inhibitors from six broad groups that are available for use in humans based on their chemical makeup (Chang 2015). Commonly used allopathic cox inhibitors include non-selective COX inhibitors like aspirin, a salicylate derivative that operates by blocking prostaglandin synthesis, inhibiting NF-kappa B, and inducing iNOS and COX inhibition (Kersley 2009). Diclofenac, an acetic acid derivative, functions by blocking VLA-4 activation (Berney 2007). Sulindac, also an acetic acid derivative, operates by inhibiting NF-kappa B (Horino et al. 2019). Ibuprofen, a propanoic acid derivative, operates through NF-kappa B inhibition (Weiser 2021). Naproxen, a propanoic acid derivative, functions through PI3/AKT inhibition (Hadi et al. 2016). Piroxicam and meloxicam are both enolic acid derivatives. They share the common mechanism of blocking β2 integrin activation (Samra et al. 2022; Ma et al. 2022). Oxaprozin, propanoic acid derivative, operates through PI3/AKT inhibition (Zhao et al. 2023). Meclofenamic acid, anthranilic acid derivative, functions by inducing L-shedding (Narsinghani and Chaturvedi 2006). Indomethacin, derived from acetic acid, operates through NF-kappa B inhibition (Janakiraman et al. 2018). Celecoxib (derived from sulfonamide) and etoricoxib (derived from carboxylic acid) are selective COX inhibitors that inhibit prostaglandin synthesis (Feng et al. 2018). Commonly used allopathic COX inhibitors include non-selective COX inhibitors like aspirin, a salicylate derivative that operates by blocking prostaglandin synthesis, inhibiting NF-Kappa B, and inducing iNOS and COX inhibition (Kersley 2009). Diclofenac, an acetic acid derivative, functions by blocking VLA-4 activation (Berney 2007). Sulindac, also an acetic acid derivative, operates by inhibiting NF-kappa B (Horino et al. 2019). Ibuprofen, a propanoic acid derivative, operates through NF-kappa B inhibition (Weiser 2021). Naproxen, a propanoic acid derivative, functions through PI3/AKT inhibition (Hadi et al. 2016). Piroxicam and meloxicam are both enolic acid derivatives. They share the common mechanism of blocking β2 integrin activation (Samra et al. 2022; Ma et al. 2022). Oxaprozin, propanoic acid derivative, operates through PI3/AKT inhibition (Zhao et al. 2023). Meclofenamic acid, an anthanilic acid derivative, functions by inducing L-shedding (Narsinghani and Chaturvedi 2006). Indomethacin, derived from acetic acid, operates through NF-kappa B inhibition (Janakiraman et al. 2018). Celecoxib (derived from sulfonamide) and etoricoxib (derived from carboxylic acid) are selective COX inhibitors that inhibit prostaglandin synthesis (Feng et al. 2018; Cheng et al. 2021).
Though COX inhibitors are effective in managing pain and inflammation, they also have numerous side effects. Common side effects of COX inhibitors include gastrointestinal symptoms such as nausea, vomiting, diarrhea, and abdominal pain. They can also cause damage to the gastrointestinal lining, leading to bleeding and ulceration, headache, dizziness, and an increased risk of heart attack or stroke (Singgih and Achmad 2020). Other side effects of COX inhibitors include renal adverse effects and hematologic side effects. Inhibition of both COX-1 and COX-2 can harm the kidneys. While this may not be a significant problem for patients with normal renal function, those with renal dysfunction are more susceptible to complications from reduced prostaglandin levels caused by COX inhibitors (Ansari 2016). Due to their antiplatelet activity, non-selective COX inhibitors are more likely to have hematologic side effects in patients with GI ulcers, von Willebrand disease, hemophilia, and thrombocytopenia and some perioperative situations (Gargya et al. 2017). Although COX inhibitors are effective in inflammation due to associated side effects in long-term use, there has been a surge in alternative therapies for managing pain and inflammation. Some of these alternatives include natural compounds (plant-derived COX inhibitors), such as ginger and turmeric, which have anti-inflammatory effects but may have fewer side effects (Sharma et al. 2021).
Methodology
This review assessed the role of inflammation and oxidative stress in rheumatoid arthritis and explored the role of phytochemicals on key signaling mechanisms involved in the disease. It provides an overview of in vitro, in vivo, and clinical studies that have investigated the underlined mechanisms and their critical targets in RA. The survey process for this study encompassed a comprehensive search across multiple esteemed scientific databases. These databases included NCBI, Springer, BioMed Central, ResearchGate, Google Scholar, Nature, Wiley Online Library, Frontiers, and ScienceDirect. The terms used during the search of the database include COX inhibitors, phytochemicals, rheumatoid arthritis, signaling pathways, oxidative stress, factors affecting inflammation, and targeting molecules of COX inhibitors. These terms were chosen based on their relevance to the topics under study. Initially, we found 310 articles that seemed to be relevant. After going through their titles and abstracts, we selected 218 papers that aligned with the objectives of our review, thereby establishing the credible scientific literature for the article.
Role of inflammation in rheumatoid arthritis and tendinitis
Inflammation is a complex biological response of the body against harmful stimuli, such as pathogens, damaged cells, or irritants. It is characterized by redness, heat, swelling, and pain in the affected area which is the body’s way of signaling the immune system to eradicate the source of injury or infection. Post pathogenic attack or infection, the body reacts by generating an inflammatory response through the release of inflammatory markers such as C-reactive protein (CRP), tumor necrosis factor (TNF), interleukin-6 (IL-6), interleukin-1 beta (IL-1β), prostaglandin E2 (PGE2), and nitric oxide (NO) (Yang et al. 2019). Recent studies confirmed the role of these inflammatory markers in RA, but there are limited reports about the involvement of pro-inflammatory markers like IL-1β in the case of tendinitis (Jomaa et al. 2020).
Rheumatoid arthritis and inflammation
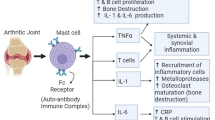
The term arthritis encompasses over 100 different disorders that affect joints with symptoms like pain, stiffness, fatigue, and deformity. This condition can also affect surrounding tissues, connective tissue in muscles, skin, and bones (Sur et al. 2021). The underlying causes can range from inflammation, degeneration, metabolism, and viral illness. Rheumatoid arthritis (RA) is the most common form of chronic inflammatory arthritis. This disease is characterized by synovial inflammation and associated tissue damage and swelling of soft tissues surrounding synovial joints (McInnes and Schett 2017). RA is considered an autoimmune disease and is associated with risk factors such as smoking, older age, positive family history, and female gender (Alieva 2016). There is evidence that patients with RA, particularly those with more severe illnesses, have a shorter life expectancy compared to the general population. The exact cause of RA remains unknown, but numerous studies suggest that a combination of genetic, environmental, and hormonal factors may play a role in its development (Mateen et al. 2016). The advancement of knowledge about the molecular and immunological processes contributing to rheumatoid arthritis (RA) has greatly improved in recent years. Elevated levels of pro-inflammatory cytokines such as C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) in both synovial tissue and plasma of RA patients have been identified as indicators of inflammation (Yang et al. 2019). The imbalance between pro-inflammatory and anti-inflammatory cytokines is a hallmark of autoimmune disorders, including RA (Uttra et al. 2019). Their concentration in blood corresponds to the severity of the inflammation. The synovial tissue is considered to be the primary site of inflammation in RA (Takeuchi 2022). The diagnosis of RA can be a challenging task due to the absence of a single diagnostic test and the variable symptoms among individuals. Symptoms may not appear until several months after the onset of joint discomfort. Furthermore, the results of hematologic and X-ray screening may still be normal even after several months of joint discomfort (Ziegelasch et al. 2017). The diagnostic criteria for definite rheumatoid arthritis (RA) were updated by the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR); according to the criteria given, there must be the presence of synovitis in at least one joint and a total score of 6 or higher from scores in four categories: the number and location of involved joints, serologic abnormality, elevated acute-phase response, and the level of rheumatoid factor (Aletaha and Smolen 2018). These updated classification criteria provide a new method for identifying individuals with early symptoms of RA and may benefit from early treatment.
Several studies have reported that IL-1 and TNF-α (tumor necrosis factor-α) are key players in the development of RA (Vasanthi et al. 2007; Unal et al. 2008). IL-1 is a major contributor to the synovial inflammation and pannus formation in RA through stimulating a variety of cells such as monocytes, macrophages, T and B lymphocytes, fibroblast-like synoviocytes, chondrocytes, and osteoclasts to contribute to the inflammatory processes (Dam and Buckner 2016). This further leads to the production of more pro-inflammatory mediators and destructive enzymes. It also increases the production of cell-adhesion molecules, cytokines, chemokines, angiogenic factors, and small inflammatory agents (prostaglandin E2 and nitric oxide) leading to inflammation (Kong et al. 2020). Several studies have reported the effectiveness of TNF-α inhibitors in treating of RA clinical trials. However, the use of TNF-α inhibitors in RA treatment can be associated with side effects such as infections and the development of malignancies. Also, the antibodies used to suppress TNF-α have been reported to decrease the production of other pro-inflammatory cytokines in synovial cells from RA patients (Amber et al. 2015). Also, interleukin-17 (IL-17) leads to activation of transcription factors like nuclear factor-kappa B and mitogen-activated protein kinase. This leads to the release of other pro-inflammatory cytokines such as IL-1, TNF-α, IL-6, IL-8, and prostaglandin-E2. IL-17 has a dual effect on cartilage, causing proteoglycan breakdown and inhibiting chondrocyte metabolism in healthy cartilage and promoting the production of metalloproteinases in chondrocytes and synoviocytes (Krueger and Brunner 2017). Interleukin-23 (IL-23) is a member of the IL-12 family. IL-23 is produced by activated dendritic cells and macrophages and triggers memory T cells, natural killer cells, macrophages, and dendritic cells. IL-23 is essential for the survival and growth of Th17 (T helper 17) cells, which secrete IL-17, IL-17F, IL-6, and TNF-α. These secreted cytokines cause inflammation in RA patients (Zaky and El-Nahrery 2016). Overproduction of these cytokines due to mutations in the TNF-α and IL-1 promoter leads to joint damage in RA patients (Voirin et al. 2020).
Tendinitis and inflammation
Tendinitis or tendonitis refers to the inflammation and pain of a tendon (a fibrous connective tissue that connects muscle to bone and enables movement). Tendons and their surrounding tissues can be damaged due to overuse and misuse, particularly in athletes and manual labor workers (Loiacono et al. 2019). Symptoms include pain and tenderness along a tendon, typically near a joint, and pain that worsens with activity. Tendonitis can also result from small tears in surrounding tissue or the slow degeneration of a tendon where it attaches to the bone. Commonly affected areas include the shoulders, elbows, hips, knees, heels, wrists, and fingers causing Tennis elbow, golfer’s elbow, and Achilles tendinitis (Mandot 2020). Treatment for tendonitis aims to relieve pain and reduce inflammation. This may involve rest and immobilization of the affected tendon, as well as non-steroidal anti-inflammatory medications. In some cases, surgical or non-surgical treatments may be necessary. Effective treatments for tendonitis may include rest, medication, and in some cases, surgery (Vaishya et al. 2021).
Scientific studies suggest that inflammation is a key factor in the development of tendinitis as in RA. Inflammatory cells, such as macrophages and neutrophils, are typically present in the affected tendons of individuals with tendinitis. These cells secrete pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), which contribute to the inflammatory process in tendinitis. The release of pro-inflammatory cytokines leads to an increase in the expression of matrix metalloproteinases (MMPs), which are enzymes that degrade the extracellular matrix (ECM) of tendons (Connizzo and Grodzinsky 2018) leading to decreased mechanical strength of the tendons and ultimately tendinitis. Additionally, the release of pro-inflammatory cytokines also leads to increased production of proteoglycans, which are large macromolecules that can disrupt the normal organization of tendon fibers and contribute to tendinitis (Brauer 2011). Another mechanism by which inflammation contributes to tendinitis is through the recruitment of additional inflammatory cells to the affected area. These cells further contribute to the inflammatory process by releasing additional pro-inflammatory cytokines and enzymes (Dean et al 2015). This results in a vicious cycle of inflammation, ECM degradation, and further recruitment of inflammatory cells, which exacerbates the severity of tendinitis (Dakin et al. 2015). The role of inflammation in tendinitis has important implications for its treatment. Anti-inflammatory drugs, such as COX inhibitors and corticosteroids, are commonly used to reduce the inflammation associated with tendinitis (Heinemeier et al. 2017). Physical therapy and rehabilitation exercises can also be used to improve the strength and function of affected tendons, reducing the risk of further episodes of tendinitis (Hak et al. 2010; Capogna et al. 2017).
Oxidative stress in inflammation: a key role
Numerous biological mechanisms are controlled by chemical reactions that involve the exchange of electrons among molecules. This alters the redox state of the molecules involved (Schafer and Buettner 2001). When the level of oxidants surpasses the cell’s antioxidant defenses, the redox balance is disrupted, resulting in either oxidative stress (a more positive redox potential) or reductive stress (a more negative redox potential). Oxidative stress is a frequent type of stress that occurs in living systems (Shao et al. 2012). Therefore, oxidative stress arises due to the overproduction of oxidizing molecules compared to the cell’s reducing abilities. The reactive oxygen species (ROS) and the reactive nitrogen species (RNS) are the common oxidants that cause oxidative stress in living systems (Pacher et al. 2007). Scientific studies suggest that ROS is positively associated with the severity of RA. Innate immune cells like macrophages and neutrophils produce ROS in the form of O2- and H2O2 (Crowley 2014). Scientific reports suggest that an increase in ROS production due to redox reactions is related to the pathophysiology of inflammation in RA (Nathan and Cunningham-Bussel 2013; Blaser et al. 2016). ROS can also modify NF-kB signaling and nuclear translocation of NF-kB can be induced by H2O2 (Kabe et al. 2005). Other components of the signaling cascade that lead to inflammation like AP-1 inducible hypoxia factor (HIF-1), and gamma-activated peroxisome proliferator receptor (PPARγ) are also induced by ROS (Espinosa-Diez et al. 2015). Scientific studies suggest that mitochondria continuously produce O2- because of the discharge of electrons from the electron transport chain (Drose and Brandt 2012). Mitochondrial ROS help in the production of inflammatory cytokines like IL-1, IL-6, and TNF-α (Bulua et al. 2011). OS is considered a pathogenic signature in RA given the fivefold increase in mitochondrial ROS production in whole blood and monocytes of RA patients compared to healthy persons (Ponist et al. 2020; García-Sánchez et al. 2020).
Other factors affecting inflammation
Heredity
There is strong evidence to suggest that genetics play a major role in the development of RA. MacGregor et al. (2000) reported the estimated heritability of RA to be ~ 60%. Different genetic loci have been identified that are associated with increased risk of RA as shown in Table 1. One of the strongest genetic risk factors for RA is associated with a set of alleles within the major histocompatibility complex (MHC) region. The MHC genes encode human leukocyte antigens (HLAs); within the MHC region, there are specific amino acid sequences in the HLA peptide binding groove that are associated with the risk of development of RA (Bang et al. 2010; Raychaudhuri et al. 2012). These are collectively referred to as shared epitope (SE). Some studies suggest SE alleles contribute ~ 40% of the genetic risk of RA (Plenge 2009; Kronzer and Davis 2021). Other genetic factors have shown a strong association with RA; as per genome-wide studies, more than 100 loci are associated with RA (Okada 2014; Messemaker et al. 2015a, b). A significant genetic association with RA has been identified in the PTPN22 gene; specifically, the polymorphism that affects its function is believed to be a major factor for RA development. However, the precise mechanism by which this polymorphism leads to RA is not fully known. Recent studies suggest that PTPN22 polymorphism may also influence the process of citrullination, which involves the conversion of arginine to citrulline. This is catalyzed by PAD (peptidylarginine deiminases) enzyme. Altered interactions between PAD and PTPN22 may lead to increased citrullination. This hypercitrullination may contribute to inflammation in RA (Chang et al. 2015). Some other genes or their products have been reported to be associated with RA including CTL4A (Plenge et al. 2005), STAT4 (Remmers et al. 2007), IL-6(Ferreira et al. 2013), and NF-kB (Spurlock et al. 2015). In addition, non-coding regions within the TRAF-C5 region have also been identified as being associated with RA. TRAF genes are involved in immune signaling pathways, and variations within TRAF-C5 region may affect the immune response and contribute to RA development (Messemaker et al. 2015a, b).
High glucose levels
Elevated glucose concentrations above the physiological normal of 22mmol/L have been found to induce a rise in the release of TNF-α and IL-6 from normal human mononuclear cells in in vitro experiments (Weyand and Goronzy 2017). There is also evidence that a 24-h incubation in a high-glucose medium increases the production of IL-6 by human monocytes separated from healthy individuals (Morohoshi et al. 2006). The chance of tendinopathy development is increased by the possibility of chronic inflammation caused by these cytokines (Ruscitti et al. 2015). Moreover, the development of RA is also significantly influenced by inflammatory cytokines like TNF-α, IL-6, and IL-1 (Movahedi et al. 2015). Therefore, high glucose levels have a significant impact on the development of inflammation in rheumatoid arthritis and tendinitis patients.
Sedentary lifestyle
A sedentary lifestyle refers to activities that involve remaining seated or reclining and require minimal energy expenditure. Examples include watching television, playing games, and spending prolonged periods being seated. A sedentary lifestyle in RA patients may be associated with increased inflammation. This could result in a vicious loop whereby the decreased physical function, increased fatigue, and increased local disease may result in sedentariness which may further increase inflammation and contribute to the severity of RA-related health outcomes (Thomsen et al. 2015). The majority of experimental conditions that cause a drop in mechanical stress result in a loss of tendon elastic characteristics in both humans and animals, except Botox-induced muscle paralysis (Fenton et al. 2017; Steultjens et al. 2022) (Fig. 1).

Hypothesized mechanism of sedentary lifestyle on rheumatoid arthritis
Gender
Women make up about two-thirds of RA patients. In the adult population, the cumulative risk of having RA is estimated to be 3.6% for women and 1.7% for males (Maranini et al. 2022). Although the contribution of hormones to the onset of RA is still debatable, estrogen’s immune system-stimulating properties may explain why RA affects women more frequently than males. Early menopause, polycystic ovarian syndrome, and pre-eclampsia are among the factors that have been connected to an elevated risk for the manifestation of RA (Alpízar-Rodríguez et al. 2016). Breastfeeding, oral contraception, and hormone replacement treatment are the things that can postpone the start of RA (Ghamarzad et al. 2016). First-degree relatives (FDRs) of RA patients with lower rates of rheumatoid factor (RF) positivity have been linked to oral contraceptive use, indicating that hormones may have an early “pre-clinical” impact on the etiology of RA (Orellana et al. 2017). Although exogenous hormone use is associated with a lowered risk for RA and may reduce endogenous hormone synthesis, the exact processes behind this link are yet unknown (Makol and Krause 2016).
Why plant-derived COX inhibitors?
The utilization of natural compounds derived from medicinal plants for the treatment of various diseases has gained substantial popularity in clinical research. Of particular interest are polyphenolic compounds, which have garnered significant attention for their ability to modulate inflammasomes (Ambriz-Pérez et al. 2016). Medicinal plants are being utilized as an alternative to allopathic COX inhibitors due to the undesirable side effects associated with their or usage, particularly on the gastrointestinal tract and renal system. The use of potent synthetic drugs is accompanied by concerns regarding their toxicity and the recurrence of symptoms upon discontinuation. As a result, there is a growing need to develop anti-inflammatory drugs derived from medicinal plants. Extensive efforts are being made to explore the potential of medicinal plants in the search for effective and safer anti-inflammatory treatments (Asenso et al. 2016). Acheflan® is an example of a phytotherapeutic agent used for the local treatment of inflammatory conditions; another phytotherapeutic agent, Daflon 500 mg®, is composed of a purified flavonoid fraction and is known for its venotonic and vasoprotective effects (Nunes et al. 2020). Consequently, the immunopharmacological properties of various plant species have revealed various extracts, fractions, and chemical classes that exhibit significant therapeutic potential. This not only offers a promising alternative for treating inflammatory processes and associated diseases but also validates their traditional use in ethnobotanical practices. Furthermore, scientific literature highlights the significant anti-inflammatory activities displayed by plant-derived molecules, with many of their mechanisms involving the inhibition of cytokines, chemokines, and adhesion molecules, as well as pathways involving arachidonic acid and nitric oxide (Hughes et al. 2017; Akbari et al. 2022) (Tables 2 and 3).
Associated target in inflammation by plant-derived COX inhibitors
Plant-derived COX inhibitors target various pathways and molecules involved in inflammation. These main targeted pathways and molecules include inducible nitric oxide synthase (iNOS), MAP kinase signaling pathway, oxidative stress, and inflammatory cytokines (Fig. 2) (Table 4).

Mechanism of action of various phytochemicals in inhibition of iNOS activation (ACA = 1-acetoxychavicol acetate (ACA), LPS = lipopolysaccharide, TLR = Toll-like receptor 4, MAPK = mitogen-activated protein kinase, NF-KB = nuclear factor-kappa B)
Inducible nitric oxide synthase
Inducible nitric oxide synthase (iNOS) is an enzyme that is involved in the production of NO (nitric oxide), a key mediator of inflammation. iNOS is one of the three isoforms of nitric oxide synthase (NOS) (Vannini et al. 2015; Kashfi et al. 2021). Unlike other forms, it produces significantly higher amounts of NO, reaching levels in the micromolecular range, and it can sustain NO production for longer periods ranging from hours to even days (Vannini et al. 2015). The interplay of pro- and anti-inflammatory cytokonins in the affected tissues of RA patients leads to the activation of iNOS (McInnes and Schett 2007). A study by Grabowski et al. (1997) suggested that CD68 + macrophages in the synovial lining and fibroblasts are the source of iNOS expression in the synovium of RA patients. In the case of tendonitis, all three isoforms of iNOS synthesize NO after tendon injury. The expression of all three isoforms was seen during shoulder surgery in tendon-injured patients (Millar et al. 2017). In the activation of iNOS gene transcription, the process involves the binding of LPS (lipopolysaccharide) to TLR4, which triggers the activation of its adaptor protein MyD88. This activation leads to the recruitment of downstream proteins like IRAK and TRAF6. Subsequently, multiple protein kinases including IkB, IKK, and MAPKs (such as p38, MAPK, JNK1/2, ERK 1/2) are activated. The activation of these protein kinases plays an important role in the activation of central transcription factors involved in iNOS gene expression, namely, NF-kB and activator protein-1 (AP-1) (Murakami and Ohigashi 2007; Wu et al. 2019). Studies by Lee et al. suggested that the iNOS promoter gene contains binding sites for NF-kB and AP-1 and these binding sites are important for the expression of iNOS induction (Lee et al. 2003; Tsai et al. 1999).
Various studies showed the therapeutic potential of phytochemicals in suppressing iNOS expression. For instance, resveratrol, a phytochemical extracted from red grapes, suppresses LPS-induced iNOS mRNA expression. This suppression results due to inhibition of IKB degradation by resveratrol thus blocking the activation of NF-KB in macrophages (Youn et al. 2009). Curcumin, a phytochemical extracted from turmeric plants, has also been reported to suppress iNOS activity (Nakatake et al. 2017). While the exact target of this agent in macrophages is not known, studies suggest that curcumin may act on several signaling pathways upstream of iNOS transcription and post transcription; these pathways include MAPK and JAK/STAT (Murakami 2009). Also, several novel phytochemicals have been isolated such as 1-acetoxychavicol acetate (ACA)—derived from Alpinia galangal (Zingiberaceae), zerumbone extracted from Zingiber zerumbet (zingiberaceae) and Auraptene and Nobiletin found in citrus fruits. These can attenuate iNOS induction in macrophages, which may help in combating the inflammation in RA as well as tendonitis (Giang et al. 2009; Murakami 2009; Murakami 2009; Kobayashi 2010).
Mitogen-activated protein kinase
Mitogen-activated protein kinase (MAPK) regulates various cellular functions in eukaryotes including cell proliferation, cell differentiation, and cell death. It consists of a series of protein kinases, MAPKKKs, MAPKKs, and MAPKs, which sequentially phosphorylate each other, finally activating several transcription factors. In mammals, MAPKs can be classified into three main classes—extracellular signal-regulated kinase (ERK), stress-activated protein kinases (SAPK), and p38 MAPK (Korb et al. 2006; (Liang and Yang 2019) (Fig. 3).

Interaction of various phytochemicals with their target molecules in MAPK pathways.(RAS = rat sarcoma protein, TRAF = tumor necrosis factor receptor-associated dactor, ASK1 = apoptosis signal-regulating kinase 1, MLK3 = mixed-lineage kinase 3, TAK1 = transforming growth factor-beta-activated kinase 1, MEKK = mitogen-activated protein kinase kinase kinase, ERK = extracellular signal-regulated kinase, JNK = Jun N-terminal kinase, AP1 = activator protein 1, SMAD = mothers against decapentaplegic homolog, MSK = mitogen- and stress-activated protein kinase, ELK = ETS-like gene, E-ts = E twenty-six, C-MYC = cellular myelocytomatosis viral oncogene)
In the synovial tissues of RA patients, all three types of MAPKs have been reported but the significant one is p38 MAPK (Thalhamer et al. 2007; Li et al. 2017). The p38 MAPK is a crucial member of the MAPK family that plays an important role in the regulation of inflammatory cytokines like- IL-1 and TNF-α. Different isoforms of p38 have been reported to play a significant role in the pathogenesis of RA by regulating various processes like migration of inflammatory cells and mediators and cytokine production. The activation of p38 MAPK cascade is initiated by various stimuli like LPS, TNF-α and IL-1 (Li et al. 2016). These ligands bind to cell surface receptors and cause conformational changes in receptors which ultimately leads to the recruitment and activation of downstream signaling proteins like TRAF. These proteins then activate the MAPKKKs (mitogen-activated protein kinase kinase kinase) like MEKK1-4, MLK, TAK-1, and ASK-1. These kinases are activated by interaction with small GTPases. Activated MAPKKKs then phosphorylate the next components in a cascade like MAPK or MAPKK (mitogen-activated protein kinase kinase). Important MAPKKs in p38 cascade are MKK-3 and MKK-6. These MAPKKs primarily activate p38 by phosphorylation of serine and tyrosine residues on p38 MAPK. Once activated, p38 MAPK phosphorylates downstream targets like ATF-2, MSK-1, Max/Myc, and ELK-1 (Li et al. 2002; Bassi et al. 2008).
The stress-activated protein kinase (SAPK) also known as the c-Jun N-terminal kinase (JNK) cascade is another member of the MAPK family. It plays an important role in various cellular and inflammatory responses. The external stimuli in the case of the JNK pathway can be stress. The MAPKKK in the case of JNK is MEKK-1 (MAPK/ERK kinase kinase-1) which can be activated by various upstream signals. Activated MAPKKK phosphorylate MAPKK which in the case of JNK are MKK4 and MKK7 (Kitanaka et al. 2017). These MAPKKs then directly activate JNK by phosphorylating serine threonine residues in its activation loop. Activated JNK translocates to the nucleus and phosphorylates transcription factors like c-Jun, smad-4, AP-1, and ELK-1 (Namba et al. 2017; Hu et al. 2018).
ERK (extracellular signal-regulated kinase) is another member of MAPK family. The extracellular signals activate surface receptors which stimulate ERK–GTPases (RAS, RAF). The activated GTPases phosphorylated MAPKKKs of ERK cascade–MEKK-1/4 which then activate MAPKKs of ERK-MEK-1/2. These MEKs are dual-specificity kinases that phosphorylate ERK. Phosphorylated ERK then translocates into the nucleus and activates transcription factors like ELK-1, Ets-1, and c-Myc (Lu and Malemud 2019; (Shang et al. 2016).
FMAPKs are considered the most promising therapeutic targets for RA. Some phytochemicals directly suppress phosphorylated MAPK complex hence stopping them from activating transcription factors leading to inflammation. These include allylpyrocatechol (APC) a phenolic compound derived from the leaves of Piper betle belonging to Piperaceae family. It has been found to suppress p38 complex and thus stop it from activating transcription factors inside the nucleus. It also has been found to inhibit the production of pro-inflammatory cytokines like TNF-α. Therefore, it helps to modulate inflammatory response in RA (De et al. 2016). Berberine (BBR) is an isoquinoline alkaloid that is extracted from various plant species belonging to the genera Berberis, Coptis, and Phellodendron of the Ranunculaceae family. It has been extensively used in RA patients for its anti-inflammatory properties. Studies suggest the suppressive effects of berberine on p-ERK, p-p38, and p-JNK (Wang et al. 2014). Cryptotanshinone (CTS) is a quinoid triterpene that is extracted from the roots of Salvia miltiorhiza of the Lamiaceae family. It has been found to possess significant anti-inflammatory and anticancer activities. It has been found that CTS suppresses ERK-1/2, JNK, and p38 and hence prevents the activation of transcription factors (Tang et al. 2010). Andrographolide (AD) is a triterpenoid compound extracted from the plant Andrographis paniculata, of the Acanthaceae family. Chemically it is [1-naphthalenyl] ethylidene} dihydroxy-4–2(3H)-furanone. AD has extensively shown anti-inflammatory and antioxidant activity. Studies have suggested that AD completely suppressed ERK signaling while it did not have any impact on p38 and JNK signaling (Li et al. 2017). Curcumin (CUR) is extracted from the roots of Curcuma longa, of the Zingiberaceae family. Studies have suggested that CUR inhibited p-p38, p-ERK, and p-JNK by preventing their translocation to the nucleus (Shang et al. 2016). Epigallocatechin-3-gallate (ECGC) is a polyphenol that is found in green tea. It has anticancerous, anti-inflammatory, and cardioprotective properties. Scientific studies suggest that ECGC inhibits the phosphorylation of MAPKs including p38, JNK, and ERK in response to TNF-α stimulation. It also inhibits the activation of transcription factors—c-Jun, AP-1, and smad-4 in JUN-k cascade. MAPKK- TAK-1 is also inhibited by ECGC (Singh et al. 2003).
NF-κB
NF-κB is the most important regulator in RA. It controls inflammation, cell survival, and cell proliferation. It is generally present in immune cells and regulates the transcription of cytokines. NF-κB activation is initiated by various stimuli including TNF-α, IL-1, and oxidative stress (Hutami et al. 2019). Mammalian NF-kB has five regulators—p105, p100, ReIA, REI-B, and C-ReI. In the active state, these regulators dimerize and activate the signaling cascade. In the inactivate state, IkB, an inhibitor, prevents the dimerization of NF-kB complex and its subsequent translocation into nucleus. NF-kB can be activated by two distinct pathways—canonical and non-canonical. But the most significant pathway in RA is canonical pathway (Fig. 4). It progresses by the dimerization of IKK proteins consisting of IKK-α, IKKβ, and IKK-γ/NEMO. NEMO (NF-kappa-B essential modulator) is a crucial regulator of NF-κB (Chakraborty et al. 2021). Scientific studies have shown that canonical pathway activation requires NEMO for phosphorylation of IkB. IkB is the inhibitor that controls regulatory unit of NF-kB. The pathway is primarily dependent on degradation of IkB that allows translocation of NF-kB complex into the nucleus and stimulate transcription. NF-kB complex in the nucleus predominantly includes p65/p50 dimers (Sarmiento Salinas et al. 2017; Choi et al. 2019).

Canonical activation of NF-kB and its targets in RA (TCR = T cell receptor, TNFR = tumor necrosis factor receptor, NF-kB = nuclear factor kappa B, IKK = inhibitor of nuclear factor kappa-B kinase, EGCG = epigallocatechin-3-gallate, GN = genistein)
Various molecules in NF-kB can be targeted to suppress RA, and numerous phytochemicals have shown the ability to effectively to inhibit activity of molecules within this pathway without causing side effects (Bacher and Schmitz 2004). These include the following: andrographolide (AD) is a triterpenoid extracted from Andrographis paniculata, of the Acanthaceae family. It has shown anti-inflammatory and antioxidant properties. Scientific studies have shown that AD prevents NF-kB signaling by inhibiting the IkB degradation and by suppressing the nuclear translocation of p65 subunit of NF-Kb (Zhai et al. 2014). Berberine (BBR) is an isoquinoline alkaloid that is extracted from various plant species belonging to the genera Berberis, Coptis, and Phellodendron, of the Ranunculaceae family. It inhibits the upregulation of AMPK (5′ AMP-activated protein kinase) and phosphorylation of p65 and IkB resulting in a negative effect on NF-kB signaling (Zhou et al. 2019). Apigenin (APG) is a flavone and is most commonly found in vegetables and fruits. It is known for its anti-inflammatory properties. It inhibits NF-kB activation inhibiting IkB degradation and phosphorylation (Xu et al. 2008). Cafeic acid (CA) is a phenol and is naturally found in various plants. It strongly inhibits the phosphorylation of P-IKKα/β and P-IκBα (Wang et al. 2017b). Celastrol (CEL) is a triterpenoid extracted from Tripterygium wilfordii, of the family Cealstraceae. It has been shown to possess anti-inflammatory activity. It suppresses the phosphorylation of IKK and IKBα and therefore inhibits the nuclear translocation of p65 (Cascão et al. 2017). CTS also inhibited the nuclear translocation of p65 (Wang et al. 2015). EGCG inhibits activation of the NF-kB pathway by inhibiting the NF-kB p65 subunit transcriptional activity without affecting IkB degradation (Lee et al. 2009). Genistein (GN) (4,5,7-trihydroxy isoflavone) is a phytoestrogen and a tyrosine kinase inhibitor. It is extracted from soybeans and has been shown to have anticancer and anti-inflammatory properties. Scientific studies have shown complete suppression of TNF-α-induced phosphorylation of p65 subunit; it also suppresses the expression levels of IKK and p65 (Li et al. 2014).
JAK-STAT pathway
The Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway is an important signaling cascade involved in the pathogenesis of RA. The cytokines that activate JAK-STAT cascade in RA include IL-6, IFN-γ, and TNF-α (Kato 2020). The JAK family consists of 4 cytoplasmic non-receptor tyrosine kinases—JAK-1, JAK-2, JAK-3, and TYK2. These kinases phosphorylate STAT proteins. The STAT proteins are transcription factors that cause the activation of target genes. STAT-3 is one of the STATs that gets activated continuously in RA. After the binding of the ligand to the receptor on the cell surface, the activation of JAK proteins is triggered. This activation leads to the autophosphorylation of JAK. The activated JAK then phosphorylates the STAT-3 proteins. Phosphorylated STAT-3 proteins undergo dimerization and translocate into the nucleus, where it binds to specific nuclear sequences and promote gene expression. One important regulator of this pathway is a family of proteins known as SOCS (suppressor of cytokine signaling). There are 8 members of the SOCS protein family and each plays a distinct role in regulating the JAK-STAT cascade. Specifically, SOCS-1 and SOCS-3 inhibit the cascade by binding to phosphorylated STAT proteins and activated proteins (Malemud 2018). Targeting this pathway has emerged as a potential therapeutic approach for managing RA. Various phytochemicals have been shown to possess the property of inhibiting this cascade by targeting the key molecules of the cascade (Malemud and Pearlman 2009; Böhmer and Friedrich 2014).
Scientific studies have shown CTS downregulated the p300-mediated acetylation of STAT-3, which is necessary for its activation, and suppressed the JAK-2-independent STAT-3 activation (Wang et al. 2017c). Curcumin suppresses the production of IFN-γ and IL-6, the key pro-inflammatory cytokines of the cascade. It also inhibited the expression of IFN-γ at the transcription level, thereby suppressing IFN-γ-stimulated STAT-1 phosphorylation and its subsequent translocation to the nucleus (Huang et al. 2012). ECGC also suppressed the nuclear translocation of p-STAT-3 proteins (Lee et al. 2016). Resveratrol (RVL) is a polyphenol that is extracted from grapes, cranberries, and peanuts. It has anticancer and anti-inflammatory properties. It inhibits the mRNA expression levels of STAT-3 (Fig. 5).

Depiction of the overview of JAK-STAT pathway and its phytochemical target in RA. (JAK-STAT = Janus kinase-signal transducers and activators of transcription, RVL = resveratrol)
Conclusion
Although COX inhibitors’ anti-inflammatory, antipyretic, and analgesic effects have been thoroughly studied, a variety of other molecular and cellular mechanisms that are still poorly understood are crucial in the etiology of inflammation. This review covers the role of various molecules in inflammation and their pathways. The role of inflammation in the etiology of rheumatoid arthritis and tendinitis has also been thoroughly covered. COX inhibitors are the widely prescribed drugs worldwide. These drugs should be provided for the shortest amount of time at the lowest dosage while being closely monitored for GI, renal, and cardiovascular damage. Many plant-derived substances are now being researched as possible anti-inflammatory drugs with the least side effects. This review also assists present and future researchers in identifying anti-inflammatory plants whose active components can be separated through a variety of separation techniques. Such a kind of research could result in the identification of novel compounds of natural origin that can be used to treat inflammatory diseases. However, more thorough research could be done to determine the real mechanism(s) of action of these phytochemical agents.
Data availability
Not applicable.
References
Ahmad S, Alam K, Hossain MM, Fatima M, Firdaus F, Zafeer MF, Arif Z, Ahmed M, Nafees KA (2016) Anti-arthritogenic and cardioprotective action of hesperidin and daidzein in collagen-induced rheumatoid arthritis. Mol Cell Biochem 423(1–2):115–127. https://doi.org/10.1007/s11010-016-2830-y
Ahmed M, Bjurholm A, Srinivasan G, Lundeberg T, Theodorsson E, Schultzberg M, Kreicbergs A (1995) Capsaicin effects on substance P and CGRP in rat adjuvant arthritis. Regul Pept 55(1):85–102. https://doi.org/10.1016/0167-0115(94)00095-f
Akbari B, Baghaei-Yazdi N, Bahmaie M, Mahdavi Abhari F (2022) The role of plant-derived natural antioxidants in reduction of oxidative stress. BioFactors 48(3):611–633. https://doi.org/10.1002/biof.1831
Aletaha D, Smolen JS (2018) Diagnosis and management of rheumatoid arthritis: a review. Jama 320(13):1360–1372. https://doi.org/10.1001/jama.2018.13103
Alieva K (2016) Think rheumatoid arthritis: causes, consequences, and management. EMJ Rheumatology 66–73. https://doi.org/10.33590/emjrheumatol/10314552
Alpízar-Rodríguez D, Pluchino N, Canny G, Gabay C, Finckh A (2016) The role of female hormonal factors in the development of rheumatoid arthritis. Rheumatology, kew318. https://doi.org/10.1093/rheumatology/kew318
Amber K, Bloom R, Mrowietz U, Hertl M (2015) TNF-α: a treatment target or cause of sarcoidosis? J Eur Acad Dermatol Venereol 29(11):2104–2111. https://doi.org/10.1111/jdv.13246
Ambriz-Pérez DL, Leyva-López N, Gutierrez-Grijalva EP, Heredia JB (2016) Phenolic compounds: natural alternative in inflammation treatment. A Review. Cogent Food & Agriculture 2(1). https://doi.org/10.1080/23311932.2015.1131412
Ansari SK (2016) COX inhibitors and renal function in rheumatoid arthritis. J Med Sci Clin Res. https://doi.org/10.18535/jmscr/v4i2.51
Arab HH, Gad AM, Fikry EM, Eid AH (2019) Ellagic acid attenuates testicular disruption in rheumatoid arthritis via targeting inflammatory signals, oxidative perturbations and apoptosis. Life Sci 239:117012. https://doi.org/10.1016/j.lfs.2019.117012
Asenso J, Yang XD, Yu J, Zhou P, Wang C, Wei W (2016) Plant-based anti-inflammatory agents: progress from Africa and China. Clinical Anti-Inflammatory & Anti-Allergy Drugs 2(1):52–66. https://doi.org/10.2174/2212703802999151230113431
Bacher S, Schmitz M (2004) The NF-kB pathway as a potential target for autoimmune disease therapy. Curr Pharm Des 10(23):2827–2837. https://doi.org/10.2174/1381612043383584
Bahar E, Kim JY, Yoon H (2017) Quercetin attenuates manganese-inducedneuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. Int J Mol Sci 18(9):1989. https://doi.org/10.3390/ijms18091989
Baito QN, Jaafar HM, Mohammad TA (2023) Piperine suppresses inflammatory fibroblast-like synoviocytes derived from rheumatoid arthritis patients Via NF-κB inhibition. Cell Immunol 391:104752. https://doi.org/10.1016/j.cellimm.2023.104752
Bang SY, Lee KH, Cho SK, Lee HS, Lee KW, Bae SC (2010) Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of RF and ACPA. Arthritis & Rheumatism, NA-NA. https://doi.org/10.1002/art.27272
Bassi R, Heads R, Marber M (2008) Mechanisms of MAPK kinase-independent p38-MAPK activation. J Mol Cell Cardiol 44(4):770–771. https://doi.org/10.1016/j.yjmcc.2008.02.142
Berney S (2007) Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Longterm (MEDAL) programme: a randomised comparison. Yearbook of Medicine 2007:47–49. https://doi.org/10.1016/s0084-3873(08)70033-6
Blaser H, Dostert C, Mak TW, Brenner D (2016) TNF and ROS crosstalk in inflammation. Trends Cell Biol 26(4):249–261. https://doi.org/10.1016/j.tcb.2015.12.002
Böhmer FD, Friedrich K (2014) Protein tyrosine phosphatases as wardens of STAT signaling. JAK-STAT 3(1):e28087. https://doi.org/10.4161/jkst.28087
Brauer S (2011) Achilles tendonitis. J Physiother 57(1):62. https://doi.org/10.1016/s1836-9553(11)70013-3
Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM (2011) Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med 208(3):519–533. https://doi.org/10.1084/jem.20102049
Cai H, Zheng Z, Sun Y, Liu Z, Zhang M, Li C (2015) The effect of curcumin and its nanoformulation on adjuvant-induced arthritis in rats. Drug Des Devel Ther 4931. https://doi.org/10.2147/dddt.s90147
Capogna BM, Shenoy K, Youm T, Stuchin SA (2017) Tendon disorders after total hip arthroplasty: evaluation and management. J Arthroplasty 32(10):3249–3255. https://doi.org/10.1016/j.arth.2017.04.015
Carretta MD, Alarcón P, Jara E, Solis L, Hancke JL, Concha II, Hidalgo MA, Burgos RA (2009) Andrographolide reduces IL-2 production in T-cells by interfering with NFAT and MAPK activation. Eur J Pharmacol 602(2–3):413–421. https://doi.org/10.1016/j.ejphar.2008.11.011
Cascão R, Vidal B, JalmariFinnilä MA, Lopes IP, Teixeira RL, Saarakkala S, Moita LF, Fonseca JE (2017) Effect of celastrol on bone structure and mechanics in arthritic rats. RMD Open 3(2):e000438. https://doi.org/10.1136/rmdopen-2017-000438
Cascão R, Vidal B, Raquel H, Neves-Costa A, Figueiredo N, Gupta V, Fonseca JE, Ferreira Moita L (2014) Potent anti-inflammatory and antiproliferative effects of gambogic acid in a rat model of antigen-induced arthritis. Mediators Inflamm 2014:1–7. https://doi.org/10.1155/2014/195327
Chakraborty D, Gupta K, Biswas S (2021) A mechanistic insight of phytoestrogens used for rheumatoid arthritis: an evidence-based review. Biomed Pharmacother 133:111039. https://doi.org/10.1016/j.biopha.2020.111039
Chandran B, Goel A (2012) A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Phytother Res 26(11):1719–1725. https://doi.org/10.1002/ptr.4639
Chang HH, Dwivedi N, Nicholas AP, Ho IC (2015) The W620 Polymorphism in PTPN22 disrupts its interaction with peptidylarginine deiminase type 4 and enhances citrullination and NETosis. Arthritis & Rheumatology 67(9):2323–2334. https://doi.org/10.1002/art.39215
Chang SS (2015) Re: Preventive effects of COX inhibitors, NO-COX inhibitors, and COX inhibitors plus difluoromethylornithine in a chemically induced urinary bladder cancer model. J Urol 193(5):1727–1727. https://doi.org/10.1016/j.juro.2015.02.029
Chen L, Qi H, Jiang D, Wang R, Chen A, Yan Z, Xiao J (2013) The new use of an ancient remedy: a double-blinded randomized study on the treatment of rheumatoid arthritis. Am J Chin Med 41(02):263–280. https://doi.org/10.1142/s0192415x13500195
Cheng BR, Chen JQ, Zhang XW, Gao QY, Li WH, Yan LJ, Zhang YQ, Wu CJ, Xing JL, Liu JP (2021) Cardiovascular safety of celecoxib in rheumatoid arthritis and osteoarthritis patients: a systematic review and meta-analysis. PLoS ONE 16(12):e0261239. https://doi.org/10.1371/journal.pone.0261239
Cheng L, Chen J, Rong X (2022) Mechanism of emodin in the treatment of rheumatoid arthritis. Evidence-Based Complementary and Alternative Medicine 2022:1–16. https://doi.org/10.1155/2022/9482570
Choi J, Park K, Park (2019) NF-b signaling pathways in osteoarthritic cartilage destruction. Cells 8(7), 734. https://doi.org/10.3390/cells8070734
Connizzo BK, Grodzinsky AJ (2018) Release of pro-inflammatory cytokines from muscle and bone causes tenocyte death in a novel rotator cuff in vitro explant culture model. Connect Tissue Res 59(5):423–436. https://doi.org/10.1080/03008207.2018.1439486
Crowley SD (2014) The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal 20(1):102–120. https://doi.org/10.1089/ars.2013.5258
Dakin SG, Martinez FO, Yapp C, Wells G, Oppermann U, Dean BJF, Smith RDJ, Wheway K, Watkins B, Roche L, Carr AJ (2015) Inflammation activation and resolution in human tendon disease. Sci Trans Med 7(311). https://doi.org/10.1126/scitranslmed.aac4269
Dam EM, Buckner JH (2016) The IL-21 signaling pathway is enhanced in RA B cells and has the potential to alter development and cytokine production in RA B cells. J Immunol 196(1_Supplement), 124.9–124.9. https://doi.org/10.4049/jimmunol.196.supp.124.9
De Figueiredo Costa AC, De Sousa LM, Alves JMDS, Góes P, Pereira KMA, Alves APNN, Vale ML, Gondim DV (2021) Anti-inflammatory and hepatoprotective effects of quercetin in an experimental model of rheumatoid arthritis. Inflammation. https://doi.org/10.1007/s10753-021-01479-y
De S, Manna A, Kundu S, De Sarkar S, Chatterjee U, Sen T, Chattopadhyay S, Chatterjee M (2016) Allylpyrocatechol attenuates collagen-induced arthritis via attenuation of oxidative stress secondary to modulation of the MAPK, JAK/STAT, and Nrf2/HO-1 pathways. J Pharmacol Exp Ther 360(2):249–259. https://doi.org/10.1124/jpet.116.238444
Dharmapatni A, Cantley M, Marino V, Perilli E, Crotti T, Smith MD, Haynes DR (2015) The X-linked inhibitor of apoptosis protein inhibitor embelin suppresses inflammation and bone erosion in collagen antibody induced arthritis mice. Mediators Inflamm. https://doi.org/10.1155/2015/564042
Dröse S, Brandt U (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748:145–169. https://doi.org/10.1007/978-1-4614-3573-0_6
Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S (2015) Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 6:183–197. https://doi.org/10.1016/j.redox.2015.07.008
Fan D, He X, Bian Y, Guo Q, Zheng K, Zhao Y, Lu C, Liu B, Xu X, Zhang G, Lu A (2016) Triptolide modulates TREM-1 signal pathway to inhibit the inflammatory response in rheumatoid arthritis. Int J Mol Sci 17(4):498. https://doi.org/10.3390/ijms17040498
Fang Z, He D, Yu B, Liu F, Zuo J, Li Y, Lin Q, Zhou X, Wang Q (2017) High-throughput study of the effects of celastrol on activated fibroblast-like synoviocytes from patients with rheumatoid arthritis. Genes 8(9):221. https://doi.org/10.3390/genes8090221
Feng X, Tian M, Zhang W, Mei H (2018) Gastrointestinal safety of etoricoxib in osteoarthritis and rheumatoid arthritis: a meta-analysis. PLoS ONE 13(1):e0190798. https://doi.org/10.1371/journal.pone.0190798
Fenton SAM, Veldhuijzen van Zanten JJCS, Duda JL, Metsios GS, Kitas GD (2017) Sedentary behaviour in rheumatoid arthritis: definition, measurement and implications for health. Rheumatology 57(2):213–226. https://doi.org/10.1093/rheumatology/kex053
Ferreira RC, Freitag DF, Cutler AJ, Howson JMM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, Pekalski ML, Chen WM, Onengut-Gumuscu S, Rich SS, Butterworth AS, Malarstig A, Danesh J, Todd JA (2013) Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet 9(4):e1003444. https://doi.org/10.1371/journal.pgen.1003444
Gao X, Kang X, Lu H, Xue E, Chen R, Pan J, Ma J (2022). Piceatannol suppresses inflammation and promotes apoptosis in rheumatoid arthritis-fibroblast-like synoviocytes by inhibiting the NF-κB and MAPK signaling pathways. Mol Med Rep 25(5). https://doi.org/10.3892/mmr.2022.12696
García-Sánchez A, Miranda-Díaz AG, Cardona-Muñoz EG (2020) The role of oxidative stress in physiopathology and pharmacological treatment with pro- and antioxidant properties in chronic diseases. Oxid Med Cell Longev 2020:1–16. https://doi.org/10.1155/2020/2082145
Gargya I, Singh B, Talnia S (2017) COX inhibitors (non- steroidal anti- inflammatory drugs)- their effects and side effects in orthodontic therapy- a review. Dental Journal of Advance Studies 05(01):008–013. https://doi.org/10.1055/s-0038-1672075
Ghamarzad Shishavan N, Pirouzpanah S, Hajialilo M, Khabbazi A, Jafarpour F, Mirtaheri E, Farrin N, Ebrahimzadeh S, Baban N, Shakiba Z, Ghahremanzadeh K (2016) Risk factors of rheumatoid arthritis development among females in north-west of Iran: a case-control study. Iran Red Crescent Med J 18(12). https://doi.org/10.5812/ircmj.26874
Giang PM, Sơn PT, Jin H, Lee JH, Lee JJ (2009) Comparative study on inhibitory activity of Zerumbone and Zerumbone 2,3-epoxide on NF-κB activation and NO production. Sci Pharm 77(3):589–596. https://doi.org/10.3797/scipharm.0907-16
Grabowski PS, Wright PK, Van ’t Hof RJ, Helfrich MH, Ohshima H, Ralston SH (1997) Immunolocalization of inducible nitric oxide synthase in synovium and cartilage in rheumatoid arthritis and osteoarthritis. Rheumatology, 36(6), 651–655. https://doi.org/10.1093/rheumatology/36.6.651
Hacker A, Satre T (2021) In acute back pain, do topical COX inhibitors relieve pain as well as oral COX inhibitors? Evidence-Based Practice 25(3):18–19. https://doi.org/10.1097/ebp.0000000000001271
Hadi MA, Raghavendra Rao N, Srinivasa Rao A (2016) Formulation and evaluation of ileo-colonic targeted matrix-mini-tablets of naproxen for chronotherapeutic treatment of rheumatoid arthritis. Saudi Pharmaceutical Journal 24(1):64–73. https://doi.org/10.1016/j.jsps.2015.03.001
Hak DJ, Sanchez A, Trobisch P (2010) Quadriceps tendon injuries. Orthopedics 33(1):40–46. https://doi.org/10.3928/01477447-20091124-20
Heinemeier KM, Øhlenschlæger TF, Mikkelsen UR, Sønder F, Schjerling P, Svensson RB, Kjaer M (2017) Effects of anti-inflammatory (COX inhibitor) treatment on human tendinopathic tissue. J Appl Physiol 123(5):1397–1405. https://doi.org/10.1152/japplphysiol.00281.2017
Horino T, Ichii O, Eguchi T, Inotani S, Matsumoto T (2019) Nonsteroidal anti-inflammatory drug-induced small bowel stricture in rheumatoid arthritis. JCR: Journal of Clinical Rheumatology, 27(3), e86–e87. https://doi.org/10.1097/rhu.0000000000001262
Hu H, Jin H, Yu L, Qu S (2018) Inhibition of ERK pathway decreases the synovial hyperplasia and angiogenesis of rheumatoid arthritis rats. European Journal of Inflammation 16:205873921879453. https://doi.org/10.1177/2058739218794531
Huang GS, Tseng CY, Lee CH, Su SL, Lee HS (2009) Effects of (−)-epigallocatechin-3-gallate on cyclooxygenase 2, PGE2, and IL-8 expression induced by IL-1β in human synovial fibroblasts. Rheumatol Int 30(9):1197–1203. https://doi.org/10.1007/s00296-009-1128-8
Huang G, Xu Z, Huang Y, Duan X, Gong W, Zhang Y, Fan J, He F (2012) Curcumin protects against collagen-induced arthritis via suppression of BAFF production. J Clin Immunol 33(3):550–557. https://doi.org/10.1007/s10875-012-9839-0
Huang M, Wang L, Zeng S, Qiu Q, Zou Y, Shi M, Xu H, Liang L (2017) Indirubin inhibits the migration, invasion, and activation of fibroblast-like synoviocytes from rheumatoid arthritis patients. Inflamm Res 66(5):433–440. https://doi.org/10.1007/s00011-017-1027-5
Huang DN, Wu FF, Zhang AH, Sun H, Wang XJ (2021) Efficacy of berberine in treatment of rheumatoid arthritis: from multiple targets to therapeutic potential. Pharmacol Res 169:105667. https://doi.org/10.1016/j.phrs.2021.105667
Hughes SD, Ketheesan N, Haleagrahara N (2017) The therapeutic potential of plant flavonoids on rheumatoid arthritis. Crit Rev Food Sci Nutr 57(17):3601–3613. https://doi.org/10.1080/10408398.2016.1246413
Hutami IR, Tanaka E, Izawa T (2019) Crosstalk between Fas and S1P1 signaling via NF-kB in osteoclasts controls bone destruction in the TMJ due to rheumatoid arthritis. Japanese Dental Science Review 55(1):12–19. https://doi.org/10.1016/j.jdsr.2018.09.004
Jabbari N, Eftekhari Z, Roodbari NH, Parivar K (2020) Evaluation of encapsulated eugenol by chitosan nanoparticles on the aggressive model of rheumatoid arthritis. Int Immunopharmacol 85:106554. https://doi.org/10.1016/j.intimp.2020.106554
Janakiraman K, Krishnaswami V, Rajendran V, Natesan S, Kandasamy R (2018) Novel nano therapeutic materials for the effective treatment of rheumatoid arthritis-recent insights. Materials Today Communications 17:200–213. https://doi.org/10.1016/j.mtcomm.2018.09.011
Javadi F, Ahmadzadeh A, Eghtesadi S, Aryaeian N, Zabihiyeganeh M, Rahimi Foroushani A, Jazayeri S (2017) The effect of quercetin on inflammatory factors and clinical symptoms in women with rheumatoid arthritis: a double-blind, randomized controlled trial. J Am Coll Nutr 36(1):9–15. https://doi.org/10.1080/07315724.2016.1140093
Javadi M, KhademHaghighian H, Goodarzy S, Abbasi M, Nassiri-Asl M (2019) Effect of curcumin nanomicelle on the clinical symptoms of patients with rheumatoid arthritis: a randomized, double-blind, controlled trial. Int J Rheum Dis 22(10):1857–1862. https://doi.org/10.1111/1756-185X.13688
Jiang LL, Li K, Lin QH, Ren J, He ZH, Li H, Shen N, Wei P, Feng F, He MF (2016) Gambogic acid causes fin developmental defect in zebrafish embryo partially via retinoic acid signaling. Reprod Toxicol 63:161–168. https://doi.org/10.1016/j.reprotox.2016.06.004
Jomaa G, Kwan CK, Fu SC, Ling SKK, Chan KM, Yung PSH, Rolf C (2020) A systematic review of inflammatory cells and markers in human tendinopathy. BMC Musculoskelet Disord 21(1). https://doi.org/10.1186/s12891-020-3094-y
Kabe Y, Ando K, Hirao S, Yoshida M, Handa H (2005) Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7(3–4):395–403. https://doi.org/10.1089/ars.2005.7.395
Kang JH, Kim CS, Han IS, Kawada T, Yu R (2007) Capsaicin, a spicy component of hot peppers, modulates adipokine gene expression and protein release from obese-mouse adipose tissues and isolated adipocytes, and suppresses the inflammatory responses of adipose tissue macrophages. FEBS Lett 581(23):4389–4396. https://doi.org/10.1016/j.febslet.2007.07.082
Kashfi K, Kannikal J, Nath N (2021) Macrophage reprogramming and cancer therapeutics: Role of iNOS-derived NO. Cells 10(11):3194. https://doi.org/10.3390/cells10113194
Kato M (2020) New insights into IFN-γ in rheumatoid arthritis: role in the era of JAK inhibitors. Immunological Medicine 43(2):72–78. https://doi.org/10.1080/25785826.2020.1751908
Kersley GD (2009) Aspirin, phenylbutazone or hormones in the treatment of rheumatoid arthritis. Acta MedicaScandinavica 162(S341):189–191. https://doi.org/10.1111/j.0954-6820.1958.tb19108.x
Khan MA, Subramaneyaan M, Arora VK, Banerjee BD, Ahmed RS (2015) Effect of Withania somnifera (Ashwagandha) root extract on amelioration of oxidative stress and autoantibodies production in collagen-induced arthritic rats. Journal of Complementary and Integrative Medicine 12(2):117–125. https://doi.org/10.1515/jcim-2014-0075
Khansai M, Phitak T, Klangjorhor J, Udomrak S, Fanhchaksai K, Pothacharoen P, Kongtawelert P (2017) Effects of sesamin on primary human synovial fibroblasts and SW982 cell line induced by tumor necrosis factor-alpha as a synovitis-like model. BMC Complement Altern Med. https://doi.org/10.1186/s12906-017-2035-2
Khayyal MT, El-Hazek RM, El-Sabbagh WA, Frank J, Behnam D, Abdel-Tawab M (2018) Micellar solubilisation enhances the antiinflammatory activities of curcumin and boswellic acids in rats with adjuvant-induced arthritis. Nutrition 54:189–196. https://doi.org/10.1016/j.nut.2018.03.055
Khojah HMJ, Ahmed S, Abdelrahman MAE, El-Hakeim EH (2018) Resveratrol as an effective adjuvant therapy in the management of rheumatoid arthritis: a clinical study. Clin Rheumatol. https://doi.org/10.1007/s10067-018-4080-8
Kim HR, Kim BM, Won JY, Lee KA, Ko HM, Kang YS, Lee SH, Kim KW (2019) Quercetin, a plant polyphenol, has potential for the prevention of bone destruction in rheumatoid arthritis. J Med Food 22(2):152–161. https://doi.org/10.1089/jmf.2018.4259
Kitanaka T, Nakano R, Kitanaka N, Kimura T, Okabayashi K, Narita T, Sugiya H (2017) JNK activation is essential for activation of MEK/ERK signaling in IL-1β-induced COX-2 expression in synovial fibroblasts. Sci Rep 7(1). https://doi.org/10.1038/srep39914
Kobayashi Y (2010) Regulatory role of nitric oxide in proinflammatory cytokine expression during the induction and resolution of inflammation. OUP Academic. https://doi.org/10.1189/jlb.0310149
Kong L, Wang L, Zhao Q, Di G, Wu H (2020) Rhodojaponin II inhibits TNF‐α‐induced inflammatory cytokine secretion in MH7A human rheumatoid arthritis fibroblast‐like synoviocytes. J Biochem Mol Toxicol 34(10). https://doi.org/10.1002/jbt.22551
Korb A, Tohidast-Akrad M, Cetin E, Axmann R, Smolen J, Schett G (2006) Differential tissue expression and activation of p38 MAPK α, β, γ, and δ isoforms in rheumatoid arthritis. Arthritis Rheum 54(9):2745–2756. https://doi.org/10.1002/art.22080
Kriplani P, Guarve K, Baghel US (2020) Helenalin: an anti-inflammatory and anti-neoplastic agent: a review. Curr Bioact Compd 16(8):1134–1146. https://doi.org/10.2174/1573407216666191226121004
Kronzer VL, Davis JM (2021) Etiologies of rheumatoid arthritis: update on mucosal, genetic, and cellular pathogenesis. Curr Rheumatol Rep 23(4). https://doi.org/10.1007/s11926-021-00993-0
Krueger JG, Brunner PM (2017) Interleukin-17 alters the biology of many cell types involved in the genesis of psoriasis, systemic inflammation and associated comorbidities. Exp Dermatol 27(2):115–123. https://doi.org/10.1111/exd.13467
Lafeber FPJG, Beukelman CJ, van den Worm E, van Roy JLAM, Vianen ME, van Roon JAG, van Dijk H, Bijlsma JWJ (1999) Apocynin, a plant-derived, cartilage-saving drug, might be useful in the treatment of rheumatoid arthritis. Rheumatology 38(11):1088–1093. https://doi.org/10.1093/rheumatology/38.11.1088
Lee AK, Sung SH, Kim YC, Kim SG (2003) Inhibition of lipopolysaccharide-inducible nitric oxide synthase, TNF-αand COX-2 expression by sauchinone effects on I-κBαphosphorylation, C/EBP and AP-1 activation. Br J Pharmacol 139(1):11–20. https://doi.org/10.1038/sj.bjp.0705231
Lee JH, Jin H, Shim HE, Kim HN, Ha H, Lee ZH (2009) Epigallocatechin-3-gallate inhibits osteoclastogenesis by down-regulating c-Fos expression and suppressing the nuclear factor-κB signal. Mol Pharmacol 77(1):17–25. https://doi.org/10.1124/mol.109.057877
Lee JY, Choi JK, Jeong NH, Yoo J, Ha YS, Lee B, Choi H, Park PH, Shin TY, Kwon TK, Lee SR, Lee S, Lee SW, Rho MC, Kim SH (2017) Anti-inflammatory effects of ursolic acid-3-acetate on human synovial fibroblasts and a murine model of rheumatoid arthritis. Int Immunopharmacol 49:118–125. https://doi.org/10.1016/j.intimp.2017.05.028
Lee SY, Jung YO, Ryu JG, Oh HJ, Son HJ, Lee SH, Kwon JE, Kim EK, Park MK, Park SH, Kim HY, Cho ML (2016) Epigallocatechin-3-gallate ameliorates autoimmune arthritis by reciprocal regulation of T helper-17 regulatory T cells and inhibition of osteoclastogenesis by inhibiting STAT3 signaling. J Leukoc Biol 100(3):559–568. https://doi.org/10.1189/jlb.3a0514-261rr
Lee YR, Lee JH, Noh EM, Kim EK, Song MY, Jung WS, Park SJ, Kim JS, Park JW, Kwon KB, Park BH (2008) Guggulsterone blocks IL-1β-mediated inflammatory responses by suppressing NF-κB activation in fibroblast-like synoviocytes. Life Sci 82(23–24):1203–1209. https://doi.org/10.1016/j.lfs.2008.04.006
Li GF, Qin YH, Du PQ (2015) Andrographolide inhibits the migration, invasion and matrix metalloproteinase expression of rheumatoid arthritis fibroblast-like synoviocytes via inhibition of HIF-1α signaling. Life Sci 136:67–72. https://doi.org/10.1016/j.lfs.2015.06.019
Li J, Liu R, Zhang P, Li J, Yue Y, Hu Y, Cheng W, Pan X (2014) Genistein suppresses tumor necrosis factor α-induced inflammation via modulating reactive oxygen species/Akt/nuclear factor ΚB and adenosine monophosphate-activated protein kinase signal pathways in human synoviocyte MH7A cells. Drug Des Devel Ther 315. https://doi.org/10.2147/dddt.s52354
Li N, Gong Z, Li X, Ma Q, Wu M, Liu D, Deng L, Pan D, Liu Q, Wei Z, Wang Q, Han L, Lin C, Chen J (2019) Betulinic acid inhibits the migration and invasion of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Int Immunopharmacol 67:186–193. https://doi.org/10.1016/j.intimp.2018.11.042
Li X, Ma J, Li Y (2016) Molecular cloning and expression determination of p38 MAPK from the liver and kidney of silver carp. J Biochem Mol Toxicol 30(5):224–231. https://doi.org/10.1002/jbt.21781
Li X, Udagawa N, Itoh K, Suda K, Murase Y, Nishihara T, Suda T, Takahashi N (2002) p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 143(8):3105–3113. https://doi.org/10.1210/endo.143.8.8954
Li ZZ, Tan JP, Wang LL, Li QH (2017) Andrographolide benefits rheumatoid arthritis via inhibiting MAPK pathways. Inflammation 40(5):1599–1605. https://doi.org/10.1007/s10753-017-0600-y
Liang YJ, Yang WX (2019) Kinesins in MAPK cascade: how kinesin motors are involved in the MAPK pathway? Gene 684:1–9. https://doi.org/10.1016/j.gene.2018.10.042
Lin N, Liu C, Xiao C, Jia H, Imada K, Wu H, Ito A (2007) Triptolide, a diterpenoid triepoxide suppresses inflammation and cartilage destruction in collagen-induced arthritis mice. Biochem Pharmacol 73(1):136–146. https://doi.org/10.1016/j.bcp.2006.08.027
Liu C, Zhang Y, Kong X, Zhu L, Pang J, Xu Y, Chen W, Zhan H, Lu A, Lin N (2013) Triptolide prevents bone destruction in the collagen-induced arthritis model of rheumatoid arthritis by targeting RANKL/RANK/OPG signal pathway. Evidence-Based Complementary and Alternative Medicine 2013:1–12. https://doi.org/10.1155/2013/626038
Liu Q, Zhao J, Tan R, Zhou H, Lin Z, Zheng M, Romas E, Xu J, Sims N (2014) Parthenolide inhibits pro-inflammatory cytokine production and exhibits protective effects on progression of collagen-induced arthritis in a rat model. Scand J Rheumatol 44(3):182–191. https://doi.org/10.3109/03009742.2014.938113
Loiacono C, Palermi S, Massa B, Belviso I, Romano V, Di Gregorio A, Sirico F, Sacco AM (2019) Tendinopathy: pathophysiology, therapeutic options, and role of nutraceutics. A Narrative Literature Review Medicina 55(8):447. https://doi.org/10.3390/medicina55080447
Lu N, Malemud CJ (2019) Extracellular signal-regulated kinase: a regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression. Int J Mol Sci 20(15):3792. https://doi.org/10.3390/ijms20153792
Makol A, Krause M (2016) Management of rheumatoid arthritis during pregnancy: challenges and solutions. Open Access Rheumat: Res Rev 23. https://doi.org/10.2147/oarrr.s85340
Ma N, Liu Y, Ling G, Zhang P (2022) Preparation of meloxicam-salicylic acid co-crystal and its application in the treatment of rheumatoid arthritis. Journal of Drug Delivery Science and Technology 74:103542. https://doi.org/10.1016/j.jddst.2022.103542
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ (2000) Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis & Rheumatism 43(1):30–37. https://doi.org/10.1002/1529-0131(200001)43:1<30::aid-anr5>3.0.co;2-b
Malemud CJ (2018) The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis 10(5–6), 117–127. https://doi.org/10.1177/1759720x18776224
Malemud C, Pearlman E (2009) Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr Signal Transduct Ther 4(3):201–221. https://doi.org/10.2174/157436209789057467
Mandot DU (2020) Evaluate efficacy of autologous platelet rich plasma injection versus conservative modalities in treatment of chronic tendenopathies. J Med Sci Clin Res 08(03). https://doi.org/10.18535/jmscr/v8i3.17
Maranini B, Bortoluzzi A, Silvagni E, Govoni M (2022) Focus on sex and gender: what we need to know in the management of rheumatoid arthritis. Journal of Personalized Medicine 12(3):499. https://doi.org/10.3390/jpm12030499
Mateen S, Zafar A, Moin S, Khan AQ, Zubair S (2016) Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. ClinicaChimicaActa 455:161–171. https://doi.org/10.1016/j.cca.2016.02.010
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7(6):429–442. https://doi.org/10.1038/nri2094
McInnes IB, Schett G (2017) Pathogenetic insights from the treatment of rheumatoid arthritis. The Lancet 389(10086):2328–2337. https://doi.org/10.1016/s0140-6736(17)31472-1
Messemaker TC, Frank-Bertoncelj M, Marques RB, Adriaans A, Bakker AM, Daha N, Gay S, Huizinga TW, Toes REM, Mikkers HMM, Kurreeman F (2015a) A novel long non-coding RNA in the rheumatoid arthritis risk locus TRAF1-C5 influences C5 mRNA levels. Genes Immun 17(2):85–92. https://doi.org/10.1038/gene.2015.54
Messemaker TC, Huizinga TW, Kurreeman F (2015b) Immunogenetics of rheumatoid arthritis: understanding functional implications. J Autoimmun 64:74–81. https://doi.org/10.1016/j.jaut.2015.07.007
Micheli L, Di Cesare Mannelli L, Mattoli L, Tamimi S, Flamini E, Garetto S, Lucci J, Giovagnoni E, Cinci L, D’Ambrosio M, Luceri C, Ghelardini C (2020) Intra-articular route for the system of molecules 14G1862 from Centella asiatica: pain relieving and protective effects in a rat model of osteoarthritis. Nutrients 12(6):1618. https://doi.org/10.3390/nu12061618
Millar NL, Murrell GAC, McInnes IB (2017) Inflammatory mechanisms in tendinopathy – towards translation. Nat Rev Rheumatol 13(2):110–122. https://doi.org/10.1038/nrrheum.2016.213
Min SY, Yan M, Kim SB, Ravikumar S, Kwon SR, Vanarsa K, Kim HY, Davis LS, Mohan C (2015) Green tea epigallocatechin-3-gallate suppresses autoimmune arthritis through indoleamine-2,3-dioxygenase expressing dendritic cells and the nuclear factor, erythroid 2-like 2 antioxidant pathway. J Inflamm 12(1). https://doi.org/10.1186/s12950-015-0097-9
Mohammad-Shahi M, Mowla K, Haidari F, Zarei M, Choghakhori R (2016) Soy milk consumption, markers of inflammation and oxidative stress in women with rheumatoid arthritis: a randomised cross-over clinical trial. Nutr Diet 73(2):139–145. https://doi.org/10.1111/1747-0080.12174
Morohoshi M, Fujisawa K, Uchimura I, Numano F (2006) The effect of glucose and advanced glycosylation end products on IL-6 production by human monocytes. Ann N Y Acad Sci 748(1):562–570. https://doi.org/10.1111/j.1749-6632.1994.tb17362.x
Movahedi, M., Beauchamp, M. E., Abrahamowicz, M., Ray, D. W., Michaud, K., Pedro, S., & Dixon, W. G. (2015). Risk of incident diabetes associated with dose and duration of oral glucocorticoid therapy in patients with rheumatoid arthritis. Arthritis & Rheumatology, n/a-n/a. https://doi.org/10.1002/art.39537
Mun SH, Kim HS, Kim JW, Ko NY, Kim DK, Lee BY, Kim B, Won HS, Shin HS, Han JW, Lee HY, Kim YM, Choi WS (2009) Oral administration of curcumin suppresses production of matrix metalloproteinase (MMP)-1 and MMP-3 to ameliorate collagen-induced arthritis: inhibition of the PKCδ/JNK/c-Jun pathway. J Pharmacol Sci 111(1):13–21. https://doi.org/10.1254/jphs.09134fp
Murakami A (2009) Chemoprevention with phytochemicals targeting inducible nitric oxide synthase. Forum of Nutr 193–203. https://doi.org/10.1159/000212751
Murakami A, Ohigashi H (2007) Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer 121(11):2357–2363. https://doi.org/10.1002/ijc.23161
Nakatake R, Hishikawa H, Matushima H, Nakamura Y, Ishizaki M, Matsui K, Kaibori M, Nishizawa M, Okumura T, Kwon AH (2017) SUN-LB315: curcumin protects liver inflammation by suppressing INOS induction in rat hepatocytes. Clin Nutr 36:S170–S171. https://doi.org/10.1016/s0261-5614(17)30648-9
Namba S, Nakano R, Kitanaka T, Kitanaka N, Nakayama T, Sugiya H (2017) ERK2 and JNK1 contribute to TNF-α-induced IL-8 expression in synovial fibroblasts. PLoS ONE 12(8):e0182923. https://doi.org/10.1371/journal.pone.0182923
Narsinghani T, Chaturvedi S (2006) QSAR analysis of meclofenamic acid analogues as selective COX-2 inhibitors. Bioorg Med Chem Lett 16(2):461–468. https://doi.org/10.1016/j.bmcl.2005.07.067
Nathan C, Cunningham-Bussel A (2013) Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol 13(5):349–361. https://doi.org/10.1038/nri3423
Ngo SNT, Addison CJ (2018) Are COX-2 selective COX inhibitors associated with less GI, renal, and cardiovascular side effects: evidence from animals treated with COX inhibitors. Annual Research & Review in Biology 29(6):1–8. https://doi.org/10.9734/arrb/2018/45152
Nunes CDR, BarretoArantes M, de Faria M, Pereira S, Leandro da Cruz L, de Souza Passos M, Pereira de Moraes L, Vieira IJC, Barros de Oliveira D (2020) Plants as sources of anti-inflammatory agents. Molecules 25(16):3726. https://doi.org/10.3390/molecules25163726
Okada Y (2014) From the era of genome analysis to the era of genomic drug discovery: a pioneering example of rheumatoid arthritis. Clin Genet 86(5):432–440. https://doi.org/10.1111/cge.12465
Orellana C, Saevarsdottir S, Klareskog L, Karlson EW, Alfredsson L, Bengtsson C (2017) Oral contraceptives, breastfeeding and the risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 76(11):1845–1852. https://doi.org/10.1136/annrheumdis-2017-211620
Owona BA, Abia WA, Moundipa PF (2020) Natural compounds flavonoids as modulators of inflammasomes in chronic diseases. Int Immunopharmacol 84:106498. https://doi.org/10.1016/j.intimp.2020.106498
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87(1):315–424. https://doi.org/10.1152/physrev.00029.2006
Pereira-Leite C, Nunes C, Jamal SK, Cuccovia IM, Reis S (2016) Nonsteroidal anti-inflammatory therapy: a journey toward safety. Med Res Rev 37(4):802–859. https://doi.org/10.1002/med.21424
Plenge RM (2009) Rheumatoid arthritis genetics: 2009 update. Curr Rheumatol Rep 11(5):351–356. https://doi.org/10.1007/s11926-009-0050-0
Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD (2005) Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. The American Journal of Human Genetics 77(6):1044–1060. https://doi.org/10.1086/498651
Poništ S, Zloh M, Bauerová K (2020, April 8) Impact of oxidative stress on inflammation in rheumatoid and adjuvant arthritis: damage to lipids, proteins, and enzymatic antioxidant defense in plasma and different tissues. IntechOpen eBooks. https://doi.org/10.5772/intechopen.89480
Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, Siminovitch KA, Bae SC, Plenge RM, Gregersen PK, de Bakker PIW (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44(3):291–296. https://doi.org/10.1038/ng.1076
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Gregersen PK (2007) STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357(10):977–986. https://doi.org/10.1056/nejmoa073003
Ritter AMV, Hernandes L, da Rocha BA, Estevão-Silva CF, Wisniewski-Rebecca ES, Cezar JDS, Caparroz-Assef SM, Cuman RKN, Bersani-Amado CA (2017) Anethole reduces inflammation and joint damage in rats with adjuvant-induced arthritis. Inflamm Res 66(8):725–737. https://doi.org/10.1007/s00011-017-1053-3
Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, Ciccia F, Alvaro S, Triolo G, Giacomelli R (2015) Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1β via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: a possible implication for therapeutic decision in these patients. Clin Exp Immunol 182(1):35–44. https://doi.org/10.1111/cei.12667
Samra MM, Hafeez H, Sadia A, Imran M, Basra MAR (2022) Synthesis, characterization, docking and biological studies of M(II) (M= Mg, Ca, Sr) piroxicam complexes. J Mol Struct 1254:132256. https://doi.org/10.1016/j.molstruc.2021.132256
Sánchez-Hidalgo, M., León-González, A. J., Gálvez-Peralta, M., González-Mauraza, N. H., & Martin-Cordero, C. (2020, May 13). d-Pinitol: a cyclitol with versatile biological and pharmacological activities. Phytochemistry Reviews, 20(1), 211–224. https://doi.org/10.1007/s11101-020-09677-6
Sarmiento Salinas FL, SantillánBenítez JG, Hernández Navarro MD, MendietaZerón H (2017) NF-κB1/IKKε gene expression and clinical activity in patients with rheumatoid arthritis. Laboratory Medicine 49(1):11–17. https://doi.org/10.1093/labmed/lmx033
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30(11):1191–1212. https://doi.org/10.1016/s0891-5849(01)00480-4
Shang W, Zhao LJ, Dong XL, Zhao ZM, Li J, Zhang BB, Cai H (2016) Curcumin inhibits osteoclastogenic potential in PBMCs from rheumatoid arthritis patients via the suppression of MAPK/RANK/c-Fos/NFATc1 signaling pathways. Mol Med Rep 14(4):3620–3626. https://doi.org/10.3892/mmr.2016.5674
Shao B, Zhu L, Dong M, Wang J, Wang J, Xie H, Zhu S (2012) DNA damage and oxidative stress induced by endosulfan exposure in zebrafish (Danio rerio). Ecotoxicology 21:1533–1540. https://doi.org/10.1007/s10646-012-0907-2
Sharma D, Chaubey P, Suvarna V (2021) Role of natural products in alleviation of rheumatoid arthritis—a review. J Food Biochem 45(4). https://doi.org/10.1111/jfbc.13673
Shavandi M, Moini A, Shakiba Y, Mashkorinia A, Dehghani M, Asar S, Kiani A (2017) Silymarin (Livergol®) decreases disease activity score in patients with rheumatoid arthritis: a non-randomized single-Arm clinical trial. Iran J Allergy Asthma Immunol 7:99–106. https://doi.org/10.1111/1747-0080.12174
Shenghao T, Yonghong H, Keqin Z, Mingmin Z, Xianyang L, Weichen Z (2005) Effects of triptolide on the expression and activity of NF-κB in synovium of collagen-induced arthritis rats. Journal of Huazhong University of Science and Technology [medical Sciences] 25(5):543–545. https://doi.org/10.1007/bf02896012
Shu C, Chen J, Lv M, Xi Y, Zheng J, Xu X (2022) Plumbagin relieves rheumatoid arthritis through nuclear factor kappa-B (NF-κB) pathway. Bioengineered 13(5):13632–13642. https://doi.org/10.1080/21655979.2022.2081756
Silvestre GFG, de Lucena RP, da Silva Alves H (2022) Cucurbitacins and the immune system: update in research on anti-inflammatory, antioxidant, and immunomodulatory mechanisms. Curr Med Chem 29(21):3774–3789. https://doi.org/10.2174/0929867329666220107153253
Singgih and Achmad (2020) A review of nonsteroidal anti-inflammatory drugs (COX inhibitors) medications in dentistry: uses and side effects. Syst Rev Pharm 11(05). https://doi.org/10.31838/srp.2020.5.43
Singh R, Ahmed S, Malemud CJ, Goldberg VM, Haqqi TM (2003) Epigallocatechin-3-gallate selectively inhibits interleukin-1 β-induced activation of mitogen activated protein kinase subgroup c-Jun N-terminal kinase in human osteoarthritis chondrocytes. J Orthop Res 21(1):102–109. https://doi.org/10.1016/s0736-0266(02)00089-x
Singh AK, Umar S, Riegsecker S, Chourasia M, Ahmed S (2016) Regulation of transforming growth factor β-activated kinase activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts: suppression of K63-linked autoubiquitination of tumor necrosis factor receptor-associated factor 6. Arthritis Rheumatol 68(2):347–358. https://doi.org/10.1002/art.39447
Spurlock CF, Tossberg JT, Olsen NJ, Aune TM (2015) Cutting edge: chronic NF-κB activation in CD4+ T cells in rheumatoid arthritis is genetically determined by HLA risk alleles. J Immunol 195(3):791–795. https://doi.org/10.4049/jimmunol.1500267
Steultjens M, Bell K, Hendry G (2022) The challenges of measuring physical activity and sedentary behaviour in people with rheumatoid arthritis. Rheumatol Adv Pract 7(1). https://doi.org/10.1093/rap/rkac101
Sun WX, Liu Y, Zhou W, Li HW, Yang J, Chen ZB (2016) Shikonin inhibits TNF-α production through suppressing PKC-NF-κB-dependent decrease of IL-10 in rheumatoid arthritis-like cell model. J Nat Med 71(2):349–356. https://doi.org/10.1007/s11418-016-1064-3
Sur LM, Gaga R, Duca E, Sur G, Lupan I, Sur D, Samasca G, Lazea C, Lazar C (2021) Different chronic disorders that fall within the term juvenile idiopathic arthritis. Life 11(5):398. https://doi.org/10.3390/life11050398
Takeuchi T (2022) Correction: Cytokines and cytokine receptors as targets of immune-mediated inflammatory diseases—RA as a role model. Inflamm Regen 42(1). https://doi.org/10.1186/s41232-022-00250-6
Tang M, Zhu W, Yang Z, He C (2019) Brucine inhibits TNF-α-induced HFLS-RA cell proliferation by activating the JNK signaling pathway. Exp Ther Med. https://doi.org/10.3892/etm.2019.7582
Tang S, Shen XY, Huang HQ, Xu SW, Yu Y, Zhou CH, Chen SR, Le K, Wang YH, Liu PQ (2010) Cryptotanshinone suppressed inflammatory cytokines secretion in RAW264.7 macrophages through inhibition of the NF-κB and MAPK signaling pathways. Inflammation 34(2), 111–118. https://doi.org/10.1007/s10753-010-9214-3
Thalhamer T, McGrath MA, Harnett MM (2007) MAPKs and their relevance to arthritis and inflammation. Rheumatology 47(4):409–414. https://doi.org/10.1093/rheumatology/kem297
Thomsen T, Beyer N, Aadahl M, Hetland ML, Løppenthin K, Midtgaard J, Esbensen BA (2015) Sedentary behaviour in patients with rheumatoid arthritis: a qualitative study. Int J Qual Stud Health Well Being 10(1):28578. https://doi.org/10.3402/qhw.v10.28578
Tian J, Chen JW, Gao JS, Li L, Xie X (2013) Resveratrol inhibits TNF-α-induced IL-1β, MMP-3 production in human rheumatoid arthritis fibroblast-like synoviocytes via modulation of PI3kinase/Akt pathway. Rheumatol Int 33(7):1829–1835. https://doi.org/10.1007/s00296-012-2657-0
Toda K (2021) Are topical non-steroidal anti-inflammatory drugs (COX inhibitors) safer than oral COX inhibitors? Or are the adverse effects of topical COX inhibitors comparable to those of oral COX inhibitors? Osteoarthritis Cartilage 29(11):1624–1625. https://doi.org/10.1016/j.joca.2021.08.010
Tsai SH, Lin-Shiau SY, Lin JK (1999) Suppression of nitric oxide synthase and the down-regulation of the activation of NFκB in macrophages by resveratrol. Br J Pharmacol 126(3):673–680. https://doi.org/10.1038/sj.bjp.0702357
Umar S, Hedaya O, Singh AK, Ahmed S (2015) Thymoquinone inhibits TNF-α-induced inflammation and cell adhesion in rheumatoid arthritis synovial fibroblasts by ASK1 regulation. Toxicol Appl Pharmacol 287(3):299–305. https://doi.org/10.1016/j.taap.2015.06.017
Unal S, Gumruk F, Aytac S, Yalnzoglu D, Gurgey A (2008) Interleukin-6 (IL-6), Tumor Necrosis Factor-α (TNF-α) Levels and IL-6, TNF-polymorphisms in children with thrombosis. J Pediatr Hematol Oncol 30(1):26–31. https://doi.org/10.1097/mph.0b013e31815b1a89
Uttra AM, Alamgeer, SM, Shabbir A, Jahan S, Bukhari IA, Assiri AM (2019) Ribesorientale: a novel therapeutic approach targeting rheumatoid arthritis with reference to pro-inflammatory cytokines, inflammatory enzymes and anti-inflammatory cytokines. J Ethnopharmacol 237, 92–107. https://doi.org/10.1016/j.jep.2019.03.019
Vaishya R, Vaish A, Ansari AH (2021) Acute painful calcific tendonitis of the shoulder. Postgrad Med J 98(1159):e2–e2. https://doi.org/10.1136/postgradmedj-2021-139719
Vannini F, Kashfi K, Nath N (2015) The dual role of iNOS in cancer. Redox Biol 6:334–343. https://doi.org/10.1016/j.redox.2015.08.009
Vasanthi P, Nalini G, Rajasekhar G (2007) Role of tumor necrosis factor-alpha in rheumatoid arthritis: a review. APLAR J Rheumatol 10(4):270–274. https://doi.org/10.1111/j.1479-8077.2007.00305.x
Venkatesha SH, Dudics S, Astry B, Moudgil KD (2016) Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog Dis 74(6), ftw059. https://doi.org/10.1093/femspd/ftw059
Verdrengh M, Jonsson IM, Holmdahl R, Tarkowski A (2003) Genistein as an anti-inflammatory agent. Inflamm Res. https://doi.org/10.1007/s00011-003-1182-8
Voirin AC, Perek N, Roche F (2020) Inflammatory stress induced by a combination of cytokines (IL-6, IL-17, TNF-α) leads to a loss of integrity on bEnd.3 endothelial cells in vitro BBB model. Brain Res 1730, 146647. https://doi.org/10.1016/j.brainres.2020.146647
Wahid S, Khan RA, Feroz Z, Ikram R (2020) Analgesic, anti-inflammatory and toxic effects of ethanol extracts of Cucumis melo and Citrullus lanatus seeds. Pak J Pharm Sci 33(3). https://doi.org/10.36721/PJPS.2020.33.3.REG.1049-1055.1
Wang J, Yang H, Li Q, Wu X, Di G, Fan J, Wei D, Guo C (2020) Novel nanomicelles based on rebaudioside A: a potential nanoplatform for oral delivery of honokiol with enhanced oral bioavailability and antitumor activity. Int J Pharm 590:119899. https://doi.org/10.1016/j.ijpharm.2020.119899
Wang L, Yang HJ, Tu R, Shen M, Liu D (2017a) In silico design of colchicine-based bioisosteric inhibitors of tubulin for the treatment of rheumatoid arthritis. Mol Med Rep 16(4):4823–4828. https://doi.org/10.3892/mmr.2017.7202
Wang S, Liu Z, Wang J, Wang Y, Liu J, Ji X, Wang X (2018a) The triptolide-induced apoptosis of osteoclast precursor by degradation of cIAP2 and treatment of rheumatoid arthritis of TNF-transgenic mice. Phytother Res 33(2):342–349. https://doi.org/10.1002/ptr.6224
Wang W, Sun W, Jin L (2017b) Caffeic acid alleviates inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes by inhibiting phosphorylation of IκB kinase α/β and IκBα. Int Immunopharmacol 48:61–66. https://doi.org/10.1016/j.intimp.2017.04.025
Wang Y, Wang S, Li Y, Jiang J, Zhou C, Li C, Li D, Lu L, Liu P, Huang M, Shen X (2015) Therapeutic effect of Cryptotanshinone on collagen-induced arthritis in rats via inhibiting nuclear factor kappa B signaling pathway. Transl Res 165(6):704–716. https://doi.org/10.1016/j.trsl.2014.12.004
Wang Y, Zhou C, Gao H, Li C, Li D, Liu P, Huang M, Shen X, Liu L (2017c) Therapeutic effect of Cryptotanshinone on experimental rheumatoid arthritis through downregulating p300 mediated-STAT3 acetylation. Biochem Pharmacol 138:119–129. https://doi.org/10.1016/j.bcp.2017.05.006
Wang Z, Chen Z, Yang S, Wang Y, Huang Z, Gao J, Tu S, Rao Z (2014) Berberine ameliorates collagen-induced arthritis in rats associated with anti-inflammatory and anti-angiogenic effects. Inflammation 37(5), 1789–1798. https://doi.org/10.1007/s10753-014-9909-y
Wang Z, Yeung S, Chen S, Moatazedi Y, Chow MS (2018b) Bioavailability of wilforlide A in mice and its concentration determination using an HPLC-APCI-MS/MS method. J Chromatogr B 1090:65–72. https://doi.org/10.1016/j.jchromb.2018.05.018
Weiser T (2021) Treatment of acute pain: comparison of the efficacy of ibuprofen lysinate and ibuprofen. Evidence for Self-Medication 1. https://doi.org/10.52778/efsm.21.0271
Weitzmann M (2011) Quercetin, a potent suppressor of NF-κB and Smad activation in osteoblasts. Int J Mol Med. https://doi.org/10.3892/ijmm.2011.749
Weyand CM, Goronzy JJ (2017) Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol 13(5):291–301. https://doi.org/10.1038/nrrheum.2017.49
Wu JJ, Yuan XM, Huang C, An GY, Liao ZL, Liu GA, Chen RX (2019) Farnesyl thiosalicylic acid prevents iNOS induction triggered by lipopolysaccharide via suppression of iNOS mRNA transcription in murine macrophages. Int Immunopharmacol 68:218–225. https://doi.org/10.1016/j.intimp.2018.12.066
Xu L, Zhang L, Bertucci AM, Pope RM, Datta SK (2008) Apigenin, a dietary flavonoid, sensitizes human T cells for activation-induced cell death by inhibiting PKB/Akt and NF-κB activation pathway. Immunol Lett 121(1):74–83. https://doi.org/10.1016/j.imlet.2008.08.004
Yan J, Yang C, He C, Yang Z, Lu CH, Chen X (2011) Andrographolide induces cell cycle arrest and apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes. Cell Biol Toxicol. https://doi.org/10.1007/s10565-011-9204-8
Yang C, Wardenaar KJ, Bosker FJ, Li J, Schoevers RA (2019) Inflammatory markers and treatment outcome in treatment resistant depression: a systematic review. J Affect Disord 257:640–649. https://doi.org/10.1016/j.jad.2019.07.045
Yang Y, Ye Y, Qiu Q, Xiao Y, Huang M, Shi M, Liang L, Yang X, Xu H (2016) Triptolide inhibits the migration and invasion of rheumatoid fibroblast-like synoviocytes by blocking the activation of the JNK MAPK pathway. Int Immunopharmacol. https://doi.org/10.1016/j.intimp.2016.10.005
Youn J, Lee JS, Na HK, Kundu JK, Surh YJ (2009) Resveratrol and piceatannol inhibit iNOS expression and NF-κ B activation in dextran sulfate sodium-induced mouse colitis. Nutr Cancer 61(6):847–854. https://doi.org/10.1080/01635580903285072
Yu WG, Shen Y, Wu JZ, Gao YB, Zhang LX (2018) Madecassoside impedes invasion of rheumatoid fibroblast-like synoviocyte from adjuvant arthritis rats via inhibition of NF- κ B-mediated matrix metalloproteinase-13 expression. Chin J Nat Med 16(5):330–338. https://doi.org/10.1016/s1875-5364(18)30064-5
Zaky DS, El-Nahrery EM (2016) Role of interleukin-23 as a biomarker in rheumatoid arthritis patients and its correlation with disease activity. Int Immunopharmacol 31:105–108. https://doi.org/10.1016/j.intimp.2015.12.011
Zeng W, Fang Y, Mo S, Shen C, Yang H, Luo G, Xiao L, Zhan R, Yan P (2023) The underling mechanisms exploration of Rubiacordifolia L. extract against rheumatoid arthritis by integrating network pharmacology and metabolomics. Drug Des Dev Ther 17:439–457. https://doi.org/10.2147/dddt.s388932
Zhai ZJ, Li HW, Liu GW, Qu XH, Tian B, Yan W, Lin Z, Tang TT, Qin A, Dai KR (2014) Andrographolide suppresses RANKL -induced osteoclastogenesis in vitro and prevents inflammatory bone loss in vivo. Br J Pharmacol 171(3):663–675. https://doi.org/10.1111/bph.12463
Zhao JH, Ma S, Li CY, Zhang HC, Zhao LJ, Zhang ZY (2023) Clinically approved small-molecule drugs for the treatment of rheumatoid arthritis. Eur J Med Chem 256:115434. https://doi.org/10.1016/j.ejmech.2023.115434
Zhou J, Yu Y, Yang X, Wang Y, Song Y, Wang Q, Chen Z, Zong S, Fan M, Meng X, Xie C, Zhou F, Liu H, Wei F (2019) Berberine attenuates arthritis in adjuvant-induced arthritic rats associated with regulating polarization of macrophages through AMPK/NF-кB pathway. Eur J Pharmacol 852:179–188. https://doi.org/10.1016/j.ejphar.2019.02.036
Zhu X, Liu Q, Wang M, Liang M, Yang X, Xu X, Zou H, Qiu J (2011) Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in fibroblasts. PLoS ONE 6(11):e27081. https://doi.org/10.1371/journal.pone.0027081
Ziegelasch M, Forslind K, Skogh T, Riklund K, Kastbom A, Berglin E (2017) Decrease in bone mineral density during three months after diagnosis of early rheumatoid arthritis measured by digital X-ray radiogrammetry predicts radiographic joint damage after one year. Arthritis Res Ther 19(1). https://doi.org/10.1186/s13075-017-1403-0
Author information
Authors and Affiliations
Contributions
A.B. drafted the manuscript. U.B., G.S., & A.B. wrote the paper and approved the final manuscript. The authors confirm that no paper mill and artificial intelligence was used.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bashir, U., Singh, G. & Bhatia, A. Rheumatoid arthritis—recent advances in pathogenesis and the anti-inflammatory effect of plant-derived COX inhibitors. Naunyn-Schmiedeberg's Arch Pharmacol 397, 5363–5385 (2024). https://doi.org/10.1007/s00210-024-02982-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-024-02982-3