
Abstract
Melatonin has the ability to intervene in the initiation, progression and metastasis of some experimental cancers. A large variety of potential mechanisms have been advanced to describe the metabolic and molecular events associated with melatonin’s interactions with cancer cells. There is one metabolic perturbation that is common to a large number of solid tumors and accounts for the ability of cancer cells to actively proliferate, avoid apoptosis, and readily metastasize, i.e., they use cytosolic aerobic glycolysis (the Warburg effect) to rapidly generate the necessary ATP required for the high metabolic demands of the cancer cells. There are several drugs, referred to as glycolytic agents, that cause cancer cells to abandon aerobic glycolysis and shift to the more conventional mitochondrial oxidative phosphorylation for ATP synthesis as in normal cells. In doing so, glycolytic agents also inhibit cancer growth. Herein, we hypothesize that melatonin also functions as an inhibitor of cytosolic glycolysis in cancer cells using mechanisms, i.e., downregulation of the enzyme (pyruvate dehydrogenase kinase) that interferes with the conversion of pyruvate to acetyl CoA in the mitochondria, as do other glycolytic drugs. In doing so, melatonin halts the proliferative activity of cancer cells, reduces their metastatic potential and causes them to more readily undergo apoptosis. This hypothesis is discussed in relation to the previously published reports. Whereas melatonin is synthesized in the mitochondria of normal cells, we hypothesize that this synthetic capability is not present in cancer cell mitochondria because of the depressed acetyl CoA; acetyl CoA is necessary for the rate limiting enzyme in melatonin synthesis, arylalkylamine-N-acetyltransferase. Finally, the ability of melatonin to switch glucose oxidation from the cytosol to the mitochondria also explains how tumors that become resistant to conventional chemotherapies are re-sensitized to the same treatment when melatonin is applied.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dysfunctional mitochondria seriously jeopardize optimal cellular and organismal health. There is a category of diseases specifically identified as being related to compromised mitochondrial physiology, the so-called “mitochondrial diseases.” Some of the conditions that fall into this category include cancer, Alzheimer disease, etc. [1,2,3,4].
One hallmark of perturbed mitochondrial physiology that contributes to altered function within cells is the generation of toxic derivatives of oxygen, i.e., reactive oxygen species (ROS) or free radicals [5,6,7]. When this occurs, it may be associated with an alteration in glucose oxidation and energy production by these cells.
Cancer is often a condition in which mitochondria exhibit markedly hindered metabolism [8,9,10]. Cancer cells of many solid tumors shift glucose oxidation from the mitochondria to the cytosol where glucose undergoes rapid metabolism to lactate (the Warburg effect) [11, 12]. This disrupts normal physiology by interfering with the function of the citric acid cycle in the mitochondrial matrix and by reducing the production of ROS (reactive oxygen species) by the electron transport chain (ETC) enzymes in the inner mitochondrial membrane; this is accompanied by a downregulation of mitochondrial ATP synthesis which is also shifted to the less efficient, but more rapid, ATP production in the cytosol [13]. These changes provide cancer cells advantages in terms of accelerated cell proliferation, avoidance of apoptosis, and more rapid metastasis. The Warburg effect occurs in some other diseases in addition to cancer [14].
Melatonin is generally known as the pineal secretory product but is not unique to this gland [15]. Recent evidence has shown melatonin is taken up by and may be produced in the mitochondria of all cells [16,17,18]. In the pineal gland, melatonin is both produced and secreted in a circadian manner with peak levels at night; in other cells mitochondrial melatonin production is neither cyclic nor it is released into the blood [19]. The pineal pool of melatonin is estimated to be less than 5% of the total melatonin produced in vertebrates [20]. The two pools of melatonin have different functions. The 24-h variation in the synthesis and discharge of pineal melatonin is associated with conveying circadian and circannual information throughout the organism, e.g., the wake/sleep cycle, seasonal reproduction, etc. [21, 22]. In contrast, melatonin produced in the mitochondria of all other cells functions in the inhibition of ROS-mediated oxidative stress and the conservation of optimal mitochondrial physiology and, via paracrine means, it possibly impacts the physiology of mitochondria in adjacent cells as well [23].
One of the earliest actions of melatonin to be identified after its discovery was its ability to inhibit the growth of certain cancer types. This ability was found to be related to the downregulation of the metabolic changes that provide them a physiological advantage by reducing tumor biomass and by limiting metastasis [24,25,26,27,28]. The following discussion reconsiders a novel action by which melatonin may inhibit the Warburg effect and function as an oncostatic agent.
Convergence of cancer, glucose oxidation and melatonin
Several years ago, in a study of xenografted human breast cancer cells growing in animals, Blask et al. [29] noted that tumors collected during the day clearly exhibited the expected cytosolic glycolysis as indicated by their high uptake of glucose and abundant release of lactate. When metabolism was measured in tumors collected at night, however, they had obviously abandoned aerobic glycolysis and switched to mitochondria glucose oxidation; the nighttime tumors metabolized only small amounts of glucose to lactate. The data suggested that the cancer cells were of the cancer phenotype (displaying the Warburg effect) during the day but they functioned as a normal cell phenotype during the night when they use mitochondrial glucose oxidation for ATP production (Fig. 1). The authors showed that this metabolic shift was a function of the nighttime rise in circulating melatonin since, when the large-amplitude nocturnal increase in melatonin was inhibited by exposing the cancer-bearing animals to light at night, the tumors continually exhibited the cancer phenotype (high glucose uptake and lactate production over the 24-h period). The authors did not provide an explanation as to the mechanisms by which the melatonin rhythm mediated the day-to-night shift from cytosolic glycolysis to mitochondrial glucose oxidation in the tumors of animals kept under an alternating light:dark cycle.

The top two panels summarize the circadian changes, or lack thereof, in glucose uptake (left) and lactate release (right) under two different lighting conditions. When tumor-bearing rats were exposed to light:dark cycle where it was dark at night (alternating white and black bars at the bottom), both the uptake of glucose and the release of lactate (black curves in panels) exhibited strong circadian rhythms with highest levels of each parameter occurring during the day, indicating that they were using cytosolic glucose oxidation to produce ATP, when circulating endogenous melatonin are lowest (black curve, right lower panel). Conversely, when tumors were collected from animals kept under a light:dark cycle where rats were exposed to dim light at night (LAN) (alternating white and red curves), both glucose and lactate remained elevated throughout the 24-h period; this indicated that the tumors were using cytosolic glucose oxidation to produce ATP. LAN was associated with a markedly diminished nocturnal melatonin increase (red curve, lower right panel). The data are double plotted for clarity. A similar rhythm was seen in 3H-thymidine uptake by the tumors (an index of DNA synthesis and cell proliferation) (data not shown). The lower left panel summarizes the growth of the tumors under the two lighting conditions. The figure was drawn and restructured from the data of Blask et al. [29]
In normal cells, glucose is abundantly taken up via glucose transporters (GLUT1-4) [30] in the cell membrane after which its metabolites enters mitochondria where it is converted to acetyl CoA by a large multienzyme complex named the pyruvate dehydrogenase complex (PDC); acetyl CoA then feeds the citric acid cycle which eventually aids the function of the electron transport chain (ETC) and ATP production. In cancer cells, pyruvate is primarily metabolized to lactate (the Warburg effect) in the cytosol rather than being converted to acetyl CoA in the mitochondria [31]. The high metabolism of glucose in the cytosol rapidly generates the necessary ATP required for the highly elevated proliferative activity of cancer cells, resists their destruction by apoptosis and enhances tumor growth and metastasis [13]. The increased cytosolic metabolism of glucose occurs in cancer cells due to the upregulation of the enzyme pyruvate dehydrogenase kinase (PDK); PDK strongly inhibits PDC interfering with the ability of the mitochondria to utilize pyruvate for acetyl CoA synthesis. This causes glucose metabolism to be shifted to the cytosol for its conversion to lactate [32].
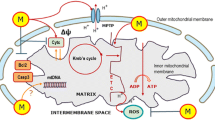
We recently proposed that the day-to-night shift of glucose metabolism in cancer cells, as reported by Blask et al. [29], is a consequence of the ability of melatonin to override the inhibitory action of cancer on the conversion of pyruvate to acetyl CoA in the mitochondrial matrix [33] (Fig. 2). As noted above, in cancer cells the mitochondrial enzyme PDK is upregulated leading to a strong inhibition of PDC, the enzyme that catalyzes the conversion of pyruvate to acetyl CoA [34]. This deprives the mitochondria of acetyl CoA, an important metabolite for normal cellular function and it shifts glucose metabolism to the cytosol. Several anti-cancer drugs being tested, e.g., dichloroacetate (DCA), enter the mitochondria and inhibit PDK allowing for the rapid upregulation of PDC which permits the cancer cells to metabolize pyruvate to acetyl CoA thereby reducing cytosolic glycolysis, converting the cells to a more normal phenotype and reducing cancer growth. Thus, like some other oncostatic drugs, we predict that melatonin, as a mitochondria-targeted molecule, also inhibits PDK and stimulates PDC; this would explain the observations of Blask et al. [29], i.e., that breast cancer cells are only capable of cytosolic aerobic glycolysis during the day when melatonin levels are low and the mitochondria are unable to metabolize pyruvate to acetyl CoA.

Diagrammatic representation of some of the mechanisms by which cancer cells determine their own growth rate and survival, especially by influencing the activity of pyruvate dehydrogenase complex (PDC) and its gate keeper enzyme, pyruvate dehydrogenase kinase (PDK), of which there are multiple isoforms. In cancer cells, several transcription factors including Wnt, Myc and hypoxia-inducible factors (HIFs) may work alone or in concert to transcriptionally promote one or more of the PDKs (the isoform of which varies among cancer cell types). The elevated activity of PDK causes the phosphorylation of serine residues in the E1α subunit of PDC, thereby inactivating the enzyme that normally converts pyruvate to acetyl CoA. With diminished acetyl CoA, the citric acid cycle is deprived of an important anaplerotic agent, mitochondrial melatonin production is inhibited and oxidative phosphorylation is compromised. Melatonin synthesis drops because acetyl CoA is a necessary co-factor for AANAT, the rate limiting enzyme in melatonin synthesis. The metabolic changes in mitochondrial function contribute to programmed cell death (apoptosis). Inhibition of PDC also reprograms glucose metabolism (the glycolytic shift) which then takes place in the cytosol (the Warburg effect) of many solid tumors (upper right). This generates large amounts of lactate and hydrogen ions (H +) which have major impacts on processes that enhance the hardiness of cancer cells, their means to reduce immune surveillance, and their ability to resist chemo- and radiotherapy. In normal cells, pyruvate is primarily shunted into the mitochondria where it is metabolized to acetyl CoA assuring an optimal function of the citric acid cycle, ATP production and melatonin synthesis. AANAT arylalkylamine N-acetyltransferase, ADP adenosine diphosphate, ASMT acetyl serotonin methyl transferase, ATS adenosine triphosphate synthase, CI-CV respiratory complexes, e− electron, LDH lactate dehydrogenase, NASN-acetyl serotonin, Pi inorganic phosphate, TRP tryptophan, 5-HT serotonin
The prediction that melatonin inhibits PDK, which is restricted to the mitochondrial matrix, would require that melatonin enter this organelle to carry out this task. The uptake of melatonin by mitochondria is consistent with a variety of published reports wherein melatonin improved the efficiency of the electron transport chain (ETC) [35], enhanced mitochondrial ATP production [15] and quenched ROS in the mitochondrial matrix [36, 37]. These beneficial actions of melatonin are an improvement over those caused by better known antioxidants, e.g., vitamin E [38, 39], even when the latter are structurally modified to allow more ready entrance into the mitochondria [40]. Recently, Acuna-Castroviejo et al. [41] showed that peripherally injected melatonin rapidly accumulates in mitochondria.
More direct evidence for the entrance of melatonin into the mitochondria of cancer cells was provided by Huo et al. [42]. This group identified two members of the solute carrier (SLC) family transporters, namely PEPT1/2, in cancer cell mitochondrial membranes which were associated with the uptake by and elevation of intramitochondrial levels of melatonin. The authors specifically used multiple cancer cell types for these studies and concluded, without proposing any mechanism, that the high levels of melatonin in mitochondria relate to the ability of this molecule to constrain cancer cell growth [42]. Here, we suggest that melatonin in mitochondria of cancer cells inhibits PDK which activates PDC and allows the entrance of pyruvate into the mitochondria and its conversion to acetyl CoA. This shifts glucose oxidation away from the cytosol and converts cancer cells to a more normal cell phenotype, as observed by Blask et al. [29] for nighttime collected breast tumors.
There is now ample evidence that mitochondria of at least normal cells also may synthesize melatonin [16, 43,44,45], which, if this also occurs in cancer cells, would be expected to inhibit PDK, so the cells would not exhibit the Warburg effect. To date, no attempt has been made to identify melatonin production or levels in cancer cell mitochondria. When attempts are made, we predict that unlike normal cells, the mitochondria of cancer cells will be incapable of or have low intrinsic melatonin synthesis.
The presumed inability of cancer cell mitochondria to synthesize melatonin, particularly during the day, is consistent with what is known about the control of the melatonin synthetic pathway. Acetyl CoA is not only an important metabolite to feed the citric acid cycle, which is situated in the mitochondrial matrix, but it is also a necessary co-factor for the rate limiting arylalkylamine N-acetyltransferase (AANAT), in the melatonin synthetic pathway [46]. In the absence of acetyl CoA, cancer cell mitochondria would not be able to produce their own melatonin, as normal cells do.
At night, however, high circulating levels of melatonin (of pineal origin) would inhibit PDK (stimulating PDC) allowing cancer cells to switch from cytosolic glycolysis in favor of mitochondrial oxidative phosphorylation (Fig. 2). This is consistent with the findings of Blask et al. [29]; during the day cancer cells use aerobic glycolysis for ATP production and at night, due to melatonin-derived via the blood (melatonin of pineal origin), they convert to a more normal cell phenotype. Thus, tumor cells only function with the cancer phenotype about half of the time; at night they display mitochondrial oxidative phosphorylation like normal cells.
A thorough search of the literature was unable to uncover other studies in which in vivo cancer cell metabolism was compared in tumors collected during the day and at night (in darkness). Perhaps this differential day:night difference also is typical of other Warburg-metabolizing solid tumors growing in vivo. This change would not be detected using cultured cancer cells since they are not exposed to a circadian melatonin rhythm.
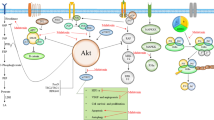
The mechanism by which melatonin may reduce PDK activity thereby allowing for the upregulation of PDC and the conversion of pyruvate to acetyl CoA in the mitochondria matrix is unresolved. Melatonin has numerous receptor-mediated actions [47] as well as functions that may be receptor-independent [48]. Mao and colleagues [49] reported that melatonin-rich blood collected from women who had taken 75 μg melatonin, when perfused onto xenografted leiomyomas growing in nude rats, caused a 57% reduction in glucose uptake and a 44% drop in lactate levels in blood collected as it was exiting the tumor. Since these changes were antagonized by the blockade of the best known melatonin receptors (MT1/MT2), S20928, Mao et al. [49] speculated that melatonin’s actions in suppressing glucose uptake and lactate release involved these receptors. Moreover, they predicted that these receptor-mediated actions involved the phosphorylation of AKT since AKT has been shown to be involved in glycolysis regulation [50]. A potential association of pAKT and PDK has not been examined.
The MT1/MT2 melatonin receptors have been identified on the cell membranes of many normal [51] and cancer cells [52]. There are also reports that document that at least the MT1 receptor is located on the mitochondrial membrane [45, 53]. Thus, the inhibitory actions of melatonin (both exogenously administered or endogenously produced) may impact aerobic glycolysis via the MT1/MT2 receptors (Fig. 3), other intracellular melatonin binding sites [54, 55] or they may be receptor-independent [38, 48].

This figure shows that melatonin likely signals via MT1/2 receptors on the cell surface or after being transported by GLUT1 and PEPT1/2 transporters into the cytosol. In the cytosol of cancer cells, melatonin may alter aerobic glycolysis by signaling through the MT1 receptor on the mitochondrial membrane, by interacting with other binding sites in the cytosol or its actions may be receptor-independent. Both endogenously produced or exogenously administered melatonin is capable of reversing the Warburg effect. GLUT1 glucose transporter 1, MT1 melatonin receptor 1, PEPT1/2 human peptide transporter 1 and 2, pO2 oxygen pressure, LA linoleic acid, 13-HODE 13-hydroxyoctadecadienoic acid, AANAT arylalkylamine N-acetyltransferase, ASMTN-acetylserotonin O-methyltransferase
Another corollary of our prediction is that cancer cell mitochondria, while being incapable of synthesizing melatonin during the day could possibly do so at night. At night, due to the shift of glucose oxidation from the cytosol to the mitochondria, acetyl CoA would be available as a co-factor for AANAT, the rate limiting enzyme in melatonin production [56]. We, thus, presume that while cancer cell mitochondria are essentially devoid of melatonin during the day, they may have high levels at night.
Drugs that reprogram glucose metabolism: glycolytics
A large number of small molecular endogenous and synthetic inhibitors of the PDKs (there are four isoforms) have been identified with the suppressive actions of these agents being executed at four binding loci. These loci include the pyruvate binding site, lipoamide binding locus, nucleotide binding site and an allosteric site [57, 58]. The first three of these are situated in the regulatory N-terminal R domain of these enzymes. The endogenous inhibitors of PDK included pyruvate, NAD+, and CoA; we have proposed that melatonin may also be a member of this group [33]. Additionally, many xenobiotic/synthetic agents capable of inhibiting the PDKs have been developed. The best known of these is dichloroacetate (DCA). DCA was used for several decades as a drug to treat type 2 diabetes and other disorders prior to its potential utility as an anti-cancer agent was discovered [59,60,61]. DCA is strongly inhibitory to the most widely distributed PDK isoform, PDK2, has roughly equal potency against PDK1 and 4 but is incapable of inhibiting PDK3, at least in the male gonad [62]. When administered orally or parenterally, it is absorbed and distributes widely including crossing the blood brain barrier (BBB), justifying its use as an investigational brain cancer inhibitor [63]. Shortly after DCA administration, tissue PDC activity is upregulated [64]. Continual dosing with DCA maintains elevated PDC activity. The rise in PDC activity is apparent by the drop in blood lactate concentrations [65].
Glycolytic agents are drugs capable of changing glucose metabolism in cancer cells so they become reliant on mitochondrial pyruvate metabolism and oxidative phosphorylation for ATP production. This switch in metabolism reduces the likelihood of cancer cell proliferation and invasiveness. Because DCA functions in reprogramming glucose metabolism from the cytosol to the mitochondria, it is classified as the prototypical glycolytic and it is the most highly tested and widely used drug of its type [63]. As a glycolytic agent, DCA stimulates PDC by releasing it from the inhibitory actions of its regulatory enzyme PDK (Fig. 4); this allows for the conversion of pyruvate to acetyl CoA in mitochondria thereby reducing cancer-promoting processes which normally are associated with cytosolic glucose metabolism to lactate [66, 67]. As a result of its actions, DCA has proven inhibitory to solid tumors that rely on the Warburg effect, a common denominator of many cancers. For example, DCA reverses the metabolic phenotype of breast cancer [68], colorectal cancer [69], esophageal squamous carcinoma cells [70], etc., as well as reducing their growth and metastasis.

This figure illustrates how the glycolytic agents, dichloroacetate (DCA) and presumably melatonin, cause the upregulation of pyruvate dehydrogenase complex (PDC) allowing for the metabolism of pyruvate to acetyl CoA in the mitochondria. In cancer cells, the pyruvate dehydrogenase kinase (PDK) is stimulated resulting in the downregulation of PDC which bolsters cytosolic glucose oxidation and high lactate production (the Warburg effect). Conversely, DCA and melatonin overcome the suppressive effect on PDK allowing for the upregulation of PDC and reprogramming pyruvate metabolism to the mitochondria and reducing the Warburg effect. AANAT arylalkylamine N-acetyltransferase, PTL pyruvate translocase, TCA tricarboxylic acid cycle
DCA has proven itself as an oncostatic agent related to its ability to switch the dysregulated cancer cell metabolism to a more normal phenotype which allows the cells to enhance their cancer cell death pathways, i.e., relieving them of their highly proliferative activity, elevating cellular apoptotic pathways, halting angiogenesis, decreasing their metastatic potential, etc. [63, 71]. Another major aspect of perturbed cancer cell metabolism is the development of resistance to conventional anti-cancer drugs [71, 72]. Since many cancers exhibit such drug resistance, identifying agents that may reverse this process is a critical area of experimental and clinical interest [71]. DCA both enhances cancer sensitivity to therapies as well as overcomes drug resistance by processes similar to those that support its inhibition of cancer growth. Ovarian cancer, which is a rapidly progressing and highly deadly cancer, exhibits increased sensitivity to cisplatin when given in conjunction with DCA [73]. Hepatocellular carcinoma which often develops resistance to sorafenib becomes responsive to the same drug when PDC is upregulated due to co-treatment with DCA [74]. Similarly, by reprogramming glucose oxidation from the cytosol to the mitochondria, DCA synergizes with cisplatin to limit HeLa cell proliferation [75]. The killing of hepatoma cells by doxorubicin is improved by DCA due to its ability to shift glucose metabolism to the mitochondria resulting in elevated ROS production which stymies cancer cell growth [76]. Likewise, taxol-resistant oral cancer cells become sensitive to the chemotherapy when it includes DCA [77]. In addition to these examples which illustrate the ability of PDC stimulation with DCA to enhance or permit a chemotherapy to carry out its prescribed actions, there are other similar studies summarized in recent reviews [71, 78].
Toxicity studies of DCA in humans have revealed sensory and motor peripheral neuropathy with long-term use [79], with age and recipient genotype being factors to consider. Overall, DCA is well tolerated when the dose is carefully controlled and consideration is given to the issues mentioned. Thus, it is identified as a relatively safe, non-toxic, anti-cancer agent [68]. The toxicity of DCA originates at several sites including the Schwann cells, which myelinate peripheral nerves, interference with the catabolism of phenylalanine and tyrosine as well as altering heme metabolism, all of which cause an elevation of damaging ROS [80]; thus, the toxicity of DCA stems primarily from the resulting oxidative stress that it induces.
Given that melatonin is a glycolytic mimetic, like DCA, which restores mitochondrial physiology in cancer cells, we predict that melatonin inhibits PDK resulting in the stimulation of PDC activity (Fig. 4). This switch allows cancer cells to abandon cytosolic glycolysis and obtain a more normal phenotype by enhancing the conversion of pyruvate to acetyl CoA in the mitochondria.
Melatonin and DCA clearly seem to employ similar mechanisms to reverse the Warburg effect and inhibit tumor growth. Which of these would actually be more useful at the clinical level as a cancer treatment has not been determined. Perhaps, however, melatonin may be an improvement since it is an endogenously produced molecule that can be administered via any route and it has a very high safety profile [27]. In comparison, DCA is a synthetic molecule which is toxic if not properly used [79]. The ability of DCA to reduce Warburg metabolism has been more extensively investigated than has melatonin’s actions in this regard. Also, whether melatonin has a specific function in inhibiting PDK3, as does DCA, remains unknown [63]. Perhaps, a more optimal cancer treatment would be the combination of melatonin and DCA since melatonin would likely reduce the toxicity of DCA given that its noxious effects involve ROS.
For decades melatonin has been recognized as having anti-cancer actions [81,82,83,84]. Moreover, two decades ago it was noted that melatonin synergizes (increases the efficacy of) chemotherapies used to treat cancer [85]. Likewise, while improving the cancer-killing activity of these highly toxic therapies, melatonin also reduced their toxicity: doxorubicin (adriamycin) [86, 87], bleomycin [88], cytarabine [89], cisplatin [90], etc. The synergistic actions of melatonin with cancer-fighting drugs, which have been confirmed in numerous subsequent studies [91,92,93,94,95,96] are reminiscent of those described for DCA when it is given in conjunction with chemotherapies. DCA and melatonin may have similar mechanisms relative to this effect. Melatonin has long been known to be a pro-oxidant in cancer cells [97,98,99], an action consistent with it forcing cancer cells to shift from cytosolic glycolysis to mitochondrial oxidative phosphorylation which increases ROS generation.
There is a plethora of studies the results of which support the conclusion that melatonin, while serving as a multifaceted antioxidant in normal cells [18, 100,101,102] is actually pro-oxidative in cancer cells. Indeed, the context specific pro-oxidant actions and the associated elevated oxidative damage have been used to explain, at least in part, the ability of melatonin to kill cancer cells [97,98,99]. Since we hypothesize that melatonin synthesis in mitochondria of cancer cells is severely compromised because of the lack of acetyl CoA, a necessary co-factor for the rate limiting enzyme (AANAT) in melatonin synthesis, when melatonin is exogenously provided and cancer cells switch from cytosolic glycolysis to oxidative phosphorylation (Fig. 2), the resulting rise in ROS would theoretically also be extinguished due to melatonin’s antioxidant actions. In normal cells, melatonin upregulates, via epigenetic mechanisms, the antioxidant enzyme superoxide dismutase 2 (SOD2) to scavenge ROS produced by this organelle [23, 37, 103]. The stimulation of SOD2 by melatonin in cancer cells seems not to occur, since the elevated ROS generated by oxidative phosphorylation go uncontested and potentially contribute to melatonin’s cancer-killing activity. Such dichotomous responses, referred to as context specificity, have been observed for other melatonin actions [104] and, to date, a satisfactory mechanistic explanation for these differential responses has yet to be provided.
Melatonin: changing sensitivity of cancer to chemotherapy
The development of multidrug resistance of tumors to oncostatic drugs may also relate to the cytosolic glycolysis that cancer cells experience. If so, melatonin, like DCA, might overcome drug resistance of Warburg-dependent cancers. The first experiments that examined this were performed by Dauchy et al. [105] using ERα + MCF-7 human breast cancer cells grown as xenografts in nude rats. They found that suppressing the endogenous nocturnal melatonin rise by exposing the cancer-bearing rats to light-at-night (LAN) caused the tumors to grow more rapidly and also rendered them insensitive to a conventional anti-cancer medication, tamoxifen, a selective estrogen receptor modulator (SERM). When exogenous melatonin was introduced to establish a nighttime rise, the breast tumors were sensitized to tamoxifen. Thus, exogenously administered melatonin was able to overcome cancer resistance to the drug resulting from endogenous melatonin depletion. The authors surmised that drug resistance was broken due to the circadian actions of melatonin. Considering the similar effects of DCA and melatonin against cancer insensitivity to chemotherapies, it also seems likely that the inhibition of glycolysis which is presumed to be achieved by melatonin (like DCA) and the re-establishment of oxidative phosphorylation was also involved. At the time of their study [105], information implicating melatonin as a Warburg effect inhibitor had not yet been suggested.
As with tamoxifen, Xiang et al. [106] reported that breast cancer insensitivity to doxorubicin was improved by melatonin treatment. The experimental design of this study was essentially identical to that of Dauchy et al. [105] except doxorubicin was used in lieu of tamoxifen. Again, dim LAN was used to deprive the cancer-bearing rats of a robust melatonin rise. The nocturnal increase was restored by giving supplemental melatonin. Whether melatonin has any benefit in defeating multidrug resistance has not been investigated. Asghari et al. [93] listed a variety of means by which cellular changes could interrupt cancer resistance to chemotherapies. One of these was for the treatment to convert cancer cells to a more normal phenotype, an action melatonin seems to possess.
Melatonin is a powerful direct ROS scavenger [107,108,109] and an indirect stimulator of antioxidant enzymes [110,111,112]. Considering that melatonin levels usually wane with age, the elevated toxicity of DCA in elderly individuals may be a result of the loss of this metabolic regulator. Likewise, light pollution which reduces at least pineal-derived melatonin [113, 114] and possibly peripherally synthesized mitochondrial melatonin [115] as well, may be a factor that increases the toxicity of chemotherapies.
Melatonin is known to reduce the toxicity of many drugs whose untoward effects relate to oxidative stress [116,117,118]. Hypoxia, which often occurs in solid tumors, induces the overexpression of HIF-1α in addition to other pathways which aid angiogenesis, cancer cell proliferation, growth and metastasis. Because of the central role that HIF-1α plays in preserving the progression and survival of tumor cells, it has often been considered a critical target for cancer therapy [119]. In an attempt to identify the role of HIF-1α in mediating the Warburg effect, Sanchez-Sanchez et al. [120] compared the actions of melatonin in an Ewing sarcoma cell line, which exhibits the Warburg effect, and with a chondrosarcoma cell line that does not display aerobic glycolysis. Whereas melatonin negated aerobic glycolysis, as indicated by the reduced lactate release from Ewing sarcoma cells and where it proved to be highly cytotoxic, melatonin had limited efficacy in killing chondrosarcoma cells. The authors speculated that melatonin’s actions in Ewing sarcoma cells was due to a downregulation of HIF-1α as had been previously reported in other cancer cell lines [121, 122].
Clinical implications for cancer patients
There are numerous epidemiology reports which strongly support the importance of the daily nighttime dark period, which allows for high blood melatonin levels, for limiting especially breast cancer growth [123,124,125]. Humans who are wittingly (night shift workers) or unwittingly (individuals whose sleeping environment is not dark) exposed to LAN are reported to have a greater propensity to develop cancer. The International Agency for Cancer Research has classified light-at-night, which is accompanied by melatonin suppression, as a potential cancer-causing factor [126]. In humans, as in animals, light exposure at night suppresses circulating melatonin concentrations due to a drop in pineal melatonin synthesis and release [127,128,129]. In the absence of a nighttime melatonin surge, as previously shown in animals [130], cancer cells may exhibit a 24/7 cancer phenotype leading to more rapid tumor growth and metastasis [29, 82]. Hospitalized or hospice care cancer patients are often exposed to a shorter period or a less dark environment due to the requirement for care/treatment. This perturbed light:dark cycle may aggravate cancer growth. This could be at least partially remedied by having cancer patients use eye shades to prevent retinal light stimulation and melatonin suppression at night. Light pollution is increasingly being considered a cancer promoting factor [125].
There are implications beyond the potential involvement of the perturbed light:dark cycle for cancer patients. The presence of a tumor seems to reduce endogenous melatonin synthesis given that cancer patients often have lowered nighttime melatonin levels [131,132,133,134] relative to those of aged-matched humans who are cancer free. If the presence of cancer interferes with melatonin synthesis, a vicious cycle could occur wherein an enlargement of a tumor may further reduce the amplitude of the melatonin rhythm thereby promoting an even more rapid cancer growth. The nighttime melatonin peak differs widely among individuals; some humans when compared with age-matched controls are relatively melatonin deficient (hypomelatoninemia) which also may elevate the likelihood of these individuals developing a rapidly growing or metastatic tumor.
Finally, advanced age is a major risk factor for many cancers. The nocturnal melatonin peak is severely attenuated in most elderly humans [135]. Healthy individuals in old age typically have a more robust nocturnal melatonin rise; associated with their improved health they also frequently have less cancer. While a cause/effect relationship has not yet been resolved for these individuals, nevertheless, it would probably be judicious for humans of all ages to strive to maintain a more normal light:dark environment to preserve a high nocturnal amplitude melatonin peak helping to possibly safeguard a more normal phenotype for cancer-prone cells. Maintaining a normal alternating light:dark cycle is becoming progressively more difficult in the urban setting where light pollution is increasing rapidly.
In addition to preserving a more normal daily dark period, other procedures may prove beneficial. Many humans, due to their lifestyle, for genetic reasons, or due to aging are severely depleted of melatonin. Melatonin has been regularly used by thousands of humans (primarily to promote sleep). It is an endogenously produced molecule that has been found to have an uncommonly high safety profile [136, 137]. No untoward effects have been reported for individuals who have used melatonin for decades. Also, many clinical trials have been performed using melatonin with only minor reported side effects over a very wide dose range, e.g., headache, sleepiness, etc., similar to those caused by placebo. In animals, no lethal dose has been identified despite attempts to do so [138]. In view of its efficacy and high safety profile, serious consideration should be given to the conduct of clinical trials related to melatonin’s potential as an oncostatic agent. Mechanisms for its potential benefits are described in this report and in other reviews [28, 82, 139, 140]. It is especially noteworthy that melatonin reduces the toxicity of chemotherapies used to treat cancer [85, 141, 142]. Thus, its use in combination with these therapies also should be performed in humans. Moreover, melatonin improves the quality of life when given to cancer patients [143,144,145]. Unfortunately, since melatonin is a naturally occurring agent, is non-patentable, and is inexpensive, the pharmaceutical industry will not support such trials and funding from other sources has proven difficult to secure.
Conclusions and perspectives
This review provides a novel perspective on how melatonin interferes with cancer cell metabolism, growth, and metastasis (Fig. 2). This cancer-inhibiting mechanism, in addition to other processes identified in earlier studies, makes it important to further test melatonin as an oncostatic agent at the clinical level. This is of special importance since cancer patients more frequently die from metastatic than from non-metastatic cancer (Fig. 5). Considering that melatonin controls tumor growth and limits their size (large tumors more readily metastasize) as well as to its direct anti-metastatic actions more than justifies tests of its efficacy. Other drugs that have actions similar to those of melatonin such as reprogramming cancer cell mitochondria are referred to as glycolytic agents. One of the best known of these is DCA, as noted above. It, as we propose for melatonin, inhibits PDK to allow upregulation of PDC resulting in acetyl CoA synthesis (Fig. 4). In addition to its utility as an oncostatic agent, DCA, because of its downregulation of PDK, also reverses the insensitivity of tumors to cancer chemotherapies. Melatonin has the same actions in rendering previously unresponsive cancers sensitive to chemotherapies, e.g., tamoxifen and doxorubicin. This is again consistent with the prediction that melatonin promotes the conversion of pyruvate to acetyl CoA in mitochondria.

This figure summarizes some of the processes cancer cells utilize to ensure their survival but compromise the survival of the host. Cytosolic glycolysis promotes the neovascularization of tumors which supplies nutrients for the rapid cancer cell proliferation and as routes of metastasis. Cancer cells also secrete vascular endothelial growth factor (VEGF) and endothelin (ET-1) to ensure the vascularization to support the rapidly growing tumor. To assist in their detachment from adjacent cells and their invasiveness, both of which are required for metastasis, cancer cells also execute other intracellular changes (not shown in this figure) and release matrix metalloproteinase-9 (MMP-9; gelatinase), one of a family of enzymes which digests connective tissue elements of the extracellular matrix allowing the cancer cells to move and invade blood vessels to more easily migrate to secondary sites. The processes summarized in this figure are not the only mechanisms that cancer cells use to improve their survival and assist in their movement. Many of the processes shown are antagonized by melatonin. Myc Myc proto-oncogene, Wnt Wnt-signaling genes (signal transduction factor), HIFs hypoxia-inducible factors, VEGF vascular endothelial growth factor, ET-1 endothelin 1, MMP metalloproteinase, Src c-Src proto-oncogene, PDKs pyruvate dehydrogenase kinases, PDC pyruvate dehydrogenase complex, OXPHOS oxidative phosphorylation, NADPH reduced nicotinamide adenine dinucleotide phosphate, ROS reactive oxygen species
Some anti-cancer drugs are useful as a treatment for several cancer types, but often they are most effective against a malignancy, possibly related to the marked heterogenicity of cancer subtypes [146]. Hence, the speculations advanced in this review may only apply to breast cancers and should be investigated relative to other Warburg-dependent cancers and diseases [14]. Also, while this review considered especially the ability of melatonin to reorient glucose metabolism, many cancers also display dysregulated fatty acid and amino acid metabolism [147, 148]. Melatonin should be tested relative to these perturbations as well in addition to the crosstalk among the glucose, fatty acid, and amino acid pathways.
Chemotherapeutic drugs often have serious side effects which limit their dose and use. For example, doxorubicin causes cardiomyocyte damage which may lead to heart failure and jeopardize life quality and survival of the patient in the long term. The co-administration of melatonin with doxorubicin significantly reduces the toxic reactions in the heart without interfering with the anti-cancer activity of the drug. Given melatonin’s ability to diminish the collateral cardiotoxicity could translate to the use of a higher dose of doxorubicin with increased antitumor activity. Numerous studies have shown that melatonin has a high safety profile and is protective against chemotherapies [136, 137, 149].
There is another consideration that supports tests of melatonin in cancer patients. In patients in the later stages of cancer, it has been noted that supplementary melatonin improves their wellbeing thereby contributing to a better life quality. This benefit alone may be adequate justification for the use of melatonin by cancer patients. As discussed herein, however, there are other compelling reasons melatonin should be tested as an integral part of the anti-cancer armamentarium. Currently, there are no on-going clinical trials of melatonin to reduce Warburg-dependent cancers in humans. This deficiency is primarily related to the lack of financial support for such studies since melatonin is a non-patentable molecule and it is inexpensive.
The hypothesis proposed here provides a working model for subsequent research on the means by which melatonin, either pineal or mitochondria-derived, influences cytosolic glucose metabolism and mitochondrial acetyl CoA production as well as its role in growth stimulation or inhibition of Warburg-dependent cancers (Fig. 2). Since there are also diseases that do not have a neoplastic component that exhibits high cytosolic glucose metabolism, the hypothesis suggested here may be applicable to those diseases as well. The proposal assumes a causal connection between melatonin and metabolic processes that support tumor growth. Based on published data, and in particular on the recent findings that melatonin is both taken up by mitochondria and likely synthesized in these organelles, it is reasonable to predict that it influences other mitochondrial processes as well. Among many considerations should be examination of the possible association of melatonin with fatty acid oxidation [150], the tricarboxylic acid cycle [151], the function of the electron transport chain [107, 152], the reverse Warburg effect [153], the ubiquitin–proteasome system [154] and on cancer stem cell survival and proliferation [155].
Finally, SIRT3, which is primarily located in the mitochondria, is a known tumor suppressor since rodents that are deficient in SIRT3 exhibit an increased propensity to develop a malignancy [153]. This mitochondrial protein deacetylase also has a major influence on the metabolic aspects of this organelle [154]. In cancer cells, the loss of SIRT3 leads to excessive reactive oxygen species (ROS) production in mitochondria, which is associated with the stabilization of HIF-1α suggesting that the deacetylase may be involved in the Warburg effect [155]. SIRT3 has been shown to be depressed in a variety of human tumors which may contribute to the accelerated growth of these neoplasms. Melatonin, in normal cells, upregulates SIRT3 activity where it has been experimentally depressed [103, 123]. How the melatonin/SIRT3 interaction plays out in cancer cells has yet to be determined but should be given consideration for examination.
While the amount of information related to melatonin as an experimental oncostatic agent is massive, the associated clinical findings are remarkably sparse primarily due to the limited tests of melatonin as a potential anticancer agent in humans. Hopefully, this will change soon.
References
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM (2016) Mitochondrial diseases. Nat Rev Dis Primers 2:16080. https://doi.org/10.1038/nrdp.2016.80
Molnar MJ, Kovacs GG (2017) Mitochondrial diseases. Handb Clin Neurol 145:147–155. https://doi.org/10.1016/B978-0-12-802395-2.00010-9
Koyano F, Yamano K, Kosako H, Tanaka K, Matsuda N (2019) Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J Biol Chem 294(26):10300–10314. https://doi.org/10.1074/jbc.RA118.006302
Shukla M, Chinchalongporn V, Govitrapong P, Reiter RJ (2019) The role of melatonin in targeting cell signaling pathways in neurodegeneration. Ann N Y Acad Sci 1443(1):75–96. https://doi.org/10.1111/nyas.14005
Atashi F, Modarressi A, Pepper MS (2015) The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 24(10):1150–1163. https://doi.org/10.1089/scd.2014.0484
Dan Dunn J, Alvarez LA, Zhang X, Soldati T (2015) Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 6:472–485. https://doi.org/10.1016/j.redox.2015.09.005
Purohit V, Simeone DM, Lyssiotis CA (2019) Metabolic regulation of redox balance in cancer. Cancers (Basel) 11:7. https://doi.org/10.3390/cancers11070955
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 13(6):472–482. https://doi.org/10.1016/j.ccr.2008.05.005
Strickaert A, Saiselet M, Dom G, De Deken X, Dumont JE, Feron O, Sonveaux P, Maenhaut C (2017) Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 36(19):2637–2642. https://doi.org/10.1038/onc.2016.411
Thomson TM, Balcells C, Cascante M (2019) Metabolic plasticity and epithelial-mesenchymal transition. J Clin Med 8:7. https://doi.org/10.3390/jcm8070967
Xu XD, Shao SX, Jiang HP, Cao YW, Wang YH, Yang XC, Wang YL, Wang XS, Niu HT (2015) Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol Res Treat 38(3):117–122. https://doi.org/10.1159/000375435
Kalyanaraman B (2017) Teaching the basics of cancer metabolism: developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol 12:833–842. https://doi.org/10.1016/j.redox.2017.04.018
Spencer NY, Stanton RC (2019) The Warburg effect, lactate, and nearly a century of trying to cure cancer. Semin Nephrol 39(4):380–393. https://doi.org/10.1016/j.semnephrol.2019.04.007
Chen Z, Liu M, Li L, Chen L (2018) Involvement of the Warburg effect in non-tumor diseases processes. J Cell Physiol 233(4):2839–2849. https://doi.org/10.1002/jcp.25998
Acuna Castroviejo D, Lopez LC, Escames G, Lopez A, Garcia JA, Reiter RJ (2011) Melatonin-mitochondria interplay in health and disease. Curr Top Med Chem 11(2):221–240. https://doi.org/10.2174/156802611794863517
Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (2013) Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin's primary function and evolution in eukaryotes. J Pineal Res 54(2):127–138. https://doi.org/10.1111/jpi.12026
Reiter RJ, Rosales-Corral S, Tan DX, Jou MJ, Galano A, Xu B (2017) Melatonin as a mitochondria-targeted antioxidant: one of evolution's best ideas. Cell Mol Life Sci 74(21):3863–3881. https://doi.org/10.1007/s00018-017-2609-7
Tan D-X, Reiter RJ (2019) Mitochondria: the birth place, battle ground and the site of melatonin metabolism in cells. Melatonin Res 2(1):44–66. https://doi.org/10.32794/mr11250011
Venegas C, Garcia JA, Escames G, Ortiz F, Lopez A, Doerrier C, Garcia-Corzo L, Lopez LC, Reiter RJ, Acuna-Castroviejo D (2012) Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res 52(2):217–227. https://doi.org/10.1111/j.1600-079X.2011.00931.x
Zhao D, Yu Y, Shen Y, Liu Q, Zhao Z, Sharma R, Reiter RJ (2019) Melatonin synthesis and function: evolutionary history in animals and plants. Front Endocrinol (Lausanne) 10:249. https://doi.org/10.3389/fendo.2019.00249
Xie Z, Chen F, Li WA, Geng X, Li C, Meng X, Feng Y, Liu W, Yu F (2017) A review of sleep disorders and melatonin. Neurol Res 39(6):559–565. https://doi.org/10.1080/01616412.2017.1315864
Reiter R, Tan D-X, Sharma R (2018) Historical perspective and evaluation of the mechanisms by which melatonin mediates seasonal reproduction in mammals. Melatonin Res 1(1):59–77. https://doi.org/10.32794/mr11250004
Reiter RJ, Rosales-Corral S, Zhou X, Tan DX (2017) Role of SIRT3/SOD2 signaling in mediating the antioxidant actions of melatonin in mitochondria. Curr Trends Endocrinol 9:45–49
Blask DE, Sauer LA, Dauchy R, Holowachuk EW, Ruhoff MS (1999) New actions of melatonin on tumor metabolism and growth. Biol Signals Recept 8(1–2):49–55. https://doi.org/10.1159/000014568
Leon-Blanco MM, Guerrero JM, Reiter RJ, Pozo D (2004) RNA expression of human telomerase subunits TR and TERT is differentially affected by melatonin receptor agonists in the MCF-7 tumor cell line. Cancer Lett 216(1):73–80. https://doi.org/10.1016/j.canlet.2004.05.003
Korkmaz A, Sanchez-Barcelo EJ, Tan DX, Reiter RJ (2009) Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res Treat 115(1):13–27. https://doi.org/10.1007/s10549-008-0103-5
Reiter RJ, Rosales-Corral SA, Tan DX, Acuna-Castroviejo D, Qin L, Yang SF, Xu K (2017) Melatonin, a full service anti-cancer agent: inhibition of initiation, progression and metastasis. Int J Mol Sci 18:4. https://doi.org/10.3390/ijms18040843
Gil-Martin E, Egea J, Reiter RJ, Romero A (2019) The emergence of melatonin in oncology: Focus on colorectal cancer. Med Res Rev 39(6):2239–2285. https://doi.org/10.1002/med.21582
Blask DE, Dauchy RT, Dauchy EM, Mao L, Hill SM, Greene MW, Belancio VP, Sauer LA, Davidson L (2014) Light exposure at night disrupts host/cancer circadian regulatory dynamics: impact on the Warburg effect, lipid signaling and tumor growth prevention. PLoS ONE 9(8):e102776. https://doi.org/10.1371/journal.pone.0102776
Hevia D, Gonzalez-Menendez P, Fernandez-Fernandez M, Cueto S, Rodriguez-Gonzalez P, Garcia-Alonso JI, Mayo JC, Sainz RM (2017) Melatonin decreases glucose metabolism in prostate cancer cells: a (13)C stable isotope-resolved metabolomic study. Int J Mol Sci 18:8. https://doi.org/10.3390/ijms18081620
Walenta S, Schroeder T, Mueller-Klieser W (2004) Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem 11(16):2195–2204. https://doi.org/10.2174/0929867043364711
Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, Mueller-Klieser W (2001) Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys 51(2):349–353. https://doi.org/10.1016/s0360-3016(01)01630-3
Reiter RJ, Sharma R, Ma Q, Rosales-Corral S, Acuna-Castroviejo D, Escames G (2019) Inhibition of mitochondrial pyruvate dehydrogenase kinase: a proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res 2(3):105–119. https://doi.org/10.32794/mr11250033
James MO, Jahn SC, Zhong G, Smeltz MG, Hu Z, Stacpoole PW (2017) Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol Ther 170:166–180. https://doi.org/10.1016/j.pharmthera.2016.10.018
Leon J, Acuna-Castroviejo D, Sainz RM, Mayo JC, Tan DX, Reiter RJ (2004) Melatonin and mitochondrial function. Life Sci 75(7):765–790. https://doi.org/10.1016/j.lfs.2004.03.003
Jou MJ, Peng TI, Reiter RJ, Jou SB, Wu HY, Wen ST (2004) Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J Pineal Res 37(1):55–70. https://doi.org/10.1111/j.1600-079X.2004.00140.x
Reiter RJ, Tan DX, Rosales-Corral S, Galano A, Jou MJ, Acuna-Castroviejo D (2018) Melatonin mitigates mitochondrial meltdown: interactions with SIRT3. Int J Mol Sci 19:8. https://doi.org/10.3390/ijms19082439
Martin M, Macias M, Escames G, Leon J, Acuna-Castroviejo D (2000) Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J 14(12):1677–1679. https://doi.org/10.1096/fj.99-0865fje
Jou MJ, Peng TI, Yu PZ, Jou SB, Reiter RJ, Chen JY, Wu HY, Chen CC, Hsu LF (2007) Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J Pineal Res 43(4):389–403. https://doi.org/10.1111/j.1600-079X.2007.00490.x
Lowes DA, Webster NR, Murphy MP, Galley HF (2013) Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth 110(3):472–480. https://doi.org/10.1093/bja/aes577
Acuña-Castroviejo D, Noguiera-Navarro M, Reiter R, Escames G (2018) Melatonin actions in the heart; more than a hormone. Melatonin Res 1:21–26. https://doi.org/10.32794/mr11250002
Huo X, Wang C, Yu Z, Peng Y, Wang S, Feng S, Zhang S, Tian X, Sun C, Liu K, Deng S, Ma X (2017) Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: an implication of the therapeutic potential. J Pineal Res 62:4. https://doi.org/10.1111/jpi.12390
Kerenyi NA, Balogh I, Somogyi E, Sotonyi P (1979) Cytochemical investigation of acetyl-serotonin-transferase activity in the pineal gland. Cell Mol Biol Incl Cyto Enzymol 25(4):259–262
He C, Wang J, Zhang Z, Yang M, Li Y, Tian X, Ma T, Tao J, Zhu K, Song Y, Ji P, Liu G (2016) Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte's quality under in vitro conditions. Int J Mol Sci 17:6. https://doi.org/10.3390/ijms17060939
Suofu Y, Li W, Jean-Alphonse FG, Jia J, Khattar NK, Li J, Baranov SV, Leronni D, Mihalik AC, He Y, Cecon E, Wehbi VL, Kim J, Heath BE, Baranova OV, Wang X, Gable MJ, Kretz ES, Di Benedetto G, Lezon TR, Ferrando LM, Larkin TM, Sullivan M, Yablonska S, Wang J, Minnigh MB, Guillaumet G, Suzenet F, Richardson RM, Poloyac SM, Stolz DB, Jockers R, Witt-Enderby PA, Carlisle DL, Vilardaga JP, Friedlander RM (2017) Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc Natl Acad Sci USA 114(38):E7997–E8006. https://doi.org/10.1073/pnas.1705768114
Hickman AB, Klein DC, Dyda F (1999) Melatonin biosynthesis: the structure of serotonin N-acetyltransferase at 2.5 A resolution suggests a catalytic mechanism. Mol Cell 3(1):23–32. https://doi.org/10.1016/s1097-2765(00)80171-9
Liu J, Clough SJ, Hutchinson AJ, Adamah-Biassi EB, Popovska-Gorevski M, Dubocovich ML (2016) MT1 and MT2 melatonin receptors: a therapeutic perspective. Annu Rev Pharmacol Toxicol 56:361–383. https://doi.org/10.1146/annurev-pharmtox-010814-124742
Hardeland R, Tan DX, Reiter RJ (2009) Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J Pineal Res 47(2):109–126. https://doi.org/10.1111/j.1600-079X.2009.00701.x
Mao L, Dauchy RT, Blask DE, Dauchy EM, Slakey LM, Brimer S, Yuan L, Xiang S, Hauch A, Smith K, Frasch T, Belancio VP, Wren MA, Hill SM (2016) Melatonin suppression of aerobic glycolysis (Warburg effect), survival signalling and metastasis in human leiomyosarcoma. J Pineal Res 60(2):167–177. https://doi.org/10.1111/jpi.12298
Ran C, Liu H, Hitoshi Y, Israel MA (2013) Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res 73(6):1831–1843. https://doi.org/10.1158/0008-5472.CAN-12-2460
Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, Cecon E, Zlotos DP (2016) Update on melatonin receptors: IUPHAR review 20. Br J Pharmacol 173(18):2702–2725. https://doi.org/10.1111/bph.13536
Li Y, Li S, Zhou Y, Meng X, Zhang JJ, Xu DP, Li HB (2017) Melatonin for the prevention and treatment of cancer. Oncotarget 8(24):39896–39921. https://doi.org/10.18632/oncotarget.16379
Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H, Kim JE, Li W, Kristal BS, Ferrante RJ, Friedlander RM (2011) The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J Neurosci 31(41):14496–14507. https://doi.org/10.1523/JNEUROSCI.3059-11.2011
Emet M, Ozcan H, Ozel L, Yayla M, Halici Z, Hacimuftuoglu A (2016) A review of melatonin, its receptors and drugs. Eurasian J Med 48(2):135–141. https://doi.org/10.5152/eurasianjmed.2015.0267
Boutin JA (2016) Quinone reductase 2 as a promising target of melatonin therapeutic actions. Expert Opin Ther Targets 20(3):303–317. https://doi.org/10.1517/14728222.2016.1091882
Klein DC (2007) Arylalkylamine N-acetyltransferase: "the Timezyme". J Biol Chem 282(7):4233–4237. https://doi.org/10.1074/jbc.R600036200
Hiromasa Y, Hu L, Roche TE (2006) Ligand-induced effects on pyruvate dehydrogenase kinase isoform 2. J Biol Chem 281(18):12568–12579. https://doi.org/10.1074/jbc.M513514200
Saunier E, Benelli C, Bortoli S (2016) The pyruvate dehydrogenase complex in cancer: an old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer 138(4):809–817. https://doi.org/10.1002/ijc.29564
Whitehouse S, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141(3):761–774. https://doi.org/10.1042/bj1410761
Kankotia S, Stacpoole PW (2014) Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta (1846) 2:617–629. https://doi.org/10.1016/j.bbcan.2014.08.005
Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, Venner P, Michelakis ED (2015) A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs 33(3):603–610. https://doi.org/10.1007/s10637-015-0221-y
Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329(Pt 1):191–196. https://doi.org/10.1042/bj3290191
Stacpoole PW (2017) Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst 109:11. https://doi.org/10.1093/jnci/djx071
Stacpoole PW (1989) The pharmacology of dichloroacetate. Metabolism 38(11):1124–1144. https://doi.org/10.1016/0026-0495(89)90051-6
Stacpoole PW (2011) The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine—both or neither? Environ Health Perspect 119(2):155–158. https://doi.org/10.1289/ehp.1002554
Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99(7):989–994. https://doi.org/10.1038/sj.bjc.6604554
De Preter G, Neveu MA, Danhier P, Brisson L, Payen VL, Porporato PE, Jordan BF, Sonveaux P, Gallez B (2016) Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 7 (3):2910–2920. https://doi.org/10.18632/oncotarget.6272
Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2010) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120(1):253–260. https://doi.org/10.1007/s10549-009-0435-9
Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 102(12):1746–1752. https://doi.org/10.1038/sj.bjc.6605701
Jia HY, Wang HN, Xia FY, Sun Y, Liu HL, Yan LL, Li SS, Jiang DC, Xu MM (2017) Dichloroacetate induces protective autophagy in esophageal squamous carcinoma cells. Oncol Lett 14(3):2765–2770. https://doi.org/10.3892/ol.2017.6562
Bhat TA, Kumar S, Chaudhary AK, Yadav N, Chandra D (2015) Restoration of mitochondria function as a target for cancer therapy. Drug Discov Today 20(5):635–643. https://doi.org/10.1016/j.drudis.2015.03.001
Zhao Y, Butler EB, Tan M (2013) Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 4:e532. https://doi.org/10.1038/cddis.2013.60
Olszewski U, Poulsen TT, Ulsperger E, Poulsen HS, Geissler K, Hamilton G (2010) In vitro cytotoxicity of combinations of dichloroacetate with anticancer platinum compounds. Clin Pharmacol 2:177–183. https://doi.org/10.2147/CPAA.S11795
Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, Liu SH, Cheng AL (2013) Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer 108(1):72–81. https://doi.org/10.1038/bjc.2012.559
Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C, Chen HZ (2011) Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int J Oncol 38(2):409–417. https://doi.org/10.3892/ijo.2010.851
Dai Y, Xiong X, Huang G, Liu J, Sheng S, Wang H, Qin W (2014) Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS ONE 9(4):e92962. https://doi.org/10.1371/journal.pone.0092962
Xie Q, Zhang HF, Guo YZ, Wang PY, Liu ZS, Gao HD, Xie WL (2015) Combination of Taxol(R) and dichloroacetate results in synergistically inhibitory effects on Taxol-resistant oral cancer cells under hypoxia. Mol Med Rep 11(4):2935–2940. https://doi.org/10.3892/mmr.2014.3080
Guaragnella N, Giannattasio S, Moro L (2014) Mitochondrial dysfunction in cancer chemoresistance. Biochem Pharmacol 92(1):62–72. https://doi.org/10.1016/j.bcp.2014.07.027
Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW (2008) Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther 324(3):1163–1171. https://doi.org/10.1124/jpet.107.134593
Stacpoole PW, Martyniuk CJ, James MO, Calcutt NA (2019) Dichloroacetate-induced peripheral neuropathy. Int Rev Neurobiol 145:211–238. https://doi.org/10.1016/bs.irn.2019.05.003
Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C, Reiter RJ (2012) Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investig Drugs 21(6):819–831. https://doi.org/10.1517/13543784.2012.681045
Hill SM, Belancio VP, Dauchy RT, Xiang S, Brimer S, Mao L, Hauch A, Lundberg PW, Summers W, Yuan L, Frasch T, Blask DE (2015) Melatonin: an inhibitor of breast cancer. Endocr Relat Cancer 22(3):R183–204. https://doi.org/10.1530/ERC-15-0030
de Almeida Chuffa LG, Seiva FRF, Cucielo MS, Silveira HS, Reiter RJ, Lupi LA (2019) Mitochondrial functions and melatonin: a tour of the reproductive cancers. Cell Mol Life Sci 76(5):837–863. https://doi.org/10.1007/s00018-018-2963-0
Li T, Yang Z, Jiang S, Di W, Ma Z, Hu W, Chen F, Reiter RJ, Yang Y (2018) Melatonin: does it have utility in the treatment of haematological neoplasms? Br J Pharmacol 175(16):3251–3262. https://doi.org/10.1111/bph.13966
Reiter RJ, Tan DX, Sainz RM, Mayo JC, Lopez-Burillo S (2002) Melatonin: reducing the toxicity and increasing the efficacy of drugs. J Pharm Pharmacol 54(10):1299–1321. https://doi.org/10.1211/002235702760345374
Morishima I, Matsui H, Mukawa H, Hayashi K, Toki Y, Okumura K, Ito T, Hayakawa T (1998) Melatonin, a pineal hormone with antioxidant property, protects against adriamycin cardiomyopathy in rats. Life Sci 63(7):511–521. https://doi.org/10.1016/s0024-3205(98)00302-6
Wahab MH, Akoul ES, Abdel-Aziz AA (2000) Modulatory effects of melatonin and vitamin E on doxorubicin-induced cardiotoxicity in Ehrlich ascites carcinoma-bearing mice. Tumori 86(2):157–162
Arslan SO, Zerin M, Vural H, Coskun A (2002) The effect of melatonin on bleomycin-induced pulmonary fibrosis in rats. J Pineal Res 32(1):21–25. https://doi.org/10.1034/j.1600-079x.2002.10796.x
Anwar MM, Mahfouz HA, Sayed AS (1998) Potential protective effects of melatonin on bone marrow of rats exposed to cytotoxic drugs. Comp Biochem Physiol A Mol Integr Physiol 119(2):493–501. https://doi.org/10.1016/s1095-6433(97)00456-x
Lopez-Gonzalez MA, Guerrero JM, Rojas F, Delgado F (2000) Ototoxicity caused by cisplatin is ameliorated by melatonin and other antioxidants. J Pineal Res 28(2):73–80. https://doi.org/10.1034/j.1600-079x.2001.280202.x
Ma C, Li LX, Zhang Y, Xiang C, Ma T, Ma ZQ, Zhang ZP (2015) Protective and sensitive effects of melatonin combined with adriamycin on ER+ (estrogen receptor) breast cancer. Eur J Gynaecol Oncol 36(2):197–202
Prieto-Dominguez N, Mendez-Blanco C, Carbajo-Pescador S, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL (2017) Melatonin enhances sorafenib actions in human hepatocarcinoma cells by inhibiting mTORC1/p70S6K/HIF-1alpha and hypoxia-mediated mitophagy. Oncotarget 8(53):91402–91414. https://doi.org/10.18632/oncotarget.20592
Asghari MH, Ghobadi E, Moloudizargari M, Fallah M, Abdollahi M (2018) Does the use of melatonin overcome drug resistance in cancer chemotherapy? Life Sci 196:143–155. https://doi.org/10.1016/j.lfs.2018.01.024
Lee JH, Yoon YM, Han YS, Yun CW, Lee SH (2018) Melatonin promotes apoptosis of oxaliplatin-resistant colorectal cancer cells through inhibition of cellular prion protein. Anticancer Res 38(4):1993–2000. https://doi.org/10.21873/anticanres.12437
Wang Q, Sun Z, Du L, Xu C, Wang Y, Yang B, He N, Wang J, Ji K, Liu Y, Liu Q (2018) Melatonin sensitizes human colorectal cancer cells to gamma-ray ionizing radiation in vitro and in vivo. Int J Mol Sci 19:12. https://doi.org/10.3390/ijms19123974
Sung GJ, Kim SH, Kwak S, Park SH, Song JH, Jung JH, Kim H, Choi KC (2019) Inhibition of TFEB oligomerization by co-treatment of melatonin with vorinostat promotes the therapeutic sensitivity in glioblastoma and glioma stem cells. J Pineal Res 66(3):e12556. https://doi.org/10.1111/jpi.12556
Leja-Szpak A, Jaworek J, Pierzchalski P, Reiter RJ (2010) Melatonin induces pro-apoptotic signaling pathway in human pancreatic carcinoma cells (PANC-1). J Pineal Res 49(3):248–255. https://doi.org/10.1111/j.1600-079X.2010.00789.x
Wang M, Xue Y, Shen L, Qin P, Sang X, Tao Z, Yi J, Wang J, Liu P, Cheng H (2019) Inhibition of SGK1 confers vulnerability to redox dysregulation in cervical cancer. Redox Biol 24:101225. https://doi.org/10.1016/j.redox.2019.101225
Zhang HM, Zhang Y (2014) Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J Pineal Res 57(2):131–146. https://doi.org/10.1111/jpi.12162
Hardeland R (2013) Melatonin and the theories of aging: a critical appraisal of melatonin's role in antiaging mechanisms. J Pineal Res 55(4):325–356. https://doi.org/10.1111/jpi.12090
Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (2016) Melatonin as an antioxidant: under promises but over delivers. J Pineal Res 61(3):253–278. https://doi.org/10.1111/jpi.12360
Galano A, Reiter RJ (2018) Melatonin and its metabolites vs oxidative stress: from individual actions to collective protection. J Pineal Res 65(1):e12514. https://doi.org/10.1111/jpi.12514
Han L, Wang H, Li L, Li X, Ge J, Reiter RJ, Wang Q (2017) Melatonin protects against maternal obesity-associated oxidative stress and meiotic defects in oocytes via the SIRT3-SOD2-dependent pathway. J Pineal Res 63:3. https://doi.org/10.1111/jpi.12431
Proietti S, Cucina A, Minini M, Bizzarri M (2017) Melatonin, mitochondria, and the cancer cell. Cell Mol Life Sci 74(21):4015–4025. https://doi.org/10.1007/s00018-017-2612-z
Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L, Anbalagan M, Hauch A, Frasch T, Rowan BG, Blask DE, Hill SM (2014) Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer Res 74(15):4099–4110. https://doi.org/10.1158/0008-5472.CAN-13-3156
Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA, Belancio VP, Mondal D, Frasch T, Blask DE, Hill SM (2015) Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res 59(1):60–69. https://doi.org/10.1111/jpi.12239
Hardeland R (2017) Melatonin and the electron transport chain. Cell Mol Life Sci 74(21):3883–3896. https://doi.org/10.1007/s00018-017-2615-9
Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, Vriend J, Tan DX, Reiter RJ (2015) Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res 59(4):403–419. https://doi.org/10.1111/jpi.12267
Álvarez-Diduk R, Galano A, Tan DX, Reiter RJ (2016) The key role of the sequential proton loss electron transfer mechanism on the free radical scavenging activity of some melatonin-related compounds. Theoret Chem Acc 135(2):38. https://doi.org/10.1007/s00214-015-1785-5
Barlow-Walden LR, Reiter RJ, Abe M, Pablos M, Menendez-Pelaez A, Chen LD, Poeggeler B (1995) Melatonin stimulates brain glutathione peroxidase activity. Neurochem Int 26(5):497–502. https://doi.org/10.1016/0197-0186(94)00154-m
Kotler M, Rodriguez C, Sainz RM, Antolin I, Menendez-Pelaez A (1998) Melatonin increases gene expression for antioxidant enzymes in rat brain cortex. J Pineal Res 24(2):83–89. https://doi.org/10.1111/j.1600-079x.1998.tb00371.x
Ortiz-Franco M, Planells E, Quintero B, Acuna-Castroviejo D, Rusanova I, Escames G, Molina-Lopez J (2017) Effect of melatonin supplementation on antioxidant status and DNA damage in high intensity trained athletes. Int J Sports Med 38(14):1117–1125. https://doi.org/10.1055/s-0043-119881
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP (1980) Light suppresses melatonin secretion in humans. Science 210(4475):1267–1269. https://doi.org/10.1126/science.7434030
Brainard GC, Hanifin JP, Warfield B, Stone MK, James ME, Ayers M, Kubey A, Byrne B, Rollag M (2015) Short-wavelength enrichment of polychromatic light enhances human melatonin suppression potency. J Pineal Res 58(3):352–361. https://doi.org/10.1111/jpi.12221
Zimmerman S, Reiter RJ (2019) Melatonin and the optics of the human body. Melatonin Res 2(1):138–160. https://doi.org/10.32794/mr11250016
Reiter RJ, Manchester LC, Tan DX (2010) Neurotoxins: free radical mechanisms and melatonin protection. Curr Neuropharmacol 8(3):194–210. https://doi.org/10.2174/157015910792246236
Haghi-Aminjan H, Asghari MH, Farhood B, Rahimifard M, Hashemi Goradel N, Abdollahi M (2018) The role of melatonin on chemotherapy-induced reproductive toxicity. J Pharm Pharmacol 70(3):291–306. https://doi.org/10.1111/jphp.12855
Haghi-Aminjan H, Farhood B, Rahimifard M, Didari T, Baeeri M, Hassani S, Hosseini R, Abdollahi M (2018) The protective role of melatonin in chemotherapy-induced nephrotoxicity: a systematic review of non-clinical studies. Expert Opin Drug Metab Toxicol 14(9):937–950. https://doi.org/10.1080/17425255.2018.1513492
Masoud GN, Li W (2015) HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5(5):378–389. https://doi.org/10.1016/j.apsb.2015.05.007
Sanchez-Sanchez AM, Antolin I, Puente-Moncada N, Suarez S, Gomez-Lobo M, Rodriguez C, Martin V (2015) Melatonin cytotoxicity is associated to Warburg effect inhibition in ewing sarcoma cells. PLoS ONE 10(8):e0135420. https://doi.org/10.1371/journal.pone.0135420
Kim KJ, Choi JS, Kang I, Kim KW, Jeong CH, Jeong JW (2013) Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J Pineal Res 54(3):264–270. https://doi.org/10.1111/j.1600-079X.2012.01030.x
Zhang Y, Liu Q, Wang F, Ling EA, Liu S, Wang L, Yang Y, Yao L, Chen X, Wang F, Shi W, Gao M, Hao A (2013) Melatonin antagonizes hypoxia-mediated glioblastoma cell migration and invasion via inhibition of HIF-1alpha. J Pineal Res 55(2):121–130. https://doi.org/10.1111/jpi.12052
Reiter RJ, Tan DX, Korkmaz A, Erren TC, Piekarski C, Tamura H, Manchester LC (2007) Light at night, chronodisruption, melatonin suppression, and cancer risk: a review. Crit Rev Oncog 13(4):303–328. https://doi.org/10.1615/critrevoncog.v13.i4.30
Erren TC, Falaturi P, Morfeld P, Knauth P, Reiter RJ, Piekarski C (2010) Shift work and cancer: the evidence and the challenge. Dtsch Arztebl Int 107(38):657–662. https://doi.org/10.3238/arztebl.2010.0657
Erren TC, Lewis P (2019) Hypothesis: ubiquitous circadian disruption can cause cancer. Eur J Epidemiol 34(1):1–4. https://doi.org/10.1007/s10654-018-0469-6
Bonde JP, Hansen J, Kolstad HA, Mikkelsen S, Olsen JH, Blask DE, Harma M, Kjuus H, de Koning HJ, Olsen J, Moller M, Schernhammer ES, Stevens RG, Akerstedt T (2012) Work at night and breast cancer–report on evidence-based options for preventive actions. Scand J Work Environ Health 38(4):380–390. https://doi.org/10.5271/sjweh.3282
Hull JT, Czeisler CA, Lockley SW (2018) Suppression of melatonin secretion in totally visually blind people by ocular exposure to white light: clinical characteristics. Ophthalmology 125(8):1160–1171. https://doi.org/10.1016/j.ophtha.2018.01.036
Stevens RG, Zhu Y (2015) Electric light, particularly at night, disrupts human circadian rhythmicity: is that a problem? Philos Trans R Soc Lond B Biol Sci 370:1667. https://doi.org/10.1098/rstb.2014.0120
Nagare R, Plitnick B, Figueiro MG (2019) Effect of exposure duration and light spectra on nighttime melatonin suppression in adolescents and adults. Light Res Technol 51(4):530–543. https://doi.org/10.1177/1477153518763003
Podolin PL, Rollag MD, Brainard GC (1987) The suppression of nocturnal pineal melatonin in the Syrian hamster: dose–response curves at 500 and 360 nm. Endocrinology 121(1):266–270. https://doi.org/10.1210/endo-121-1-266
Chang WP, Lin CC (2017) Relationships of salivary cortisol and melatonin rhythms to sleep quality, emotion, and fatigue levels in patients with newly diagnosed lung cancer. Eur J Oncol Nurs 29:79–84. https://doi.org/10.1016/j.ejon.2017.05.008
de Castro TB, Bordin-Junior NA, de Almeida EA, de Campos Zuccari DAP (2018) Evaluation of melatonin and AFMK levels in women with breast cancer. Endocrine 62(1):242–249. https://doi.org/10.1007/s12020-018-1624-2
Li W, Kwok CC, Chan DC, Ho AW, Ho CS, Zhang J, Wing YK, Wang F, Tse LA (2019) Disruption of sleep, sleep-wake activity rhythm, and nocturnal melatonin production in breast cancer patients undergoing adjuvant chemotherapy: prospective cohort study. Sleep Med 55:14–21. https://doi.org/10.1016/j.sleep.2018.11.022
Veiga ECA, Simoes R, Valenti VE, Cipolla-Neto J, Abreu LC, Barros EPM, Sorpreso ICE, Baracat MCP, Baracat EC, Soares Junior JM (2019) Repercussions of melatonin on the risk of breast cancer: a systematic review and meta-analysis. Rev Assoc Med Bras (1992) 65(5):699–705. https://doi.org/10.1590/1806-9282.65.5.699
Scholtens RM, van Munster BC, van Kempen MF, de Rooij SE (2016) Physiological melatonin levels in healthy older people: a systematic review. J Psychosom Res 86:20–27. https://doi.org/10.1016/j.jpsychores.2016.05.005
Andersen LP, Gogenur I, Rosenberg J, Reiter RJ (2016) The safety of melatonin in humans. Clin Drug Investig 36(3):169–175. https://doi.org/10.1007/s40261-015-0368-5
Gringras P, Nir T, Breddy J, Frydman-Marom A, Findling RL (2017) Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry 56(11):948 e944–957 e944. https://doi.org/10.1016/j.jaac.2017.09.414
Barchas J, DaCosta F, Spector S (1967) Acute pharmacology of melatonin. Nature 214(5091):919–920. https://doi.org/10.1038/214919a0
Ma Z, Yang Y, Fan C, Han J, Wang D, Di S, Hu W, Liu D, Li X, Reiter RJ, Yan X (2016) Melatonin as a potential anticarcinogen for non-small-cell lungcancer. Oncotarget 7(29):46768–46784. https://doi.org/10.18632/oncotarget.8776
Chao YH, Wu KH, Yeh CM, Su SC, Reiter RJ, Yang SF (2019) The potential utility of melatonin in the treatment of childhood cancer. J Cell Physiol 234(11):19158–19166. https://doi.org/10.1002/jcp.28566
Govender J, Loos B, Marais E, Engelbrecht AM (2014) Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: a review of the protective role of melatonin. J Pineal Res 57(4):367–380. https://doi.org/10.1111/jpi.12176
Guven C, Taskin E, Akcakaya H (2016) Melatonin prevents mitochondrial damage induced by doxorubicin in mouse fibroblasts through Ampk-Ppar gamma-dependent mechanisms. Med Sci Monit 22:438–446. https://doi.org/10.12659/msm.897114
Lissoni P, Chilelli M, Villa S, Cerizza L, Tancini G (2003) Five years survival in metastatic non-small cell lung cancer patients treated with chemotherapy alone or chemotherapy and melatonin: a randomized trial. J Pineal Res 35(1):12–15. https://doi.org/10.1034/j.1600-079x.2003.00032.x
Innominato PF, Lim AS, Palesh O, Clemons M, Trudeau M, Eisen A, Wang C, Kiss A, Pritchard KI, Bjarnason GA (2016) The effect of melatonin on sleep and quality of life in patients with advanced breast cancer. Support Care Cancer 24(3):1097–1105. https://doi.org/10.1007/s00520-015-2883-6
Maria S, Samsonraj RM, Munmun F, Glas J, Silvestros M, Kotlarczyk MP, Rylands R, Dudakovic A, van Wijnen AJ, Enderby LT, Lassila H, Dodda B, Davis VL, Balk J, Burow M, Bunnell BA, Witt-Enderby PA (2018) Biological effects of melatonin on osteoblastt/osteoclast cocultures, bone, and quality of life: Implications of a role for MT2 melatonin receptors, MEK1/2, and MEK5 in melatonin-mediated osteoblastogenesis. J Pineal Res 64:3. https://doi.org/10.1111/jpi.12465
Monaco ME (2017) Fatty acid metabolism in breast cancer subtypes. Oncotarget 8(17):29487–29500. https://doi.org/10.18632/oncotarget.15494
Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr (2013) Cellular fatty acid metabolism and cancer. Cell Metab 18(2):153–161. https://doi.org/10.1016/j.cmet.2013.05.017
Li Z, Zhang H (2016) Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci 73(2):377–392. https://doi.org/10.1007/s00018-015-2070-4
Nordlund JJ, Lerner AB (1977) The effects of oral melatonin on skin color and on the release of pituitary hormones. J Clin Endocrinol Metab 45(4):768–774. https://doi.org/10.1210/jcem-45-4-768
Jin JX, Lee S, Taweechaipaisankul A, Kim GA, Lee BC (2017) Melatonin regulates lipid metabolism in porcine oocytes. J Pineal Res 62:2. https://doi.org/10.1111/jpi.12388
Cipolla-Neto J, Amaral FGD (2018) Melatonin as a hormone: new physiological and clinical insights. Endocr Rev 39(6):990–1028. https://doi.org/10.1210/er.2018-00084
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2017) Mitochondrial bioenergetics decay in aging: beneficial effect of melatonin. Cell Mol Life Sci 74(21):3897–3911. https://doi.org/10.1007/s00018-017-2619-5
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17(1):41–52. https://doi.org/10.1016/j.ccr.2009.11.023
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35(12):669–675. https://doi.org/10.1016/j.tibs.2010.07.003
Bell EL, Emerling BM, Ricoult SJ, Guarente L (2011) SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30(26):2986–2996. https://doi.org/10.1038/onc.2011.37
Author information
Authors and Affiliations
Contributions
RJR conceived the idea and, after soliciting input from all co-authors (RS, QM, SRC and LGC), wrote the preliminary version of the report. The preliminary report was read by all co-authors who provided comments and suggestions for revisions after which changes were incorporated into additional versions of the manuscript by RJR. Figures were prepared and revised by RS, QM, SRC and LGC. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Reiter, R.J., Sharma, R., Ma, Q. et al. Melatonin inhibits Warburg-dependent cancer by redirecting glucose oxidation to the mitochondria: a mechanistic hypothesis. Cell. Mol. Life Sci. 77, 2527–2542 (2020). https://doi.org/10.1007/s00018-019-03438-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03438-1