
Abstract
The oncostatic properties of melatonin as they directly or indirectly involve epigenetic mechanisms of cancer are reviewed with a special focus on breast cancer. Five lines of evidence suggest that melatonin works via epigenetic processes: (1) melatonin influences transcriptional activity of nuclear receptors (ERα, GR and RAR) involved in the regulation of breast cancer cell growth; (2) melatonin down-regulates the expression of genes responsible for the local synthesis or activation of estrogens including aromatase, an effect which may be mediated by methylation of the CYP19 gene or deacetylation of CYP19 histones; (3) melatonin inhibits telomerase activity and expression induced by either natural estrogens or xenoestrogens; (4) melatonin modulates the cell cycle through the inhibition of cyclin D1 expression; (5) melatonin influences circadian rhythm disturbances dependent on alterations of the light/dark cycle (i.e., light at night) with the subsequent deregulation of PER2 which acts as a tumor suppressor gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction. Cancer: genetic and epigenetic mechanisms
The term epigenetics describes the study of heritable alterations in gene expression that occur in the absence of changes in genome sequence. This can be contrasted with genetics, which deals with the transmission of information based on differences in DNA sequence. The traditional view that gene and environment interactions control disease susceptibility can now be expanded to include epigenetic reprogramming as a key determinant in the origin of human disease [1]. The classic concept of carcinogenesis describes a multistage process consisting of three distinct phases: initiation, promotion, and progression. Mutations are clearly implicated in initiation. Most mutations are sporadic and their prevalence increases with age. Other mutations are inherited. Regardless, a mutation may result in activation of oncogenes or inactivation of tumor suppressor genes [2].
Gene silencing or inactivation of a tumor suppressor gene may be not only due to a mutation, but also a result of a translocation or inhibition of transcription. Inactivation of genes that regulate cell proliferation and death is a critical part of the neoplastic process. One important mechanism that may lead to inhibition of transcription is gene silencing through epigenetic alterations such as acquisition of promoter methylation and changes in chromatin structure. Activation of oncogenes may also be a result of epigenetic changes through post-translational modifications in histone acetylation or DNA methylation [3].
The epigenome (the overall epigenetic state of a cell) can be transmitted from parent to daughter cell maintaining a specific epigenotype within cell lineages. Thus, the phenotype is a result of the genotype (the specific DNA sequence) and the epigenotype. The genotype must exist in a particular chromatin configuration, the epigenotype, which allows a secondary level of fine control over gene expression. The epigenotype shows far greater plasticity than the genotype, and it has been speculated that epigenetic errors could be a major contributor to human diseases [4]. Epigenotype is generally accepted to be less stable than the genetic system and more sensitive to environmental [5], nutritional [6] and chemical toxicants which behave as endocrine disruptors [7, 8] and which may contribute to the development of abnormal phenotypes. For example, dietary supplements such as folate or several vitamins that affect the activity of methylating enzymes can be important in the incidence of colon cancer [9, 10]. A methyl-deficient diet has been shown to induce liver cancer associated with enhanced expression of c-ras, c-myc or c-fos oncogenes [10, 11]. Epigenetic memory of cells can be passed onto subsequent generations and can transfer the perturbed epigenome upon unaffected or normal genetic sequences. In contrast to gene mutations, epigenetic changes occur at a higher frequency than genetic changes, are limited at defined regions of a gene, and are reversible upon pharmacological treatment [12].
Since the genome contains information in two forms, genetic and epigenetic, initial studies focused on human cancers and rapidly revealed that most of human cancers are related to epigenetic aberrations (i.e., epimutations) [3] including epigenetic silencing of tumor suppressor genes because of hypermethylation and oncogenes because of demethylation [13, 14]. To date, numerous tumor suppressor genes have been found to undergo hypermethylation in cancer [15, 16]. Such epimutations rarely appear in healthy tissue indicating that epigenetic therapies may have high tumor specificity. Currently, two DNA methyl transferases (DNMT) inhibitors received US Food and Drug Administration approval for the treatment of myelodysplastic syndrome. 5-azacytidine (Vidaza®) and its derivative decitabine (Dacogen®) are now being marketed [17] and several presently available drugs are under extensive clinical investigation [18].
DNA methylation and histone acetylation as the best characterized epigenetic markers
Our current understanding of epigenetic gene regulation involves basically two classes of molecular mechanisms: DNA methylation and histone modifications [19, 20]. A variety of enzymes are involved in this process including most importantly DNA methyltransferases (DNMTs), histone deacetylases (HDACs) and histone acetyl transferases (HATs) [21].
DNMTs add a methyl group to the cytosine ring to form methyl cytosine. This modification takes place only on a cytosine that precedes a guanosine in the DNA sequence, called the CpG dinucleotide. GpG sites are not randomly distributed in the genome. The GpG-reach regions (known as GpG islands) are mainly present on the promoter region of genes [22, 23]. These CpG islands in gene promoter regions are usually unmethylated in normal cells and allow for active gene transcription. In cancer cells, CpG islands that are normally unmethylated may become methylated, resulting in silencing of important genes, such as inactivation of tumor suppressor genes. At the same time, CpG dinucleotides in other regions may become unmethylated, leading to diminished transcriptional repression of normally silenced genes such as oncogenes [22, 23].
DNA methylation is mediated by several proteins. As noted, DNMTs add methyl groups to the cytosines in CpG dinucleotides. Three active DNMTs have been recognized in humans and are designated DNMT1, DNMT3a, and DNMT3b. Each DNMT may have a specific role in the methylation process, or may act in association with another methyltransferase [19].
DNA methylation occurs in a complex chromatin network and can be influenced by several modifications in the structure of histones [24]. Chromatin is a nucleoprotein complex consisting of a basic repeating unit known as the nucleosome. A single nucleosome contains two turns of DNA wrapped around a core histone octamer comprising the histones H2A, H2B, H3 and H4. Nucleosomes represent the first level of compaction in chromatin, restricting access to enzymes involved in DNA metabolism. In addition to these basic components, linker histones and a variety of non-histone proteins are incorporated to generate a fully functional genome within a higher-order chromatin structure.
Indeed, the transcriptional status of all genes (silent, repressed or active) is determined by its chromatin environment and many molecular responses to toxicants involve alterations in gene expression that are elicited via changes in the chromatin structure of target genes [4]. The steady state level of histone acetylation is regulated by the HATs and HDACs. HATs are responsible for the addition of acetyl groups that stabilize open chromatin structures, while the HDACs deacetylate histones, and are thus responsible for resetting chromatin into a close conformation. Open chromatin is transcriptionally active, whereas condensed chromatin is transcriptionally inactive. Reduced or abnormal histone acetylation has been found in many types of cancer including breast cancer [23–25].
Major epigenetic alterations in breast cancer
Hypermetylation of CpG-island promoter can influence genes involved in DNA repair, cell cycle control, apoptosis, angiogenesis, cell-to-cell interactions, etc, all of which are involved in cancer development [25] (Fig. 1). In reference to breast cancer, hypermetylation of CpG islands occurs in DNA repair genes including BRCA1, genes encoding BRCA1 binding protein (SRBC), tumor suppressor genes like CDH1, CDH13, genes involved in cell adherence and the metastasic process (CDH1-E-cadherin, CDH13-H-cadherin), cell cycle inhibitor genes (p16 ink4a), genes coding tyrosine kinases (SYK) or metabolic enzymes (GSTP1), genes coding estrogen, progesterone and prolactin receptors (ER, PR PRLR), proapoptotic genes (TMS1) and histone/protein metyltransferase (RIZ1), as well as global genomic hypomethylation. Collectively, these changes constitute the epigenetic aberrations [25, 26].

Hypermetylation of CpG islands in promoter regions of genes involved in breast cancer. Modified from Esteller [25]
Melatonin and epigenetics of breast cancer
Breast cancer is a complex disease that results from a multi-stage process involving deregulation of a number of different signaling cascades. It is also the most frequent non-skin cancer to affect women worldwide, and remains a major public health burden. As previously summarized, in breast cancer multiple genes are methylated compared to non-cancerous tissue. DNA methylation, acquired over time, leads to silencing of genes that are critical in several pathways. For example, genes involved in growth (estrogen receptor, progesterone receptor) metastasis (TIMPs), and limitless replicative potential (cyclin D, pl6, BRCA1) can be altered in breast cancers [12, 26]. Methylation of CpG islands within the specific promoter region usually leads to the silencing of gene expression, as is frequently observed in tumor-suppressor genes in tumors [27]. In contrast, the global hypomethylation seen in a number of cancers, such as breast and cervical cancer, is related to an increase in the grade of malignancy [28]. A growing number of findings indicate aberrant gene expression in breast cancer cells that, in some cases has been associated with alterations in the expression, the activity or/and the recruitment of HATs and HDACs. For example, the functional activity of ERα is an important promoter of human breast cancer, and is regulated by acetylation. The exact role of ERα acetylation and the deacetylation may be involved in both hormone-sensitive and hormone-insensitive breast cancer [29].
Melatonin is a secretory product of the pineal gland discovered just 50 years ago [30]. The number of physiological functions attributed to this indoleamine is so diverse than the adjective “pleiotropic” is frequently used to refer to the nature of its actions. Among melatonin’s properties is its ability to inhibit the growth of mammary cancer cells both in vitro and in vivo; this suppressive effect is especially apparent in reference to ER positive tumors [31, 32]. Cohen et al. [33] proposed in 1978 a possible relationship between pineal function and mammary carcinogenesis by considering the possible antiestrogenic actions of melatonin. This hypothesis has since been examined in numerous epidemiologic and experimental studies. To the epidemiologic studies belongs the demonstration of a decreased nocturnal plasma melatonin peak in patients with ER positive breast cancer [34] as well as the assessment of low risk of breast cancer among blind women [35, 36] and the inverse association between breast cancer incidence and degree of visual impairment [37]. In the visually impaired individuals, the total or partial suppression of the light input would be expected to result in increased circulating melatonin levels which could explain the low incidence of tumours. More recently, it has been demonstrated, in a prospective case-control study in nurses, that higher melatonin levels, as measured in first morning urine, are associated with a lower risk of breast cancer [38]. On the contrary, the high incidence of breast cancer among women exposed to light during night, such as shift workers [39], and those exposed to low frequency electromagnetic fields [40], could be explained by the reduced melatonin synthesis under these environmental conditions. The epidemiologic evidence is reinforced by the experimental demonstration that low intensity light exposure during nocturnal darkness, with the subsequent reduction of melatonin synthesis and secretion, enhanced the growth of previously induced rat mammary tumors [41] or MCF-7 cell xenografts in nude mice [42]. The experimental studies, carried out with different animal models of mammary carcinogenesis and breast cancer cell lines have confirmed that melatonin, in vivo, reduces the incidence and growth of chemically induced, or spontaneous mammary tumors in rodents, whereas in vitro, at concentrations corresponding to the physiological levels present in human blood during the night (1 nM), melatonin inhibits proliferation, increases expression of p53 and reduces the invasiveness of the estrogen-responsive MCF-7 human breast cancer cells [31, 32, 43–46].
In the next subsequent sections the oncostatic properties of melatonin directly or indirectly related with epigenetic mechanisms of cancer will be reviewed with a special focus on breast cancer.
Nuclear receptors (NRs) and co-regulators
Members of the NRs or ligand-dependent transcription factors play a multitude of essential roles in development, homeostasis, reproduction, and immune function [47]. NRs regulate transcription by several mechanisms and can both activate and inhibit gene expression [48]. The NRs include steroidal transcription factors such as the estrogen (ER) glucocorticoid (GR), thyroid hormone receptor (TR), liver X receptor (LXR), frasenoid X receptor (FXR), vitamin D receptor (VDR), retinoid acid receptor (RAR), retinoid X receptor (RXR) and peroxisome proliferators-activated receptors (PPARs) [49, 50].
Co-regulators (co-repressors and co-activators), those that interact directly with NRs, exist in large steady-state complexes with multiple secondary co-coregulator partners. Each component may possess multiple enzymatic capabilities such as acetyltransferase, deacetylase methyltransferase ultimately making these complexes versatile enzymatic machines for regulating gene expression. The activity of co-regulators is directly affected by its methylation, acetylation, or other modifications of their status, forming a posttranslational modification code. This code then controls the co-regulator’s transcriptional activity and transcription factor preferences [51].
Co-regulator-mediated modulation of gene expression targets histones in the chromatin that surround genes [52] generated by a ‘‘histone code’’ in virtually all parts of DNA [53]. Thereby, co-regulators lead to a variety of biological responses that are distinct from targeting histones and further epigenetic modification of DNA by altering the methylation levels. In this manner, co-regulators can be differentially coded, allowing for an extremely large degree of combinational complexity. It is estimated that ~25,000 human genes exist, of which differential splicing introduces more diversity, yielding ~125,000 different coding transcripts and potential proteins [54]. Co-regulators are “master regulators” responsible for establishing homeostasis for cells, tissues, and metabolism and development throughout the life. There is no doubt that co-regulator dysfunction is not restricted to rare genetic conditions, but instead is involved in numerous human diseases [53, 54].
Half of the NRs are so-called “orphan” receptors because the identity of their ligand, if any, is unknown. The definition of orphan receptors is a loose and paradoxical one because, by definition, orphan receptors are receptors for which no ligand is known. The term “receptor” itself implies that a physiological ligand should exist, even though there is still no consensus in the field as to whether this will be true for all orphan NRs. Because the absence of proof is not the proof of absence, it is extremely difficult to demonstrate that a given orphan NR truly has no endogenous ligand. Complicating the issue is the fact that once a natural ligand has been discovered for an orphan NR, the receptor is no longer considered to be an orphan, despite the fact that it may retain structural and functional features more similar to the other orphan NRs than to the classic SR and TR. Two prime examples are the PPARs and RXRs, which were discovered as orphan NRs, but which are now clearly considered to be liganded receptors, although the precise identity of their physiological, endogenous ligands is somewhat controversial [49]. Some of receptors belonging to RXRs are no longer orphaned. Evidence of a genomic action of melatonin via nuclear RZR/ROR receptors was initially suggested in 1994 [55]. Subsequent studies have detected the nuclear melatonin receptor (NMRs; RXRs) mRNA transcripts by using in situ hybridization neuronal tissue [56–59] including pineal gland [60] and many other tissues as well [59, 61–63].
In the unligated state, PPARs associate with a multicomponent complex containing co-repressors with HDAC activity [64]. The nuclear co-repressor complex inhibits transcription by deacetylating histones [65]. Upon ligand binding, PPARs recruit co-activators that lead to histone acetylase activity and initiate a sequence of events that induces gene transcription processes [66, 67]. RXR can regulate transcription in a heterodimeric complex and generally does not involve gene transcription. Through its role as a required heterodimeric partner, RXRs control the function of many other NRs, thus integrating a unique transcriptional network dependent on RXR responses [68, 69]. RXR forms heterodimers with virtually all NRs including GR, ER, TR, PPAR, VDR, LXR and FXR. Both in vitro and in vivo approaches have revealed that NRs require RXR as a heterodimerization partner for their function. NRs can activate transcription as monomers and/or dimers with the RXR. Ligand-activated NRs dissociate from co-repressors and recruit co-activator proteins, which promote transcriptional activation [54, 70–72].
Regarding nuclear and epigenetic involvement in breast cancer in the context of melatonin, it is remarkable that transcriptional activity of ERα, GR and RAR receptors, which are involved in the regulation of breast cancer cell growth [73], are modulated by melatonin [74]. In the same context, PPARγ agonists significantly inhibit breast cancer growth [75] and RXR agonists potentiate the antiproliferative and apoptotic effects of PPARγ agonists [76].
Eck-Enriquez et al. [77] demonstrated that MCF-7 cells treated sequentially with melatonin and all-trans-retinoic acid (atRA) resulted in enhanced sensitivity to the apoptotic effects of atRA, which did not appear to be due to increased expression of the RAR, but rather to enhanced transcriptional activity.
Sharma et al. [78] provide direct evidence of epigenetic actions for melatonin including NRs, co-regulators and histone acetylating enzymes. In this study, melatonin significantly increased mRNA expression for various HDAC isoforms and increased histone H3 acetylation in neural stem cell lines; also DNMT inhibitory actions of melatonin may suggest an epigenetic regulation at NR/co-regulator level rather than selective enzymatic inhibition or activation. The apparent nuclear harmony possibly through heterodimerization of melatonin-liganded RXR may open new avenues in both the pathogenesis of breast cancer and therapeutic advantages of melatonin in combination with certain NRs agonists and epigenetic modifiers.
Estrogens (E) play an important role in the development of breast cancer. They act on nuclear ER receptors and induce the expression of E-dependent genes. Furthermore, prolonged exposure to estradiol (E2), which exerts effects on epithelial cells either directly or via stromal cells, allows cells to propagate heritable changes including DNA methylation [26]. Thus, for more than a century, suppression of estrogenic actions has been a therapeutic tool in breast cancer therapy. Melatonin counteracts the effects of E2 and xenoestrogens on breast cancer cell proliferation, invasiveness and telomerase activity [46, 79–82], augments the sensitivity of MCF-7 to other anti-estrogens such as tamoxifen [43], and down-regulates the expression of proteins, growth factors, and proto-oncogens regulated by estrogens [45, 83]. These antiestrogenic effects depend on the interaction of melatonin with the ERα. The nature of this interaction has been investigated for recent years. Melatonin decreases the expression of ERα and inhibits the binding of the E2-ER complex to the estrogen response element on DNA [84–86]. These effects depend on melatonin binding to specific melatonin (MT1) membrane receptors [87–91], also found in human breast tissue, both normal and tumorous [92]. The role of calmodulin as a mediator of the interactions of melatonin has been recently demonstrated [93, 94]. Interestingly, whereas melatonin is a specific inhibitor of E2-induced ERα-mediated transcriptional activation, it does not inhibit transactivation of ERβ which does not interact with calmodulin [95]. The loss of ER expression indicates a poor prognosis for a significant number of breast cancer patients [26]. Recent results show that p53 up-regulates ERα genes expression and pRb2/p130 has also an important role in the transcriptional regulation of the ERα promoters [26, 96, 97]. These findings suggest that both p53 and pRb2/p130 may be targets for the development of novel therapeutic strategies in the treatment of ER-negative breast tumors, by re-establishing ER expression in ER-negative breast cancer [26]. The combination of demethylating agents and HDAC inhibitors has also been demonstrated to be synergic in the re-expression of ERα in the ER negative breast cancer cells [26].
Breast cancer incidence among women with gross cystic breast disease (GCBD) is to 2–7 times higher than that of the general population although the mechanism for this increase remains unexplained. Burch et al. [98] investigated 142 breast cyst fluid (BCF) samples were collected from 93 women with GCBD [98]. They observed the lowest growth index was among BCF samples with elevated concentrations of both melatonin and estrogens when added to MCF-7 cell line medium suggesting a beneficial coexistence of estrogens and melatonin, possibly at the NR level.
Approximately one third of breast cancers do not express the ERα. These tumors exhibit a greater aggressiveness and do not respond to endocrine therapy with estrogens as a target. The loss of ER expression is the result of the hypermethylation of CpG islands within the ERa promoter [26]. Treatment of ER-negative human breast cancer cells with methyltranferase or histone deacetylase inhibitors leads to a partial demetylation of the ER CpG and re-expression of ER mRNA as well as synthesis of functional ER proteins [26, 99].
Breast cancer and aromatase enzyme complex: epigenetic involvement and role of melatonin
In the postmenopausal period, estrogens are synthesized in mammary tissue by the enzyme complex known as aromatase encoded by the CYP19 gene, from the androgenic precursors of adrenal origin (Fig. 2). Therefore aromatase is crucial, especially for postmenopausal breast cancer development as well as endometrial, ovarian and uterine disorders [100]. Breast tumors from postmenopausal women maintain a high estrogen content, even though the plasma concentrations of estradiol decrease to very low values [101]. One pathway for in situ synthesis involves the conversion of androstenedione to estrone/estradiol, catalyzed by aromatase and it may play a role in postmenopausal breast cancer development [102]. Aromatase is sufficient to maintain preneoplastic changes without circulating estrogens in breast tissue [103]. Other enzymes involved in the local biotransformation of esteroids are the reluctant isoforms of 17beta-hydroxysteroid dehydrogenases (17β-HSD) which catalyze the conversion of the relatively weak estrone (E1), androstenedione and 5-androstenedione into the more potent estradiol (E2), testosterone and 5-dihydrotestosterone, whereas the oxidative isoforms catalyze the formation of steroids of low activity, thus exerting a protective role in different tissues including the mammary gland [104, 105]. Finally, estrogen sulfatases (STS) convert estrogen sulfates into E1 and E2 and estrogen sulfotransferases (EST) catalyze the conversation of estrogens into their sulfates, thus providing protection from excessive estrogenic effects [104, 105] .

Pathways of interconversion of steroids and enzymes involved. From Gonzalez et al. [110]
Sanchez-Barcelo et al. [106, 107] recently demonstrate that melatonin modulates local estrogen biosynthesis by reducing aromatase expression and activity in MCF-7 human breast cancer cells as well as in glioma cells, and enhances the antiaromatase activity of aminoglutethimide [108]. In vivo, melatonin inhibits the growth of DMBA-induced mammary tumors by limiting the tumoral aromatase activity and the local synthesis of estrogens [109]. Melatonin, at physiological (1 nM) concentrations, also reduces the synthesis of biologically active estrogens in MCF7 cells, through the inhibition of STS and 17βHSD1 and the stimulation of EST, the enzyme responsible for the formation of the biologically inactive estrogen sulfates [110]. Although the exact mechanism as to how melatonin down-regulates aromatase expression at the transcriptional level in MCF-7 cells is unknown, recent evidence suggests epigenetic involvement. Izawa et al. [111] reported that the up-regulation of aromatase gene in endometrial cells may be ascribed to a disorder of methylation status within CpG islands in aromatase genes. When endometrial cells were treated with 5-aza-deoxycytidine (a DNMT inhibitor), aromatase mRNA expression was markedly enhanced depending on the same proximal promoters as those in endometrial cells [111]. They also found that treatment with a DNMT inhibitor enhanced aromatase expression in the eutopic endometrium [111]. If melatonin has epigenetic actions on the aromatase gene, melatonin causes methylation of the CYP19 gene or deacetylation of CYP19 histones and leads to gene silencing remains unknown.
The crucial transcription factor nuclear factor-κB (NF-κB) is involved in CYP19 activation by inducing several pro-inflammatory molecules including tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), cyclooxygenase (COX-2) and prostaglandin E2 (PGE2) [112]. Positive correlations have been detected between TNF-α, COX-2 and aromatase expression in human breast cancer tissue [113]. In mice genetically engineered to overexpress COX-2 in the mammary gland, increased levels of PGE2 and aromatase were found [114]. Subaramaiah et al. [115] reported that p300, a co-regulator which has HAT activity, is important in the regulation of CYP19 leading to increased aromatase expression via this pathway.
Of interest is that melatonin acts on every molecule in this pathway through its nuclear actions. As recently documented by Esposito et al. among others [116–118], melatonin suppresses NF-κB binding to DNA via a NRs cross-talk and decreases TNF-α, iNOS, COX-2 and PGE2 levels. Furthermore, Deng et al. [119] revealed that melatonin inhibits p300 HAT activity and abrogated p300-augmented COX-2 and iNOS expression. Melatonin suppresses macrophage COX-2 and iNOS synthase expression by inhibiting p52 acetylation and binding to DNA. This experiment suggests that melatonin causes deacetylation and leads to iNOS and COX-2 gene silencing. One mechanism by which PPARγ and RXR heterodimer inhibit breast cancer growth is also by means of the same pathway [76]. Fan et al. [120] reported that activation of PPARγ and RXR receptor inhibits aromatase transcription via NF-κB. Although not confirmed, PPARγ agonists and melatonin may act through in this pathway as well as suggesting that melatonin may be a “gene silencer” through modulation of histone acetylation and DNA methylation.
Telomere length, telomerase and melatonin
Telomeres, the physical ends of linear chromosomes, play an important role in maintaining the chromosome integrity and stability as well as in DNA replication [121]. During the cell division cycle, these structures are progressively shortened since DNA polymerase cannot replicate the terminal end of chromosomes. To overcome this loss of DNA, telomerase, a multi-component ribonucleoprotein, replenishes telomeric DNA by synthesizing the G-strand, whereas the C-strand is synthesized by the normal DNA replication machinery [121]. Human telomerase is basically composed of: a catalytic subunit (hTERT: human telomerase reverse transcriptase) which, by using the chromosome end as a primer, drives the synthesis of the G-reach strand of telomeric DNA with a RNA subunit acting as a template (hTR: human telomerase RNA) [121]. The hTERT subunit is the rate-limiting determinant of telomerase enzyme activity [121].
Activation of telomerase plays an important role in human cancer development, providing the mechanism for an unlimited neoplastic cell division capacity [121]. Telomerase activity is observed in 85–90% of all cancers, whereas it is absent in most differentiated tissues [122]. In relation to breast cancer, telomerase activation is a relatively earlier event in the carcinogenic process [123], and the expression of hTERT closely correlates with telomerase activity thus serving as an indicator of telomerase activation [124].
Telomerase activity is also under control of epigenetic regulation [125, 126]. Increasing evidence indicates that chromatin modifications are important regulators of mammalian telomeres. The suppression of epigenetic regulators such as DNA- and histone-methyltransferases induces the loss of the control of telomeric length with the subsequent telomere elongation [125–127]. Guilleret and Benhattar [128] reported that 5-azacytidine (a DNMT inhibitor) reduces hTERT expression, telomerase activity and shortens telomeres in a human cervical adenocarcinoma cell line. Interestingly, hTERT is likely to be regulated by methylation, and hypermethylation of hTERT promoter is likely to be necessary for its expression [128]. This preliminary finding has recently been confirmed by Renaud et al. [129]. They revealed that hTERT expression is induced when the hTERT CpG island is sufficiently hypermethylated.
Similarly, melatonin inhibits telomerase activity in MCF-7 breast cancer cells and decreases mRNA levels of hTERT, as well as hTR [80]. Furthermore, melatonin inhibits the hTERT expression induced by either natural estrogens (17β-estradiol) or metalloestrogens (cadmium) in MCF-7 human breast cancer cells [81, 82]. Interestingly, this action of melatonin resembles that of DNMT inhibitors [130] e.g., tea polyphenol (−)-epigallocatechin-3-O-gallate (EGCG) [131], a compound that reduces cellular proliferation and induces apoptosis of MCF-7 cells [132]. Like melatonin, EGCG significantly decreases hTERT mRNA with the down-regulation of hTERT gene expression in MCF-7 cells seemingly being largely due to epigenetic alterations [132]. Interestingly, Leon-Blanco et al. [133] observed a significant decrease in the RNA levels of hTERT after treatment with an GP 52608 (an agonist of melatonin nuclear receptors), while treatment with S 20098 (an agonist of the melatonin membrane receptors) had opposite effects. This study strongly indicates that nuclear and/or epigenetic, but not membrane receptor-mediated actions of melatonin, are responsible for the decrement in the RNA levels of hTERT in the MCF-7 cell line. Furthermore, EGCG down-regulates ERs function in MCF-7 cells [134] in a similar manner to that of melatonin’s modulation of ERs in these cells [32, 135].
The link between histone deacetylation and telomerase activity has been studied in normal as well tumor cells. Mukhopadhyay et al. [136] demonstrated that trichostatin A (TSA), a HDAC inhibitor, induces hTERT expression and activates telomerase activity in normal lung cells compared to non-small-cell lung cancer, and that hTERT is directly correlated to histone acetylation in normal cells. However, in prostate cancer cells, TSA treatment decreases hTERT expression and telomerase activity [137]. The increase in hTERT expression following TSA treatment has also been reported in other cancer cell lines including Ha1-IM, SiHa and Hela cells [138]. It is likely that, in cancer cells, histones of telomerase are already acetylated and active. Therefore, inhibition of HDAC may either have no effect [136] or may augment telomerase activation [138].
To reduce telomerase activity in cancer cells but not in normal cells, inhibition of DNA methylation, as obtained in the melatonin and EGCG studies, seems feasible, since telomerase histones are already acetylated in cancer cells. Moreover, several reports indicate that DNMT inhibitors (e.g., EGCG) reactivate some methylation-silenced tumor suppressor genes in human colon cancer HT-29 cells, prostate cancer PC3 cells and esophageal cancer KYSE 150 cells [139].
Cyclin d1 connection in the pathogenesis of breast cancer
Class D cyclins (D1, D2 and D3), each of which binds cyclin-dependent kinase, associates with, and regulates activity of, transcription factors, co-activators and co-repressors that govern histone acetylation and chromatin remodeling proteins. It is cyclin D1 over-expression that is predominantly associated with tumorigenesis and cellular metastases. In human cancer, over-expression of cyclin D1 is one of the most commonly observed alterations in the transit through the G phase of the cell cycle. In a large majority of breast cancer cases, cyclin D1 is over-expressed and its levels correlate with a negative prognosis [140].
Cyclin D1 forms physical associations with more than 30 transcription factors or transcriptional co-regulators. Several NRs, including GR, ERα, and PPARγ, bind directly to cyclin D1 and both basal and ligand-dependent transactivation of NRs is regulated by cyclin D1 [141]. Not surprisingly, cyclin D1 is a co-activator of ERα transcription and a co-repressor of GR and PPARγ [142]. A key event for the anti-proliferative effects of anti-estrogens appears to be the down-regulation of cyclin D1 [143].
Santos et al. [144] reported that the repression of cyclin D1 expression by HDAC inhibitors (butyrate and SAHA) was stronger than that found with a pure anti-estrogen (ICI 182.780). This study also revealed that HDAC inhibitors down-regulate ER levels and transcription of ER target genes in breast cancer cells. Apart from similar effects of HDAC inhibitors including downregulation of ER levels and transcription of ER target genes in breast cancer cells, melatonin has been shown to inhibit transcription of cyclin D1 expression supporting the epigenetic efficacy of melatonin on hormone sensitive breast [145] and prostate cancer [146]. Several years ago, it was reported that melatonin significantly increases the duration of the cell cycle of human breast cancer cells [147].
Epigenetic actions of melatonin via the circadian system
There is increasing interest in the possible role of environmental factors that can alter normal endocrine function, often referred to as ‘endocrine disruptors’, in the etiology of cancer. Of particular interest is the potential influence of exposure to light-at-night (LAN), and sleep disruption as occurs in night shift workers, on endocrine function and the regulation of hormones, e.g., melatonin, that are important in the etiology of some types of cancer [148]. It is important take into consideration the role of light as the main zeitgeber of the circadian system and that circadian rhythm disruptions may be epigenetic causes of cancer [149]. Persons who engage in night-shift work are subjected to the influence of both factors, and exhibit altered hormone profiles, most importantly decreased melatonin production and a disrupted melatonin rhythm [38]; these changes could increase the risk of hormone-related diseases, including breast and prostate cancer (see Section “Melatonin and epigenetics of breast cancer” of this article). A recent meta-analysis [150], and on-going studies in Denmark and Seattle have recently found a strong relation between night-shift and increased breast cancer [151, 152]. Moreover, melatonin-depleted blood from postmenopausal women exposed to LAN stimulates growth of rat hepatomas and MCF-7 xenografts in comparison to the effects of night-collected blood with normally elevated melatonin [149]. It has also been shown that MCF-7 cell proliferation was greater in presence of sera obtained from breast cancer women than with sera from healthy women [153], although the authors did not find differences in the melatonin concentrations between the two sera, probably because melatonin levels were only measured in morning samples. Interestingly, the serum concentration of melatonin in breast cancer women decreased after mastectomy, thus suggesting that the presence of the tumor could induce changes in the secretory pattern of this hormone, as was previously described for PRL and GH.
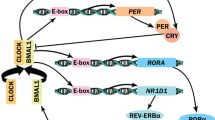
The mammalian clock machinery includes, among others, three period proteins (PER1, PER2, and PER3), two cryptochromes (CRY1 and CRY2), CLOCK and two BMAL proteins. In mammals, the central elements of the circadian machinery, CLOCK and BMAL1, heterodimerize and involve transcriptional/translational feedback loops with negative and positive limbs [154]. Recent mammalian clock gene studies uncovered molecular clocks in many brain regions and peripheral tissues synchronized by the master clock [155]. Currently, it is thought that transcription of PER and CRY genes are driven by accumulating CLOCK-BMAL1 heterodimers. At the same time, PER2 up-regulates the levels of BMAL1 mRNA leading to the formation of CLOCK-BMAL1 heterodimers, which drive PER2 and CRY2 transcription and restart the cycle [156].
Doi et al. [157] reported that the CLOCK is a HAT suggesting a strong epigenetic involvement. Hirayama et al. [158] revealed that CLOCK also acetylates its own partner BMAL1 and CLOCK-mediated acetylation contributes in multiple ways to the time-dependent regulation of circadian physiology. The compelling links that exist between the circadian cycle and metabolism suggest that the HAT function of CLOCK may be controlled by the redox state of cells and as well as by the light/dark cycle [159].
At present, the mechanism by which the circadian clock influences tumor growth is not fully understood. Recently, the circadian clock gene PER2, which helps to synchronize mammalian organisms with environmental light, has been reported to function as a tumor suppressor gene. Fu et al. [160] observed the development of spontaneous lymphomas and teratomas in PER2 knockout mice at only 6 months of age. Thirty per cent of mutant mice died before the age of 16 months. Disruption of the PER2 gene in mice abolishes the response of all core circadian genes to gamma radiation whereas in wild-type mice, clock genes are induced rapidly, suggesting they may be involved in DNA damage response. Furthermore, a number of cell cycle and checkpoint proteins were deregulated in these PER2 mutant mice including cyclin D1. Experimental evidence demonstrates the importance of circadian clock genes, in particular PER2, as regulators of the cell cycle and, therefore, cancer progression. Based on the role of PER2 in cancer development and the clear epidemiologic connection between circadian disruption (e.g., LAN and night-shift work) and the risk of breast cancer development in women, Xiang et al. [161] clearly demonstrated the tumor suppressive nature of PER2 as evinced by inhibition of cell growth, induction of apoptosis, reduced colony formation and growth in MCF-7 cells. Gery et al. [162] reported that PER2 links the circadian cycle to the ERα signaling network. While suppression of PER2 stabilizes ERα, binding enhances ERα degradation. PER2 itself was found to be estrogen inducible in these cells, suggesting a feedback mechanism to attenuate stimulation by estrogen. In addition, over-expression of PER2 in breast cancer cells leads to significant growth inhibition, loss of clonogenic ability and apoptosis.
Since consistent evidence reveals that PER2 is a circadian tumor suppressor gene, existence of this gene is welcome. However, its circadian nature suggests that PER2 should fluctuate throughout a 24 h period depending on melatonin levels and the light-dark cycle. Boivin et al. [163] reported that circadian clock genes do oscillate in human peripheral blood mononuclear cells. Peak clock gene expression was observed mostly during the usual time of activity and light exposure. Transcript levels of PER1-3 were found to correlate positively and significantly with the level of plasma melatonin. Peak clock gene expression followed the highest melatonin concentration by a maximum of 9 h [163]. Cajochen et al. [164] developed a non-invasive method to measure and quantify human circadian PER2 gene expression in oral mucosal samples and found that PER2 gene expression is both light and melatonin dependent. Consistent with this, evening exposure to blue light had a strong stimulatory effect on expression of PER2 in humans [164]. Because of this, the circadian rhythmicity of PER2 gene is perturbed and may not function as a tumor suppressor gene.
The visible light spectrum covers a wavelength range from approximately 400 nM (violet) to 700 nM (red); blue light has a wavelength close to the lower visible range (around 500 nM). From the studies of Provencio et al. [165], it is known that circadian photoreception depends on the ganglion cell layer of the retina. In these cells, photopigments different than opsins have been described as involved in this non-visual pathway (the so-called “non-image-forming system”) which carries photoperiodic information to the suprachiasmatic nuclei of the hypothalamus. These photopigments are melanopsins [165] and chryptocrhomes [166, 167] and they respond to a narrow band of blue wavelengths. Thus, the human circadian system is sensitive to non-visual effects of ocular light at short wavelengths [168], and maximal response to light for melatonin suppression [169, 170] and circadian phase shifting [171] is obtained between 446 and 483 nm (violet-blue). Acute (early) effect of light on melatonin, alertness, thermoregulation, and heart rate is blue-shifted, such that short wavelength of light at 460 nm induces a greater melatonin suppressive, alerting, hyperthermic and tachycardic effect than light at 550 nm [164, 172].
As a consequence, under a physiologic light-dark cycle, peak melatonin level is followed by PER expression and higher PER levels persevere until dusk [173]. Night-shift work and LAN as well as several indoor instruments, e.g., personal computers and TV can further prolong PER gene expression. Use of TV and/or computers in the evening is endemic in Western societies, and they emit a blue light mimicking “circadian high noon”. Watching TV at night was recently shown to be associated with lower urinary melatonin metabolite concentrations [174]. Finally, Chen et al. [175] also found, in 55 cases of breast cancer, deregulated expression of the three PER genes in more than 95% of the breast cancer cells in comparison with the nearby non-cancer cells. More recently Gery et al. [162] demonstrated that PER2 links the circadian cycle to the ERα signaling network. Thus, suppression of PER2 levels leads to ERα stabilization whereas its over-expression induces ERα degradation. Furthermore, PER2 itself is estrogen inducible in breast cancer cells; this fact suggesting a feedback mechanism which attenuates the stimulatory effects of the estrogens [162]. Interestingly, the PER gene deregulation is not caused by genetic mutations but by methylation of the PER1 and PER2 promoter [175], i.e., by epigenetic mechanisms. The methylation status of PER2 has a strong correlation with c-erB2 expression [175], and the over-expression of this oncogene may result in a more aggressive tumor phenotype. Whether methylation of PER2 influences the c-erB2 expression, or vice versa, needs to be clarified [175].
Concluding remarks
Environmental factors are among the major determinants of epigenetic changes related to carcinogenesis. In regard to this, there is an obvious epidemiologic connection between the frequency of breast cancer and LAN and/or night-shift strongly relate to reduced melatonin production and a disrupted melatonin rhythm. Furthermore, the context of the current review, we speculate that postmenopausally estrogen usage increases breast cancer risk since its antagonist, melatonin, is physiologically reduced due to aging. In other words, the postmenopausal period may mimic night-shift work or LAN; thus high estrogen levels in the presence of attenuated circulating melatonin concentrations increases the likelihood of breast cancer. The studies carried out on breast cyst fluid from women with gross cystic breast disease demonstrated an inverse relationship between its concentration of melatonin and estrogen and its ability to induce proliferation of MCF-7 breast cancer cells. This fact suggests a beneficial co-existence between estrogen and melatonin possibly at the NRs level. These observations coupled with those coming from a variety of laboratories demonstrated the oncostatic properties of melatonin expose the interaction of melatonin with the estrogen signaling pathway as potentially one of the main mechanisms involved in development of breast cancer. The interaction with other signaling pathways has also been proven. Thus, MCF-7 cells treated sequentially with melatonin and atRA enhanced the apoptotic effects of atRA, which did not appear to be due to increased expression of the RAR, but rather to enhanced transcriptional activity. Similar effects were also obtained with PPARγ and melatonin.
The oncostatic effects of melatonin that are of a direct epigenetic nature have been demonstrated in studies unrelated to cancer cell proliferation. These studies reported that melatonin significantly elevates mRNA expression for various HDAC isoforms and significantly increases histone H3 acetylation in neural stem cells. This apparently dual function of melatonin as reported in this study as well as the DNMT inhibitory action of melatonin suggests an epigenetic regulation at the NR/co-regulator level rather than selective enzymatic inhibition or activation. The apparent nuclear harmony possibly through heterodimerization of melatonin-liganded RXR may open new avenues in both the pathogenesis of breast cancer and therapeutic advantages of melatonin in combination with certain NRs agonists and epigenetic modifiers.
References
Bronner C, Chataigneau T, Schini-Kerth VB, Landry Y (2007) The “Epigenetic Code Replication Machinery”, ECREM: a promising drugable target of the epigenetic cell memory. Curr Med Chem 14:2629–2641. doi:10.2174/092986707782023244
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70. doi:10.1016/S0092-8674(00)81683-9
Ducasse M, Brown MA (2006) Epigenetic aberrations and cancer. Mol Cancer 5:60. doi:10.1186/1476-4598-5-60
Jiang YH, Bressler J, Beaudet AL (2004) Epigenetics and human disease. Annu Rev Genom Hum Genet 5:479–510. doi:10.1146/annurev.genom.5.061903.180014
McLachlan JA (2001) Environmental signaling: what embryos and evolution teach us about endocrine disrupting chemicals. Endocr Rev 22:319–341. doi:10.1210/er.22.3.319
Gallou-Kabani C, Junien C (2005) Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 54:1899–1906. doi:10.2337/diabetes.54.7.1899
Bombail V, Moggs JG, Orphanides G (2004) Perturbation of epigenetic status by toxicants. Toxicol Lett 149:51–58. doi:10.1016/j.toxlet.2004.01.003
McLachlan JA, Burow M, Chiang TC, Li SF (2001) Gene imprinting in developmental toxicology: a possible interface between physiology and pathology. Toxicol Lett 120:161–164. doi:10.1016/S0378-4274(01)00295-8
Giovanucci E, Stampfer MJ, Colditz GA et al (1993) Folate, methionine, and alcohol intake and risk of colorectal adenoma. J Natl Cancer Inst 85:875–884. doi:10.1093/jnci/85.11.875
Janisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254. doi:10.1038/ng1089
Dizik M, Christman JK, Wainfan E (1991) Alterations in expression and methylation of specific genes in livers of rats fed a cancer promoting methyl-deficient diet. Carcinogenesis 12:1307–1312. doi:10.1093/carcin/12.7.1307
Widschwendter M, Jones PA (2002) DNA methylation and breast carcinogenesis. Oncogene 21:5462–5482. doi:10.1038/sj.onc.1205606
Stearns V, Zhou Q, Davidson NE (2007) Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest 25:659–665. doi:10.1080/07357900701719234
Esteller M (2007) Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet; 16 Spec No 1:R50–59
Das PM, Singal R (2004) DNA methylation and cancer. J Clin Oncol 22:4632–4642. doi:10.1200/JCO.2004.07.151
Garinis GA, Patrinos GP, Spanakis NE, Menounos PG (2002) DNA hypermethylation: when tumour suppressor genes go silent. Hum Genet 111:115–127. doi:10.1007/s00439-002-0783-6
Brueckner B, Kuck D, Lyko F (2007) DNA methyltransferase inhibitors for cancer therapy. Cancer J 13:17–22. doi:10.1097/PPO.0b013e31803c7245
Peedicayil J (2006) Epigenetic therapy—a new development in pharmacology. Indian J Med Res 123:17–24
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402. doi:10.1093/hmg/9.16.2395
Yang XJ, Seto E (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26:5310–2318. doi:10.1038/sj.onc.1210599
Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C (2007) Cancer genetics of epigenetic genes. Hum Mol Genet 16 Spec No 1: R28–49
Esteller M (2000) Epigenetic lesions causing genetic lesions in human cancer: promoter hypermethylation of DNA repair genes. Eur J Cancer 36:2294–2300. doi:10.1016/S0959-8049(00)00303-8
Jacinto FV, Esteller M (2007) Mutator pathways unleashed by epigenetic silencing in human cancer. Mutagenesis 22:247–253. doi:10.1093/mutage/gem009
Fraga MF, Ballestar E, Villar-Garea et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400. doi:10.1038/ng1531
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159. doi:10.1056/NEJMra072067
Giacinti L, Claudio PP, Lopez M, Giordano A (2006) Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist 11:1–8. doi:10.1634/theoncologist.11-1-1
Szyf M, Pakneshan P, Rabbani SA (2004) DNA methylation and breast cancer. Biochem Pharmacol 68:1187–1197. doi:10.1016/j.bcp. 2004.04.030
Ehrlich M (2002) DNA methylation in cancer: too much, but also too little. Oncogene 21:5400–5413. doi:10.1038/sj.onc.1205651
Popov VM, Wang C, Shirley LA et al (2007) The functional significance of nuclear receptor acetylation. Steroids 72:221–230. doi:10.1016/j.steroids.2006.12.001
Lerner AB, Case JD, Takahashi Y (1960) Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands. J Biol Chem 235:1992–1997
Cos S, Sanchez-Barcelo EJ (2000) Melatonin and mammary pathological growth. Front Neuroendocrinol 21:133–170. doi:10.1006/frne.1999.0194
Sanchez-Barcelo EJ, Cos S, Mediavilla D, Martinez-Campa C, Gonzalez A, Alonso-Gonzalez C (2005) Melatonin-estrogen interactions in breast cancer. J Pineal Res 38:217–222. doi:10.1111/j.1600-079X.2004.00207.x
Cohen M, Lippman M, Chabner B (1978) Role of pineal gland in the aetiology and treatment of breast cancer. Lancet 2:814–816. doi:1016/S0140-6736(78)9259-6
Tamarkin L, Danforth D, Lichter A et al (1982) Decreased nocturnal plasma melatonin peak in patients with estrogen receptor positive breast cancer. Science 216:1003–1005. doi:10.1126/science.7079745
Coleman MP, Reiter RJ (1992) Breast cancer, blindness and melatonin. Eur J Cancer 28:501–503. doi:10.1016/S0959-8049(05)80087-5
Kliukiene J, Tynes T, Andersen A (2001) Risk of breast cancer among Norwegian women with visual impairment. Br J Cancer 84:397–399. doi:10.1054/bjoc.2000.1617
Verkasalo PK, Pukkala E, Stevens RG, Ojamo M, Rudanko SL (1999) Inverse association between breast cancer incidence and degree of visual impairment in Finland. Br J Cancer 80:1459–1460. doi:10.1038/sj.bjc.6690544
Schernhammer ES, Hankinson SE (2005) Urinary melatonin levels and breast cancer risk. J Natl Cancer Inst 97:1084–1087
Kheifets LI, Matkin CC (1999) Industrialization, electromagnetic fields, and breast cancer risk. Environ Health Perspect 107(Suppl 1):145–154. doi:10.2307/3434479
Brainard GC, Kavet R, Kheifets LI (1999) The relationship between electromagnetic field and light exposures to melatonin and breast cancer risk: a review of the relevant literature. J Pineal Res 26:65–100. doi:10.1111/j.1600-079X.1999.tb00568.x
Cos S, Mediavilla D, Martinez-Campa C, Gonzalez A, Alonso-Gonzalez C, Sanchez-Barcelo EJ (2006) Exposure to light-at-night increases the growth of DMBA- induced mammary adenocarcinomas in rats. Cancer Lett 235:266–271. doi:10.1016/j.canlet.2005.04.025
Blask DE, Dauchy RT, Sauer LA, Krause JA, Brainard GC (2003) Growth and fatty acid metabolism of human breast cancer (MCF-7) xenografts in nude rats: impact of constant light-induced nocturnal melatonin suppression. Breast Cancer Res Treat 79:313–320. doi:10.1023/A:1024030518065
Wilson ST, Blask DE, Lemus-Wilson AM (1992) Melatonin augments the sensitivity of MCF-7 human breast cancer cells to tamoxifen in vitro. J Clin Endocrinol Metab 75:669–670. doi:10.1210/jc.75.2.669
Blask DE, Wilson ST, Zalatan F (1997) Physiological melatonin inhibition of human breast cancer cell growth in vitro: evidence for a glutathione-mediated pathway. Cancer Res 57:1909–1914
Mediavilla MD, Güezmez A, Ramos S, Kothari L, Garijo F, Sánchez Barceló EJ (1997) Effects of melatonin on mammary gland lesions in transgenic mice overexpressing N-ras proto-oncogene. J Pineal Res 22:86–94. doi:10.1111/j.1600-079X.1997.tb00308.x
Cos S, Fernandez R, Güezmes A, Sanchez-Barcelo EJ (1998) Influence of melatonin on invasive and metastatic properties of MCF-7 human breast cancer cells. Cancer Res 58:4383–4390
Mangelsdorf DJ, Thummel C, Beato M et al (1995) The nuclear receptor superfamily: the second decade. Cell 83:835–839. doi:10.1016/0092-8674(95)90199-X
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14:121–141
Lu NZ, Wardell SE, Burnstein KL et al (2006) International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol Rev 58:782–797. doi:10.1124/pr.58.4.9
Benoit G, Cooney A, Giguere V et al (2006) International Union of Pharmacology LXVI. Orphan nuclear receptors. Pharmacol Rev 58:798–836. doi:10.1124/pr.58.4.10
Lonard DM, Lanz RB, O’Malley BW (2007) Nuclear receptor coregulators and human disease. Endocr Rev 28:575–587. doi:10.1210/er.2007-0012
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080. doi:10.1126/science.1063127
Chen J, Kinyamu HK, Archer TK (2006) Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol Endocrinol 20:1–13. doi:10.1210/me.2005-0192
Lonard DM, O’Malley BW (2007) Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell 27:691–700. doi:10.1016/j.molcel.2007.08.012
Becker-Andre M, Wiesenberg I, Schaeren-Wiemers N et al (1994) Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J Biol Chem 269:28531–28534
Park HT, Baek SY, Kim BS, Kim JB, Kim JJ (1996) Developmental expression of ‘RZR beta, a putative nuclear-melatonin receptor’ mRNA in the suprachiasmatic nucleus of the rat. Neurosci Lett 217:17–20. doi:10.1016/S0304-3940(96)13060-3
Park HT, Kim YJ, Yoon S, Kim JB, Kim JJ (1997) Distributional characteristics of the mRNA for retinoid Z receptor beta (RZR beta), a putative nuclear melatonin receptor, in the rat brain and spinal cord. Brain Res 747:332–337. doi:10.1016/S0006-8993(96)01320-0
Agez L, Laurent V, Pevet P, Masson-Pevet M, Gauer F (2007) Melatonin affects nuclear orphan receptors mRNA in the rat suprachiasmatic nuclei. Neuroscience 144:522–530. doi:10.1016/j.neuroscience.2006.09.030
Naji L, Carrillo-Vico A, Guerrero JM, Calvo JR (2004) Expression of membrane and nuclear melatonin receptors in mouse peripheral organs. Life Sci 74:2227–2236. doi:10.1016/j.lfs.2003.08.046
Baler R, Coon S, Klein DC (1996) Orphan nuclear receptor RZRbeta: cyclic AMP regulates expression in the pineal gland. Biochem Biophys Res Commun 220:975–978. doi:10.1006/bbrc.1996.0517
Garcia-Maurino S, Gonzalez-Haba MG, Calvo JR et al (1997) Melatonin enhances IL-2, IL-6, and IFN-gamma production by human circulating CD4+ cells: a possible nuclear receptor-mediated mechanism involving T helper type 1 lymphocytes and monocytes. J Immunol 159:574–581
Bordji K, Grillasca JP, Gouze JN et al (2000) Evidence for the presence of peroxisome proliferator-activated receptor (PPAR) alpha and gamma and retinoid Z receptor in cartilage. PPARgamma activation modulates the effects of interleukin-1beta on rat chondrocytes. J Biol Chem 275:12243–12250. doi:10.1074/jbc.275.16.12243
Smirnov AN (2001) Nuclear melatonin receptors. Biochemistry (Mosc) 66(1):19–26. doi:10.1023/A:1002821427018
Nolte RT, Wisely GB, Westin S et al (1998) Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395:137–143. doi:10.1038/25931
Horlein AJ, Naar AM, Heinzel T et al (1995) Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377:397–404. doi:10.1038/377397a0
Chawla A, Lee CH, Barak Y et al (2003) PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci USA 100:1268–1273. doi:10.1073/pnas.0337331100
Xu L, Glass CK, Rosenfeld MG (1999) Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev 9(2):140–147. doi:10.1016/S0959-437X(99)80021-5
Mukherjee R, Davies PJ, Crombie DL et al (1997) Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 386:407–410. doi:10.1038/386407a0
Mukherjee R, Jow L, Croston GE, Paterniti JR (1997) Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem 272:8071–8076. doi:10.1074/jbc.272.4.2346
McKenna NJ, Lanz RB, O’Malley BW (1999) Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344. doi:10.1210/er.20.3.321
McKenna NJ, O’Malley BW (2002) Minireview: nuclear receptor coactivators—an update. Endocrinology 143:2461–2465. doi:10.1210/en.143.7.2461
McKenna NJ, O’Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474. doi:10.1016/S0092-8674(02)00641-4
Schneider SM, Offterdinger M, Huber H, Grunt TW (1999) Involvement of nuclear steroid/thyroid/retinoid receptors and of protein kinases in the regulation of growth and of c-erbB and retinoic acid receptor expression in MCF-7 breast cancer cells. Breast Cancer Res Treat 58:171–181. doi:10.1023/A:1006377006816
Kiefer TL, Lai L, Yuan L, Dong C, Burow ME, Hill SM (2005) Differential regulation of estrogen receptor alpha, glucocorticoid receptor and retinoic acid receptor alpha transcriptional activity by melatonin is mediated via different G proteins. J Pineal Res 38:231–239. doi:10.1111/j.1600-079X.2004.00198.x
Jarrar MH, Baranova A (2007) PPARgamma activation by thiazolidinediones (TZDs) may modulate breast carcinoma outcome: the importance of interplay with TGFbeta signalling. J Cell Mol Med 11:71–87. doi:10.1111/j.1582-4934.2007.00003.x
Crowe DL, Chandraratna RA (2004) A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res 6:R546–R555. doi:10.1186/bcr913
Eck-Enriquez K, Kiefer TL, Spriggs LL, Hill SM (2000) Pathways through which a regimen of melatonin and retinoic acid induces apoptosis in MCF-7 human breast cancer cells. Breast Cancer Res Treat 61:229–239. doi:10.1023/A:1006442017658
Sharma R, Ottenhof T, Rzeczkowska PA, Niles LP (2008) Epigenetic targets for melatonin: induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J Pineal Res (Epub ahead of print). doi:10.1111/j.1600-079X.2008.00587.x
Blask DE, Hill SM (1968) Effects of melatonin on cancer: studies on MCF-7 human breast cancer cells in culture. J Neural Transm Suppl 21:433–449
Leon-Blanco MM, Guerrero JM, Reiter RJ, Calvo JR, Pozo D (2003) Melatonin inhibits telomerase activity in the MCF-7 tumor cell line both in vivo and in vitro. J Pineal Res 35:204–211. doi:10.1034/j.1600-079X.2003.00077.x
Martinez-Campa C, Alonso-Gonzalez C, Mediavilla MD et al (2006) Melatonin inhibits both ER alpha activation and breast cancer cell proliferation induced by a metalloestrogen, cadmium. J Pineal Res 40:291–296. doi:10.1111/j.1600-079X.2006.00315.x
Martinez-Campa C, Alonso-Gonzalez C, Mediavilla MD, Cos S, Gonzalez A, Sanchez-Barcelo EJ (2008) Melatonin down-regulates hTERT expression induced by either natural estrogens (17β-estradiol) or metalloestrogens (cadmium) in MCF-7 human breast cancer cells. Cancer Lett (Epub ahead of print). doi:10.1016/j.canlet.2008.04.001
Molis TM, Spriggs LL, Jupiter Y, Hill SM (1995) Melatonin modulation of estrogen-regulated proteins, growth factors, and proto-oncogenes in human breast cancer. J Pineal Res 18:93–103. doi:10.1111/j.1600-079X.1995.tb00146.x
Lawson NO, Wee BE, Blask DE, Castles CG, Spriggs LL, Hill SM (1992) Melatonin decreases estrogen receptor expression in the medial preoptic area of inbred (LSH/SsLak) golden hamsters. Biol Reprod 47:1082–1090. doi:10.1095/biolreprod47.6.1082
Molis TM, Spriggs LL, Hill SM (1994) Modulation of estrogen receptor mRNA expression by melatonin in MCF-7 human breast cancer cells. Mol Endocrinol 8:1681–1690. doi:10.1210/me.8.12.1681
Rato AG, Pedrero JG, Martinez MA, del Rio B, Lazo PS, Ramos S (1999) Melatonin blocks the activation of estrogen receptor for DNA binding. FASEB J 13:857–868
Baldwin WS, Barrett JC (1998) Melatonin: receptor-mediated events that may affect breast and other steroid hormone-dependent cancers. Mol Carcinog 21:149–155. doi:10.1002/(SICI)1098-2744(199803)21:3≤149::AID-MC1≥3.0.CO;2-H
Yuan L, Collins AR, Dai J, Dubocovich ML, Hill SM (2002) MT(1) melatonin receptor verexpression enhances the growth suppressive effect of melatonin in human breast cancer cells. Mol Cell Endocrinol 192:147–156. doi:10.1016/S0303-7207(02)00029-1
Jones MP, Melan MA, Witt-Enderby PA (2000) Melatonin decreases cell proliferation and transformation in a melatonin receptor-dependent manner. Cancer Lett 151:133–143. doi:10.1016/S0304-3835(99)00394-8
Ram PT, Dai J, Yuan L et al (2002) Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett 179:141–150. doi:10.1016/S0304-3835(01)00873-4
Collins A, Yuan L, Kiefer TL, Cheng Q, Lai L, Hill SM (2003) Overexpression of the MT1 melatonin receptor in MCF-7 human breast cancer cells inhibits mammary tumor formation in nude mice. Cancer Lett 189:49–57. doi:10.1016/S0304-3835(02)00502-5
Dillon DC, Easley SE, Asch BB et al (2002) Differential expression of high-affinity melatonin receptors (MT1) in normal and malignant human breast tissue. Am J Clin Pathol 118:451–458. doi:10.1309/1T4V-CT1G-UBJP-3EHP
Garcia Pedrero JM, Del Rio B, Martínez-Campa C, Muramatsu M, Lazo PS, Ramos S (2002) Calmodulin is a selective modulator of estrogen receptors. Mol Endocrinol 16:947–960. doi:10.1210/me.16.5.947
Dai J, Inscho EW, Yuan L, Hill SM (2002) Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J Pineal Res 32(2):112–119. doi:10.1034/j.1600-079x.2002.1844.x
del Río B, García Pedrero JM, Martínez-Campa C, Zuazua P, Lazo PS, Ramos S (2004) Melatonin, an endogenous-specific inhibitor of estrogen receptor alpha via calmodulin. J Biol Chem 279:38294–38302. doi:10.1074/jbc.M403140200
Macaluso M, Cinti C, Russo G, Russo A, Giordano A (2003) pRb2/p130–E2F4/5- HDAC1-SUV39H1–p300 and pRb2/p130–E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene 22:3511–3517. doi:10.1038/sj.onc.1206578
Martin MB, Angeloni SV, Garcia-Morales P (2004) Regulation of estrogen receptor-alpha expression in MCF-7 cells by taxol. J Endocrinol 180:487–496. doi:10.1677/joe.0.1800487
Burch JB, Walling M, Rush A et al (2007) Melatonin and estrogen in breast cyst fluids. Breast Cancer Res Treat 103:331–341. doi:10.1007/s10549-006-9372-z
Yang X, Ferguson AT, Nass SJ et al (2000) Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res 60:6890–6894
Bulun SE, Lin Z, Imir G et al (2005) Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev 57:359–383. doi:10.1124/pr.57.3.6
van Landeghem AA, Poortman J, Nabuurs M, Thijssen JH (1985) Endogenous concentration and subcellular distribution of estrogens in normal and malignant human breast tissue. Cancer Res 45:2900–2906
Bulun SE, Zeitoun K, Sasano H, Simpson ER (1999) Aromatase in aging women. Semin Reprod Endocrinol 17:349–358. doi:10.1055/s-1999-7720
Tekmal RR, Kirma N, Gill K, Fowler K (1999) Aromatase overexpression and breast hyperplasia, an in vivo model—continued overexpression of aromatase is sufficient to maintain hyperplasia without circulating estrogens, and aromatase inhibitors abrogate these preneoplastic changes in mammary glands. Endocr Relat Cancer 6:307–314. doi:10.1677/erc.0.0060307
Suzuki T, Miki Y, Nakamura Y et al (2005) Sex steroid-producing enzymes in human breast cancer. Endocr Relat Cancer 12:701–720
Pasqualini JR, Chetrite GS (2005) Recent insight on the control of enzymes involved in estrogen formation and transformation in human breast cancer. J Steroid Biochem Mol Biol 93:221–236. doi:10.1016/j.jsbmb.2005.02.007
Cos S, Martinez-Campa C, Mediavilla MD, Sanchez-Barcelo EJ (2005) Melatonin modulates aromatase activity in MCF-7 human breast cancer cells. J Pineal Res 38:136–142. doi:10.1111/j.1600-079X.2004.00186.x
González A, Martínez-Campa C, Mediavilla MD, Alonso-González C, Sánchez-Barceló EJ, Cos S (2007) Inhibitory effects of pharmacological doses of melatonin on aromatase activity and expression in rat glioma cells. Br J Cancer 97:755–760. doi:10.1038/sj.bjc.6603935
Martinez-Campa C, Gonzalez A, Mediavilla MD, Alonso-Gonzalez C, Sanchez-Barcelo EJ, Cos S (2005) Melatonin enhances the inhibitory effect of aminoglutethimide on aromatase activity in MCF-7 human breast cancer cells. Breast Cancer Res Treat 94:249–254. doi:10.1007/s10549-005-9006-x
Cos S, Gonzalez A, Güezmes A et al (2006) Melatonin inhibits the growth of DMBA-induced mammary tumors by decreasing the local biosynthesis of estrogens through the modulation of aromatase activity. Int J Cancer 118:274–278. doi:10.1002/ijc.21401
Gonzalez A, Cos S, Martinez-Campa C et al (2008) Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J Pineal Res (Epub ahead of print). doi:10.1111/j.1600-079X.2008.00559.x
Izawa M, Harada T, Taniguchi F, Ohama Y, Takenaka Y, Terakawa N (2008) An epigenetic disorder may cause aberrant expression of aromatase gene in endometriotic stromal cells. Fertil Steril 89:1390–1396
Cai Z, Kwintkiewicz J, Young ME, Stocco C (2007) Prostaglandin E2 increases cyp19 expression in rat granulosa cells: implication of GATA-4. Mol Cell Endocrinol 263:181–189. doi:10.1016/j.mce.2006.09.012
Irahara N, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S (2006) Quantitative analysis of aromatase mRNA expression derived from various promoters (I.4, I.3, PII and I.7) and its association with expression of TNF-alpha, IL-6 and COX-2 mRNAs in human breast cancer. Int J Cancer 118:1915–1921. doi:10.1002/ijc.21562
Subbaramaiah K, Howe LR, Port ER et al (2006) HER-2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase-2-dependent mechanism. Cancer Res 66:5504–5511. doi:10.1158/0008-5472.CAN-05-4076
Subbaramaiah K, Hudis C, Chang SH, Hla T, Dannenberg AJ (2008) EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells. Evidence of a BRCA1 and p300 exchange. J Biol Chem 283:3433–3444. doi:10.1074/jbc.M705409200
Esposito E, Iacono A, Muia C et al (2008) Signal transduction pathways involved in protective effects of melatonin in C6 glioma cells. J Pineal Res 44:78–87
Dong WG, Mei Q, Yu JP, Xu JM, Xiang L, Xu Y (2003) Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J Gastroenterol 9:1307–1311
Mrnka L, Hock M, Rybova M, Pacha J (2008) Melatonin inhibits prostaglandin E2- and sodium nitroprusside-induced ion secretion in rat distal colon. Eur J Pharmacol 581:164–170. doi:10.1016/j.ejphar.2007.11.031
Deng WG, Tang ST, Tseng HP, Wu KK (2006) Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108:518–5124. doi:10.1182/blood-2005-09-3691
Fan W, Yanase T, Morinaga H et al (2005) Activation of peroxisome proliferator-activated receptor-gamma and retinoid X receptor inhibits aromatase transcription via nuclear factor-kappaB. Endocrinology 146:85–92. doi:10.1210/en.2004-1046
Moon IK, Jarstfer MB (2007) The human telomere and its relationship to human disease, therapy, and tissue engineering. Front Biosci 12:4595–4620. doi:10.2741/2412
Shay JW, Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33:787–791. doi:10.1016/S0959-8049(97)00062-2
Shpitz B, Zimlichman S, Zemer R (1999) Telomerase activity in ductal carcinoma in situ of the breast. Breast Cancer Res Treat 58:65–69. doi:10.1023/A:1006394209922
Kirkpatrick KL, Clark G, Ghilchick M, Newbold RF, Mokbel K (2003) hTERT mRNA expression correlates with telomerase activity in human breast cancer. Eur J Surg Oncol 29:321–326. doi:10.1053/ejso.2002.1374
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8:299–309. doi:10.1038/nrg2047
Lai SR, Phipps SM, Liu L, Andrews LG, Tollefsbol TO (2005) Epigenetic control of telomerase and modes of telomere maintenance in aging and abnormal systems. Front Biosci 10:1779–1796. doi:10.2741/1661
Gonzalo S, Jaco I, Fraga MF et al (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8:416–424. doi:10.1038/ncb1386
Guilleret I, Benhattar J (2003) Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res 289:326–334. doi:10.1016/S0014-4827(03)00281-7
Renaud S, Loukinov D, Abdullaev Z et al (2007) Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res 35:1245–1256. doi:10.1093/nar/gkl1125
Korkmaz A, Reiter RJ (2008) Epigenetic regulation: a new research area for melatonin? J Pineal Res 44:41–44
Lee WJ, Shim JY, Zhu BT (2005) Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol 68:1018–1030. doi:10.1124/mol.104.008367
Berletch JB, Liu C, Love WK, Andrews LG, Katiyar SK, Tollefsbol TO (2008) Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J Cell Biochem 103:509–519. doi:10.1002/jcb.21417
Leon-Blanco MM, Guerrero JM, Reiter RJ, Pozo D (2004) RNA expression of human telomerase subunits TR and TERT is differentially affected by melatonin receptor agonists in the MCF–7 tumor cell line. Cancer Lett 216:73–80. doi:10.1016/j.canlet.2004.05.003
Farabegoli F, Barbi C, Lambertini E, Piva R (2007) (−)-Epigallocatechin-3-gallate downregulates estrogen receptor alpha function in MCF-7 breast carcinoma cells. Cancer Detect Prev 31:499–504. doi:10.1016/j.cdp. 2007.10.018
Cos S, Gonzalez A, Martinez-Campa C, Mediavilla MD, Alonso-Gonzalez C, Sanchez-Barcelo EJ (2006) Estrogen-signaling pathway: a link between breast cancer and melatonin oncostatic actions. Cancer Detect Prev 30:118–128. doi:10.1016/j.cdp. 2006.03.002
Mukhopadhyay NK, Gordon GJ, Maulik G et al (2005) Histone deacetylation is directly involved in desilencing the expression of the catalytic subunit of telomerase in normal lung fibroblast. J Cell Mol Med 9:662–669. doi:10.1111/j.1582-4934.2005.tb00496.x
Suenaga M, Soda H, Oka M et al (2002) Histone deacetylase inhibitors suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells. Int J Cancer 97:621–625. doi:10.1002/ijc.10082
Hou M, Wang X, Popov N (2002) The histone deacetylase inhibitor trichostatin A derepresses the telomerase reverse transcriptase (hTERT) gene in human cells. Exp Cell Res 274:25–34. doi:10.1006/excr.2001.5462
Fang MZ, Wang Y, Ai N et al (2003) Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 63:7563–7570
Garcia V, Garcia JM, Pena C (2008) Free circulating mRNA in plasma from breast cancer patients and clinical outcome. Cancer Lett 263:312–320. doi:10.1016/j.canlet.2008.01.008
Fu M, Wang C, Li Z, Sakamaki T, Pestell RG (2004) Minireview: cyclin D1: normal and abnormal functions. Endocrinology 145:5439–5447. doi:10.1210/en.2004-0959
Fu M, Rao M, Bouras T et al (2005) Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem 280:16934–16941. doi:10.1074/jbc.M500403200
Cicatiello L, Addeo R, Sasso A (2004) Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol 24:7260–7274. doi:10.1128/MCB.24.16.7260-7274.2004
De Los Santos M, Martinez-Iglesias O, Aranda A (2007) Anti-estrogenic actions of histone deacetylase inhibitors in MCF-7 breast cancer cells. Endocr Relat Cancer 14:1021–1028. doi:10.1677/ERC-07-0144
Cini G, Neri B, Pacini A et al (2005) Antiproliferative activity of melatonin by transcriptional inhibition of cyclin D1 expression: a molecular basis for melatonin-induced oncostatic effects. J Pineal Res 39:12–20. doi:10.1111/j.1600-079X.2004.00206.x
Siu SW, Lau KW, Tam PC, Shiu SY (2002) Melatonin and prostate cancer cell proliferation: interplay with castration, epidermal growth factor, and androgen sensitivity. Prostate 52:106–122. doi:10.1002/pros.10098
Cos S, Recio J, Sánchez-Barceló EJ (1996) Modulation of the length of the cell cycle time of MCF-7 human breast cancer cells by melatonin. Life Sci 58:811–816. doi:10.1016/0024-3205(95)02359-3
Reiter RJ, Tan DX, Korkmaz A et al (2007) Light at night, chronodisruption, melatonin suppression, and cancer risk: a review. Crit Rev Oncog 13:303–328
Blask DE, Brainard GC, Dauchy RT et al (2005) Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res 65:11174–11184. doi:10.1158/0008-5472.CAN-05-1945
Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES (2005) Night work and breast cancer risk: a systematic review and meta-analysis. Eur J Cancer 41:2023–2032. doi:10.1016/j.ejca.2005.05.010
Davis S, Mirick DK (2006) Circadian disruption, shift work and the risk of cancer: a summary of the evidence and studies in Seattle. Cancer Causes Control 17:539–545. doi:10.1007/s10552-005-9010-9
Hansen J (2006) Risk of breast cancer after night- and shift work: current evidence and ongoing studies in Denmark. Cancer Causes Control 17:531–537. doi:10.1007/s10552-005-9006-5
Cos S, Alvarez A, Mediavilla MD, Bartsch C, Bartsch H, Sanchez-Barcelo EJ (2000) Influence of serum from healthy or breast tumor-bearing women on the growth of MCF-7 human breast cancer cells. Int J Mol Med 5:651–656
Hirayama J, Sassone-Corsi P (2005) Structural and functional features of transcription factors controlling the circadian clock. Curr Opin Genet Dev 15:548–556. doi:10.1016/j.gde.2005.07.003
Yoo SH, Yamazaki S, Lowrey PL et al (2004) PERIOD2: luciferase real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 101:5339–53346. doi:10.1073/pnas.0308709101
Kume K, Zylka MJ, Sriram S et al (1999) mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98:193–205. doi:10.1016/S0092-8674(00)81014-4
Doi M, Hirayama J, Sassone-Corsi P (2006) Circadian regulator CLOCK is a histone acetyltransferase. Cell 125:497–508. doi:10.1016/j.cell.2006.03.033
Hirayama J, Sahar S, Grimaldi B et al (2007) CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450:1086–1090. doi:10.1038/nature06394
Hirayama J, Cho S, Sassone-Corsi P (2007) Circadian control by the reduction/oxidation pathway: catalase represses light-dependent clock gene expression in the zebrafish. Proc Natl Acad Sci USA 104:15747–15752. doi:10.1073/pnas.0705614104
Fu L, Pelicano H, Liu J, Huang P, Lee C (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111:41–50. doi:10.1016/S0092-8674(02)00961-3
Xiang S, Coffelt SB, Mao L, Yuan L, Cheng Q, Hill SM (2008) Period-2: a tumor suppressor gene in breast cancer. J Circadian Rhythms 6:4. doi:10.1186/1740-3391-6-4
Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP (2007) The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene 26:7916–7920. doi:10.1038/sj.onc.1210585
Boivin DB, James FO, Wu A, Cho-Park PF, Xiong H, Sun ZS (2003) Circadian clock genes oscillate in human peripheral blood mononuclear cells. Blood 102:4143–4145. doi:10.1182/blood-2003-03-0779
Cajochen C, Jud C, Munch M, Kobialka S, Wirz-Justice A, Albrecht U (2006) Evening exposure to blue light stimulates the expression of the clock gene PER2 in humans. Eur J Neurosci 23:1082–1086. doi:10.1111/j.1460-9568.2006.04613.x
Provencio I, Rollag MD, Castrucci AM (2002) Photoreceptive net in the mammalian retina. This mesh of cells may explain how some blind mice can still tell day from night. Nature 415(6871):493. doi:10.1038/415493a
Miyamoto Y, Sancar A (1998) Vitamin B2-based blue-light photoreceptors in the retinohypothalamic tract as the photoactive pigments for setting the circadian clock in mammals. Proc Natl Acad Sci USA 95:6097–6102. doi:10.1073/pnas.95.11.6097
Selby CP, Thompson C, Schmitz TM, Van Gelder RN, Sancar A (2000) Functional redundancy of cryptochromes and classical photoreceptors for nonvisual ocular photoreception in mice. Proc Natl Acad Sci USA 97:14697–14702. doi:10.1073/pnas.260498597
Berson DM, Dunn FA, Takao M (2002) Phototransduction by retinal ganglion cells that set the circadian clock. Science 295:1070–1073. doi:10.1126/science.1067262
Brainard GC, Hanifin JP, Greeson JM et al (2001) Action spectrum for melatonin regulation in humans: evidence for a novel circadian photoreceptor. J Neurosci 21:6405–6412
Wright HR, Lack LC (2001) Effect of light wavelength on suppression and phase delay of the melatonin rhythm. Chronobiol Int 18:801–808. doi:10.1081/CBI-100107515
Lockley SW, Brainard GC, Czeisler CA (2003) High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab 88:4502–4505. doi:10.1210/jc.2003-030570
Cajochen C, Munch M, Kobialka S et al (2001) High sensitivity of human melatonin, alertness, thermoregulation, and heart rate to short wavelength light. J Clin Endocrinol Metab 90:1311–1. doi:10.1210/jc.2004-0957
Reppert SM, Weaver DR (2001) Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63:647–676. doi:10.1146/annurev.physiol.63.1.647
Salti R, Tarquini R, Stagi S et al (2006) Age-dependent association of exposure to television screen with children’s urinary melatonin excretion? Neuroendocrinol Lett 27:73–80
Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG (2005) Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis 26:1241–1246. doi:10.1093/carcin/bgi075
Acknowledgements
This work is supported in part by a grant from the Spanish Ministry of Education and Science (SAF2007-62762).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Korkmaz, A., Sanchez-Barcelo, E.J., Tan, DX. et al. Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res Treat 115, 13–27 (2009). https://doi.org/10.1007/s10549-008-0103-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-008-0103-5