
Abstract
The long-recognized fact that oxidative stress within mitochondria is a hallmark of mitochondrial dysfunction has stimulated the development of mitochondria-targeted antioxidant therapies. Melatonin should be included among the pharmacological agents able to modulate mitochondrial functions in cancer, given that a number of relevant melatonin-dependent effects are triggered by targeting mitochondrial functions. Indeed, melatonin may modulate the mitochondrial respiratory chain, thus antagonizing the cancer highly glycolytic bioenergetic pathway of cancer cells. Modulation of the mitochondrial respiratory chain, together with Ca2+ release and mitochondrial apoptotic effectors, may enhance the spontaneous or drug-induced apoptotic processes. Given that melatonin may efficiently counteract the Warburg effect while stimulating mitochondrial differentiation and mitochondrial-based apoptosis, it is argued that the pineal neurohormone could represent a promising new perspective in cancer treatment strategy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Mitochondrion: a metabolic-regulatory factory
Mitochondrial studies have gained momentum during the last decade. It is likely that this growing attention for mitochondrial biology could chiefly be attributed to the fact that mitochondria represent actually the physical ‘hub’, where many signaling pathways converge in modulating a number of critical biological functions, including cell death control, metabolism, and even differentiation [1].
Mitochondria are bio-energetic and biosynthetic organelles that take up substrates from the cytoplasm and use them to drive fatty acid oxidation (FAO), glucose oxidative metabolism along the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC). As a result, mitochondria produce ATP and synthesize amino acids, lipids, nucleotides, and iron sulfur clusters, as well as NADPH [2]. In addition, reactive oxygen species (ROS) are generated in mitochondria as a byproduct of the ETC. Under normal physiological conditions, ROS can activate signal transduction pathways, including MAPK/ERK kinases and Hypoxia-Inducible Factor (HIF). Above a threshold value, increased ROS production triggers apoptosis [3].
Mitochondria participate in regulating apoptotic processes also through modulation of several pro- and antiapoptotic proteins—including Bcl-2, Bax, and Bak proteins—via the release of cytochrome c from the mitochondrial intermembrane space. In turn, cytochrome c release activates caspase-dependent processes in the cytosol, leading then to cell death [4].
Downstream of the above-mentioned alterations, mitochondria selectively regulate Ca2+ release, thus contributing to the modulation of a wide array of cellular functions [5]. Those findings evidence a tight regulated mitochondria-cell cross talk, involving also epigenetic mechanisms through which mitochondria can actually modulate gene expression pattern [6] in a cell and tissue-specific-dependent manner, thus providing a reliable explanation for tissue-specific alterations in mitochondrial diseases [7]. Conversely, an imbalance in mitochondrial activity, especially if the oxidative/reductive mitochondrial metabolism is involved, can severely impair mitochondria function and contribute to the onset of different diseases, including cancer [8].
Melatonin and mitochondria
Melatonin seems to have a special relationship with mitochondria. Recent publications in the field principally rely on three lines of evidence—the relationship among oxidative stress in mitochondria and the scavenging activity of melatonin; the presence of high amount of melatonin in these organelles; and the existence of a circadian/seasonal variations in mitochondria structure and functions—suggesting a physiological role of the pineal hormone on mitochondrial function [9, 10].
Mitochondria likely synthesize melatonin at high rates, as melatonin levels within mitochondria are 100-fold higher than that in the plasma [11]. Enzymes for melatonin synthesis have been found in mitochondria obtained from pinealocytes and oocytes [12, 13]. It is worth noting that mitochondria participate also in melatonin catabolism by producing N1-acetyl-N2-formyl-5-methoxykynuramin (AFMK). Several lines of evidence suggest that melatonin displays a protective function in these cells, namely, by counteracting oxidative-reductive stresses and improving several functions, including synthetic pathways and maturation processes in the oocyte [14].
Mitochondria also display circadian rhythms meet the fluctuations in energy supply and cellular requirements. Mitochondria are indeed involved in continuous remodeling processes—including mitochondrial biogenesis, fission, and fusion—recognized as being part of the so-called mitochondrial dynamics [15]. Circadian oscillations of both structure and functions in mitochondria are supported by the clock proteins Period1 and Period2 (Per1 and Per2), both of which are tightly regulated by melatonin. Mitochondrial dynamics is significantly impaired in mice lacking Per1/2 [16], whereas melatonin increases Per1/2 expression both in suprachiasmatic nucleus as well as in peripheral cells [17, 18].
Mitochondrial fission/fusion, a process chiefly responsible for the generation of ‘young’ mitochondria while eliminating the old/damaged ones, displays a strong association with melatonin circadian rhythms in both pinealocytes and non-neural cells [19]. Melatonin principally down-regulates the translocation to the outer mitochondrial membrane of several proteins linked to the fission process (such as the mitochondrial fission 1 protein and the dynamin-related protein 1), thus reducing fission [20]. Conversely, melatonin enhances fusion processes by modulating mitofusins and optic atrophy protein 1. Even if is still unclear if melatonin up- or down-regulated those proteins [21, 22], there is no doubt that melatonin plays a relevant role in that process. By analogy with autophagy, a similar process has been described in mitochondria, i.e., mitophagy. Accordingly, mitophagy is mandatory for ensuring mitochondrial turnover and it seems that melatonin positively regulates it [23], through the modulation of an AMPK-dependent process leading to the inhibition of the mTOR pathway [24].
It is worth noting that melatonin is in higher concentrations in mitochondria, where free radicals are maximally generated. This leads to the speculation that melatonin plays a special role in antagonizing the oxidative stress occurring within mitochondria, thus protecting them from inappropriate ROS concentrations [25]. Furthermore, besides its ability in scavenging directly ROS, melatonin increases the SOD and catalase activities and induces the expression and activity of glutathione peroxidase (GPx) and glutathione reductase (GRd). In addition, melatonin stimulates the rate limiting enzyme in glutathione synthesis—γ-glutamylcysteine synthase—thus regulating the redox cycle of glutathione [26,27,28]. Similarly, melatonin reduces the expression and activity of the inducible nitric oxide synthase (iNOS) and, thereby, decreases the levels of NO and peroxynitrite [29].
The antioxidant activity of melatonin in the mitochondria has been specifically demonstrated in vivo, given that melatonin-treated rats showed a significant increase in the activities of C-I and C-IV components of the mitochondrial respiratory chain measured in isolated mitochondria [30]. Moreover, melatonin specifically counteracts the oxidative damage induced by ruthenium on mitochondria respiratory chain components. Those effects are mirrored by results obtained in vitro by treating with melatonin mitochondria exposed to oxidative agents. Oxidative stress (induced by incubation of mitochondria with t-butyl hydroperoxide) depletes the mitochondrial GSH pool and inhibits both GPx and GRd activities [31]. Melatonin antagonizes these effects by restoring basal levels of GSH and the normal activities of both GPx and GRd. It is noteworthy that other well-known antioxidant compounds (like N-acetyl cysteine and vitamins E and C) were unable to exert any significant effect on t-butyl hydroperoxide-induced oxidative stress in mitochondria, while melatonin induces also a significant increase in C-I and C-IV components of the mitochondrial respiratory chain [32].
The reported effects of melatonin on the mitochondria respiratory chain complexes are of utmost physiological significance given that melatonin overall improves the respiratory chain efficiency during oxidative phosphorylation, as reflected by elevated ATP synthesis, in both normal mitochondria or in mitochondria exposed to oxidative-damaging agents such as cyanide [33, 34]. In these experiments, mitochondria actively concentrate melatonin, which maintained the respiratory control ratio and the efficiency of oxidative phosphorylation and ATP synthesis while increasing the activity of the respiratory complexes. In the meantime, melatonin decreased oxygen consumption in the presence of ADP as well as the membrane potential. Consequently, melatonin inhibits the production of O2 − and H2O2. Overall, those data highlight the pivotal role of melatonin in safeguarding mitochondrial bioenergetics homeostasis (reviewed in [35]).
The widespread activities of melatonin on mitochondria have suggested that the neurohormone could also target the mitochondrial DNA (mtDNA). Indeed, melatonin prevents oxidative degradation of mtDNA in several tissues [36], while a direct effect of melatonin on mitochondrial genome expression in adipocytes of the Siberian hamster has been reported [37]. Furthermore, melatonin increases the expression of the mRNAs for subunits I, II, and III of complex IV in both in vivo and in vitro [38].
Mitochondria and metabolism in cancer
Deregulated mitochondria function contributes significantly in hindering cell metabolism, chiefly by shifting glucose degradation from an aerobic towards an anaerobic glycolytic pathway. In turn, dysregulation of mitochondrial function characterized by Krebs cycle defects has been associated with over-production of ROS, which may participate in oncogenic signaling and tumor progression by irreversible modification of DNA and oxidation of proteins [39, 40].
A first hint that mitochondria may play an important role in cancer cell biology was reported in the 1920s when it was shown that cancer cells constitutively up-regulate glycolytic degradation of glucose, even in the presence of abundant oxygen. The biochemist Otto Warburg suggested that cancer causation might be related to an altered metabolism, i.e., a shift in energy production from oxidative phosphorylation to glycolysis, even if in presence of normal oxygen levels [41]. However, the remarkable progress in molecular biology achieved thereafter has left no room for approaches that are anything but gene-based. Hence, the “metabolic theory” was viewed as a generic “epiphenomenon” and it was rapidly discarded. Yet, due to the meaningful insights provided by metabolomics and the recent studies on mitochondrial function in cancer, Warburg’s theory has unexpectedly enjoyed a resurrection in the last decade [42]. Accordingly, the metabolic phenotype acquired by transformed cancer cells cannot be thought as a ‘simple’ byproduct of cancer development, and is now considered a relevant property that can be efficiently exploited for widespread clinical applications [43].
Given that anaerobic conversion of glucose (glycolysis) to lactic acid is substantially less efficient than complete oxidation to CO2 and H2O, tumor cells need to sustain elevated ATP production by increasing glucose flux through an enhanced conversion of glucose to glucose-6-phosphate. This characteristic provides the biochemical rationale for tumor imaging with 2-fluoro-2-deoxy-d-glucose-positron emission tomography (FDG-PET), a technique now widely used in radiological studies. PET investigation revealed a significant increased uptake of glucose in both primary and metastatic cancers, showing a direct correlation between tumor aggressiveness and the rate of glucose utilization [44]. These results reinforced the relevance of metabolic studies in cancer as they have moved the “glycolytic phenotype” from a laboratory oddity to mainstream oncology.
Alterations in cancer metabolism, however, are not only relevant for diagnostic purposes, but also in drug discovery. Macromolecule synthesis from glucose and glycogenic precursors are critical pathways: by revealing disease specific metabolic shifts, metabolomic studies could identify the key-metabolic steps involved in controlling growth and/or apoptosis, and thus, acting as potential new targets for therapeutic intervention [45].
Proliferating and tumor-derived cells frequently display an elevated aerobic glycolysis with an up-regulated expression of glycolytic enzymes and typically maintain this metabolic phenotype in culture even under normoxic conditions. This implies that the interplay existing in normal cells between mitochondrial respiration and glycolytic flux, by which high O2 values inhibit the latter process (the so-called Pasteur effect) [46], is lost in cancer cells. Glycolysis is inefficient in terms of ATP production, as it generates only two ATP molecules per molecule of glucose, whereas complete oxidation of one glucose molecule by oxidative phosphorylation generates up to 36 ATP molecules. Yet, despite its low efficiency in ATP yield per molecule of glucose, aerobic glycolysis can generate more ATP than oxidative phosphorylation by producing ATP at a faster rate [47]. Therefore, an inefficient but faster pathway for ATP production may be preferred to meet the high demands of dividing cells.
This mechanism is of strategic relevance under conditions of hypoxia or fluctuating oxygen availability in which mitochondria cannot generate enough ATP. In those conditions, aerobic glycolysis may give cancers a significant growth advantage [48]. In turn, high glycolytic fluxes are coupled to high lactate levels, mainly produced via the glycolytic pathway and partially obtained through the degradation of glutamine and serine (glutaminolysis and serinolysis) [49]. The conversion of pyruvate to lactate is carried out by lactate dehydrogenase (LDH), since the LDH-A isoform is strongly up-regulated in cancer tissues. Lactate production is essential for the recycling of NAD+ in the absence of functional mitochondrial cytoplasmic NADH shuttles, due to reduced oxidative phosphorylation.
An increase in LDH-A levels is essential for proliferating cells, as LDH-A suppression not only drives cancer cells towards a mitochondrial oxidative phenotype, but also impairs cancer cell proliferation both in vitro and in vivo [50]. Glycolytic activity seems indeed to correlate with the degree of tumor malignancy: glycolysis is faster and oxidative phosphorylation is slower in highly undifferentiated and fast-growing tumors than in slow-growing cancers or in normal cells [51]. This pattern is apparent, namely, in breast cancer cells. Non-invasive MCF7 cells have much lower aerobic glucose consumption rates when compared to the highly invasive MDA-MB231 mammary cancer cell line [52]. The high rate of glucose consumption correlates with both malignancy growth and response to therapy [53], while a high level of lactate (and choline phospholipids metabolites) has been proposed as a predictor of malignant evolution [54]. Accordingly, cancer cells, which exhibit enzymatic deficiencies in their oxidative capacity, are more malignant than those that have active oxidative phosphorylation [55].
Glucose degradation into lactate allows the cell to avoid oxygen consumption while producing ATP. Wherever oxygen reacts with iron containing proteins, e.g., complexes of mitochondrial respiratory chain, ROS, such as the O2 −, peroxide anions, and hydroxyl radicals (·OH), can be generated. Interaction of ROS with cellular macromolecules (DNA, proteins, and lipids) under steady-state conditions can lead to oxidative damage if the antioxidant defenses are not fully efficient. Hence, one can hypothesize that transition to aerobic glycolysis serves as a means to minimize the production of ROS in cells during the critical phases of enhanced biosynthesis and cell division [56].
The consensus view, however, ‘over-production’ of ROS is unfavorable for cells, is an overly simplistic statement as ROS accomplish several other tasks and may act as second messengers in mammalian cells [57]. Notably, several genes are activated in response to alterations in ROS concentration including those for protein kinases [58], tyrosine kinases, and growth factors [59]. Therefore, a perturbed redox state, because of prevalent glycolytic metabolism coupled with reduced O2 availability, can affect gene expression as well as enzymatic reactions, thus favoring the emergence of abnormal phenotypes. Indeed, an imbalance in the redox metabolism and mitochondrial respiratory functions has been implicated in the etiology and pathology of cancer [60].
An ultimate critical consequence of a high glycolytic phenotype is increased tumor cell acid production and consequently increased release of H+ in the surrounding milieu. In turn, acidification of the microenvironment allows cancer cell to become more invasive and more competitive for space and substrate utilization [61]. In addition, rapidly growing cancer cells require enhanced glutaminolytic capacities, which are consequently driven towards synthetic processes, such as nucleic acid synthesis through oxidative and non-oxidative pentose pathways [62]. It has been shown that the glycolysis-derived carbons are used mainly for intracellular synthetic reactions, i.e., fatty acids and nucleic acid ribose synthesis through glutaminolysis and the non-oxidative pentose-cycle [63] This is an unexpected feature of cancer metabolism in that the high level of ‘aerobic glycolysis’ was initially thought to be explained solely by the increasing energy demand of tumor cells. Those results provided, therefore, a timely reason to revisit an old question—why do tumor cells glycolyse?—giving new provocative answers and insights [64].
In addition to supporting nucleotide biosynthesis, glycolysis is also a source of carbon for lipid precursors [65]. Citrate molecules expelled from tumor mitochondria accumulate into the cytosol owing to a defect in the transformation of citrate into 2-oxoglutarate. This enhanced cytosolic release is a prerequisite for de novo tumor lipogenesis [66]. In the cytosol, citrate is cleaved by ATP-citrate lyase to acetyl-CoA (AcCoA), leading to oxaloacetate (OAA) and AcCoA, which is further carboxylated for incorporation into fatty acids and cholesterol, an essential molecule required for de novo membranogenesis [67]. De novo lipogenesis is an absolute requirement for highly proliferating cells as inhibition of fatty acid synthase activity has been shown to kill cancer cells and hinder the growth of tumors in xenograft models [68].
Melatonin and mitochondrial respiration
The collective data suggest a critical role of mitochondria in regulating glucose and energy metabolism in cancer cells. As a result, an in depth investigation of the role of mitochondria in cancer could likely reveal novel approaches to targeted therapy. Some attempts have been already made in targeting glutamine metabolism and aspartate synthesis, while metformin, commonly used in the treatment of diabetes as an inhibitor of the mitochondrial complex I, has shown having anticancer activity [69]. Furthermore, cancer can be inhibited or even induced to differentiate by ‘normalizing’ the mitochondrial metabolism. Breast cancer cells, growing within an embryonic morphogenetic milieu (constituted by protein egg’s extract), were induced to recover a ‘normal’ oxidative metabolism [70]. In the treated cancer cells, glycolytic fluxes diminished, with a parallel decrease in lactate, glutathione, and glutamine levels. Namely, MDA-MB231 cell line, characterized by a truly glycolytic phenotype, after 72 h of culture in the embryonic environment, underwent a complete metabolic reversion. A parallel change was observed for the mitochondria shape: when cells experienced a transition from a glycolytic phenotype into an oxidative metabolism, mitochondria lost their ‘condensed’ structure evolving into an orthodox conformation [71]. Indeed, reversion of the glycolytic phenotype (by forcing cells to grow in a glucose-free, glutamine/pyruvate-containing medium) induces loss of many cancer stemness features and restores sensitivity to drugs, as evidenced by studies on pluripotent cancer stem cells [72], where glycolytic phenotype, mitochondrial dysfunction, pluripotency, and resistance to apoptosis are tightly inter-connected. In that study, normalization of the metabolomic profile was associated with profound changes in mitochondria morphology, biogenesis, and physiology along the differentiation process, culminating with the establishment of a long filamentous, polarized, and active network. This finding provides clear evidence that both mitochondrial as well as cell membrane morphology are critically linked to energy metabolism and to cancer development.
The glycolytic phenotype is also associated with resistance to anticancer treatment. It is worth of noting that highly glycolytic cancer cells showed to be resistant to both the antiproliferative and pro-apoptotic effects of melatonin. Indeed, melatonin did not significantly modify the behavior of glycolytic embryonal carcinoma cells, whereas cancer cells relying preferentially to oxidative phosphorylation for ATP synthesis (particularly cells grown up in a galactose medium) demonstrated to be significantly modulated by melatonin [73]. In addition, low-malignant breast cancer MCF-7 cells has been shown to decrease their ATP production as well as their viability upon melatonin treatment [74].
The concentration of melatonin required for triggering an anticancer effect has been shown to be strictly dependent on the energetic supply of the medium in which cells are cultured: the more differentiated is the metabolomic fingerprint of the cancer cell, the more active will be melatonin in promoting an antiproliferative and/or pro-apoptotic effect. A similar association has also been reported between full-differentiated mitochondrial morphology and melatonin potency. Namely, the highest efficacy of melatonin has been observed in embryonal cancer cells growing in a galactose medium and presenting a more open conformation of the mitochondrial permeability transition pore, a condition required to destabilize the mitochondrial membrane potential and to foster the release of cytochrome c [73].
Those data seem to suggest that melatonin may display an anticancer effect only in those cancerous cells primarily relying on the mitochondrial respiratory chain for ATP production. Indeed, in prolactinoma cancer cells overexpressing the respiratory complexes (namely, complexes I, III, and IV) under β-estradiol stimulation, melatonin induces a strong inhibition on the complex I–IV activity while promoting an increased release of ROS, thus leading to mitochondrial shrinkage and apoptosis [75]. Similarly, an indole derivative of melatonin (5,6-dihydroxytryptamine) has been demonstrated to enhance the mitochondrial complex III, thus ultimately leading to the destruction of mitochondrial function [76]. Accordingly, in isolated mitochondria, the main melatonin catabolite 6-hydroxymelatonin (6-OHM) has been demonstrated to up-regulate the complex III activity, thus leading to increased production of ROS [77]. Mechanistically, these effects seem to involve specifically some component of the complex III, such as Qi and Qn, given that antimycin A—a specific inhibitor that binds to both Qi and Qn—almost completely antagonizes ROS increase induced by melatonin in leukemic cells [78]. Therefore, it has been surmised that melatonin could differently modulate the activity of complex III in both normal and cancerous cells through an allosteric regulation [79]. Furthermore, melatonin and its main metabolite (6-OHM) have been shown to induce a relevant cytotoxic effect in breast MCF-7 [80] and in leukemic HL-60 cells [81], mechanistically attributable to increased release of ROS, including H2O2.
It is tempting to speculate that differences in cancer cell sensitivity to melatonin may also depend on the specific metabolomic fingerprint of each cancer cell type. Indeed, millimolar concentrations of melatonin decrease S-phase population and trigger apoptosis in colon cancer cells, while the same concentrations only reduce the proportion of cells in G2/M phase in both osteosarcoma and leukemia cells, without any effect on cell death [82, 83]. Definitively, these data suggest that those effects depend on the overall metabolic and differentiation state of the cancer cells.
The findings reported above indicate that melatonin anticancer activities are strongly dependent of the functioning of the respiratory chain, given that only by enhancing the activity of respiratory complexes melatonin could induce a relevant increase in ROS release and hence a consequent up-regulation of apoptosis.
The reported melatonin effect on cancerous cells is opposite to what is reported in normal cells exposed to a number of experimental stressors, where melatonin prevents the mitochondrial transition pore, stabilizes the mitochondrial membrane potential and modulates the activity of the mitochondrial respiratory chain, overall reducing ROS production. In normal cells, melatonin improves the activity of complexes I-IV, down-regulates the levels of complex III [84], and antagonizes the respiratory chain damage induced by rotenone [85]. In these conditions, melatonin decreases the oxygen consumption [86] and optimizes the respiratory chain functioning [87] by stabilizing the electron transfer, preventing the electron leakage [88] and thus increasing the respiratory control index [89]. These paradoxical results are indeed not surprising, as the anticancer effects displayed by melatonin have been already reported to be strictly context dependent: they have been observed only in some types of cancer cells and not in normal cells [90].
These data are in support for a ‘normalizing’ effect exerted by melatonin on glucose degradation through the phosphorylating-oxidative pathway. Indeed, a very recent paper demonstrated that in leiomyosarcoma, both aerobic glycolysis and linoleic acid uptake—two metabolic hallmark of malignancy—were markedly suppressed after treatment with even low pharmacological doses of melatonin [91]. Proliferative activity of cancer cells was also significantly reduced, while the addition of S20928, a nonselective melatonin antagonist, reversed these melatonin inhibitory effects. That finding confirms previous results in which melatonin has been shown to induce apoptosis in Ewing sarcoma cells with high glycolytic metabolism, while having no effect on cancerous cells with normal glucose metabolism, like chondrosarcoma [92]. In this study, melatonin has a general effect on glucose metabolism, as the pineal hormone induces a decrease in glucose uptake, lactate levels, and LDH activity, further confirming that aerobic glycolysis is essential for the survival of Ewing sarcoma cells. In the absence of external supply, cells are obliged to obtain glucose from the degradation of glycogen. Hence, melatonin inhibition of glucose uptake could likely promote the breakdown of glycogen stores observed in Ewing sarcoma cells, possibly due to an attempt to obtain energy and maintain cell viability.
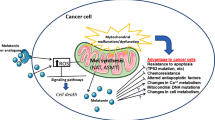
The blockage of glycogen breakdown conversely enhances the toxicity of melatonin and further enhances melatonin-induced apoptosis. Yet, in the above-referred study, melatonin effects on glucose metabolism have been primarily ascribed to the modulation of hypoxia-inducible factor 1 (HIF). Indeed, melatonin inactivates HIF in Ewing sarcoma cells, which could account for the decrease in aerobic glycolysis. HIF is essential for cancer cells to exhibit the Warburg effect, since it increases the activity of the vast majority of the enzymes involved in aerobic glycolysis even under normoxic conditions [93]. Even if no direct effects on mitochondria and, namely, on the activity of the respiratory chain have been recorded, a concomitant effect of melatonin in this experimental model cannot be discarded. Conclusively, melatonin seems to modulate glucose metabolism by acting both on mitochondrial function and on some key enzymatic processes involved in regulating glucose metabolism, such as HIF (Fig. 1).

Melatonin effects on mitochondria. Melatonin (M)—produced within the mitochondria and imported from the cytosol—enhances the disclosure of mitochondrial permeability transition pore (MPTP), thus modifying the mitochondrial potential (∆Ψ). Melatonin improves the electron transport chain (ETC) and the overall Kreb’s cycle activity, leading to enhanced oxidative glucose metabolism. Moreover, melatonin modulates reactive oxygen species (ROS) production in a context-dependent manner. Similarly, melatonin increases or decreases apoptosis (respectively, in cancer or normal cells) by influencing antiapoptotic (Bcl-2) or pro-apoptotic (caspase-3, Casp3; cytochrome c, Cytc) effectors. Eventually, a direct action of melatonin on mitochondrial DNA (mtDNA) activity has been proposed
Melatonin effects on the function of mitochondria from cancer cells may also depend on the subtle modulation of Ca2+ release. In fact, oscillation of Ca2+ differs significantly in both stem and somatic cells, as well as during cell cycle phase progression. Namely, Ca2+ increases during cell differentiation; meanwhile, its values are low in cancer stem cells [94]. Accordingly, the effect of melatonin on free Ca2+ was also dissimilar in cancer stem cells and somatic cancer cells, respectively [73]. During melatonin treatment, cancer stem cells showed a slight increase in Ca2+ without any observable effect on cell viability; in differentiated, low glycolytic cancer cells grown on glutamine/pyruvate-containing medium, melatonin arrested cell cycle at S-phase, and decreased both cell viability and free Ca2+ levels. It is worth of noting that mitochondria in differentiated cancer cells seem to retain with difficulty calcium ions; meanwhile, high Ca2+ levels are required to support mitochondrial maturation [72]. Thus, the melatonin-dependent calcium-releasing effect [95] could play a role in mediating its anticancer effects on neoplastic cells with high-oxidizing mitochondrial metabolism.
Mitochondria, melatonin, and apoptosis
Mitochondria sustain a privileged role in the apoptotic process, namely, within the so-called intrinsic pathway [96]. A second mitochondria-derived activator of caspases—such SMACs and Diablo—are released into the cytosol following the increase in permeability of the mitochondrial membranes. In turn, SMAC binds to inhibitor apoptotic proteins (including IAPs, Bcl-2, and Bcl-xl), thereby deactivating them. Namely, antiapoptotic proteins counteract cytosolic factors such as Bax and Bak, preventing them from opening the mitochondrial outer membrane permeabilization pore [97]. Antiapoptotic factors also suppress the activity of caspases, a group of cysteine proteases that are ultimately responsible for the cell death [98]. In this manner, modulation of mitochondria function antagonizes apoptotic activation. In contrast, mitochondria can release cytochrome c in the cytosol through the so-called mitochondrial apoptosis-induced channel. Once released, the cytochrome c binds to the apoptotic protease activating factor-1 (Apaf-1) and further to pro-caspase-9, to constitute a complex known as apoptosome responsible for the cleavage of caspase-9. Once activated, caspase-9 activates in turn caspase-3, thus reinforcing the apoptotic process. Thereby, a pivotal hub of the machinery involved in executing the apoptotic pathway can considered to be dependent on mitochondrial permeability.
Melatonin modulates both pro- and antiapoptotic processes within the mitochondria according to cell types [99]. As previously reported, melatonin-dependent effects on apoptosis should be regarded as context dependent: melatonin usually inhibits apoptosis in normal cells while promoting it in cancerous cells. Mechanistically, how melatonin displays such a paradoxical action is still a matter of debate. Differences in cell metabolism, in their enzymatic activities, in the intracellular redox status, or in network modulation may offer an explanation that has yet to be uncovered.
Melatonin activates both the intrinsic and extrinsic apoptotic pathways in a wide range of cancer cells. While in a number of cases, melatonin triggers apoptosis by targeting several cytosolic pathways—including calmodulin, p53/MDM2, and PI3K/Akt/Erk pathways [100]—in other situations, melatonin seems to influence directly mitochondria-dependent apoptotic processes by interfering with ROS, cytochrome c release, and antiapoptotic proteins (reviewed in [101]). Namely, melatonin is able to induce de novo synthesis of the apoptosis-inducing factor (AIF) precursor protein. After being imported into mitochondria, the mitochondrial localizing sequence contained in AIF 67-kDa is cleaved, resulting in the accumulation of the mature 57-kDa form of the AIF protein. Consequently, activated-AIF is translocated into the nucleus, where AIF triggers an early, caspase-3-independent type of cell death [102]. This pathway has a pivotal role in melatonin-induced apoptosis in both embryonal carcinoma and low-malignant breast cancer cells (MCF-7) [73, 103].
Despite the well-known antioxidant properties sustained by melatonin in normal tissues during many metabolic/hypoxic stresses [104], melatonin increases ROS in several cancer cell lines [105, 106]. ROS, in turn, promote apoptosis by upregulating several pro-apoptotic effectors [107]. It has been argued that differential pro-oxidant or antioxidant effects of melatonin in cancerous versus normal cells may be dependent on the intracellular and intramitochondrial redox status. Indeed, melatonin is likely to enhance ROS production in both cases. However, the increase in ROS is nullified by the concomitant increase in glutathione induced in normal cells by the pineal neurohormone [107]. Yet, as cancer cells are generally unable to increase GSH availability in response to increased oxidizing stresses, the net result of melatonin stimulation is an increase in ROS and thus in ROS-mediated cell death [108].
Conclusion
The long-recognized fact that oxidative stress in mitochondria is a hallmark of mitochondrial dysfunction has stimulated the development of mitochondria-targeted antioxidant therapies [109]. Based on pre-clinical studies and safety in phase 1 clinical trials in humans, phase 2 trials are now ongoing for mitochondria-targeted antioxidant molecules including MitoQ (ubiquinone mesylate, NCT02597023), the peptide SS-31 (d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2, NCT02245620), and other compounds [109]. Other mitochondrial components, particularly energy exchange systems and the pro-apoptotic function of the permeability transition pore, constitute new potential targets for mitochondrial medicine [110].
Blockage of glycolytic metabolism currently constitutes a major target to antagonize cancer growth and a number of experimental treatments based on those assumptions are already tested in pre-clinical studies. These include genetic or pharmacological inhibition of glycolytic enzymes or LDH [111], as well as the use of non-metabolizable glucose analogues [112, 113] Many of these approaches have already demonstrated their usefulness in blocking tumor progression and/or in enhancing apoptosis, mainly through increase in mitochondrial-based ROS production [114].
Melatonin should be included among the pharmacological agents able to modulate mitochondria functions in cancer, given that a number of relevant melatonin-dependent effects are triggered by targeting mitochondria functions [115]. Indeed, melatonin may modulate the mitochondrial respiratory chain, thus antagonizing the cancer highly glycolytic bioenergetics pathway. Modulation of the ETC, altogether with Ca2+ release and mitochondrial apoptotic effectors, may hence enhance spontaneous or drug-induced apoptotic processes. The mitochondria-dependent programmed cell death is likely to involve specifically AIF, a key-factor of early-apoptosis. Consequently, the mechanism of caspase-3-independent cell death and stimulation of mitochondrial differentiation and metabolism, with consequent disruption of the Warburg effect, may represent a promising new perspective when targeting resistant cancer cells.
References
Pagliarini DJ, Rutter J (2013) Hallmarks of a new era in mitochondrial biochemistry. Genes Dev 27:2615–2627. doi:10.1101/gad.229724.113
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698. doi:10.1038/nrc3365
Sullivan LB, Chandel NS (2014) Mitochondrial reactive oxygen species and cancer. Cancer Metab 2:17. doi:10.1186/2049-3002-2-17
Moldoveanu T, Follis AV, Kriwacki RW, Green DR (2014) Many players in BCL-2 family affairs. Trends Biochem Sci 39:101–111. doi:10.1016/j.tibs.2013.12.006
Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol 15:529
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770. doi:10.1038/nature07107
Hamalainen RH, Manninen T, Koivumaki H, Kislin M, Otonkoski T, Suomalainen A (2013) Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc Natl Acad Sci USA 110(38):E3622–E3630. doi:10.1073/pnas.1311660110
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148:1145–1159. doi:10.1016/j.cell.2012.02.035
Tan DX, Manchester LC, Qin L, Reiter RJ (2016) Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci 17(12):E2124. doi:10.3390/ijms17122124
Raza H, John A, Brown EM, Benedict S, Kambal A (2008) Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol Appl Pharmacol 226(2):161–168
Acuna-Castroviejo D, Escames G, Venegas C, Diaz-Casado ME, Lima-Cabello E, Lopez LC, Rosales-Corral S, Tan DX, Reiter RJ (2014) Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci 71:2997–3025. doi:10.1007/s00018-014-1579-2
Coelho LA, Peres R, Amaral FG, Reiter RJ, Cipolla-Neto J (2015) Daily differential expression of melatonin-related genes and clock genes in rat cumulus-oocyte complex: changes after pinealectomy. J Pineal Res 58:490–499. doi:10.1111/jpi.12234
Sakaguchi K, Itoh MT, Takahashi N, Tarumi W, Ishizuka B (2013) The rat oocyte synthesises melatonin. Reprod Fertil Dev 25:674–682. doi:10.1071/RD12091
Adriaens I, Jacquet P, Cortvrindt R, Janssen K, Smitz J (2006) Melatonin has dose-dependent effects on folliculogenesis, oocyte maturation capacity and steroidogenesis. Toxicology 228:333–343. doi:10.1016/j.tox.2006.09.018
Horbay R, Bilyy R (2016) Mitochondrial dynamics during cell cycling. Apoptosis 21:1327–1335. doi:10.1007/s10495-016-1295-5
Neufeld-Cohen A, Robles MS, Aviram R, Manella G, Adamovich Y, Ladeuix B, Nir D, Rousso-Noori L, Kuperman Y, Golik M, Mann M, Asher G (2016) Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc Natl Acad Sci USA 113:E1673–E1682. doi:10.1073/pnas.1519650113
Hardeland R, Madrid JA, Tan DX, Reiter RJ (2012) Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res 52:139–166. doi:10.1111/j.1600-079X.2011.00934.x
Kandalepas PC, Mitchell JW, Gillette MU (2016) Melatonin signal transduction pathways require E-box-mediated transcription of Per1 and Per2 to reset the SCN clock at dusk. PLoS One 11:e015. doi:10.1371/journal.pone.0157824
Hsiao CW, Peng TI, Peng AC, Reiter RJ, Tanaka M, Lai YK, Jou MJ (2013) Long-term Aβ exposure augments mCa2+-independent mROS-mediated depletion of cardiolipin for the shift of a lethal transient mitochondrial permeability transition to its permanent mode in NARP cybrids: a protective targeting of melatonin. J Pineal Res 54:107–125. doi:10.1111/jpi.12004
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256. doi:10.1091/mbc.12.8.2245
Suwanjang W, Abramov AY, Charngkaew K, Govitrapong P, Chetsawang B (2016) Melatonin prevents cytosolic calcium overload, mitochondrial damage and cell death due to toxically high doses of dexamethasone-induced oxidative stress in human neuroblastoma SH-SY5Y cells. Neurochem Int 97:34–41. doi:10.1016/j.neuint.2016.05.003
Pei H, Du J, Song X, He L, Zhang Y, Li X, Qiu C, Zhang Y, Hou J, Feng J, Gao E, Li D, Yang Y (2016) Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic Biol Med 97:408–417. doi:10.1016/j.freeradbiomed.2016.06.015
Coto-Montes A, Boga JA, Rosales-Corral S, Fuentes-Broto L, Tan DX, Reiter RJ (2012) Role of melatonin in the regulation of autophagy and mitophagy: a review. Mol Cell Endocrinol 361:12–23. doi:10.1016/j.mce.2012.04.009
Kang JW, Hong JM, Lee SM (2016) Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res 60:383–393. doi:10.1111/jpi.12319
Reiter RJ, Tan DX, Qi W, Manchester LC, Karbownik M, Calvo JR (2000) Pharmacology and physiology of melatonin in the reduction of oxidative stress in vivo. Biol Signals Recept 9:160–171
Kotler M, Rodriguez C, Sainz RM, Antolin I, Menendez-Pelaez A (1998) Melatonin increases gene expression for antioxidant enzymes in rat brain cortex. J Pineal Res 24(2):83–89
Urata Y, Honma S, Goto S, Todoroki S, Iida T, Cho S, Honma K, Kondo T (1999) Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic Biol Med 27:838–847
Okatani Y, Wakatsuki A, Kaneda C (2000) Melatonin increases activities of glutathione peroxidase and superoxide dismutase in fetal rat brain. J Pineal Res 28(2):89–96. doi:10.1034/j.1600-079X.2001.280204.x
Cuzzocrea S, Costantino G, Caputi AP (1998) Protective effect of melatonin on cellular energy depletion mediated by peroxynitrite and poly (ADP-ribose) synthetase activation in a non-septic shock model induced by zymosan in the rat. J Pineal Res 25(2):78–85
Martín M, Macías M, Escames G, Reiter RJ, Agapito MT, Ortiz GG, Acuña-Castroviejo D (2000) Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J Pineal Res 28:242–248. doi:10.1034/j.1600-079X.2000.280407.x
Liu H, Kehrer JP (1996) The reduction of glutathione disulfide produced by t-butyl hydroperoxide in respiring mitochondria. Free Radic Biol Med 20(3):433–442
Martin M, Macias M, Escames G, Leon J, Acuna-Castroviejo D (2000) Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J 14:1677–1679. doi:10.1096/fj.99-0865fje
López A, García JA, Escames G, Venegas C, Ortiz F, López LC, Acuña-Castroviejo D (2009) Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J Pineal Res 46:188–198. doi:10.1111/j.1600-079X.2008.00647.x
Martin M, Macias M, Leon J, Escames G, Khaldy H, Acuna-Castroviejo D (2002) Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int J Biochem Cell Biol 34(4):348–357. doi:10.1016/S1357-2725(01)00138-8
Leon J, Acuña-Castroviejo D, Sainz RM, Mayo JC, Tan DX, Reiter RJ (2004) Melatonin and mitochondrial function. Life Sci 75(7):765–790. doi:10.1016/j.lfs.2004.03.003
Acuña-Castroviejo D, Escames G, López LC, Hitos AB, León J (2005) Melatonin and nitric oxide: two required antagonists for mitochondrial homeostasis. Endocrine 27:159–168. doi:10.1385/ENDO:27:2:159
Prunet-Marcassus BL, Ambid N, Viguerie-Bascands L, Pénicaud L, Casteilla L (2001) Evidence for a direct effect of melatonin on mitochondrial genome expression of Siberian hamster brown adipocytes. J Pineal Res 30:108–115. doi:10.1034/j.1600-079X.2001.300206.x
Acuña-Castroviejo D, Escames G, López León J, Carazo A, Khaldy H (2003) Mitochondrial regulation by melatonin and its metabolites. Adv Exp Med Biol 527:549–557
Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N (2005) A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res 65:203–209
Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394:694–697. doi:10.1038/29331
Warburg O (1926) Ǘber den Stoffwechsel der Tumoren. Springer, Berlin
Garber K (2004) Energy boost: the Warburg effect returns in a new theory of cancer. J Natl Cancer Inst 96:1805–1806. doi:10.1093/jnci/96.24.1805
Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707. doi:10.1016/j.cell.2008.08.021
Kunkel M, Reichert TE, Benz P, Lehr HA, Jeong JH, Wieand S, Bartenstein P, Wagner W, Whiteside TL (2003) Overexpression of Glut-1 and increased metabolism in tumours are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer 97:1015–1024. doi:10.1002/cncr.11159
Kroemer G, Pouyssegur J (2008) Tumour cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13:472–482. doi:10.1016/j.ccr.2008.05.005
Crabtree HG (1929) Observations on the carbohydrate metabolism of tumours. Biochem J 23:536–545
Pfeiffer T, Schuster S, Bonhoeffer S (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science 292:504–507. doi:10.1126/science.1058079
Postovit LM, Adams MA, Lash GE et al (2002) Oxygen-mediated regulation of tumor cell invasiveness. Involvement of a nitric oxide signaling pathway. J Biol Chem 277:35730–35737. doi:10.1074/jbc.M204529200
Lobo C, Ruiz-Bellido MA, Aledo JC, Márquez J, Núñez De Castro I, Alonso FJ (2000) Inhibition of glutaminase expression by antisense mRNA decreases growth and tumorigenicity of tumour cells. Biochem J 348:257–261. doi:10.1042/bj3480257
Fantin VR, St-Pierre J, Leder P (2006) Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9:425–434. doi:10.1016/j.ccr.2006.04.023
Krieg RC, Knuechel R, Schiffmann E, Liotta LA, Petricoin EF III, Herrmann PC (2004) Mitochondrial proteome: cancer-altered metabolism associated with cytochrome c oxidase subunit level variation. Proteomics 4:2789–2795. doi:10.1002/pmic.200300796
Schomack PA, Gilles RJ (2003) Contributions of cell metabolism and H+ diffusion to the acidic pH of tumours. Neoplasia 5:135–145
Smith TA (2001) The rate-limiting step for tumor [18F]fluoro-2-deoxy-d-glucose (FDG) incorporation. Nucl Med Biol 28:1–4. doi:10.1016/S0969-8051(00)00177-3
Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, Mueller-Klieser W (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res 60:916–921
Soderberg K, Nissinen E, Bakay B (1980) The energy charge in wild-type and respiration deficient Chinese hamster cell mutants. J Cell Physiol 103:169–172. doi:10.1002/jcp.1041030121
Gilbert DL, Colton CA (1999) An overview of reactive oxygen species. In: Gilbert DL, Colton CA (eds) Reactive oxygen species in biological systems. Kluwer Academic-Plenum Publishers, New York
Wenger RH (2000) Mammalian oxygen sensing, signalling and gene regulation. J Exp Biol 203:1253–1263
Burdon RH (1996) Control of cell proliferation by reactive oxygen species. Biochem Soc Trans 24:1028–1032
Nose K (2000) Role of reactive oxygen species in the regulation of physiological functions. Biol Pharm Bull 23:897–903
Enns GM (2003) The contribution of mitochondria to common disorders. Mol Genet Metab 80:11–26. doi:10.1016/j.ymgme.2003.08.009
Gatenby RA, Gawlinski ET (1996) A reaction-diffusion model of cancer invasion. Cancer Res 56:5745–5753
Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E (2002) Pyruvate kinase type M2: a crossroad in the tumor metabolome. Br J Nutr 87(Suppl 1):S23–S29
Richardson AD, Yang C, Osterman A, Smith JW (2008) Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res Treat 110:297–307. doi:10.1007/s10549-007-9732-3
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899. doi:10.1038/nrc1478
Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7:763–777. doi:10.1038/nrc2222
Parlo RA, Coleman PS (1984) Enhanced rate of citrate export from cholesterol-rich hepatoma mitochondria. The truncated Krebs cycle and other metabolic ramifications of mitochondrial membrane cholesterol. J Biol Chem 259:9997–10003
Menendez JA, Colomer R, Lupu R (2005) Why does tumour-associated fatty acid synthase (oncogenic antigen 519) ignore dietary fatty acids? Med Hypotheses 64:342–349. doi:10.1016/j.mehy.2004.07.022
Lupu R, Menendez JA (2006) Pharmacological inhibitors of fatty acid synthase (FASN)-catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Curr Pharm Biotechnol 7:483–493. doi:10.2174/138920106779116928
Pollak M (2013) Potential applications for biguanides in oncology. J Clin Invest 123:3693–3700. doi:10.1172/JCI67232
D’Anselmi F, Valerio M, Cucina A, Galli L, Proietti S, Dinicola S, Pasqualato A, Manetti C, Ricci G, Giuliani A, Bizzarri M (2011) Metabolism and cell shape in cancer: a fractal analysis. Int J Biochem Cell Biol 3:1052–1058. doi:10.1016/j.biocel.2010.05.002
Alirol E, Martinou JC (2006) Mitochondria and cancer: is there a morphological connection? Oncogene 25:4706–4716. doi:10.1038/sj.onc.1209600
Vega-Naredo I, Loureiro R, Mesquita KA, Barbosa IA, Tavares LC, Branco AF, Erickson JR, Holy J, Perkins EL, Carvalho RA, Oliveira PJ (2014) Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell Death Differ 21:1560–1574. doi:10.1038/cdd.2014.66
Loureiro R, Magalhães-Novais S, Mesquita KA, Baldeiras I, Sousa IS, Tavares LC, Barbosa IA, Oliveira PJ, Vega-Naredo I (2015) Melatonin antiproliferative effects require active mitochondrial function in embryonal carcinoma cells. Oncotarget 6:17081–17096. doi:10.18632/oncotarget.4012
Scott AE, Cosma GN, Frank AA, Wells RL, Gardner HS Jr (2001) Disruption of mitochondrial respiration by melatonin in MCF-7 cells. Toxicol Appl Pharmacol 171:149–156. doi:10.1006/taap.2000.9115
Wang BQ, Yang QH, Xu RK, Xu JN (2013) Elevated levels of mitochondrial respiratory complexes activities and ATP production in 17-β-estradiol-induced prolactin-secretory tumor cells in male rats are inhibited by melatonin in vivo and in vitro. Chin Med J (Engl) 126:4724–4730
Tutton PJ, Barkla DH (1977) Cytotoxicity of 5,6-dihydroxytryptamine in dimethylhydrazine-induced carcinomas of rat colon. Cancer Res 37:1241–1244
Klemm HP, Baumgarten HG, Schlossberger HG (1980) Polarographic measurements of spontaneous and mitochondria-promoted oxidation of 5,6- and 5,7-dihydroxytryptamine. J Neurochem 35:1400–1408
Perdomo J, Cabrera J, Estévez F, Loro J, Reiter RJ, Quintana J (2013) Melatonin induces apoptosis through a caspase-dependent but reactive oxygen species-independent mechanism in human leukemia Molt-3 cells. J Pineal Res 55:195–206. doi:10.1111/jpi.12062
Zhang HM, Zhang Y, Zhang BX (2011) The role of mitochondrial complex III in melatonin-induced ROS production in cultured mesangial cells. J Pineal Res 50:78–82. doi:10.1111/j.1600-079X.2010.00815.x
Shellard SA, Whelan RD, Hill BT (1989) Growth inhibitory and cytotoxic effects of melatonin and its metabolites on human tumour cell lines in vitro. Br J Cancer 60:288–290
Sakano K, Oikawa S, Hiraku Y, Kawanishi S (2004) Oxidative DNA damage induced by a melatonin metabolite, 6-hydroxymelatonin, via a unique non-O-quinone type of redox cycle. Biochem Pharmacol 68:1869–1878. doi:10.1016/j.bcp.2004.06.016
Liu L, Xu Y, Reiter RJ (2013) Melatonin inhibits the proliferation of human osteosarcoma cell line MG-63. Bone 55:432–438. doi:10.1016/j.bone.2013.02.021
Hong Y, Won J, Lee Y, Lee S, Park K, Chang KT (2014) Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J Pineal Res 56:264–274. doi:10.1111/jpi.12119
Zhang H, Zhang HM, Wu LP, Tan DX, Kamat A, Li YQ, Katz MS, Abboud HE, Reiter RJ, Zhang BX (2011) Impaired mitochondrial complex III and melatonin responsive reactive oxygen species generation in kidney mitochondria of db/db mice. J Pineal Res 51:338–344. doi:10.1111/j.1600-079X.2011.00894.x
Sousa SC, Castilho RF (2005) Protective effect of melatonin on rotenone plus Ca2+-induced mitochondrial oxidative stress and PC12 cell death. Antioxid Redox Signal 7:1110–1116. doi:10.1089/ars.2005.7.1110
Reyes-Toso CF, Rebagliati IR, Ricci CR, Linares LM, Albornoz LE, Cardinali DP, Zaninovich A (2006) Effect of melatonin treatment on oxygen consumption by rat liver mitochondria. Amino Acids 31:299–302. doi:10.1007/s00726-005-0280-z
Zavodnik IB, Lapshina EA, Cheshchevik VT, Dremza IK, Kujawa J, Zabrodskaya SV, Reiter RJ (2011) Melatonin and succinate reduce rat liver mitochondrial dysfunction in diabetes. J Physiol Pharmacol 62:421–427
Poeggeler B, Sambamurti K, Siedlak SL, Perry G, Smith MA, Pappolla MA (2010) A novel endogenous indole protects rodent mitochondria and extends rotifer lifespan. PLoS One 5:e10206. doi:10.1371/journal.pone.0010206
Okatani Y, Wakatsuki A, Reiter RJ, Miyahara Y (2002) Hepatic mitochondrial dysfunction in senescence-accelerated mice: correction by long-term, orally administered physiological levels of melatonin. J Pineal Res 33:127–133. doi:10.1034/j.1600-079X.2002.02109.x
Proietti S, Cucina A, Reiter RJ, Bizzarri M (2013) Molecular mechanisms of melatonin’s inhibitory actions on breast cancers. Cell Mol Life Sci 70:2139–2157. doi:10.1007/s00018-012-1161-8
Mao L, Dauchy RT, Blask DE, Dauchy EM, Slakey LM, Brimer S, Yuan L, Xiang S, Hauch A, Smith K, Frasch T, Belancio VP, Wren MA, Hill SM (2016) Melatonin suppression of aerobic glycolysis (Warburg effect), survival signalling and metastasis in human leiomyosarcoma. J Pineal Res 60:167–177. doi:10.1111/jpi.12298
Sanchez-Sanchez AM, Antolin I, Puente-Moncada N, Suarez S, Gomez-Lobo M, Rodriguez C, Martin V (2015) Melatonin cytotoxicity is associated to warburg effect inhibition in ewing sarcoma cells. PLoS One 10:e0135420. doi:10.1371/journal.pone.0135420
Stubbs M, Griffiths JR (2010) The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv Enzym Regul 50:44–55. doi:10.1016/j.advenzreg.2009.10.027
Resende RR, Adhikari A, da Costa JL, Lorencon E, Ladeira MS, Guatimosim S, Kihara AH, Ladeira LO (2010) Influence of spontaneous calcium events on cell-cycle progression in embryonal carcinoma and adult stem cells. Biochim Biophys Acta 1803:246–260. doi:10.1016/j.bbamcr.2009.11.008
Dai J, Inscho EW, Yuan L, Hill SM (2002) Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J Pineal Res 32:112–119. doi:10.1034/j.1600-079x.2002.1844.x
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15:2922–2933
Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW (2006) Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta 1762:191–201. doi:10.1016/j.bbadis.2005.07.002
Fesik SW, Shi Y (2001) Controlling the caspases”. Science 294:1477–1478. doi:10.1126/science.1062236
Sainz RM, Mayo JC, Rodriguez C, Tan DX, Lopez-Burillo S, Reiter RJ (2003) Melatonin and cell death: differential actions on apoptosis in normal and cancer cells. Cell Mol Life Sci 60:1407–1426. doi:10.1007/s00018-003-2319-1
Proietti S, Cucina A, Dobrowolny G, D’Anselmi F, Dinicola S, Masiello MG, Pasqualato A, Palombo A, Morini V, Reiter RJ, Bizzarri M (2014) Melatonin down-regulates MDM2 gene expression and enhances p53 acetylation in MCF-7 cells. J Pineal Res 57:120–129. doi:10.1111/jpi.12150
Bizzarri M, Proietti S, Cucina A, Reiter RJ (2013) Molecular mechanisms of the pro-apoptotic actions of melatonin in cancer: a review. Expert Opin Ther Targets 17:1483–1496. doi:10.1517/14728222.2013.834890
Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K (2005) Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J 24:1375–1386. doi:10.1038/sj.emboj.7600614
Cucina A, Proietti S, D’Anselmi F, Coluccia P, Dinicola S, Frati L, Bizzarri M (2009) Evidence for a biphasic apoptotic pathway induced by melatonin in MCF-7 breast cancer cells. J Pineal Res 46:172–180. doi:10.1111/j.1600-079X.2008.00645.x
Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 51:1–16. doi:10.1111/j.1600-079X.2011.00916.x
Büyükavci M, Ozdemir O, Buck S, Stout M, Ravindranath Y, Savaşan S (2006) Melatonin cytotoxicity in human leukemia cells: relation with its pro-oxidant effect. Fundam Clin Pharmacol 20:73–79. doi:10.1111/j.1472-8206.2005.00389.x
Osseni RA, Rat P, Bogdan A, Warnet JM, Touitou Y (2000) Evidence of prooxidant and antioxidant action of melatonin on human liver cell line HepG2. Life Sci 68:387–399. doi:10.1016/S0024-3205(00)00955-3
Matés JM, Segura JA, Alonso FJ, Márquez J (2008) Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Arch Toxicol 82:273–299. doi:10.1007/s00204-008-0304-z
Sánchez-Sánchez AM, Martín V, García-Santos G, Rodríguez-Blanco J, Casado-Zapico S, Suarez-Garnacho S, Antolín I, Rodriguez C (2011) Intracellular redox state as determinant for melatonin antiproliferative vs. cytotoxic effects in cancer cells. Free Radic Res 45:1333–1341. doi:10.3109/10715762.2011.623700
Picard M, Wallace DC, Burelle Y (2016) The rise of mitochondria in medicine. Mitochondrion 30:105–116. doi:10.1016/j.mito.2016.07.003
Wang W, Karamanlidis G, Tian R (2016) Novel targets for mitochondrial medicine. Sci Transl Med 8:326rv3. doi:10.1126/scitranslmed.aac7410
Pelicano H, Martin DS, Xu RH, Huang P (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25:4633–4646. doi:10.1038/sj.onc.1209597
Dwarakanath BS (2009) Cytotoxicity, radiosensitization, and chemosensitization of tumor cells by 2-deoxy-d-glucose in vitro. J Cancer Res Ther 5(Suppl 1):S27–S31. doi:10.4103/0973-1482.55137
Cardaci S, Desideri E, Ciriolo MR (2012) Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J Bioenerg Biomembr 44:17–29. doi:10.1007/s10863-012-9422-7
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107:2037–2042. doi:10.1073/pnas.0914433107
Pacini N, Borziani F (2016) Oncostatic-cytoprotective effect of melatonin and other bioactive molecules: a common target in mitochondrial respiration. Int J Mol Sci 17:341. doi:10.3390/ijms17030341
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Proietti, S., Cucina, A., Minini, M. et al. Melatonin, mitochondria, and the cancer cell. Cell. Mol. Life Sci. 74, 4015–4025 (2017). https://doi.org/10.1007/s00018-017-2612-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2612-z