
Abstract
Metabolic reprogramming is widely observed during cancer development to confer cancer cells the ability to survive and proliferate, even under the stressed, such as nutrient-limiting, conditions. It is famously known that cancer cells favor the “Warburg effect”, i.e., the enhanced glycolysis or aerobic glycolysis, even when the ambient oxygen supply is sufficient. In addition, deregulated anabolism/catabolism of fatty acids and amino acids, especially glutamine, serine and glycine, have been identified to function as metabolic regulators in supporting cancer cell growth. Furthermore, extensive crosstalks are being revealed between the deregulated metabolic network and cancer cell signaling. These exciting advancements have inspired new strategies for treating various malignancies by targeting cancer metabolism. Here we review recent findings related to the regulation of glucose, fatty acid and amino acid metabolism, their crosstalk, and relevant cancer therapy strategy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Energy metabolism reprogramming, which fuels fast cell growth and proliferation by adjustments of energy metabolism, has been considered as an emerging hallmark of cancer [1]. It is well established that normal cells get energy via first glycolysis in the cytosol that is followed by mitochondrial oxidative phosphorylation under aerobic conditions. When oxygen is scarce, the cells rely on glycolysis rather than oxygen-consuming mitochondrial metabolism for energy supply. However, cancer cells prefer to perform glycolysis in the cytosol even in the presence of oxygen, a phenomenon first observed by Otto Warburg [2, 3] and now famously known as “Warburg effect” or “aerobic glycolysis”. It is suggested that although efficiency in ATP production per molecule of glucose is much lower via glycolysis, the yield rate is much faster than that in the oxidative phosphorylation [4] and, hence, it meets fast growth and proliferation demand of cancer cells. Such reprogramming of glucose metabolism has been validated within many tumors, and increased glycolysis facilitates biosynthesis of biomass (e.g., nucleotides, amino acids and lipids) by providing glycolytic intermediates as raw material [5].
Besides the dysregulation of glucose metabolism, metabolic reprogramming in cancer cells has been characterized by aberrant lipid metabolism, amino acids metabolism, mitochondrial biogenesis, and other bioenergetic metabolism pathways. Investigation on these energy metabolism reprogramming would uncover fundamental molecular events of malignancy and help to find better ways to diagnose and treat cancer. In this paper, we review recent findings related to the regulation of glucose, fatty acid and amino acid metabolism reprogramming, their crosstalk in cancer cells, as well as therapeutic strategy based on the understanding of the reprogramming mechanism.
Regulation of glucose metabolism in cancer cells
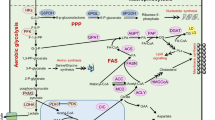
As a major nutrient to fuel cell growth, glucose metabolism contains glycolysis pathway, pentose phosphate pathway (PPP), and serine synthesis pathway (SSP) in the cytoplasm and TCA cycle in the mitochondria. Glycolysis is a central pathway of glucose metabolism and the metabolite pyruvate can be converted into lactate as the end product for extracellular secretion, or enter into the mitochondria for TCA cycle and oxidative phosphorylation. Glycolysis pathway can be branched to the pentose phosphate pathway and the serine synthesis pathway via its metabolic intermediary. In addition, the hexosamine biosynthetic pathway is also a glucose metabolism pathway branched from glycolysis via fructose-6-phosphate, which not only contributes to protein modification by providing final product of UDP-GlcNAc, but also coordinates growth factor-induced glucose and glutamine metabolism [6]. Recent results reveal alterations or reprogramming of these pathways in cancer cells.
Regulation of glucose metabolism by oncogenes, tumor suppressors or non-coding RNAs
Mounting evidence shows that alterations of oncogenes and tumor suppressors, via regulation of key metabolic enzyme effecters, are responsible for metabolic reprogramming [7]. The proto-oncogene cMyc has been documented to render metabolic shift to glycolysis in cancer cells and regulate most of the glycolytic enzymes [8, 9]. An early study has demonstrated that cMyc also induces genes involved in mitochondrial structure and function and stimulates mitochondrial biogenesis [10]. Besides, one of our recent studies showed that cMyc drives SSP in cancer cells under nutrient deprivation conditions [11]. Hypoxia-inducible factor-1(HIF-1), another key oncogene, is a master regulator in regulating glucose metabolism in cancer cells under hypoxic microenvironment, which is typical during cancer development. It has been documented that HIF-1α activates the expression of multiple glycolytic enzymes, including GLUT1, GLUT3, HK1, HK2, GAPDH, PGK1, PKM2, LDHA and PDK1 [12–15]. HIF-1 not only increases glucose uptake and lactate production, but also blocks TCA cycle and oxidative phosphorylation in mitochondria [15]. However, the tumor suppressor p53 decreases glucose uptake in cells by inhibiting transcription expression of GLUT1 and GLUT4 [16], or plays a key role in negative regulation of glycolysis under normoxic conditions or hypoxic conditions through its targets [17]. Mdm2, a downstream effector of p53, has recently been identified as a direct binding partner and ubiquitin ligase for PGAM in cultured cells and in vitro, attenuating the Warburg effect via ubiquitination and degradation of PGAM [18]. TIGAR (TP53-induced glycolysis and apoptosis regulator), another p53-inducible gene, lowers fructose-2,6-bisphosphate levels in cells and results in an inhibition of glycolysis, but functions through the pentose phosphate pathway to decrease intracellular ROS levels [19]. It is worth mentioning that a growing body of evidence points to the pivotal roles of sirtuin-family deacetylases in regulation of cancer progression and metabolism. It has been reported that several mouse models with SIRT1, SIRT2, SIRT3, SIRT4 or SIRT6 knockout are more prone to developing tumors [20, 21]. Sirtuins can control glucose metabolism not only by regulating transcriptional activity of cMyc and HIF-1, two drivers of metabolic reprogramming in cancer [22], but also by deacetylating and affecting the activity of glycolytic enzyme directly [23–25]. The mitochondrial deacetylase SIRT3 also promotes mitochondrial metabolism by deacetylating and activating enzymes involved in the TCA cycle and fatty acid oxidation [26]. In addition, mounting evidence shows microRNAs are involved in regulation of the Warburg effect, in particular through interplay with oncogenes/tumor suppressors such as cMyc, HIF-1 and P53 [7, 27]. A long noncoding RNA (LncRNA) PCGEM1 (prostate cancer gene expression marker 1), has been recently shown to promote glucose uptake for aerobic glycolysis via cMyc activation [28]. Here we summarize the recent evidence showing the alterations of glucose metabolism by oncogenes, tumor suppressors, or non-coding RNAs.
Glycolysis
The enhancement of aerobic glycolysis facilitates anabolic metabolism of cancer cells by providing more carbon intermediates for the biosynthesis of nucleotides, amino acids and lipids [29]. The enzymes involved in glycolysis include hexokinase, phosphofructokinase, aldolase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, phosphoglycerate mutase, enolase, pyruvate kinase and lactate dehydrogenase (Fig. 1). Alterations of oncogenes and tumor suppressors drive cancer cells to aerobic glycolysis through regulation of metabolic enzymes [30].

Regulation of glucose metabolism in cancer cells. Glucose metabolism contains glycolysis, pentose phosphate pathway (PPP), serine synthesis pathway (SSP) in the cytoplasm and TCA cycle in the mitochondrion. These pathways are generally altered in tumor cells. Please see more detail in the text. GLUT glucose transporter, G6P glucose-6-phosphate, F6P fructose-6-phosphate, F1,6P fructose-1,6-bisphosphate, GA3P glyceraldehyde 3-phosphate, 1,3-DPG 1,3-disphosphoglycerate, 3PG 3-phospho-glycerate, 2PG 2-phospho-glycerate, PEP phosphoenolpyruvate, R5P ribose-5-phosphate, HK hexokinase, PFK phosphofructokinase, ALDOA aldolase A, fructose-bisphosphate, GAPDH glyceraldehyde-3-phosphate dehydrogenase, PGK1 phosphoglycerate kinase 1, PGAM1 phosphoglycerate mutase 1, ENO1 alpha-enolase, PKM2 pyruvate kinase isozyme type 2, LDHA lactate dehydrogenase A, PDK1 pyruvate dehydrogenase kinase 1, PDH pyruvate dehydrogenase, G6PD glucose-6-phosphate dehydrogenase, 6PGL 6-phosphogluconolactone, 6PGD 6-phosphogluconate dehydrogenase, TKT transketolase, PHGDH phosphoglycerate dehydrogenase, PSAT1 phosphoserine aminotransferase 1, PSPH phosphoserine phosphatase, SHMT serine hydroxymethyltransferase, TCA cycle tricarboxylic acid cycle
Glucose transporters mediate glucose transport across the plasma membrane, which is the first step of glycolysis. GLUT1, glucose transporter 1, was transactivated by oncogenes cMyc, KRas and HIF-1α, but inhibited by tumor suppressor p53 [31]. GLUT1 is a direct target of miR-22 in breast cancer cells, and the ectopic expression of miR-22 inhibits cell proliferation and invasion by targeting GLUT1 [32]. It has been reported that activated Akt enhances glucose uptake by promoting and maintaining GLUT1 cell surface localization, as well as attenuating GLUT1 internalization [33, 34]. Expression of PTEN and GLUT1 on the plasma membrane of thyroid cancer cells negatively correlates and lack of PTEN increases the membrane expression of GLUT1 and glucose uptake [35], indicating that PTEN is also a negative regulator of GLUT1. In addition, a recent study shows that GLUT1 can be potential prognosis marker for patients with colorectal cancer by predicting survival after resection of colorectal cancer liver metastasis [36]. GLUT3, another type of glucose transporter, is activated by IKK/NF-κB axis, which is suppressed by p53 [37]. Activation of NF-κB by loss of p53 results in enhanced GLUT3 expression and aerobic glycolysis.
Hexokinase (HK) is the first rate-limiting enzyme in the glucose metabolism pathway. Glucose across the plasma membrane mediated by glucose transporters is rapidly phosphorylated by hexokinase (primarily HK II) bound to the outer mitochondrial membrane via the voltage-dependent anion channel, which allows direct access to HK II of ATP generated by the ATP synthase and transported across the inner mitochondrial membrane by the adenine nucleotide translocator [38]. HK1 and HK2 are direct targets of miR-34a, and p53-inducible miR-34a repressed glycolysis and enhanced mitochondrial respiration [39]. Another microRNA, miR-143, regulates HK2 expression in cancer cells via a specific recognition motif located in its 3′-UTR [40]. Recently, Wang et al. reported that HK2-mediated Warburg effect is required for cell transformation and tumorigenesis [41]. They found that HK2 protein expression is elevated in PTEN/p53-deficient prostate cancer cells, PTEN deletion increases HK2 mRNA translation through the activation of the AKT-mTORC1-4EBP1 axis, and p53 loss enhances HK2 mRNA stability through the inhibition of miR-143 biogenesis [41]. Recently, a study also show LncRNA UCA1 promotes glycolysis in bladder cancer cells by inducing HK2, through mTOR-mediated activation of STAT3 and repression of miR-143 [42].
Aldolase catalyzes the reversible conversion of fructose-1, 6-bisphosphate to glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. Aldolase a (ALDOA) is highly expressed in lung squamous cell carcinoma (LSCC), and its expression level is correlated with LSCC metastasis, grades, differentiation status, survival rates and prognosis of LSCC patients [43]. Another recent study reports an important role of ALDOA in osteosarcoma progression and metastasis [44]. In human melanoma cells, ANGPTL4 (Angiopoietin-like 4) upregulates ALDOA expression at the transcriptional level through a PKC-dependent mechanism, and ALDOA is a critical mediator of the promoting effect of ANGPTL4 on melanoma cell invasion [45].
Although remarkably increased GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression has been observed in a subset of cancers [46], the cancer-related mechanism involved in GAPDH regulation remains largely unknown [47]. p53 and NO enhance GAPDH gene expression, but the molecular mechanism is not clear [13]. A recent study reports that acetylation at lysine 254 (K254) increases GAPDH enzyme activity in response to glucose, which promotes tumor cell proliferation [48]. Furthermore, acetylation of GAPDH (K254) is reversibly regulated by the acetyltransferase PCAF and the deacetylase HDAC5. Besides mediating an elevation in glycolysis, GAPDH can also induce autophagy of damaged mitochondria, both of which cooperate to protect cells from caspase-independent cell death [49].
It has been reported that PGAM1 (phosphoglycerate mutase 1) has a higher expression level and enzymatic activity in the lung, colon, liver and breast carcinoma tissues than those of adjacent normal tissues [50], and knockdown of PGAM1 decreased glycolytic rates and cell proliferation rates in cancer cells and attenuates lung cancer cell growth in xenograft nude mice [51]. Acetylated PGAM1 displays enhanced activity, although C-terminal lysine deacetylation mediated by SIRT1, a NAD+-dependent deacetylase, attenuates its catalytic activity [23]. SIRT2, another NAD+-dependent deacetylase, has been reported to deacetylate PGAM at lysines 100/106/113/138 in its central region in HCT116 cells, decrease the enzymatic activity of PGAM and inhibit cell proliferation [24]. However, another study reported that PGAM2 is acetylated at lysine 100 in HEK293T and A549 cells, and SIRT2 is responsible for the deacetylation of PGAM2 and stimulates its activity [25]. The opposite deacetylation effect of SIRT2 on the PGAM activity may be attributed to different cell lines used in these studies.
Five active lactate dehydrogenase (LDH) isoforms (LDH-1 through LDH-5) exist in human tissues, each of which is a homo- or heterotetramer assembled from two types of subunit LDHA and LDHB. The LDHA catalyzes the conversion of pyruvate and NADH into l-lactate and NAD in the final step of anaerobic glycolysis, whereas LDHB protein favors the reversible conversion of lactate and pyruvate. Dang’s group has documented that inhibition of LDHA by a small-molecule inhibitor (FX11) restrains glycolysis and shunts pyruvate into the mitochondrion [52]. Moreover, FX11 slow the growth of human lymphoma and pancreatic cancer xenografts indicates that LDHA is required for tumor maintenance and progression. A recent study shows that inhibition of LDHA by oxamate in gastric cancer cells decreased the lactate production and may have a negative effect on cell growth and tumor invasion [53]. LDHA is also directly targeted by miR-34a, which resensitizes colon cancer cells to 5-fluorouracil [54]. Compared to LDHA, the study for LDHB has attracted less attention. LDHB is overexpressed in triple-negative breast cancer and essential for tumor growth [55]. A tumor suppressor, drs (down-regulated by v-src), has been validated to regulate glucose metabolism by inhibiting expression of LDHB [56].
Pentose phosphate pathway (PPP)
The pentose phosphate pathway (PPP), branching from glycolysis, is also a major pathway for glucose catabolism (Fig. 1). The PPP includes two phases: the oxidative branch and the nonoxidative branch. Besides converted into fructose-6-phosphate (F6P) in the glycolytic pathway, glucose-6-phosphate (G6P) can also be dehydrogenated by glucose-6-phosphate dehydrogenase (G6PD) in the oxidative branch of the PPP, which produces not only ribose-5-phosphate (R5P) but also NAPDH [57]. R5P is an important precursor to many macromolecules, such as nucleotides. Cancer cells have high reactive oxygen species (ROS) levels for their accelerated metabolism, which renders them more susceptible to oxidative stress-induced cell death [58]. NPDPH plays a critical role in the reductive biosynthesis and protects cells from ROS. In the nonoxidative branch of PPP, glycolytic intermediates, such as fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P), can be converted into R5P, and vice versa. The PPP flux is generally enhanced in cancer cells by increased activity of G6PD to meet the bioenergy requirement for growth and proliferation. Recent studies have shown that the PPP is regulated by numerous factors, including oncogenes and tumor suppressors [59–61]. The tumour suppressor p53 protein inhibits the PPP by directly binding to G6PD and preventing the formation of the active dimer [59]. Similar to p53, the phosphatase and tensin homologue (PTEN) suppresses glucose consumption and biosynthesis through the PPP by binding to G6PD and suppressing its enzymatic activity [60]. However, oncoproteins, including PI3K, mTORC1 and K-rasG12D, stimulate glycolysis and PPP [57, 61]. In addition, some metabolic enzymes in the glycolysis pathway, such as HK2, PFK1, and PKM2, also impact the oxidative or nonoxidative branch of PPP by balancing glycolytic and PPP flux [62–64]. A recent study demonstrated that PKM2 modulates esophageal squamous cell carcinoma chemotherapy response by regulating the PPP and PKM2 inhibition restored cisplatin sensitivity by inactivating PPP [65].
Serine synthesis pathway (SSP)
About 10 % of the 3-phosphoglycerate generated from glycolysis in cancer cells is oxidized by PHGDH (phosphoglycerate dehydrogenase) and NAD to 3-phosphohydroxypyruvate, a precursor for de novo serine synthesis pathway (SSP) [66] (Fig. 1). Subsequently, PSAT1 (phosphoserine aminotransferase 1) and PSPH (phosphoserine phosphatase) convert 3-phosphohydroxypyruvate into serine [67]. Through the reaction catalyzed by SHMT (serine hydroxymethyltransferase), serine is further converted into glycine, which fuels one-carbon metabolism by conferring a major source of methyl groups [67]. Our recent study validates that cMyc transactivates the expression of those enzymes involved in serine de novo biosynthesis and activates serine biosynthesis pathway (SSP) [11]. Our findings have demonstrated that cMyc-mediated PSPH expression and SSP activation led to elevated glutathione (GSH) production, cell cycle progression and nucleic acid synthesis, which are essential for cell survival and proliferation especially under nutrient-deprived conditions. As the final rate-limiting enzyme of the SSP pathway, PSPH is vital for the tumorigenesis capacity of cMyc in vitro and in vivo [11].
TCA cycle
Mitochondria have been recognized as a central hub for bioenergetics and biosynthesis. Using multiple carbon fuels including glucose, amino acid such as glutamine, and fatty acids, mitochondria produce ATP and the intermediates needed for macromolecule biosynthesis, which are required for tumorigenesis. Our early studies showed that, by inhibiting cMyc activity through MXL1-dependent transcription repression or proteasome-dependent degradation, HIF-1 negatively regulates mitochondrial biogenesis and O2 consumption in renal carcinoma cells lacking the von Hippel–Lindau tumor suppressor (VHL) [68]. Glucose-derived pyruvate in glycolysis can be converted to acetyl-CoA by pyruvate dehydrogenase (PDH), and then acetyl-CoA enters the TCA cycle within mitochondria and produce NADH and FADH2. Under hypoxic condition, except activating glycolytic genes and promoting glycolysis, HIF-1 also suppresses metabolism through the TCA cycle by directly trans-activating PDK1, which inactivates PDH and inhibits conversion of pyruvate to acetyl-CoA [69]. However, the contribution of PDK1 to metabolic switch may depend on a different molecular basis in cancer cells under normal oxygen conditions. Recently, we demonstrate that Lin28A and Lin28B enhance, but let-7 suppresses aerobic glycolysis in cancer cells, and PDK1 expression is critical for Lin28A- and Lin28B-mediated cancer proliferation both in vitro and in vivo [70]. We further uncover the molecular mechanism that Lin28/let-7 axis regulates cancer cell aerobic glycolysis by targeting PDK1, but in a hypoxia- or HIF-1-independent manner.
Regulation of fatty acid metabolism in cancer cells
Although received less attention compared to aerobic glycolysis, elevated lipid synthesis has been recognized as another important aberration of metabolism required for carcinogenesis recently [71]. A fatty acid is a carboxylic acid with a long aliphatic chain, which mostly occurs in even numbers of carbons and can be either saturated or unsaturated. Fatty acids are required for energy storage, membrane synthesis and production of signaling molecules [72]. Here we focus on some new studies about aberrant regulation of fatty acid anabolism and catabolism in cancers (Fig. 2).

Fatty acid anabolism and catabolism in cancer cells. Both fatty acid anabolism and catabolism are dysregulated in cancer cells. Please see more detail in the text. FA fatty acid, CIC citrate carrier, CPT1 carnitine palmitoyl transferase 1, ACLY ATP citrate lyase, ACC acetyl-CoA carboxylase, MCD malonyl-CoA decarboxylase, FASN fatty acid synthase, ACS acetyl-CoA synthetase
Regulation of fatty acid anabolism
Fatty acid (FA) biosynthesis is frequently increased in cancer cells to satisfy the requirement of lipids for synthesis of membranes and signaling molecules, and cancer cells usually obtain higher lipid accumulation in the form of lipid droplets than that of normal cells. Citrate, produced via TCA cycle in the mitochondria, is exported across the inner mitochondrial membrane into the cytosol by the transport protein citrate carrier (CIC), and then feeds into de novo FA synthesis. CIC levels are elevated in various human cancer cell lines and its activity is required for tumor proliferation in vitro and tumorigenesis in vivo [73]. Chemical inhibition of CIC not only reduces cytoplasmic citrate levels, but also limits breast cancer cell viability effectively [74].
ATP citrate lyase (ACLY) is the first rate-limiting enzyme involved in de novo lipogenesis and links glucose metabolism and FA metabolism by converting citrate to oxaloacetate and acetyl-CoA, which feeds into FA synthesis in the cytoplasm. By converting glucose-derived citrate into acetyl-CoA, ACLY is also required for increasing histone acetylation in mammalian cells in response to growth factor stimulation [75]. Expression of ACLY is upregulated in a number of cancers, including colorectal cancer [76], breast cancer [77], glioblastoma [78] and ovarian cancer [79]. Overexpression of ACLY promotes but knockdown of ACLY inhibits tumor cell growth [80–82]. Acetylation of ACLY stabilizes this enzyme by inhibiting its ubiquitylation and degradation and promotes de novo lipid synthesis and tumor cell proliferation, but deacetylation of ACLY by deacetylase, such as SIRT2, destabilizes ACLY [83]. Except its function in de novo lipogenesis, ACLY has been found to regulate cellular senescence. ACLY can inhibit AMPK activity by physically interacting with the catalytic subunit of AMPK, and the activation of AMPK under ACLY knockdown conditions may result in p53 activation, ultimately leading to cellular senescence [84].
Acetyl-CoA carboxylase (ACC) catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, which is another rate-limiting step in fatty acid synthesis. There are two ACC forms in the human genome, ACC1 and ACC2. ACC1 is located in the cytosol and is highly expressed in lipogenic tissues, but ACC2 is imbedded in the mitochondrial membrane and primarily locates in oxidative tissues [85]. It is ACC1 that is responsible for the rate-limiting step of de novo fatty acid synthesis by converting acetyl-CoA to malonyl-CoA. ACC2 may be involved in the regulation of fatty acid oxidation, rather than fatty acid biosynthesis. AMPK, a central metabolic sensor that is directly phosphorylated and activated by tumor suppressor LKB1 [86], strongly inhibits fatty acid synthesis by phosphorylating and inactivating ACC1 [87]. Conversely, MCD (malonyl-CoA decarboxylase) catalyzes the breakdown of malonyl-CoA to acetyl-CoA and carbon dioxide. Balance between fatty acid synthesis and oxidation is regulated in part by the cellular levels of the metabolite malonyl-CoA. As the mitochondrial sirtuin, SIRT4 directly binds, deacetylates, and inhibits malonyl-CoA decarboxylase (MCD). Consequently, SIRT4 represses fatty acid oxidation and promoting lipid anabolism [88].
Fatty acid synthase (FASN) catalyzes the terminal steps in the de novo biogenesis of fatty acids. Under the function of FASN, palmitate was formed from malonyl-CoA and acetyl-CoA substrates via successive condensation reactions and further processed into saturated fatty acids in the presence of NADPH. The expression of FASN is over-expressed in many types of cancers, and high expression and activity of FASN provide a survival advantage in cancer cells [89]. A recent study reports that in ovarian cancer and its precursor cells, FASN is a metabolic marker of cell proliferation rather than a marker of malignancy [90]. Although lack of FASN expression in quiescent normal cells further shows it is a useful target for future drug development [90]. Knobloch et al. has shown that FASN is highly active in adult neural stem and progenitor cells (NSPCs) and conditional deletion of FASN in mouse NSPCs impairs adult neurogenesis [91]. Therefore, side effect of FASN inhibition on brain and neurons should be deliberated when we address the cancer therapy potential of FASN inhibitors.
SREBP1 (Sterol regulatory element binding protein 1), the basic helix loop helix leucine zipper (bHLHLZ) family of transcription factors, regulates lipogenic processes by activating a wide array of genes involved in fatty acid and triglyceride biosynthesis, including ACLY, ACC1 and FASN [92]. Expression and activity of SREBP1, as well as transcription of its target genes, are repressed by overexpression of the NAD+-dependent protein deacetylase SIRT6, leading to reduced triglyceride levels in hepatocytes [93]. However, SIRT1 activates SREBP1 and lipogenesis and promotes endometrial tumor growth [94]. Although SREBP1 activity was suggested to be regulated by mTORC1 and contributes to Akt-dependent lipogenesis and cell growth [95], a recent study reveals that mTORC2 also positively regulates SREBP1 stability and lipogenesis [96].
Acetate fuels fatty acid synthesis
Acetyl-CoA is a central node in carbon metabolism and is primarily generated in the mitochondria through glycolysis, lipid catabolism and amino acid metabolism [97]. As a vital intermediate of carbon sources, acetyl-CoA can be utilized for the synthesis of nucleotides, fatty acids, cholesterol and glutamate, or further oxidation via the TCA cycle for ATP production. In mammalian cells cultured under nutrient-unlimited conditions, acetyl-CoA used for lipid synthesis is primarily converted from mitochondria-derived citrate. Many cancer cells are highly glycolytic and preferentially convert pyruvate into lactate, leading to failure of most pyruvate to enter the TCA cycle for the synthesis of citrate that is transported to the cytoplasm for ACLY-mediated production of acetyl-CoA. A new study from Tu’s lab [98] illustrates that in highly glycolytic or hypoxic cancer cells, acetate is captured as a carbon source of acetyl-CoA. The nucleocytosolic ACSS2 (acyl-CoA synthetases 2), but not mitochondrial positioning ACSS1 and ACSS3, is the major enzyme required for the incorporation of acetate into lipids and histones and support tumor growth and survival. Loss of ACSS2 exhibits a significant reduction in tumor burden in adult mice model with liver cancer, and high expression of ACSS2 is associated with poor overall survival in triple negative breast cancer. Their study indicates that acetate uptake mediated by ACSS2 supports tumor cell growth and survival under nutrient-limiting conditions. In the same issue of Cell, the study of the Bachoo lab also provides a potential mechanistic link between ACSS2 activity and in vivo acetate oxidation in tumors [99]. Using 13C-NMR analysis, they have demonstrated that glucose contributes less than 50 % of the carbons to the acetyl-CoA pool in brain tumors resected from patients with glioblastomas and brain metastases [100]. Further in this study, they find that besides [1, 6-13C] glucose, primary and metastatic mouse orthotopic brain tumors have the capacity to oxidize [1, 2-13C] acetate in the citric acid cycle simultaneously, but the tumors do not oxidize [U-13C] glutamine [99]. ACSS2 correlates with acetate metabolism in brain tumors, and 13C-acetate fails to label citric acid cycle intermediates in ACSS2 knockout mouse embryo fibroblasts. The finding that an array of cancers are addicted in acetate uptake mediated by ACSS2 may qualify ACSS2 as a potential and valuable target for cancer therapy.
Regulation of the mevalonate pathway
Besides used in de novo fatty acid synthesis, acetyl-coA can also be utilized as substrate for the mevalonate pathway. The mevalonate pathway is an important metabolic pathway that plays a key role in multiple cellular processes by synthesizing sterol isoprenoids, such as cholesterol, and non-sterol isoprenoids, such as heme-A, ubiquinone and dolichol. In addition, the metabolites farnesyl pyrophosphate and geranylgeranyl pyrophosphate can serve as prenyl donors for a posttranslational modification at the C-terminus of various cellular proteins, which is defined as protein prenylation. AMPK, a major cellular kinase, phosphorylates and inactivates HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis [101]. SREBPs activate the genes encoding nearly every key enzyme in both the fatty acid and sterol biosynthetic pathways [102]. Tumor suppressor pRb downregulates many genes involved in protein isoprenylation by the way of E2F-dependent transcription control, and inactivation of pRb induces aberrant expression of its target genes including farnesyl diphosphate synthase, most of prenyltransferases, and their upstream regulators SREBPs, leading to enhanced isoprenylation and activation of N-Ras, which plays a role in Rb-Deficient C cell carcinogenesis of the thyroid [103]. Recently, it was reported that mutant p53 interact with SREBPs and is recruited to the promoters of multiple genes encoding mevalonate pathway enzymes, including HMG-CoA reductase, in a SREBP-dependent way [104]. By increasing expression of the genes in the mevalonate pathway, mutant p53 may affect 3D morphology of breast cancer cells.
Regulation of fatty acid catabolism
Unlike well-characterized glycolysis and fatty acid synthesis pathway, changes of fatty acid oxidation (FAO; also known as β-oxidation) in cancer cells still remain largely unknown. Fatty acid oxidation pathway occurs in the mitochondria, but accumulating studies show that fatty acid can also be oxidized by autophagic pathway in the cytoplasm [105]. Fatty acids need to be transformed to acyl-CoA before they enter into the subsequent metabolism, including anabolism or catabolism. Acyl-CoA synthetase, also known as fatty acid-CoA ligase, located on the endoplasmic reticulum and mitochondrial outer membrane, catalyzes the conversion of fatty acids to FA-CoA in the presence of ATP, CoA and Mg2+ [106]. Acyl-CoA synthases can be classified into very long-chain acyl-CoA synthases (ACSVL), long-chain acyl-CoA synthases (ACSL), medium-chain acyl-CoA synthases (ACSM) and shortchain acyl-CoA synthases (ACSS), according to the carbon chain length of the fatty acid they catalyze [106]. ACSL4, one of the ACSL family members, express markedly higher in the cancerous tissues than in the adjacent non-cancerous liver tissues of hepatocellular carcinoma patients, and is involved in tumorigenesis, probably by both cAMP and p38 MAPK pathways [107]. High levels expression of ACSL4 is also observed in breast cancer cells and this promotes tumor growth both in vitro and in vivo [108].
CPT1 (carnitine palmitoyltransferase 1) is responsible for the transfer of free FA-CoA across the mitochondrial membrane mediated by carnitine for subsequent oxidation. By repeated multi-round reaction of enzyme-catalyzed dehydrogenation, hydration, dehydrogenation and thiolysis, fatty acid can be processed by means of β-oxidation, which produced acetyl-CoA, NADH, and FADH2 in each cycle. Acetyl-CoA generated by FAO enters into TCA cycle for synthesis of citrate, and NADH and FADH2 enter the electron transport chain to produce ATP. Recently, we identified that by inhibiting the expression of MCAD and LCAD (the medium- and long-chain acyl-CoA dehydrogenases), HIF-1α suppresses fatty acids β-oxidation in cancer cells under hypoxia [109].
Regulation of amino acid metabolism in cancer cells
Cancer cells have an increased requirement for amino acids to meet their rapid proliferation. Amino acids can be divided into two classes: nonessential amino acids, such as glutamate, glutamine, serine, glycine, and proline; and essential amino acids, such as arginine, leucine, and methionine. In addition to being utilized as substrates for protein synthesis, amino acids have been documented by mounting studies that they function as metabolites and metabolic regulators in supporting cancer cell growth, among which research on glutamine, serine and glycine has been focused (Fig. 3).

Amino acid metabolism in cancer cells and its crosstalk with other metabolism pathways. Amino acids synthesis, utilization, and involvement in other metabolism pathways are usually changed in cancer cells. Please see more detail in the text. α-KG α-ketoglutarate, GSA glutamic semialdehyde, P5C pyrroline-5-carboxylate, GLS glutaminase, GLUD1 glutamate dehydrogenase 1, ASS argininosuccinate synthetase, ASL argininosuccinate lyase, ADI arginine deiminase, IDH1 isocitrate dehydrogenase-1, ACO1 aconitase 1, SSR serine racemase. Dashed arrows represent indirect effects or serial reactions
Glutamine
Increased glutamine metabolism is a common metabolic alteration in cancer and the importance of glutamine as nutrient is considered second only to glucose in cancer. As the most abundant free amino acid, glutamine participates in an array of pathways in energy generation, macromolecular synthesis and signal transmission in cancer cells by donating its nitrogen and carbon. Glutamine is imported into cytoplasm via transporters (e.g., SLC1A5 or ASCT2) and glutamine catabolism begins with its conversion to glutamate, catalyzed by the glutaminase (GLS) [110, 111]. Glutaminase expression and glutamine metabolism were activated by oncogenic transcription factor cMyc in cancer cells [112, 113]. A study also demonstrated that GLS2 is directly bound and induced by tumor suppressor p53 at the transcriptional level, and controls glutamine metabolism and GSH antioxidant capacity to decrease intracellular ROS levels [114]. Glutamate can be transferred into mitochondria and converted to α-ketoglutarate through oxidative deamination by glutamate dehydrogenase (GLUD1). Glutamate can also be transformed to α-ketoglutarate by transamination either in the cytoplasm or mitochondria, producing nonessential amino acids (e.g., serine) at the same time. Subsequently, α-ketoglutarate in the mitochondrion is utilized as a TCA cycle intermediate for energy recycling. Son et al. reported that in pancreatic cancer, oncogenic KRAS also regulates reprogramming of glutamine metabolism, through the transcriptional upregulation of aspartate transaminase (GOT1) and repression of GLUD1 [115]. Using [U-13C, 15N]-glutamine as the tracer, Le et al. found that cMyc induces glutamine to glutamate conversion that persists in hypoxia, and glutamine contributed significantly to citrate carbons [116]. Furthermore, glutamine-derived fumarate, malate, and citrate were significantly increased when glucose is deprived, which suggests that glutamine drives glucose-independent TCA cycle. Increased use of glutamine for mitochondrial-dependent bio-energy production and cellular biosynthesis is a key feature of many tumor cells. A recent study demonstrated that silencing of LKB1 (liver kinase B1), a serine/threonine kinase and tumor suppressor that couples bioenergetics to cell growth control through regulation of mTOR activity, increases glucose and glutamine consumption in tumor cells [117]. Many cancer cells utilize acetyl-CoA mainly converted from glucose metabolism product pyruvate [118], but a pathway in which acetyl-CoA was generated from glutamine downstream of GDH will be activated upon glucose deprivation. A recent study from DeBerardinis’s group showed that import of pyruvate into the mitochondria suppresses GDH and glutamine-dependent acetyl-CoA formation. Nonetheless, blockade of the mitochondrial pyruvate transport activates GDH and redirects glutamine metabolism to generate both oxaloacetate and acetyl-CoA, inducing glutamine-dependent lipid synthesis [119].
Serine and glycine
Although partially derived from glucose metabolism, serine can also be obtained by extracellular uptake. Serine and glycine are linked in biosynthesis, and together refuel as essential precursors for the synthesis of building blocks including proteins, nucleic acids, and lipids that are crucial to cancer proliferation. Both de novo synthesis of serine derived from 3-phosphoglycerate and imported serine can be further converted to glycine under the catalyzation of SHMTs, which are direct transcriptional targets of c-Myc [120]. Glycine can also be transformed from threonine by threonine dehydrogenase and glycine C-acetyltransferase [67]. Subsequently, glycine provides methyl groups for one-carbon metabolism required by the synthesis of nucleic acids, proteins and lipids, as well as DNA methylation. However, Labuschagne et al. recently found that exogenous glycine cannot replace serine to support cancer cell proliferation because cancer cells selectively consumes exogenous serine, which is converted into intracellular glycine and one-carbon units for building nucleotides [121]. Moreover, uptake of exogenous glycine without serine loses the ability to support nucleotide synthesis [121]. Their study suggests that cancer cell proliferation is supported by serine rather than glycine consumption.
Proline
Proline is a unique proteinogenic secondary amino acid and stored in collagen, the most abundant protein in the body [122]. Proline is interconvertible with glutamate, in which Δ1-pyrroline-5-carboxylate (P5C) and glutamic-γ-semialdehyde (GSA) are utilized as intermediates. Proline dehydrogenase (oxidase) (PRODH/POX), which catalyzes proline to P5C and functions as a mitochondrial tumor suppressor, is induced by p53 and PPARγ but suppressed by miR-23b* and cMyc [123]. Furthermore, GSA derived from glutamate or proline can be converted into ornithine, which serves as a precursor for arginine synthesis in the Urea cycle [122]. A recent study indicates that proline metabolism is involved in aggressive phenotype of cancer [124]. Nishio’s team found that overexpression of the ORAOV1 (oral cancer overexpressed 1) gene in ESCC (esophageal squamous cell cancer) cell lines enhances cellular growth and colony formation. The possible mechanism is that ORAOV1 binds to pyrroline-5-carboxylate reductase (PYCR) and influences its activity. As a result, PYCR converts P5C back to proline, leading to increased intracellular proline level and reduced ROS (reactive oxygen species) production, thereby promoting tumor progression [124].
Arginine
Arginine is an essential amino acid and many types of solid tumor cells died rapidly in culture medium with arginine deprivation [125]. Arginine participates in many important cellular metabolic pathways including urea cycle, biosyntheses of nitric oxide, nucleotides, proline and glutamate [126]. Argininosuccinate synthetase (ASS) catalyzes the synthesis of argininosuccinate from l-citrulline and aspartic acid and is the rate-limiting enzyme for the de novo biosynthesis of arginine. As a next step, argininosuccinate lyase (ASL) converts argininosuccinate into l-Arginine and fumaric acid, and the latter links arginine metabolism to glucose-generated energy metabolism via TCA cycle. ASS is deficient in some human cancers, especially in malignant melanoma and hepatocellular carcinoma, which are unable to synthesize arginine and are therefore susceptible to arginine deprivation therapy using arginine-degrading enzymes [127]. This character was utilized for the treating of this subset of cancers, because these cancer cells would die when they are exposed to recombinant arginine-degrading enzymes (arginine deiminase or arginase).
Crosstalk among glucose, fatty acid, and amino acid metabolism, as well as cancer cell signaling
Crosstalk among glucose, fatty acid, and amino acid metabolism
In most tumor cells, glucose is the major lipogenic substrate. Fatty acid synthesis relies on citrate efflux from the mitochondria, but the TCA cycle does not function as a carbon sink. Glucose-derived pyruvate is converted to acetyl-CoA by PDH and entry into the TCA cycle to be transformed to citrate, which is exported from the mitochondria to the cytosol and utilized for de novo fatty acid biosynthesis [118]. In LNCaP prostate cancer cells, androgen can enhance the utilization of glucose for de novo lipid synthesis by increasing the expression of HK2 (hexokinase 2) and the cardiac isoform of PFKFB2 (6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 2) [128]. This study supports the notion that the metabolites from glycolysis are the main carbon sources for lipid synthesis.
Besides glucose, glutamine can also replenish TCA cycle intermediates by deriving α-ketoglutarate and contributes to the production of citrate by forward flux through the TCA cycle [118], which is mitochondrial glutamine oxidation pathway. However, glutamine-derived α-ketoglutarate can also be converted to citrate by the reversal of the TCA cycle reactions catalyzed by isocitrate dehydrogenase-1 (IDH1) and aconitase 1 (ACO1) in the cytoplasm [129, 130] (Fig. 3), which is a process of glutamine-dependent reductive carboxylation. Hypoxia can shift glutamine metabolism from oxidation to reductive carboxylation. Sun et al. identify the mechanism by which HIF stabilization under hypoxia promotes E3 ubiquitin ligae SIAH2 targeted ubiquitination and proteolysis of the E1 subunit of the α-KGDH (α-ketoglutarate dehydrogenase), leading to reduced α-KGDH activity and mitochondrial oxidation of glutamine to succinate (standard TCA cycle reaction catalyzed by α-KGDH). Furthermore, HIF-dependent change in glutamine metabolism shifts the fate of α-ketoglutarate from energy production to the production of lipids and promotes the growth of tumors in vivo [131].
Relationship also exists between glucose and amino acid metabolism. Gottlieb’s group found that serine is an allosteric activator of PKM2 whose activity in cells is reduced in response to serine deprivation [132]. By binding to and activating PKM2, serine supports aerobic glycolysis and lactate production, which are critical for cancer cell growth and survival. On the contrary, Thompson’s group showed that PKM2 expression contributes to the de novo serine synthetic pathway [133]. PKM2-expressing cells can maintain mTORC1 (mammalian target of rapamycin complex 1) activity and proliferate in serine-depleted medium by promoting endogenous serine synthesis. Another glycolytic enzyme, GAPDH, is also related to serine metabolism. l-serine can be converted to d-serine by SSR (serine racemase). GAPDH inhibits d-serine production by direct binding and inhibiting SRR activity or by exposing SRR to a glycolytic metabolite, NADH [134]. Glycolytic flux decreased by 2-deoxyglucose, an inhibitor of hexokinase, results in impaired interaction between SRR and GAPDH. Subsequently, free SRR facilitates d-serine generation.
Interaction between metabolic alterations and cancer signaling pathways
It has been well documented that anabolic reprogramming is induced by multiple signaling pathway, such as PI3K/Akt signaling and Ras/ERK signaling pathway [135]. Conversely, emerging studies demonstrate that metabolic enzymes participate in modulation of cell signaling pathway, as well as function regulation of transcription factors. Here we list some representative researches about the function of metabolic enzymes beyond the well recognized catalyzation in the glycolysis flux.
HK2, a critical glycolytic enzyme, activates G1/S checkpoint through regulating the protein level and T14 phosphorylation of CDK2 in cancer-associated fibroblasts [136]. Phosphofructokinase (PFK) is the second rate-limiting enzyme in glycolysis and is a key mediator of glycolytic flux in cancer progression [137]. However, PFK also regulates YAP/TAZ transcriptional activity by binding the YAP/TAZ transcriptional cofactors TEADs and promoting their functional and biochemical cooperation with YAP/TAZ [138].
As the third rate-limiting enzyme of glycolysis, pyruvate kinase M2 (PKM2) is commonly upregulated in many human cancers. It has been reported that activation of PKM2 by growth factor signaling via PI3K/AKT/mTOR axis is critical for aerobic glycolysis and tumor growth [139]. Nevertheless, there is some controversy about the function of PKM2 is oncogenic. Despite the expression of protein, the enzyme activity of PKM2 can be decreased in rapidly dividing cancer cells [140]. Moreover, some studies have shown that PKM2 activation inhibits xenograft tumor growth [141] and PKM2 loss in a mouse model of breast cancer accelerates tumor formation [142]. Besides a metabolic role, emerging studies have revealed the role of PKM2 in gene transcription and its ability to act as protein kinase. It has been documented that PKM2 plays a key role in stabilizing HIF-1α and regulates HIF-1α-dependent genes [143]. PKM2 activating transcription of HIF-1α by phosphorylating STAT3 at Y705 (tyrosine 705) may promote liver cancer cell proliferation [144]. Another study shows that overexpression of PKM2, but not PKM1, promotes colorectal cancer cell migration and cell adhesion by regulating STAT3-associated signalling [145]. PKM2 is overexpressed in HCC cell lines and tissues, and PKM2 knockdown inhibits HCC cell proliferation and induces apoptosis [146]. Bim, a member of Bcl-2 family proteins which possesses diverse pro-apoptotic activity, is considered as an essential factor for PKM2-depletion-induced apoptosis because PKM2 knockdown attenuates the degradation of Bim. Although it has not been ascertained that regulation of Bim expression by PKM2 is direct or indirect [146]. Besides located in the cytoplasm as a glycolytic enzyme and located in the nucleus as a protein kinase, PKM2 can also be secreted outside from the cell. Yang et al. demonstrated that secreted PKM2 from colon cancer cells promotes cell migration by activating PI3K/Akt and then stimulates Wnt/β-catenin signaling pathway [147].
Accumulating evidence uncovers alternative function of aldolase beyond glycolytic metabolism. Aldolase depletion reduces the proliferation of several transformed cell lines, but does not affect glycolytic flux or intracellular ATP concentration, revealing that aldolase affects cancer cell proliferation through a non-glycolytic mechanism [148]. Aldolase also activates the canonical Wnt signaling pathway by forming a complex with GSK-3β that disrupts the Axin-GSK-3β interaction [149]. Fu et al. reported recently that in non-small cell lung cancer, alpha-enolase (ENO1) promotes cell glycolysis, proliferation, migration, and invasion through activating FAK/PI3K/AKT pathway and its downstream signals [150].
Cancer therapy by targeting cancer metabolic reprogramming
Altered tumor metabolism can be utilized as a target for cancer therapy, which aims to the development of small-molecule inhibitors of metabolic enzymes for treating many types of malignancies. One of the metabolic changes exhibited by tumor cells is an increase in glucose metabolism. It provoked the exploration of the drugs that target key metabolic enzymes to block or attenuate the glycolytic flux, which may lead to the death of cancer cells but have minimal adverse effects on normal cells. Besides glycolytic rate-limiting enzymes, glucose transporters and other metabolic enzymes have also been selected as targets for drug screening [151]. However, most of the promising drugs targeting glucose metabolism for anticancer therapy are still at the stage of development, based on cell line or animal data, including inhibitor of GLUT1 (WZB117) [152], GLUT4 (silybin) [153], HK2 (methyljasmonate) [154], PFKFB3 (1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one) [155], and LDHA (3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid) [52]. Except inhibitors of metabolic enzyme, activators of metabolic enzyme can also be utilized for potential treatment of cancer. Different from other metabolic enzymes, increased pyruvate kinase activity impair tumor growth [141]. Two small molecules, TEPP-46 and DASA-58, can specifically activate PKM2 in cells by promoting PKM2 tetramer formation and preventing inhibition by phosphotyrosine signaling, alter metabolism in cultured cells, and inhibit xenograft tumor growth [141]. Combination of therapeutic agents, as well as therapeutic agent with radiotherapy, may exhibit a synergetic anticancer therapeutic effect [154, 156].
Tumor cells rely on de novo synthesis of fatty acids for energy metabolism and membrane production that sustains fast growth and proliferation. It carves out the way to the research aimed to target lipogenic enzymes. Hydroxycitrate is an enzymatic inhibitor of ACLY. It has been demonstrated that hydroxycitrate treatment mimics the effects of ACLY knockdown and downregulates the stemness of caner stem cells [157]. ACC1 and ACC2 inhibition has been utilized for the treatment of metabolic diseases such as type 2 diabetes and dyslipidemia, as well as potential cancer therapeutic. Although a large number of ACC inhibitors have been reported by multiple research groups, only one compound developed by Pfizer company, PF-05175157, has been moved into human clinical trials [85]. FASN is up-regulated in many cancers but do not express in most normal non-fat tissues, which makes it a valuable target for drug discovery. Although various FASN inhibitors have been identified, none of them have been advanced to clinical use [158]. Recently, Zhang’s group identified that proton pump inhibitors (PPIs), which are FDA-approved therapeutics for treatment of a variety of acid-related disease, effectively repress the thioesterase activity of human FASN, inhibit proliferation, and induce apoptosis of pancreatic cancer cells [159]. Their study provides theory basis for repositioning PPIs as anticancer therapeutics.
Many types of tumor display addiction to glutamine, the most abundant amino acid in plasma. GLS converts glutamine to glutamate, which is converted into the TCA cycle intermediate α-ketoglutarate by glutamate dehydrogenase or aminotransferases [160]. One specific GLS inhibitor, the small molecule 968, blocks the GLS activity and inhibits oncogenic transformation in preclinical models of cancer, without affecting normal cells [161]. A recent study reports that aminooxyacetate, an aminotransferase inhibitor, targets glutamine metabolism in breast cancer and induces ER stress pathway leading to cell growth inhibition and apoptosis [162]. Notably, aminooxyacetate has shown antitumor effect in cancer models and acceptable toxicity profile in small clinical trials. These studies suggest aminooxyacetate as an appealing therapeutic agent for further clinical drug development targeting glutamine metabolism. Besides inhibition of glutamine metabolic enzymes, decrease of glutamine uptake also blocks cancer growth and tumor development. A recent study of Wang et al. shows chemical (benzylserine) or shRNA-mediated inhibition of ASCT2 (SLC1A5) function, which is highly expressed in prostate cancer patient samples, decreases glutamine uptake and cell growth, as well as fatty acid synthesis [163]. Furthermore, knockdown of ASCT2 in prostate cancer xenografts significantly inhibits tumor growth and metastasis in vivo. Therefore, it may provide new avenues for therapeutic intervention by targeting key glutamine transporter that regulates cancer proliferation and metabolism. Another kind of important amino acids involved in cancer metabolism is serine and glycine, whose metabolism promotes tumorigenesis and malignancy [164]. Serine and glycine are biosynthetically linked, and together provide the essential precursors for the synthesis of proteins, nucleic acids, and lipids that are crucial to cancer cell growth, especially through the participation of glycine in one-carbon metabolism. So far, several drugs targeting enzymes that catalyze the generation of tetrahydrofolate involved in one-carbon metabolism, including methotrexate and Pemetrexed against dihydrofolate reductase, have been clinically approved in multiple cancers [67]. Nevertheless, the development of drugs for targeting of PHGDH, PSAT and PSPH in anti-serine biosynthesis and for targeting of SHMT1 and SHMT2 in anti-glycine biosynthesis is in preclinical study.
Although some drugs can be applied to multiple cancers, most of them are suitable for only one specific type of cancer for the heterogeneity of cancer. Reprogramming of glucose, fatty acid and amino acid metabolism endows cancer cells with ability of growth and survival, even under the limitation of one kind of nutrient supplying. Design of combination therapies using multiple metabolic inhibitors may help to improve the anticancer efficacy [165]. The basic thought of cancer therapy would be carried forward through understanding more about the basic biology of these energy metabolism pathways and their crosstalk in regulating tumorigenesis.
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314
Warburg O (1956) On respiratory impairment in cancer cells. Science 124(3215):269–270
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27:441–464
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033
Wellen KE et al (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24(24):2784–2799
Gao P, Sun L, He X, Cao Y, Zhang H (2012) MicroRNAs and the Warburg effect: new players in an old arena. Curr Gene Ther 12(4):285–291
Kim JW et al (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24(13):5923–5936
Mikawa T et al (2015) Dysregulated glycolysis as an oncogenic event. Cell Mol Life Sci 72(10):1881–1892
Li F et al (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25(14):6225–6234
Sun L et al (2015) cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res 25(4):429–444
Graven KK, Yu Q, Pan D, Roncarati JS, Farber HW (1999) Identification of an oxygen responsive enhancer element in the glyceraldehyde-3-phosphate dehydrogenase gene. Biochim Biophys Acta 1447(2–3):208–218
Zhang JY et al (2015) Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med 12(1):10–22
Kress S et al (1998) Expression of hypoxia-inducible genes in tumor cells. J Cancer Res Clin Oncol 124(6):315–320
Huang D, Li C, Zhang H (2014) Hypoxia and cancer cell metabolism. Acta Biochim Biophys Sin 46(3):214–219
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E (2004) The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 64(7):2627–2633
Zhang C et al (2014) Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget 5(14):5535–5546
Mikawa T et al (2014) Senescence-inducing stress promotes proteolysis of phosphoglycerate mutase via ubiquitin ligase Mdm2. J Cell Biol 204(5):729–745
Bensaad K et al (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1):107–120
Yuan H, Su L, Chen WY (2013) The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther 6:1399–1416
German NJ, Haigis MC (2015) Sirtuins and the Metabolic Hurdles in Cancer. Curr Biol 25(13):R569–R583
Zwaans BM, Lombard DB (2014) Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Model Mech 7(9):1023–1032
Hallows WC, Yu W, Denu JM (2012) Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem 287(6):3850–3858
Tsusaka T et al (2014) Deacetylation of phosphoglycerate mutase in its distinct central region by SIRT2 down-regulates its enzymatic activity. Genes Cells 19(10):766–777
Xu Y et al (2014) Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res 74(13):3630–3642
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13(4):225–238
Sun L, He X, Cao Y, Gao P, Zhang H (2014) MicroRNAs and energy metabolism in cancer cells, chapter 4. In: Babashah S (ed) MicroRNAs: key regulators of oncogenesis. Springer, Switzerland, pp 84–95
Hung CL et al (2014) A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci USA 111(52):18697–18702
Vander Heiden MG et al (2011) Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 76:325–334
Dang CV (2012) Links between metabolism and cancer. Genes Dev 26(9):877–890
Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17(4):351–359
Chen B et al (2015) miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett 356(2 pt B):410–417
Rathmell JC et al (2003) Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23(20):7315–7328
Wieman HL, Wofford JA, Rathmell JC (2007) Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18(4):1437–1446
Morani F et al (2014) PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol 53(2):247–258
Goos JA et al (2015) Glucose transporter 1 (SLC2A1) and vascular endothelial growth factor A (VEGFA) predict survival after resection of colorectal cancer liver metastasis. Ann Surg. doi:10.1097/SLA.0000000000001109
Kawauchi K, Araki K, Tobiume K, Tanaka N (2008) p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 10(5):611–618
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25(34):4777–4786
Kim HR, Roe JS, Lee JE, Cho EJ, Youn HD (2013) p53 regulates glucose metabolism by miR-34a. Biochem Biophys Res Commun 437(2):225–231
Peschiaroli A et al (2013) miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene 32(6):797–802
Wang L et al (2014) Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep 8(5):1461–1474
Li Z, Li X, Wu S, Xue M, Chen W (2014) Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci 105(8):951–955
Du S et al (2014) Fructose-bisphosphate aldolase a is a potential metastasis-associated marker of lung squamous cell carcinoma and promotes lung cell tumorigenesis and migration. PLoS One 9(1):e85804
Long F, Cai X, Luo W, Chen L, Li K (2014) Role of aldolase A in osteosarcoma progression and metastasis: in vitro and in vivo evidence. Oncol Rep 32(5):2031–2037
Sun Y, Long J, Zhou Y (2014) Angiopoietin-like 4 promotes melanoma cell invasion and survival through aldolase A. Oncol Lett 8(1):211–217
Guo C, Liu S, Sun MZ (2013) Novel insight into the role of GAPDH playing in tumor. Clin Transl Oncol 15(3):167–172
Ramos D et al (2015) Deregulation of glyceraldehyde-3-phosphate dehydrogenase expression during tumor progression of human cutaneous melanoma. Anticancer Res 35(1):439–444
Li T et al (2014) Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem 289(6):3775–3785
Colell A et al (2007) GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129(5):983–997
Jiang X, Sun Q, Li H, Li K, Ren X (2014) The role of phosphoglycerate mutase 1 in tumor aerobic glycolysis and its potential therapeutic implications. Int J Cancer 135(9):1991–1996
Hitosugi T et al (2012) Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22(5):585–600
Le A et al (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107(5):2037–2042
Liu X et al (2015) Effects of the suppression of lactate dehydrogenase A on the growth and invasion of human gastric cancer cells. Oncol Rep 33(1):157–162
Li X, Zhao H, Zhou X, Song L (2015) Inhibition of lactate dehydrogenase A by microRNA-34a resensitizes colon cancer cells to 5-fluorouracil. Mol Med Rep 11(1):577–582
McCleland ML et al (2012) An integrated genomic screen identifies LDHB as an essential gene for triple-negative breast cancer. Cancer Res 72(22):5812–5823
Tambe Y, Hasebe M, Kim CJ, Yamamoto A, Inoue H (2015) The drs tumor suppressor regulates glucose metabolism via lactate dehydrogenase-B. Mol Carcinog. doi:10.1002/mc.22258
Jiang P, Du W, Wu M (2014) Regulation of the pentose phosphate pathway in cancer. Protein Cell 5(8):592–602
Nogueira V, Hay N (2013) Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res 19(16):4309–4314
Jiang P et al (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13(3):310–316
Hong X et al (2014) PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 63(10):1635–1647
Patra KC, Hay N (2014) The pentose phosphate pathway and cancer. Trends Biochem Sci 39(8):347–354
Patra KC et al (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24(2):213–228
Yi W et al (2012) Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337(6097):975–980
Anastasiou D et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334(6060):1278–1283
Fukuda S et al (2015) Pyruvate kinase M2 modulates esophageal squamous cell carcinoma chemotherapy response by regulating the pentose phosphate pathway. Ann Surg Oncol. doi:10.1245/s10434-015-4522-3
DeBerardinis RJ (2011) Serine metabolism: some tumors take the road less traveled. Cell Metab 14(3):285–286
Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G (2014) Serine and glycine metabolism in cancer. Trends Biochem Sci 39(4):191–198
Zhang H et al (2007) HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11(5):407–420
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3):177–185
Ma X et al (2014) Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat Commun 5:5212
Swierczynski J, Hebanowska A, Sledzinski T (2014) Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol 20(9):2279–2303
Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr (2013) Cellular fatty acid metabolism and cancer. Cell Metab 18(2):153–161
Catalina-Rodriguez O et al (2012) The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 3(10):1220–1235
Ozkaya AB, Ak H, Atay S, Aydin HH (2015) Targeting mitochondrial citrate transport in breast cancer cell lines. Anti-Cancer Agents Med Chem 15(3):374–381
Wellen KE et al (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324(5930):1076–1080
Zhou Y et al (2013) ATP citrate lyase mediates resistance of colorectal cancer cells to SN38. Mol Cancer Ther 12(12):2782–2791
Szutowicz A, Kwiatkowski J, Angielski S (1979) Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br J Cancer 39(6):681–687
Beckner ME et al (2010) Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer 126(10):2282–2295
Wang Y et al (2012) Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep 27(4):1156–1162
Zaidi N, Swinnen JV, Smans K (2012) ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 72(15):3709–3714
Hanai J et al (2012) Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol 227(4):1709–1720
Zong H, Zhang Y, You Y, Cai T, Wang Y (2015) Decreased Warburg effect induced by ATP citrate lyase suppression inhibits tumor growth in pancreatic cancer. Med Oncol 32(3):85
Lin R et al (2013) Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 51(4):506–518
Lee JH et al (2015) ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J 282(2):361–371
Bourbeau MP, Bartberger MD (2015) Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J Med Chem 58(2):525–536
Shaw RJ et al (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101(10):3329–3335
Carling D, Zammit VA, Hardie DG (1987) A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 223(2):217–222
Laurent G et al (2013) SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell 50(5):686–698
Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7(10):763–777
Hopperton KE, Duncan RE, Bazinet RP, Archer MC (2014) Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp Cell Res 320(2):302–310
Knobloch M et al (2013) Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 493(7431):226–230
Sun Y et al (2015) SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol 36(6):4133–4141
Elhanati S et al (2013) Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep 4(5):905–912
Lin L et al (2014) SIRT1 promotes endometrial tumor growth by targeting SREBP1 and lipogenesis. Oncol Rep 32(6):2831–2835
Porstmann T et al (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8(3):224–236
Li S, Oh YT, Yue P, Khuri FR, Sun SY (2015) Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene. doi:10.1038/onc.2015.123
Lyssiotis CA, Cantley LC (2014) Acetate fuels the cancer engine. Cell 159(7):1492–1494
Comerford SA et al (2014) Acetate dependence of tumors. Cell 159(7):1591–1602
Mashimo T et al (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159(7):1603–1614
Maher EA et al (2012) Metabolism of [U-13 C] glucose in human brain tumors in vivo. NMR Biomed 25(11):1234–1244
Zhang X et al (2015) Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J Lipid Res 56(5):963–971
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109(9):1125–1131
Shamma A et al (2009) Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 15(4):255–269
Freed-Pastor WA et al (2012) Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148(1–2):244–258
Singh R, Cuervo AM (2012) Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012:282041
Yan S et al (2015) Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol 21(12):3492–3498
Liang YC et al (2005) Involvement of fatty acid-CoA ligase 4 in hepatocellular carcinoma growth: roles of cyclic AMP and p38 mitogen-activated protein kinase. World J Gastroenterol 11(17):2557–2563
Wu X et al (2013) Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One 8(10):e77060
Huang D et al (2014) HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep 8(6):1930–1942
Hassanein M et al (2013) SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res 19(3):560–570
Mates JM et al (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13(4):514–534
Gao P et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458(7239):762–765
Wise DR et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105(48):18782–18787
Suzuki S et al (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 107(16):7461–7466
Son J et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105
Le A et al (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15(1):110–121
Faubert B et al (2014) Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci USA 111(7):2554–2559
DeBerardinis RJ et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104(49):19345–19350
Yang C et al (2014) Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell 56(3):414–424
Nikiforov MA et al (2002) A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol Cell Biol 22(16):5793–5800
Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD (2014) Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7(4):1248–1258
Phang JM, Liu W, Hancock CN, Fischer JW (2015) Proline metabolism and cancer: emerging links to glutamine and collagen. Curr Opin Clin Nutr Metab Care 18(1):71–77
Phang JM, Liu W, Hancock C, Christian KJ (2012) The proline regulatory axis and cancer. Front Oncol 2:60
Togashi Y et al (2014) Frequent amplification of ORAOV1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ROS production. Oncotarget 5(10):2962–2973
Scott L, Lamb J, Smith S, Wheatley DN (2000) Single amino acid (arginine) deprivation: rapid and selective death of cultured transformed and malignant cells. Br J Cancer 83(6):800–810
Kuo MT, Savaraj N, Feun LG (2010) Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget 1(4):246–251
Feun LG, Kuo MT, Savaraj N (2015) Arginine deprivation in cancer therapy. Curr Opin Clin Nutr Metab Care 18(1):78–82
Moon JS et al (2011) Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J 433(1):225–233
Metallo CM et al (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481(7381):380–384
Mullen AR et al (2012) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481(7381):385–388
Sun RC, Denko NC (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19(2):285–292
Chaneton B et al (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491(7424):458–462
Ye J et al (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA 109(18):6904–6909
Suzuki M et al (2015) Glycolytic flux controls d-serine synthesis through glyceraldehyde-3-phosphate dehydrogenase in astrocytes. Proc Natl Acad Sci USA 112(17):E2217–E2224
Gomes AP, Blenis J (2015) A nexus for cellular homeostasis: the interplay between metabolic and signal transduction pathways. Curr Opin Biotechnol 34C:110–117
Hu JW, Sun P, Zhang DX, Xiong WJ, Mi J (2014) Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal 26(10):2210–2216
Al Hasawi N, Alkandari MF, Luqmani YA (2014) Phosphofructokinase: a mediator of glycolytic flux in cancer progression. Crit Rev Oncol Hematol 92(3):312–321
Enzo E et al (2015) Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J 34(10):1349–1370
Sun Q et al (2011) Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 108(10):4129–4134
Vander Heiden MG et al (2010) Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329(5998):1492–1499
Anastasiou D et al (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8(10):839–847
Israelsen WJ et al (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155(2):397–409
Palsson-McDermott EM et al (2015) Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21(1):65–80
Dong T et al (2015) Pyruvate kinase M2 affects liver cancer cell behavior through up-regulation of HIF-1alpha and Bcl-xL in culture. Biomed Pharmacother 69:277–284
Yang P, Li Z, Fu R, Wu H, Li Z (2014) Pyruvate kinase M2 facilitates colon cancer cell migration via the modulation of STAT3 signalling. Cell Signal 26(9):1853–1862
Hu W et al (2015) Pyruvate kinase M2 prevents apoptosis via modulating Bim stability and associates with poor outcome in hepatocellular carcinoma. Oncotarget 6(9):6570–6583
Yang P et al (2015) Secreted pyruvate kinase M2 facilitates cell migration via PI3K/Akt and Wnt/beta-catenin pathway in colon cancer cells. Biochem Biophys Res Commun 459(2):327–332
Ritterson Lew C, Tolan DR (2012) Targeting of several glycolytic enzymes using RNA interference reveals aldolase affects cancer cell proliferation through a non-glycolytic mechanism. J Biol Chem 287(51):42554–42563
Caspi M et al (2014) Aldolase positively regulates of the canonical Wnt signaling pathway. Mol Cancer 13:164
Fu QF et al (2015) Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol 8(1):22
Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G (2013) Metabolic targets for cancer therapy. Nat Rev Drug Discov 12(11):829–846
Liu Y et al (2012) A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 11(8):1672–1682
Zhan T, Digel M, Kuch EM, Stremmel W, Fullekrug J (2011) Silybin and dehydrosilybin decrease glucose uptake by inhibiting GLUT proteins. J Cell Biochem 112(3):849–859
Klippel S et al (2012) Methyljasmonate displays in vitro and in vivo activity against multiple myeloma cells. Br J Haematol 159(3):340–351
Clem BF et al (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 12(8):1461–1470
Zhang D et al (2014) 2-Deoxy-d-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett 355(2):176–183
Hanai JI, Doro N, Seth P, Sukhatme VP (2013) ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis 4:e696
Pandey PR, Liu W, Xing F, Fukuda K, Watabe K (2012) Anti-cancer drugs targeting fatty acid synthase (FAS). Recent Pat Anti Cancer Drug Discov 7(2):185–197
Fako VE, Wu X, Pflug B, Liu JY, Zhang JT (2015) Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human fatty acid synthase. J Med Chem 58(2):778–784
Weinberg SE, Chandel NS (2015) Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 11(1):9–15
Wang JB et al (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18(3):207–219
Korangath P et al (2015) Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin Cancer Res 21(14):3263–3273
Wang Q et al (2015) Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol 236(3):278–289
Jain M et al (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336(6084):1040–1044
Schwartz L et al (2013) Tumor regression with a combination of drugs interfering with the tumor metabolism: efficacy of hydroxycitrate, lipoic acid and capsaicin. Invest New Drugs 31(2):256–264
Acknowledgments
This work was supported in part by National Basic Key Research Program of China (2014CB910600 and 2012CB910104), National Natural Science Foundation of China (31171358, 31371429, 81372215, 81572714 and 31301069), Specialized Research Fund for the Doctoral Program of Higher Education of China (20133402110020, 20133402120008), the Fundamental Research Funds for the Central Universities of China (WK2070000065, WK2060190018, WK2070000034), Anhui Provincial Natural Science Foundation (1408085MC42), and the Major/Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology (CX2070000104).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, Z., Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 73, 377–392 (2016). https://doi.org/10.1007/s00018-015-2070-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2070-4