
Abstract
Glioblastoma (GBM) is the most common primary brain tumors in adults. Despite aggressive multimodality therapies, GBM unfortunately remains among the most resistant cancers to treatment. In the past, traditional chemotherapy which works by impeding DNA synthesis or cell metabolism has been used to try and slow the progression of GBM with little success. Recently, research has become more focused into the development of targeted therapies in which drugs (small molecules or antibodies) effect specific molecular and genetic alterations in GBM attempting to inhibit and deregulate cell signaling pathways. The Cancer Genome Atlas (TCGA) GBM project has provided an in depth description of the distinct molecular and genetic alterations in GBM stimulating interest in the development of targeted molecular therapies. While the results of targeted therapy studies to date have failed to improve the overall survival of GBM patients, there continues to be enthusiasm in this approach with numerous clinical trials currently underway. Hopefully, knowledge from the previous failed trials will help provide further insight and assist future clinicians in designing new novel targeted treatments to overcome these barriers.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Glioblastoma
- targeted therapy
- The Cancer Genome Atlas
- Retinoblastoma pathway
- p53 pathway
- Receptor tyrosine kinase pathway
4.1 Recent Advances for Targeted Therapies in Glioblastoma
Gliobastoma multiforme (GBM) is the most common malignant primary brain tumor in adults (Davis et al. 2001; Cloughesy et al. 2014). Currently, 10,000 new cases of GBM are diagnosed each year in the United States, and approximately 100,000 new cases are diagnosed yearly worldwide (Davis et al. 2001; Cloughesy et al. 2014; Porter et al. 2010; Ohgaki and Kleihues 2005). Patients often initially undergo surgical resection to provide symptomatic relief and confirm a pathologic diagnosis. However, surgery is not curative as the tumor cells invade surrounding normal brain tissue rendering a complete resection of the tumor impossible (Cloughesy et al. 2014). Following a maximal safe resection, the standard of care treatment for newly diagnosed GBM consists of cytotoxic chemotherapy with daily temozolomide and concurrent radiation therapy for 6 weeks, followed by 6–12 cycles of adjuvant temozolomide (Masui et al. 2012; Stupp et al. 2005). Despite aggressive multimodality therapies, GBM unfortunately remains among the most resistant cancers to treatment leading to a median survival of around 16 months (Stupp et al. 2005). Several potential reasons have been proposed to explain GBMs resistance to treatment including the genetic heterogeneity of the tumor, elaborate signaling pathways, and difficulties with designing drugs capable of crossing the blood brain barrier (Tanaka et al. 2013). In the past, traditional chemotherapy which works by impeding DNA synthesis or cell metabolism has been most often used to try and slow the progression of GBM with little success. Recently, research has become more focused into the development of targeted therapies in which drugs (small molecules or antibodies) effect specific molecular and genetic alterations in GBM attempting to inhibit and deregulate cell signaling pathways. This chapter will explore current targeted therapies and how they relate to the aberrant signaling pathways in GBM.
4.2 The Cancer Genome Atlas
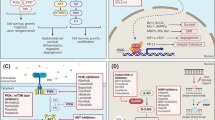
GBM was one of the first cancers studied by The Cancer Genome Atlas (TCGA) Research Network, a collaboration between the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI) . The TCGAs key aims were to identify changes in each cancer’s genome and understand how these changes interact to drive the disease, thereby laying the foundation for improved cancer prevention, early detection, and treatment (Cancer Genome Atlas Research Network 2008; Bredel et al. 2011; Parsons et al. 2008; Verhaak et al. 2010). The TCGA GBM project was conducted in two phases and developed a genome wide map of the genetic, epigenetic, and transcriptomic changes, as well as proteomic changes in over 500 GBM samples (Cancer Genome Atlas Research Network 2008; Brennan et al. 2013). Based on molecular typing and gene expression profiles, four distinct subtypes of GBM were found which are the classical, mesenchymal, neural, and proneural aubtypes (Freije et al. 2004; Gravendeel et al. 2009; Li et al. 2009; Nigro et al. 2005; Vitucci et al. 2011; Jue and McDonald 2016). The Classical subtype is associated with Endothelial growth factor receptor (EGFR) amplification, concomitant chromosome 7 amplification and chromosome 10 loss, and focal deletions of 9p encompassing cyclin-dependent kinase Inhibitor 2A (CDKN2A) . Tumor protein p53 (TP53) mutations, while common in GBM, are not seen in the classical subtype (Jue and McDonald 2016). The Mesenchymal subtype is characterized by deletions and mutations in Neurofibromin 1 (NF1) and Phosphatase and tensin homolog (PTEN) genes (Jue and McDonald 2016). The Neural subtype exhibits expression of neuronal markers and displays various mutations and copy number alterations including amplification of EGFR and deletion of PTEN (Jue and McDonald 2016). The Proneural subtype exhibits an oliogodendrocytic expression signature and features mutations of the isocitrate dehydrogenase 1(IDH 1) gene (Jue and McDonald 2016). The proneural subtype is associated with younger age and prolonged survival time, given the IDH1 mutation, as IDH1 mutations are frequently seen in lower grade gliomas and secondary gliomas (Verhaak et al. 2010; Jue and McDonald 2016). The TCGA analysis further identified three key molecular pathways for tumorigenesis: the p53 tumor suppressor and Retinoblastoma (RB) pathways , and the receptor tyrosine kinases (RTKs) signaling pathway (Fig. 4.1).

Critical signaling pathways altered in malignant gliomas. Primary sequence alterations and significant copy number changes for components of the (a) RTK/RAS/PI3K, (b) p53 and (c) Rb signalling pathways are shown. Red indicates activating genetic alterations. Conversely, blue indicates inactivating alterations. For each altered component of a particular pathway, the nature of the alteration and the percentage of tumours affected are indicated. Boxes contain the final percentages of glioblastomas with alterations in at least one known component gene of the designated pathway. Abbreviation: RTK receptor tyrosine kinase (Permission obtained from Nature Publishing Group © The Cancer Genome Atlas Research Network (2008))
4.3 Tumor Protein P53 Signaling Pathway
Tumor protein p53 is a well-known tumor suppressor gene and transcription factor involved in the coordination of cell responses that are involved in processes such as apoptosis, DNA repair, neovascularization, and metabolism (Bogler et al. 1995; Matlashewski et al. 1984; May and May 1999). p53 has been found mutated in 37.5% and 58% of untreated and treated GBM samples, according to the TCGA (Cancer Genome Atlas Research Network 2008). Disruptions in the p53 pathway are achieved by disruptions in genes that regulate its function, including Mouse double minute homolog (MDM) 2/4 and the tumor suppressor protein alternate reading frame (ARF) in 70% of GBM samples (Cancer Genome Atlas Research Network 2008). A complex that can suppress p53 function is the MDM2-MDM4 heterocomplex through the exertion of degradative control. MDM2-MDM4 protein amplification may represent a possible mechanism that gliomas escape p53 restricted growth (Herman et al. 2011; Reifenberger et al. 1993; Riemenschneider et al. 1999). Inactivation of CDKN2a can also dysregulate the p53 signaling pathway. CDKN2a encodes two proteins (p16INK4a and p14ARF) which are tumor suppressors and are negative regulators of the cell cycle (Ruas and Peters 1998). p16INK4a and p14ARF are deleted in approximately 55% of GBMs (Cancer Genome Atlas Research Network 2008; Schmidt et al. 1994). An encoded protein product, p14ARF, was found to promote degradation of the p53 repressor and lead to stabilization and accumulation of p53. Loss of p14ARF results in suppression of p53 and provides a mechanism for tumorigenesis (Kamijo et al. 1997, 1998; Zhang et al. 1998). CDKN2a also encodes for p16INK4a which is a protein that inhibits CDK4/6 association with cyclin D. When associated, this forms a complex that promotes G1/S transition through activation of downstream mediators . This process is involved in phosphorylating retinoblastoma protein and facilitating the release of bound E2F, a G1/S transcription factor. If p16INK4a is lost, then CDK4/6 and cyclin D can associate and the G1/S transition occurs freely. In patients with wild-type pRB, CDK4/6 is a target for inhibition (Bastien et al. 2015).
4.4 Retinoblastoma (Rb) Pathway
The Rb protein is encoded by the Rb gene located on chromosome 13q14.1-q14.2. The function of the protein is to prevent unwanted cell growth by inhibiting cell cycle progression until the cell is to undergo mitosis, and at that point the Rb protein becomes phosphorylated by Cyclin D, CDK4, and CDK6, which inactivates it and allows for cell cycle progression (Murphree and Benedict 1984). Typically what occurs is a homozygous deletion of CDKN2A which produces a loss of p16INK4a, a suppressor of CDK4. This leads to a dysregulation of Rb signaling (Murphree and Benedict 1984; Ohgaki and Kleihues 2009; Lin et al. 2013).
4.5 Receptor Tyrosine Kinases Pathway
Recurring molecular alterations have recently been identified in GBM, leading to a better understanding of the pathways that become disrupted in this disease. Frequently seen are gene amplifications and deletions, with deletions most often in chromosomes 1, 9, and 10 and amplifications in chromosomes 7 and 12 (Bello et al. 1994; James et al. 1991; Reifenberger et al. 1995; Rey et al. 1987). Amplifications of a gene can cause an upregulation of various oncogenes while deletions can target tumor suppressors (Purow and Schiff 2009). These are mediated by receptor tyrosine kinases (RTKs) , which are also key targets for deregulation in cancers (Zwick et al. 2001). Examples of RTKs in GBM include vascular endothelial growth factor (VEGFR) , EGFR, Platelet-derived growth factor receptor (PDGFR) , and hepatocyte growth factor receptor (MET) (Blume-Jensen and Hunter 2001). Mutations in these receptors act to relieve auto-inhibitory constraints to prevent degradation (Blume-Jensen and Hunter 2001). Growth factors and RTKs are typically strong candidates for therapeutic targets because mutations here are driver mutations critical for oncogenesis and because kinase receptors are targets for inhibitors that block kinase activation. Moreover, there has been success in other solid tumors (such as erlotinib in lung cancer) and in utilizing RTKs as a target.
4.5.1 VEGFR
A feature of high grade gliomas is angiogenesis, which may be attributed to high levels of VEGF-A in and around the tumor. Bevacizumab is a humanized monoclonal antibody which functions to bind VEGF-A ligand and alter binding to endothelial cells (Ferrara et al. 2005). Studies of bevacizumab have shown high radiographic response rate, prolonged progression-free survival (PFS) , and reduced glucocorticoid requirements, all of which led to approval by the Food and Drug administration for patients with GBM (Gilbert et al. 2014). However, several phase III studies have indicated that despite the improved radiographic response, as well as PFS, there is no significant overall survival benefit (Gilbert et al. 2014; Chinot et al. 2014). Both of these studies showed a prolonged PFS with bevacizumab, however the Avaglio trial indicated that health-related quality of life was stable prior to progression while the RTOG-0825 indicated overall quality of life based on symptom burden was significantly worse in some domains with bevacizumab (Gilbert et al. 2014; Chinot et al. 2014). A post-hoc subgroup analysis of the Avaglio study identified a 4.3 month potential increase in median survival with the addition of bevacizumab for IDH1 wild-type GBM in the proneural subgroup (Sandmann et al. 2015). Phase III studies have not demonstrated a survival benefit in recurrent GBM for bevacizumab. The phase II BELOB trial initially suggested a benefit with combined bevacizumab and lomustine, however the phase III trial EORTC 26101 recently underwent an interim analysis and found no overall survival benefit with the addition of bevacizumab to lomustine compared to lomustine alone for recurrent GBM (Taal et al. 2014). Cediranib, another anti-angiogenic agent whose mechanism of action is as a VEGF receptor tyrosine kinase inhibitor, has been tested in phase III trials with GBM and were found to be ineffective in altering overall outcome (Swartz et al. 2014). Additionally, other VEGF inhibitors such as Pazopanib, Sorafenib, Nintedanib, Sunitib, Vandetanib, Aflibercept, Vatalanib, and Cabozantinib have also failed to improve survival in glioblastoma patients (Table 4.1).
4.5.2 EGFR
In GBM there is evidence EGFR plays an important role in oncogenesis and tumor biology (Swartz et al. 2014). EGFR is amplified in approximately 50% of GBM samples and is associated with an active, mutant form of the EGFRvIIIL receptor, which when overexpressed enhances GBM cell growth and contributes to GBM pathogenesis (Jaros et al. 1992; Nishikawa et al. 1994; Schlegel et al. 1994). EGFRvIII, has been identified in 30% of newly diagnosed GBM and is characterized by deletion of exons 2–7 (Gan et al. 2009). The receptor is rendered active and thereby enhances the tumorigenicity by promoting tumor cell migration, conferring protection from radiation and temozolomide, and secreting EGFRvIII-bound oncosomes onto plasma membranes of neighboring cells (Prados et al. 2015).
Furthermore, EGFR can stimulate increased signaling through the RAS, RAF, MEK, MAP, and mTOR pathways, as well as downregulating cell cycle inhibitor proteins (Mao et al. 2012; Nishikawa et al. 2004). A set of studies found that increased levels of EGFR and EGFRvIII co-activates the RTK MET which leads to ligand-independent activation of the EGFR receptor (Huang et al. 2007; Jo et al. 2000; Stommel et al. 2007). This suggests that a mechanism for tumor cells to reduce dependence on either RTK for downstream signaling exists due to the interaction between the c-MET and EGFR/EGFRvIII signaling pathways. Studies have suggested that RTK MET overexpression may correlate with a shorter median survival time (Kong et al. 2009; Koochekpour et al. 1997; Lamszus et al. 1999).
EGFR remains an elusive target for therapy in GBM as initial studies have suggested activity of some EGFR inhibitors in molecular subsets of GBM but larger studies failed to replicate these results (Haas-Kogan et al. 2005; Mellinghoff et al. 2005; Reardon et al. 2014).
Rindopepimut is a tumor vaccine which is a conjugate of peptides that span the EGFRvIII mutation site with an immunogenic carrier protein keyhole limpet hemocyanin. Phase I and II trials in newly diagnosed GBM patients treated with rindopepimut along with temozolomide indicated a PFS of 10–15 months and overall survival of 22–26 months, which was improved compared to historical controls of 6 and 15 months respectively (Swartz et al. 2014). Another multicenter trial with rindopepimut in newly diagnosed GBM, the ACTIVATE trial, demonstrated an immune response after vaccination. In ACTIVATE, 43% of those treated had a positive humoral response, and the majority of patients with relapse had lost all of the EGFRvIII expression, a phenomenon known as antigen escape which suggested that the immune system successfully targeted EGFRvIII-expressing cells (Swartz et al. 2014). Furthermore a similar response in recurrent disease was noted in the ReACT trial, where rindopepimut reportedly caused an immune response and significantly prolonged survival when administered with bevacizumab (Phillips et al. 2016). However, in the phase III study ACT IV, rindopepimut combined with temozolomide did not increase overall survival in newly diagnosed EGFRvIII-positive GBM (Reardon et al. 2015; Inman 2016).
Therapy remains ongoing to identify new targets and mechanisms, such as ABT-414, which is a conjugate of a potent microtubule inhibitor and a monoclonal antibody against a tumor-selective EGFR epitope found in EGFR wild-type-overexpressing tumors and EGFRvIII mutant-expressing tumors. Preclinical studies have indicated that ABT-414 is selective and a phase I study, as well as a phase III study are ongoing (Gan et al. 2015; Phillips et al. 2016). Unfortunately, other EGFR inhibitors such as Gefitinib, Erlotinib, Cetuximab, Nimotuzumab, and Lapatinib, have similarly failed to improve survival in glioblastoma patients (Table 4.1).
4.5.3 PDGFR
Platelet derived growth factor receptors (PDGFR) are cell surface receptors for members of the platelet derived growth factor family and signal through the alpha and beta platelet derived growth factor receptor tyrosine kinases (Matsui et al. 1989). Chromosome 7p22 contains the PDGFR alpha gene which is amplified in approximately 13% of GBM samples (Cancer Genome Atlas Research Network 2008; Stenman et al. 1992). Multiple aberrations in expression of PDGFR have been observed, including overexpression, amplification, mutations, and truncations, however point mutations are exclusively seen in GBM (Alentorn et al. 2012).
An inhibitor of PDGFR is Imatinib mesylate, which also functions to inhibit bcr-abl and c-kit tyrosine kinases and is beneficial in the treatment of CML and in GI stromal tumors (Buchdunger et al. 2000; Druker et al. 2001; Demetri et al. 2002). Imatinib however has shown minimal activity in recurrent gliomas as well as newly diagnosed GBM (Wen et al. 2006; Razis et al. 2009). Others studies have been performed looking at the addition of hydroxyurea to imatinib in recurrent malignant gliomas and found that it failed to show any meaningful anti-tumor activity (Reardon et al. 2005, 2009a; Dresemann et al. 2010; Desjardins et al. 2007).
Another drug that has been studied is dastinib , which is an inhibitor of PDGFR, SRC, bcr-abl, c-Kit, and EphA2 receptors. The study was conducted in patients with recurrent GBM and found that dastinib was ineffective with no radiographic responses (Lassman et al. 2015). Another phase 1 trial of dastinib in combination with CCNU found hematological toxicities, which limited the amount of exposure available for both therapeutic agents, in contrast to a trial with dastinib and erlotinib which was much better tolerated (Reardon et al. 2012; Franceschi et al. 2012).
4.5.4 PI3K/AKT/PTEN/mTOR
Stimulation of the PI3K/AKT/PTEN/mTOR pathway enhances growth via activation of receptor tyrosine kinases. This occurs via the regulation of cell division, proliferation, differentiation, metabolism, and survival (Carnero et al. 2008; Morgensztern and McLeod 2005). Several genomic alterations in GBM activate this pathway, most often of which is the amplification of EGFR (Peraud et al. 1997; Watanabe et al. 1996; Wong et al. 1992). Other alterations leading to activation of this pathway include lesions in PIK3R1/PIK3CA and mutations or deletions of AKT, which occur in 84% of GBM cell lines and/or PTEN mutations, which occur in 30–44% of high-grade gliomas (Wang et al. 1997, 2004; Tohma et al. 1998; Teng et al. 1997; Koul 2008). PTEN typically inhibits the AKT pathway, therefore deletion of PTEN leads to activation of this pathway (Liu et al. 1997; Li et al. 1997; Stambolic et al. 1998). PTEN also facilitates the degradation of EGFR, which leads to termination of EGFR signaling, leading to an explanation for why PTEN confers resistance to epidermal growth factor inhibitors in vitro (Vivanco et al. 2010; Bianco et al. 2003).
mTOR is a master nutrient and energy sensor which regulates processes including transcription, protein synthesis, as well as other cellular functions including proliferation, cell motility survival, and anabolism (Hay and Sonenberg 2004; Kim et al. 2002; Sarbassov et al. 2004, 2005; Facchinetti et al. 2008; Ikenoue et al. 2008). mTOR is made of two different complexes—mammalian target of rapamycin complex 1 mTORC1 and mTORC2—and acts as a regulator of PI3K upstream and as its effector downstream (Akhavan et al. 2010). Inhibitors of mTOR have been developed, including Sirolimus, a mTORC1 inhibitor, which leads to reduced expression of neural stem cell progenitor markers and neurosphere formation in GBM (Sami and Karsy 2013; Sunayama et al. 2010). Another mTOR inhibitor, Dactolisib, a potent dual PI3K-mTOR inhibitor has shown potential benefit as a radiosensitizer for GBMs in preclinical studies (Fan et al. 2010; Mukherjee et al. 2012). Regrettably, mTOR inhibitors examined to date including Sirolimus, Everolimus and Temsirolimus have not prolonged overall survival in glioblastoma patients (Table 4.1).
4.5.5 RAS/RAF/MEK/MAP (ERK) Kinase
The RAS/RAF/MEK/MAP kinase pathway mediates cellular responses via growth, migration, apoptosis, proliferation, differentiation, and cell survival. This pathway can be activated by EGFR and PDGFR via signal transmission through indirect associations with cytosolic mediator proteins growth factor receptor-bound protein 2 (GRB2) and son of sevenless (SOS) to RAS. RAS then stimulates RAF, which activates MAPK and MEK (Moodie et al. 1993; Thomas et al. 1992).
RAS has been found to be upregulated in GBM samples, and activation is typically through loss of the NF-1 gene, which encodes a tumor suppressor protein and is a negative regulator of RAS and mTOR signaling in astrocytes (Banerjee et al. 2011; Nissan et al. 2014; Dasgupta et al. 2005). NF-1 mutation has been implicated in prior studies in glioma tumorigenesis, specifically noting that the homozygous loss of NF-1 in glial cells has been shown to develop fully malignant astrocytomas with a p53-null background (Alcantara Llaguno et al. 2009; Zhu et al. 2005; Kwon et al. 2008). Hyper activation of protein kinase C also causes increased degradation of NF-1, which also puts patients at an increased risk of developing gliomas (McGillicuddy et al. 2009). NF-1 mutations are a defining feature of the mesenchymal GBM subtype, and therefore this subgroup may potentially be good candidates for agents that target NF-1 driven pathways (Cancer Genome Atlas Research Network 2008; Phillips et al. 2006). Loss of NF-1 function leads to enhanced RAS activity, which will lead to increased RAS/RAF/MEK/MAPK pathway activation, leading to the hypothesis that MEK inhibitors were good therapeutic targets. Two MEK inhibitors, PD0325901 and AZD6244, both appeared effective against NF-1 deficient GBM cells dependent on RAF/MEK signaling in preclinical studies (See et al. 2012). Furthermore, this study showed that MEK inhibitor-resistant NF-1 deficient cells could be re-sensitized to MEK inhibitors with the co-application of dual PI3K-mOTR inhibitor PI-103, also suggesting that NF-1 deficient GBM patients may respond to a MEK inhibitor based chemotherapy (See et al. 2012). Drugs that have been tested that target RAS specifically (Tipifarnib, Lonafarnib) have not shown any improvement in overall survival for GBM patients (Table 4.1).
4.6 IDH1/IDH2 Mutations
The identifications of mutations in the metabolic enzymes IDH 1 and 2 is one of the most important discoveries that has led to a remodeling of our understanding of gliomas, including GBMS (Parsons et al. 2008). The majority of tumor samples in this study bore this mutation and were classified as secondary GBMs, suggesting that IDH 1 and IDH 2 mutations can serve as a genetic marker for this type of GBM. When IDH 1 and 2 were mutated, the result was enzymes with a neomorphic function, meaning that the mutant enzymes acquired the ability to catalyze the NADPH-dependent reduction of alpha-KG to the R-enantiomer of 2-hydroxyglutarate (2-HG) (Dang et al. 2010). This study showed that IDH 1 and 2 mutants had high levels of 2-HG, which is also found in primary IDH 1 mutant gliomas, and is also found in AML patients (Gross et al. 2010; Ward et al. 2010). IDH 1 and 2 mutant expression results in inhibition of alpha-KG-dependent dioxygenases by 2-HG. The enzymes that are dependent on this regulate physiological processes including hypoxia sensing, histone demethylation, and changes in DNA methylation (Loenarz and Schofield 2008). The glioma CpG island methylator phenotype (C-CIMP) is a distinctive and nearly invariable feature of IDH 1 and 2 mutant gliomas that has been studied to indicate that gliomas with this mutant expression are correlated with better prognosis (Baysan et al. 2012; Noushmehr et al. 2010). IDH 1 and 2 mutant gliomas are detected with the IDH-R132H antibody as well as with DNA sequencing of antibody-negative cases, which provides more accurate diagnosis and prediction of patient outcomes and prognosis.
IDH 1 and 2 mutational status also provides a prognostic marker in patients with lower grade gliomas and GBMs, as well as providing insights about the origin of gliomas (Parsons et al. 2008; Hartmann et al. 2009; Sanson et al. 2009; Weller et al. 2009; Yan et al. 2009). IDH 1 and 2 wild-type GBMs typically exhibit a characteristic pattern of genetic changes associated with primary GBMs such as a gain of chromosome 7, loss of chromosome 10, and amplification of EGFR which are not seen in IDH 1 and 2 mutant GBMs.
The importance of IDH mutation is eminent as the new 2016 WHO Classification of tumors of the central nervous system now defines glioblastoma based upon IDH status (Louis et al. 2016). Currently, several phase I studies examining IDH mutation inhibitors in advanced malignancies including glioblastoma are underway. However, as IDH is thought to be an early driver mutation in glioma, it remains unknown the potential impact of its inhibition in recurrent glioblastoma (Watanabe et al. 2009; Mandel et al. 2016).
4.7 Discussion
The Cancer Genome Atlas analysis revealed multiple molecular pathways and potential therapeutic genetic targets ushering in a new era for glioblastoma treatment. Bevacizumab, an anti-VEGF agent, has displayed an increased time to progression and improved imaging response but disappointingly has been unsuccessful in increasing patient overall survival. Furthermore, it remains an area of debate whether bevacizumab has any anti-tumor benefit (Kruser et al. 2016). Despite our increased knowledge of these tumors, our ability to divide them into molecular subgroups, and several promising therapeutic targets, every targeted therapy examined to date unfortunately has failed to demonstrate any benefit in overall survival.
Several possible explanations for this lack of success have been proposed including lack of tumor dependence on the pathway targeted, inadequate CNS penetration of the drug, intratumoral heterogeneity, and clonal evolution.
In regards to lack of pathway dependence, evidence suggests that tumors may be able upregulate a different pathway when another pathway is inhibited. This has been seen in the use of EGFR inhibitors, where treatment has demonstrated the lack of ability to change targets downstream like Akt and can even upregulate the PI3K/Akt pathway (Hegi et al. 2011; Chakravarti et al. 2002). Another possible area of concern is that many of the mutations targeted are essential for early tumor development and may be subsequently superseded by secondary pathways of tumor growth (Lee et al. 2012). Additionally, mutations present in a tumor on initial presentation may change or no longer be expressed on disease recurrence (van den Bent et al. 2015).
Drug penetrance is also major concern as it is frequently difficult to determine how well therapeutic agents cross the blood brain barrier in brain tumor patients. Due to the eloquent location, it is often unfeasible to obtain tissue samples at recurrence making it impossible to assess how much of a chemotherapeutic agent is being delivered to its intended target. It is also possible that a targeted agent may fail, not due to being a poor genetic target, but rather because the drug is not reaching that target in adequate quantity to cause the desired or intended treatment effect.
Additionally, tumor heterogeneity and clonal evolution is an issue that may affect targeted therapies. GBM’s are heterogenous in nature, and it is possible that when one cell group is inhibited via a targeted therapy to one pathway, another is left to proliferate unabated as their development is uninhibited.
While the results of targeted therapy studies to date in glioblastoma have been disappointing, there continues to be enthusiasm in this approach with numerous clinical trials currently underway. Hopefully, knowledge from the previous failed trials will help provide further insight and assist future clinicians in designing new novel targeted treatments to overcome these barriers.
References
Akhavan, D., T.F. Cloughesy, and P.S. Mischel. 2010. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-Oncology 12: 882–889.
Alcantara Llaguno, S., J. Chen, C.H. Kwon, et al. 2009. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15: 45–56.
Alentorn, A., Y. Marie, C. Carpentier, et al. 2012. Prevalence, clinico-pathological value, and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro-Oncology 14: 1393–1403.
Banerjee, S., N.R. Crouse, R.J. Emnett, S.M. Gianino, and D.H. Gutmann. 2011. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proceedings of the National Academy of Sciences of the United States of America 108: 15996–16001.
Bastien, J.I., K.A. McNeill, and H.A. Fine. 2015. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer 121: 502–516.
Batchelor, T.T., D.G. Duda, E. di Tomaso, et al. 2010. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Journal of Clinical Oncology 28: 2817–2823.
Batchelor, T.T., P. Mulholland, B. Neyns, et al. 2013. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. Journal of Clinical Oncology 31: 3212–3218.
Baysan, M., S. Bozdag, M.C. Cam, et al. 2012. G-cimp status prediction of glioblastoma samples using mRNA expression data. PLoS One 7: e47839.
Bello, M.J., J. Vaquero, J.M. de Campos, et al. 1994. Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. International Journal of Cancer 57: 172–175.
Bianco, R., I. Shin, C.A. Ritter, et al. 2003. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 22: 2812–2822.
Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signalling. Nature 411: 355–365.
Bogler, O., H.J. Huang, P. Kleihues, and W.K. Cavenee. 1995. The p53 gene and its role in human brain tumors. Glia 15: 308–327.
Brandes, A.A., R. Stupp, P. Hau, et al. 2010. EORTC study 26041-22041: phase I/II study on concomitant and adjuvant temozolomide (TMZ) and radiotherapy (RT) with PTK787/ZK222584 (PTK/ZK) in newly diagnosed glioblastoma. European Journal of Cancer 46: 348–354.
Bredel, M., D.M. Scholtens, A.K. Yadav, et al. 2011. NFKBIA deletion in glioblastomas. The New England Journal of Medicine 364: 627–637.
Brennan, C.W., R.G. Verhaak, A. McKenna, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477.
Brown, P.D., S. Krishnan, J.N. Sarkaria, et al. 2008. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. Journal of Clinical Oncology 26: 5603–5609.
Buchdunger, E., C.L. Cioffi, N. Law, et al. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. The Journal of Pharmacology and Experimental Therapeutics 295: 139–145.
Butowski, N., S.M. Chang, K.R. Lamborn, et al. 2011. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro-Oncology 13: 1331–1338.
Cancer Genome Atlas Research Network. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068.
Carnero, A., C. Blanco-Aparicio, O. Renner, W. Link, and J.F. Leal. 2008. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Current Cancer Drug Targets 8: 187–198.
Chakravarti, A., J.S. Loeffler, and N.J. Dyson. 2002. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Research 62: 200–207.
Chakravarti, A., M. Wang, H.I. Robins, et al. 2013. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. International Journal of Radiation Oncology, Biology, Physics 85: 1206–1211.
Chang, S.M., P. Wen, T. Cloughesy, et al. 2005. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investigational New Drugs 23: 357–361.
Chheda, M.G., P.Y. Wen, F.H. Hochberg, et al. 2015. Vandetanib plus sirolimus in adults with recurrent glioblastoma: Results of a phase I and dose expansion cohort study. Journal of Neuro-Oncology 121: 627–634.
Chinot, O.L., W. Wick, W. Mason, et al. 2014. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. The New England Journal of Medicine 370: 709–722.
ClinicalTrialsgov. 2016. Bevacizumab and Lomustine for Recurrent GBM. Identifier: NCT01290939.
Cloughesy, T.F., P.Y. Wen, H.I. Robins, et al. 2006. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: A North American Brain Tumor Consortium Study. Journal of Clinical Oncology 24: 3651–3656.
Cloughesy, T.R.J., J. Drappatz, et al. 2011. A phase II trial of everolimus in patients with recurrent glioblastoma multiforme. Neuro-Oncology 13: 42–43.
Cloughesy, T.F., W.K. Cavenee, and P.S. Mischel. 2014. Glioblastoma: From molecular pathology to targeted treatment. Annual Review of Pathology 9: 1–25.
Cohen, M.H., Y.L. Shen, P. Keegan, and R. Pazdur. 2009. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. The Oncologist 14: 1131–1138.
Dang, L., S. Jin, and S.M. Su. 2010. IDH mutations in glioma and acute myeloid leukemia. Trends in Molecular Medicine 16: 387–397.
Dasgupta, B., W. Li, A. Perry, and D.H. Gutmann. 2005. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Research 65: 236–245.
Davis, F.G., V. Kupelian, S. Freels, B. McCarthy, and T. Surawicz. 2001. Prevalence estimates for primary brain tumors in the United States by behavior and major histology groups. Neuro-Oncology 3: 152–158.
de Groot, J.F., M.R. Gilbert, K. Aldape, et al. 2008. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. Journal of Neuro-Oncology 90: 89–97.
de Groot, J.F., K.R. Lamborn, S.M. Chang, et al. 2011. Phase II study of aflibercept in recurrent malignant glioma: A North American Brain Tumor Consortium study. Journal of Clinical Oncology 29: 2689–2695.
Demetri, G.D., M. von Mehren, C.D. Blanke, et al. 2002. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. The New England Journal of Medicine 347: 472–480.
Desjardins, A., J.A. Quinn, J.J. Vredenburgh, et al. 2007. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. Journal of Neuro-Oncology 83: 53–60.
Desjardins, A., D.A. Reardon, K.B. Peters, et al. 2011. A phase I trial of the farnesyl transferase inhibitor, SCH 66336, with temozolomide for patients with malignant glioma. Journal of Neuro-Oncology 105: 601–606.
Dresemann, G., M. Weller, M.A. Rosenthal, et al. 2010. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. Journal of Neuro-Oncology 96: 393–402.
Druker, B.J., M. Talpaz, D.J. Resta, et al. 2001. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England Journal of Medicine 344: 1031–1037.
Facchinetti, V., W. Ouyang, H. Wei, et al. 2008. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. The EMBO Journal 27: 1932–1943.
Fan, Q.W., C. Cheng, C. Hackett, et al. 2010. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Science Signaling 3: ra81.
Ferrara, N., K.J. Hillan, and W. Novotny. 2005. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochemical and Biophysical Research Communications 333: 328–335.
Field, K.M., J. Simes, A.K. Nowak, et al. 2015. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro-Oncology 17: 1504–1513.
Franceschi, E., G. Cavallo, S. Lonardi, et al. 2007. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). British Journal of Cancer 96: 1047–1051.
Franceschi, E., R. Stupp, M.J. van den Bent, et al. 2012. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro-Oncology 14: 1503–1510.
Freije, W.A., F.E. Castro-Vargas, Z. Fang, et al. 2004. Gene expression profiling of gliomas strongly predicts survival. Cancer Research 64: 6503–6510.
Friedman, H.S., M.D. Prados, P.Y. Wen, et al. 2009. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Journal of Clinical Oncology 27: 4733–4740.
Galanis, E., J.C. Buckner, M.J. Maurer, et al. 2005. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. Journal of Clinical Oncology 23: 5294–5304.
Galanis, E., S.K. Anderson, J.M. Lafky, et al. 2013. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clinical Cancer Research 19: 4816–4823.
Gan, H.K., A.H. Kaye, and R.B. Luwor. 2009. The EGFRvIII variant in glioblastoma multiforme. Journal of Clinical Neuroscience 16: 748–754.
Gan, H.K., P. Kumthekar, A.B. Lassman, et al. 2015. ATNT-01ABT-414 MONO- OR COMBINATION THERAPY WITH TEMOZOLOMIDE (TMZ) RECHALLENGE IN PATIENTS WITH RECURRENT GLIOBLASTOMA (GBM) AND AMPLIFIED EPIDERMAL GROWTH FACTOR RECEPTOR (EGFR): A PHASE I STUDY. Neuro-Oncology 17: v10.
Gerstner, E.R., A.F. Eichler, S.R. Plotkin, et al. 2011. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. Journal of Neuro-Oncology 103: 325–332.
Gilbert, M.R., J.J. Dignam, T.S. Armstrong, et al. 2014. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England Journal of Medicine 370: 699–708.
Gravendeel, L.A., M.C. Kouwenhoven, O. Gevaert, et al. 2009. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Research 69: 9065–9072.
Gross, S., R.A. Cairns, M.D. Minden, et al. 2010. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. The Journal of Experimental Medicine 207: 339–344.
Haas-Kogan, D.A., M.D. Prados, T. Tihan, et al. 2005. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. Journal of the National Cancer Institute 97: 880–887.
Hainsworth, J.D., T. Ervin, E. Friedman, et al. 2010. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer 116: 3663–3669.
Hainsworth, J.D., K.C. Shih, G.C. Shepard, G.W. Tillinghast, B.T. Brinker, and D.R. Spigel. 2012. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clinical Advances in Hematology & Oncology 10: 240–246.
Hartmann, C., J. Meyer, J. Balss, et al. 2009. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathologica 118: 469–474.
Hasselbalch, B., U. Lassen, S. Hansen, et al. 2010. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro-Oncology 12: 508–516.
Hay, N., and N. Sonenberg. 2004. Upstream and downstream of mTOR. Genes & Development 18: 1926–1945.
Hegi, M.E., A.C. Diserens, P. Bady, et al. 2011. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib – A phase II trial. Molecular Cancer Therapeutics 10: 1102–1112.
Herman, A.G., M. Hayano, M.V. Poyurovsky, et al. 2011. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discovery 1: 312–325.
Huang, P.H., A. Mukasa, R. Bonavia, et al. 2007. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proceedings of the National Academy of Sciences of the United States of America 104: 12867–12872.
Ikenoue, T., K. Inoki, Q. Yang, X. Zhou, and K.L. Guan. 2008. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO Journal 27: 1919–1931.
Inman S. 2016. Rindopepimut misses OS endpoint in phase III glioblastoma trial. OncLive. http://www.onclive.com/web-exclusives/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial.
Iwamoto, F.M., K.R. Lamborn, H.I. Robins, et al. 2010. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro-Oncology 12: 855–861.
James, C.D., J. He, E. Carlbom, M. Nordenskjold, W.K. Cavenee, and V.P. Collins. 1991. Chromosome 9 deletion mapping reveals interferon alpha and interferon beta-1 gene deletions in human glial tumors. Cancer Research 51: 1684–1688.
Jaros, E., R.H. Perry, L. Adam, et al. 1992. Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki-67 labelling in brain tumours. British Journal of Cancer 66: 373–385.
Jo, M., D.B. Stolz, J.E. Esplen, K. Dorko, G.K. Michalopoulos, and S.C. Strom. 2000. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. The Journal of Biological Chemistry 275: 8806–8811.
Jue, T.R., and K.L. McDonald. 2016. The challenges associated with molecular targeted therapies for glioblastoma. Journal of Neuro-Oncology 127: 427–434.
Kamijo, T., F. Zindy, M.F. Roussel, et al. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91: 649–659.
Kamijo, T., J.D. Weber, G. Zambetti, F. Zindy, M.F. Roussel, and C.J. Sherr. 1998. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proceedings of the National Academy of Sciences of the United States of America 95: 8292–8297.
Karavasilis, V., V. Kotoula, G. Pentheroudakis, et al. 2013. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. Journal of Neurology 260: 1469–1480.
Kim, D.H., D.D. Sarbassov, S.M. Ali, et al. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175.
Kong, D.S., S.Y. Song, D.H. Kim, et al. 2009. Prognostic significance of c-Met expression in glioblastomas. Cancer 115: 140–148.
Koochekpour, S., M. Jeffers, S. Rulong, et al. 1997. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Research 57: 5391–5398.
Koul, D. 2008. PTEN signaling pathways in glioblastoma. Cancer Biology & Therapy 7: 1321–1325.
Kreisl, T.N., L. Kim, K. Moore, et al. 2009a. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. Journal of Clinical Oncology 27: 740–745.
Kreisl, T.N., A.B. Lassman, P.S. Mischel, et al. 2009b. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). Journal of Neuro-Oncology 92: 99–105.
Kreisl, T.N., S. Kotliarova, J.A. Butman, et al. 2010. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro-Oncology 12: 181–189.
Kreisl, T.N., K.A. McNeill, J. Sul, F.M. Iwamoto, J. Shih, and H.A. Fine. 2012. A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro-Oncology 14: 1519–1526.
Kreisl, T.N., P. Smith, J. Sul, et al. 2013. Continuous daily sunitinib for recurrent glioblastoma. Journal of Neuro-Oncology 111: 41–48.
Krishnan, S., P.D. Brown, K.V. Ballman, et al. 2006. Phase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: Results of North Central Cancer Treatment Group protocol N0177. International Journal of Radiation Oncology, Biology, Physics 65: 1192–1199.
Kruser, T.J., M.P. Mehta, and K.R. Kozak. 2016. Identification of patients who benefit from bevacizumab in high-grade glioma-an easy question turned difficult: Treat the scan or the patient? Journal of Clinical Oncology 34: 1281–1282.
Kwon, C.H., D. Zhao, J. Chen, et al. 2008. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Research 68: 3286–3294.
Lamszus, K., J. Laterra, M. Westphal, and E.M. Rosen. 1999. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. International Journal of Developmental Neuroscience 17: 517–530.
Lassen, U., M. Sorensen, T.B. Gaziel, B. Hasselbalch, and H.S. Poulsen. 2013. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer Research 33: 1657–1660.
Lassman, A.B., S.L. Pugh, M.R. Gilbert, et al. 2015. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro-Oncology 17: 992–998.
Lee, E.Q., J. Kuhn, K.R. Lamborn, et al. 2012. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-Oncology 14: 1511–1518.
Li, J., C. Yen, D. Liaw, et al. 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275: 1943–1947.
Li, A., J. Walling, S. Ahn, et al. 2009. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Research 69: 2091–2099.
Lin, F., M.C. de Gooijer, D. Hanekamp, D. Brandsma, J.H. Beijnen, and O. van Tellingen. 2013. Targeting core (mutated) pathways of high-grade gliomas: Challenges of intrinsic resistance and drug efflux. CNS Oncology 2: 271–288.
Liu, W., C.D. James, L. Frederick, B.E. Alderete, and R.B. Jenkins. 1997. PTEN/MMAC1 mutations and EGFR amplification in glioblastomas. Cancer Research 57: 5254–5257.
Loenarz, C., and C.J. Schofield. 2008. Expanding chemical biology of 2-oxoglutarate oxygenases. Nature Chemical Biology 4: 152–156.
Louis, D.N., A. Perry, G. Reifenberger, et al. 2016. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathologica 131: 803–820.
Ma, D.J., E. Galanis, S.K. Anderson, et al. 2015. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 17: 1261–1269.
Mandel, J.J., D. Cachia, D. Liu, et al. 2016. Impact of IDH1 mutation status on outcome in clinical trials for recurrent glioblastoma. Journal of Neuro-Oncology 129: 147–154.
Mao, H., D.G. Lebrun, J. Yang, V.F. Zhu, and M. Li. 2012. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investigation 30: 48–56.
Masui, K., T.F. Cloughesy, and P.S. Mischel. 2012. Review: Molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathology and Applied Neurobiology 38: 271–291.
Matlashewski, G., P. Lamb, D. Pim, J. Peacock, L. Crawford, and S. Benchimol. 1984. Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. The EMBO Journal 3: 3257–3262.
Matsui, T., M. Heidaran, T. Miki, et al. 1989. Isolation of a novel receptor cDNA establishes the existence of two PDGF receptor genes. Science 243: 800–804.
May, P., and E. May. 1999. Twenty years of p53 research: Structural and functional aspects of the p53 protein. Oncogene 18: 7621–7636.
McGillicuddy, L.T., J.A. Fromm, P.E. Hollstein, et al. 2009. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 16: 44–54.
Mellinghoff, I.K., M.Y. Wang, I. Vivanco, et al. 2005. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. The New England Journal of Medicine 353: 2012–2024.
Moodie, S.A., B.M. Willumsen, M.J. Weber, and A. Wolfman. 1993. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 260: 1658–1661.
Morgensztern, D., and H.L. McLeod. 2005. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anti-Cancer Drugs 16: 797–803.
Muhic, A., H.S. Poulsen, M. Sorensen, K. Grunnet, and U. Lassen. 2013. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. Journal of Neuro-Oncology 111: 205–212.
Mukherjee, B., N. Tomimatsu, K. Amancherla, C.V. Camacho, N. Pichamoorthy, and S. Burma. 2012. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia 14: 34–43.
Murphree, A.L., and W.F. Benedict. 1984. Retinoblastoma: Clues to human oncogenesis. Science 223: 1028–1033.
Neyns, B., J. Sadones, E. Joosens, et al. 2009. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Annals of Oncology 20: 1596–1603.
Neyns, B., J. Sadones, C. Chaskis, et al. 2011. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. Journal of Neuro-Oncology 103: 491–501.
Nghiemphu, P.L., P.Y. Wen, K.R. Lamborn, et al. 2011. A phase I trial of tipifarnib with radiation therapy, with and without temozolomide, for patients with newly diagnosed glioblastoma. International Journal of Radiation Oncology, Biology, Physics 81: 1422–1427.
Nghiemphu, P.L., A. Lai, R.M. Green, D.A. Reardon, and T. Cloughesy. 2012. A dose escalation trial for the combination of erlotinib and sirolimus for recurrent malignant gliomas. Journal of Neuro-Oncology 110: 245–250.
Nigro, J.M., A. Misra, L. Zhang, et al. 2005. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Research 65: 1678–1686.
Nishikawa, R., X.D. Ji, R.C. Harmon, et al. 1994. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America 91: 7727–7731.
Nishikawa, R., T. Sugiyama, Y. Narita, F. Furnari, W.K. Cavenee, and M. Matsutani. 2004. Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma. Brain Tumor Pathology 21: 53–56.
Nissan, M.H., C.A. Pratilas, A.M. Jones, et al. 2014. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Research 74: 2340–2350.
Noushmehr, H., D.J. Weisenberger, K. Diefes, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522.
Ohgaki, H., and P. Kleihues. 2005. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. Journal of Neuropathology and Experimental Neurology 64: 479–489.
———. 2009. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Science 100: 2235–2241.
Pan, E., D. Yu, B. Yue, et al. 2012. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. Journal of Neuro-Oncology 110: 111–118.
Parsons, D.W., S. Jones, X. Zhang, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812.
Peereboom, D.M., D.R. Shepard, M.S. Ahluwalia, et al. 2010. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. Journal of Neuro-Oncology 98: 93–99.
Peereboom, D.M., M.S. Ahluwalia, X. Ye, et al. 2013. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro-Oncology 15: 490–496.
Peraud, A., K. Watanabe, K.H. Plate, Y. Yonekawa, P. Kleihues, and H. Ohgaki. 1997. p53 mutations versus EGF receptor expression in giant cell glioblastomas. Journal of Neuropathology and Experimental Neurology 56: 1236–1241.
Phillips, H.S., S. Kharbanda, R. Chen, et al. 2006. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9: 157–173.
Phillips, A.C., E.R. Boghaert, K.S. Vaidya, et al. 2016. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Molecular Cancer Therapeutics 15: 661–669.
Porter, K.R., B.J. McCarthy, S. Freels, Y. Kim, and F.G. Davis. 2010. Prevalence estimates for primary brain tumors in the United States by age, gender, behavior, and histology. Neuro-Oncology 12: 520–527.
Prados, M.D., K.R. Lamborn, S. Chang, et al. 2006. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro-Oncology 8: 67–78.
Prados, M.D., S.M. Chang, N. Butowski, et al. 2009. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. Journal of Clinical Oncology 27: 579–584.
Prados, M.D., S.A. Byron, N.L. Tran, et al. 2015. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro-Oncology 17: 1051–1063.
Purow, B., and D. Schiff. 2009. Advances in the genetics of glioblastoma: Are we reaching critical mass? Nature Reviews. Neurology 5: 419–426.
Raizer, J.J., L.E. Abrey, A.B. Lassman, et al. 2010. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 12: 95–103.
Razis, E., P. Selviaridis, S. Labropoulos, et al. 2009. Phase II study of neoadjuvant imatinib in glioblastoma: Evaluation of clinical and molecular effects of the treatment. Clinical Cancer Research 15: 6258–6266.
Reardon, D.A., M.J. Egorin, J.A. Quinn, et al. 2005. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. Journal of Clinical Oncology 23: 9359–9368.
Reardon, D.A., J.A. Quinn, J.J. Vredenburgh, et al. 2006. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clinical Cancer Research 12: 860–868.
Reardon, D.A., G. Dresemann, S. Taillibert, et al. 2009a. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. British Journal of Cancer 101: 1995–2004.
Reardon, D.A., M.J. Egorin, A. Desjardins, et al. 2009b. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer 115: 2188–2198.
Reardon, D.A., A. Desjardins, J.J. Vredenburgh, et al. 2010. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. Journal of Neuro-Oncology 96: 219–230.
Reardon, D.A., J.J. Vredenburgh, A. Desjardins, et al. 2011a. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. Journal of Neuro-Oncology 101: 57–66.
Reardon, D.A., J.J. Vredenburgh, A. Coan, et al. 2011b. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. Journal of Neuro-Oncology 105: 621–627.
Reardon, D.A., J.J. Vredenburgh, A. Desjardins, et al. 2012. Phase 1 trial of dasatinib plus erlotinib in adults with recurrent malignant glioma. Journal of Neuro-Oncology 108: 499–506.
Reardon, D.A., M.D. Groves, P.Y. Wen, et al. 2013. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clinical Cancer Research 19: 900–908.
Reardon, D.A., P.Y. Wen, and I.K. Mellinghoff. 2014. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro-Oncology 16 Suppl 8: viii7–vii13.
Reardon, D.A., A. Desjardins, J. Schuster, et al. 2015. IMCT-08ReACT: LONG-TERM SURVIVAL FROM A RANDOMIZED PHASE II STUDY OF RINDOPEPIMUT (CDX-110) PLUS BEVACIZUMAB IN RELAPSED GLIOBLASTOMA. Neuro-Oncology 17: v109.
Reifenberger, G., L. Liu, K. Ichimura, E.E. Schmidt, and V.P. Collins. 1993. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Research 53: 2736–2739.
Reifenberger, G., J. Reifenberger, K. Ichimura, and V.P. Collins. 1995. Amplification at 12q13-14 in human malignant gliomas is frequently accompanied by loss of heterozygosity at loci proximal and distal to the amplification site. Cancer Research 55: 731–734.
Rey, J.A., M.J. Bello, J.M. de Campos, M.E. Kusak, C. Ramos, and J. Benitez. 1987. Chromosomal patterns in human malignant astrocytomas. Cancer Genetics and Cytogenetics 29: 201–221.
Rich, J.N., D.A. Reardon, T. Peery, et al. 2004. Phase II trial of gefitinib in recurrent glioblastoma. Journal of Clinical Oncology 22: 133–142.
Riemenschneider, M.J., R. Buschges, M. Wolter, et al. 1999. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Research 59: 6091–6096.
Ruas, M., and G. Peters. 1998. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochimica et Biophysica Acta 1378: F115–F177.
Sami, A., and M. Karsy. 2013. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumour Biology 34: 1991–2002.
Sandmann, T., R. Bourgon, J. Garcia, et al. 2015. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide: Retrospective analysis of the AVAglio trial. Journal of Clinical Oncology 33: 2735–2744.
Sanson, M., Y. Marie, S. Paris, et al. 2009. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. Journal of Clinical Oncology 27: 4150–4154.
Sarbassov, D.D., S.M. Ali, D.H. Kim, et al. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology 14: 1296–1302.
Sarbassov, D.D., D.A. Guertin, S.M. Ali, and D.M. Sabatini. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101.
Sarkaria, J.N., E. Galanis, W. Wu, et al. 2010. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clinical Cancer Research 16: 5573–5580.
Sathornsumetee, S., A. Desjardins, J.J. Vredenburgh, et al. 2010. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro-Oncology 12: 1300–1310.
Schlegel, J., G. Stumm, K. Brandle, et al. 1994. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. Journal of Neuro-Oncology 22: 201–207.
Schmidt, E.E., K. Ichimura, G. Reifenberger, and V.P. Collins. 1994. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Research 54: 6321–6324.
See, W.L., I.L. Tan, J. Mukherjee, T. Nicolaides, and R.O. Pieper. 2012. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Research 72: 3350–3359.
Soffietti, R., E. Trevisan, L. Bertero, et al. 2014. Bevacizumab and fotemustine for recurrent glioblastoma: A phase II study of AINO (Italian Association of Neuro-Oncology). Journal of Neuro-Oncology 116: 533–541.
Solomon, M.T., J.C. Selva, J. Figueredo, et al. 2013. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: Results from a randomized, double blind trial. BMC Cancer 13: 299.
Stambolic, V., A. Suzuki, J.L. de la Pompa, et al. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95: 29–39.
Stenman, G., F. Rorsman, K. Huebner, and C. Betsholtz. 1992. The human platelet-derived growth factor alpha chain (PDGFA) gene maps to chromosome 7p22. Cytogenetics and Cell Genetics 60: 206–207.
Stommel, J.M., A.C. Kimmelman, H. Ying, et al. 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318: 287–290.
Stupp, R., W.P. Mason, M.J. van den Bent, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine 352: 987–996.
Sunayama, J., A. Sato, K. Matsuda, et al. 2010. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro-Oncology 12: 1205–1219.
Swartz, A.M., Q.J. Li, and J.H. Sampson. 2014. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 6: 679–690.
Taal, W., H.M. Oosterkamp, A.M. Walenkamp, et al. 2014. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. The Lancet Oncology 15: 943–953.
Tanaka, S., D.N. Louis, W.T. Curry, T.T. Batchelor, and J. Dietrich. 2013. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nature Reviews. Clinical Oncology 10: 14–26.
Teng, D.H., R. Hu, H. Lin, et al. 1997. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Research 57: 5221–5225.
Thiessen, B., C. Stewart, M. Tsao, et al. 2010. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemotherapy and Pharmacology 65: 353–361.
Thomas, S.M., M. DeMarco, G. D’Arcangelo, S. Halegoua, and J.S. Brugge. 1992. Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell 68: 1031–1040.
Tohma, Y., C. Gratas, W. Biernat, et al. 1998. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. Journal of Neuropathology and Experimental Neurology 57: 684–689.
Uhm, J.H., K.V. Ballman, W. Wu, et al. 2011. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. International Journal of Radiation Oncology, Biology, Physics 80: 347–353.
van den Bent, M.J., Y. Gao, M. Kerkhof, et al. 2015. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology 17: 935–941.
Verhaak, R.G., K.A. Hoadley, E. Purdom, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110.
Vitucci, M., D.N. Hayes, and C.R. Miller. 2011. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. British Journal of Cancer 104: 545–553.
Vivanco, I., D. Rohle, M. Versele, et al. 2010. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proceedings of the National Academy of Sciences of the United States of America 107: 6459–6464.
Wang, S.I., J. Puc, J. Li, et al. 1997. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Research 57: 4183–4186.
Wang, H., W. Zhang, H.J. Huang, W.S. Liao, and G.N. Fuller. 2004. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Laboratory Investigation 84: 941–951.
Ward, P.S., J. Patel, D.R. Wise, et al. 2010. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17: 225–234.
Watanabe, K., O. Tachibana, K. Sata, Y. Yonekawa, P. Kleihues, and H. Ohgaki. 1996. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathology 6: 217–223; discussion 223–214.
Watanabe, T., S. Nobusawa, P. Kleihues, and H. Ohgaki. 2009. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. The American Journal of Pathology 174: 1149–1153.
Weller, M., J. Felsberg, C. Hartmann, et al. 2009. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: A prospective translational study of the German Glioma Network. Journal of Clinical Oncology 27: 5743–5750.
Wen, P.Y., W.K. Yung, K.R. Lamborn, et al. 2006. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clinical Cancer Research 12: 4899–4907.
Wen, P.Y.P.M., D. Schiff, D.A. Reardon, T. Cloughesy, T. Mikkelsen, T. Batchelor, J. Drappatz, M.C. Chamberlain, and J.F. De Groot. 2010. Phase II study of XL184 (BMS 907351), an inhibitor of MET, VEGFR2, and RET, in patients (pts) with progressive glioblastoma (GB) [abstract]. Journal of Clinical Oncology 28: 181s.
Wick, W., V.K. Puduvalli, M.C. Chamberlain, et al. 2010. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. Journal of Clinical Oncology 28: 1168–1174.
Wick, W., J.P. Steinbach, M. Platten, et al. 2013. Enzastaurin before and concomitant with radiation therapy, followed by enzastaurin maintenance therapy, in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation. Neuro-Oncology 15: 1405–1412.
Wong, A.J., J.M. Ruppert, S.H. Bigner, et al. 1992. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences of the United States of America 89: 2965–2969.
Yan, H., D.W. Parsons, G. Jin, et al. 2009. IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine 360: 765–773.
Yung, W.K., J.J. Vredenburgh, T.F. Cloughesy, et al. 2010. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro-Oncology 12: 1061–1070.
Yust-Katz, S., D. Liu, Y. Yuan, et al. 2013. Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer 119: 2747–2753.
Zhang, Y., Y. Xiong, and W.G. Yarbrough. 1998. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92: 725–734.
Zhu, Y., F. Guignard, D. Zhao, et al. 2005. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8: 119–130.
Zwick, E., J. Bange, and A. Ullrich. 2001. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocrine-Related Cancer 8: 161–173.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Youssef, M., Mandel, J., Chowdhary, S., Kesari, S. (2017). Recent Advances for Targeted Therapies in Glioblastoma. In: Somasundaram, K. (eds) Advances in Biology and Treatment of Glioblastoma. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-319-56820-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-56820-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56819-5
Online ISBN: 978-3-319-56820-1
eBook Packages: MedicineMedicine (R0)